Oncolytic virus therapy: A new era of cancer treatment at dawn (original) (raw)
Abstract
Oncolytic virus therapy is perhaps the next major breakthrough in cancer treatment following the success in immunotherapy using immune checkpoint inhibitors. Oncolytic viruses are defined as genetically engineered or naturally occurring viruses that selectively replicate in and kill cancer cells without harming the normal tissues. T‐Vec (talimogene laherparepvec), a second‐generation oncolytic herpes simplex virus type 1 (HSV‐1) armed with GM‐CSF, was recently approved as the first oncolytic virus drug in the USA and Europe. The phase III trial proved that local intralesional injections with T‐Vec in advanced malignant melanoma patients can not only suppress the growth of injected tumors but also act systemically and prolong overall survival. Other oncolytic viruses that are closing in on drug approval in North America and Europe include vaccinia virus JX‐594 (pexastimogene devacirepvec) for hepatocellular carcinoma, GM‐CSF‐expressing adenovirus CG0070 for bladder cancer, and Reolysin (pelareorep), a wild‐type variant of reovirus, for head and neck cancer. In Japan, a phase II clinical trial of G47∆, a third‐generation oncolytic HSV‐1, is ongoing in glioblastoma patients. G47∆ was recently designated as a “Sakigake” breakthrough therapy drug in Japan. This new system by the Japanese government should provide G47∆ with priority reviews and a fast‐track drug approval by the regulatory authorities. Whereas numerous oncolytic viruses have been subjected to clinical trials, the common feature that is expected to play a major role in prolonging the survival of cancer patients is an induction of specific antitumor immunity in the course of tumor‐specific viral replication. It appears that it will not be long before oncolytic virus therapy becomes a standard therapeutic option for all cancer patients.
Keywords: Clinical trial, G47∆, herpes simplex virus, oncolytic immunotherapy, oncolytic virus
Oncolytic virus therapy has recently been recognized as a promising new therapeutic approach for cancer treatment. An oncolytic virus is defined as a genetically engineered or naturally occurring virus that can selectively replicate in and kill cancer cells without harming the normal tissues. In contrast to gene therapy where a virus is used as a mere carrier for transgene delivery, oncolytic virus therapy uses the virus itself as an active drug reagent.
The concept of oncolytic virus therapy has existed for some time (Fig. 1). Tumor regression has often been observed during or after a naturally acquired, systemic viral infection.1, 2 In 1949, 22 patients with Hodgkin's disease were treated with sera or tissue extracts containing hepatitis virus.3 Between 1950 and 1980, many clinical trials were performed in attempts to treat cancer with wild type or naturally attenuated viruses, including hepatitis. West Nile fever, yellow fever, dengue fever and adenoviruses.4 However, these viruses were not deemed useful as therapeutics reagents because, in those days, there was no known method to control the virulence and yet retain viral replication in cancer cells.
Figure 1.

Milestones of oncolytic virus therapy development.
It is now recognized, because protection mechanisms against viral infection (e.g. interferon‐beta signal pathway) are impaired in the majority of cancer cells,5 that most viruses can replicate to a much greater extent in cancer cells than in normal cells. Therefore, getting a virus to replicate in cancer cells is not a problem: What is difficult is making a virus not replicate in normal cells at all, while retaining its replication capability in cancer cells. Attempts to achieve cancer cell‐specific replication have been undertaken either by selecting a virus that is non‐virulent in humans or by engineering the virus genome (Fig. 2). Representing the former strategy is Reolysin, a wild‐type variant of reovirus that exhibits oncolytic properties in cells with activated Ras signaling with limited virulence in normal human cells. The latter strategy is, however, better suited to achieving strict control of viral replication. In 1991, Martuza et al.6 demonstrated that a genetically engineered herpes simplex virus type I (HSV‐1) with a mutation in the thymidine kinase (TK) gene replicated selectively in cancer cells and was useful for treating experimental brain tumors. Their findings opened up a whole new area of oncolytic virus development that involves designing and constructing the viral genome. During the past two decades of thriving development, probably the most important finding regarding oncolytic virus therapy was that a systemic tumor‐specific immunity is efficiently induced in the course of oncolytic activities.7, 8 This phenomenon is now widely recognized as the common feature for all oncolytic virus therapy that is expected to play a major role in prolonging the survival of cancer patients (Fig. 3).
Figure 2.
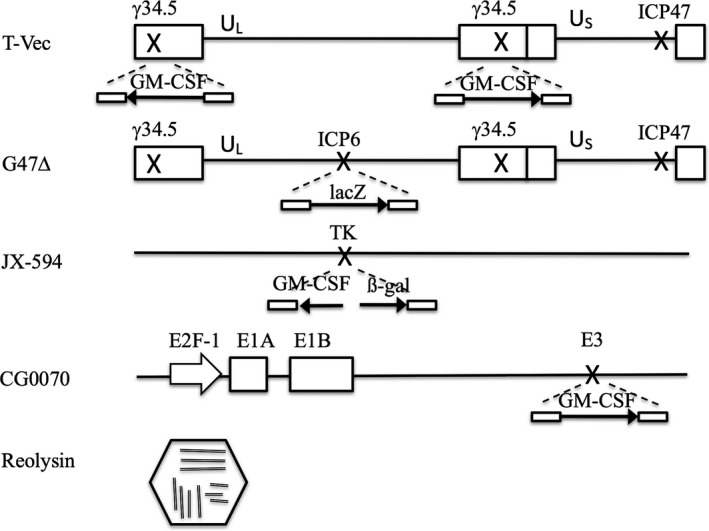
Structures of major oncolytic viruses. Boxes represent inverted repeat sequences flanking the long (UL) and short (US) unique sequences of HSV‐1 DNA in T‐Vec and G47∆. T‐Vec has an insertion of human GM‐CSF in both copies of the γ_34.5_ gene and a deletion in the α_47_ gene. G47∆ has a deletion in both copies of the γ_34.5_ gene, a deletion in the α_47_ gene, and an insertion of the lacZ coding sequence in the ICP6 locus. JX‐594 has an insertion of human GM‐CSF and lacZ transgenes in the TK locus. Reolysin has a segmented genome composed of ten segments of double stranded RNA and a double shell of capsid.
Figure 3.
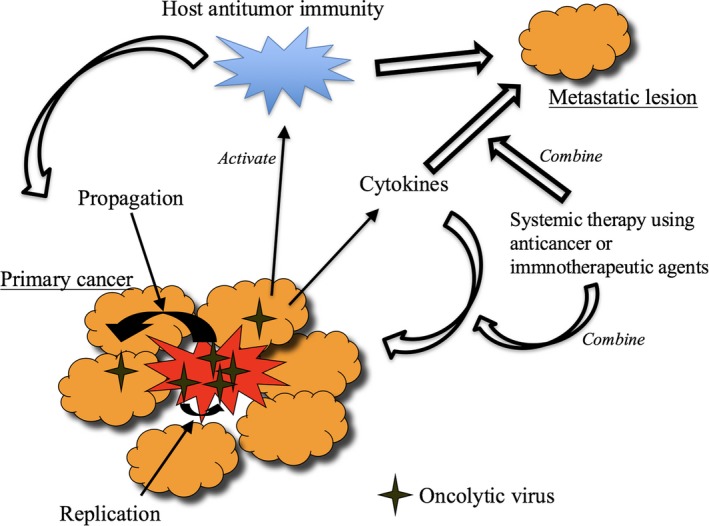
Mechanisms of action of oncolytic virus therapy. Local replication of oncolytic virus induces specific antitumor immunity in the course of its oncolytic activities that act on remote lesions. A combination with immune checkpoint inhibitors or chemotherapy may enhance the efficacy of oncolytic virus therapy. Arming oncolytic viruses with immunostimulatory gene(s) or cancer therapeutic genes may also be beneficial.
To date, two genetically engineered oncolytic viruses have been approved for marketing as drugs. One is Oncorine (H101, the same construct as ONYX‐015),9 an _E1B_‐deleted adenovirus, which was approved in China for head and neck cancer and esophagus cancer in 2005.10, 11 The use and clinical data of Oncorine is so far limited to China. The other is T‐Vec (talimogene laherparepvec, IMLYGIC, formerly OncoVEXGM‐CSF), which was approved for melanoma by the FDA in the USA in October 2015 and was subsequently approved in Europe in January 2016 and in Australia in May 2016 (Fig. 1).12, 13 Many clinical trials using T‐Vec are currently performed worldwide by the pharmaceutical company in order to expand its application and also to expand countries for marketing. This review focuses on those oncolytic viruses under development that are likely to become treatment options in the near future (Table 1).
Table 1.
Summary of major oncolytic viruses under clinical development
| Virus | Gene modification | Gene insertion | Target disease | Company | Status | |
|---|---|---|---|---|---|---|
| T‐Vec (Imlygic, talimogene laherparepvec) | HSV‐1 | γ34.5, α47 | Human GM‐CSF | Unresected stage IIIB to IV melanoma | Amgen | The drug is approved in the USA in 2015 and in Europe in 2016 |
| G47∆ | HSV‐1 | γ34.5, ICP6, α47 | lacZ | Glioblastoma | Investigator‐initiated | A phase II study started in 2015. It was designated as Sakigake breakthrough therapy by MHLW of Japan |
| JX‐594 (Pexa‐vec, pexastimogene devacirepvec) | Vaccinia virus | Thymidine kinase | Human GM‐CSF, lacZ | Advanced stage hepatocellular carcinoma | Sillajen | A phase III started in 2015 |
| CG0070 | Adenovirus | E2F‐1 promoter /E1A gene | Human GM‐CSF | Non‐muscle invasive bladder cancer after BCG failure | Cold Genesys | A phase II/III randomized controlled trial is ongoing in patients with bladder cancer |
| Reolysin (pelareorep) | Reovirus | None | Metastatic and/or recurrent head and neck cancer | Oncolytics Biotech | A phase III is completed. It received an orphan drug designation from FDA |
Genetically engineered oncolytic viruses
With the development of modern techniques of genetic engineering and increasing knowledge regarding the functions and structures of viral genes, designing and manipulating the viral genome to create a non‐pathogenic virus has become the standard method for oncolytic virus development. Typically, DNA viruses are used for this strategy.
T‐Vec
T‐Vec is a double‐mutated HSV‐1 with deletions in the γ_34.5_ and α_47_ genes, and the human granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) gene inserted into the deleted γ_34.5_ loci.14 The deletion in the γ_34.5_ genes is mainly responsible for cancer‐selective replication and attenuation of pathogenicity.15, 16, 17 Because the γ_34.5_ gene functions to negate the host cell's shut‐off of protein synthesis upon viral infection,18 inactivation of γ_34.5_ renders the virus unable to replicate in normal cells. However, because cancer cells are in defect of the shut‐off response, γ_34.5_‐deficient HSV‐1 can still replicate in cancer cells.19 The α_47_ gene functions to antagonize the host cell's transporter associated with antigen presentation; therefore, the deletion of the gene precludes the downregulation of MHC class I expression, which should enhance the antitumor immune responses.20, 21, 22 The deletion in the α_47_ gene also results in immediate early expression of the neighbor US11 gene, which results in enhanced viral replication in cancer cells.23 The GM‐CSF expression was intended to enhance the antitumor immunity induction, although convincing preclinical evidence has not been shown.
The safety of T‐Vec was tested in a phase I study in patients with various metastatic tumors, including breast, head/neck and gastrointestinal cancers, and malignant melanoma. Overall, intralesional administration of the virus was well tolerated by patients.14 Although no complete or partial responses were observed, stable disease was observed in several patients, and most tumor biopsies showed tumor necrosis. T‐Vec was further tested in phase II studies in patients with metastatic melanoma.24 A single arm phase II study resulted in an overall response rate of 26%, with responses in both injected and uninjected lesions, including visceral lesions. An increase in CD8+ T cells and a reduction in CD4+FoxP3+ regulatory T cells were detected in biopsy samples of regressing lesions.25 A randomized phase III trial was performed in patients with unresected stage IIIB–IV melanoma (OPTiM; NCT00769704).13 A total of 436 patients were randomly assigned in a 2:1 ratio to intralesional T‐Vec or subcutaneous GM‐CSF treatment arms. T‐Vec was administered at a concentration of 108 plaque forming units (pfu)/mL injected into 1 or more skin or subcutaneous tumors on Days 1 and 15 of each 28‐day cycle for up to 12 months, while GM‐CSF was administered at a dose of 125 μg/m²/day subcutaneously for 14 consecutive days followed by 14 days of rest, in 28‐day treatment cycles for up to 12 months. At the primary analysis, 290 deaths had occurred (T‐Vec, n = 189; GM‐CSF, n = 101). The durable response rate (objective response lasting continuously ≥6 months) was significantly higher in the T‐Vec arm (16.3%) compared with the GM‐CSF arm (2.1%). The overall response rate was also higher in the T‐Vec arm (26.4 vs 5.7%). The most common adverse events with T‐Vec were fatigue, chills and pyrexia, but the only grade 3 or 4 treatment‐related adverse event, occurring in over 2% of patients, was cellulitis (T‐Vec, n = 6; GM‐CSF, n = 1). There were no fatal treatment‐related adverse events. At the time of publication, median overall survival (OS) was 23.3 months for the T‐Vec arm versus 18.9 months for the GM‐CSF arm (hazard ratio, 0.79; P = 0.051),13 but the difference in OS became significant (P = 0.049) by the time of drug application. The treatment benefit in OS was more obviously significant when T‐Vec was used as the first‐line treatment, and in the subgroup of patients with stage IIIB, IIIC or IVM1.13 This phase III trial was the first to prove that local intralesional injections with an oncolytic virus can not only suppress the growth of injected tumors but also prolong the OS, supposedly via induction of systemic antitumor immunity. Based on this observation, several clinical trials of T‐Vec in combination with systemic administration with immune check point inhibitors are ongoing.
G47∆
G47Δ is a triple‐mutated third‐generation oncolytic HSV‐1 that was developed by Todo et al. by adding another deletion mutation to the genome of G207, a second generation HSV‐1.26, 27 G47∆ was developed to strengthen the antitumor efficacy while retaining the safety features of G207, mainly through enhancing the capability to elicit specific antitumor immunity.27 Two of the mutations of G47Δ are created in the γ_34.5_ and α_47_ genes, the same genes that T‐Vec utilizes. G47∆ further has an insertion of the Escherichia coli LacZ gene inactivating the ICP6 gene. The ICP6 gene encodes the large subunit of ribonucleotide reductase (RR) that is essential for viral DNA synthesis.28, 29 When ICP6 is inactivated, HSV‐1 can replicate only in proliferating cells that express high enough levels of host RR to compensate for the deficient viral RR. Because of the three manmade mutations in the genome, G47∆ should be much attenuated and, therefore, safer in normal tissues than those with two mutations such as G207 and T‐Vec. Furthermore, because the immediate‐early expression of US11 caused by the deletion within the α_47_ gene prevents the premature termination of protein synthesis that slows the growth of γ_34.5_‐deficient HSV‐1 strains such as G207, G47∆ shows augmented replication capability in cancer cells, resulting in having a wider therapeutic window than any other oncolytic HSV‐1.
G47Δ demonstrated a greater replication capability and a higher antitumor efficacy than G207.27 G47∆ exhibited efficacy in basically all in vivo solid tumor models tested, including glioma, breast cancer,30 prostate cancer,31, 32, 33 schwannoma,34 nasopharyngeal carcinoma,35 hepatocellular carcinoma,36 colorectal cancer,37 malignant peripheral nerve sheath tumor38 and thyroid carcinoma.39 G47∆ has been shown to kill cancer stem cells derived from human glioblastoma efficiently.40
G47∆ is currently the only third generation HSV‐1 to be tested in humans.27, 41 Following the phase I–IIa study in patients with recurrent glioblastoma that was conducted in Japan and successfully completed in 2014, a phase II study started in 2015 in patients with residual or recurrent glioblastoma (UMIN000015995). G47∆ (1 × 109 pfu) is injected stereotactically into the brain tumor twice within 2 weeks and then every 4 weeks, for a maximum six times. In February 2016, G47∆ was designated as a “Sakigake” breakthrough therapy drug by the Ministry of Health, Labour and Welfare of Japan (MHLW). “Sakigake” is a Japanese word meaning “ahead of the world.” This new system by the Japanese government provides the designated drug candidate, namely G47∆, with an early assessment and priority reviews by the Pharmaceuticals and Medical Devices Agency of Japan (PMDA), and therefore should allow its fast‐tracked drug approval by MHLW.
Besides the clinical trials in glioblastoma, we have just completed a single arm phase I study in patients with castration‐resistant prostate cancer, in which 3 × 108 pfu of G47∆ was injected into the prostate using a transrectal ultrasound‐guided transperineal technique (UMIN000010463). Dose escalation was planned in three cohorts, with patients receiving G47∆ twice in the first cohort, three times in the second and four times in the third. The treatment was well tolerated by patients, with no severe adverse events attributable to G47∆ observed to date. A phase I study has been ongoing in patients with recurrent olfactory neuroblastoma since 2013 (UMIN000011636).
JX‐594
JX‐594 (pexastimogene devacirepvec, Pexa‐Vec) is a genertically engineered vaccinia virus that has a mutation in the TK gene, conferring cancer cell‐selective replication, and an insertion of the human GM‐CSF gene, augmenting the antitumor immune response. JX‐594 also has a LacZ gene insertion as a marker.42, 43, 44 The advantages of using vaccinia virus include intravenous stability for delivery, strong cytotoxicity and extensive safety experience as a live vaccine.42 In a phase I study, intralesional injection of primary or metastatic liver tumors with JX‐594 was generally well tolerated in the context of JX‐594 replication, GM‐CSF expression and systemic dissemination. Direct hyperbilirubinemia was the dose‐limiting toxicity.45 High dose JX‐594 was used for a dose‐escalation phase I trial to test the feasibility of intravenous delivery.46 A randomized phase II dose‐finding trial was performed in patients with hepatocellular carcinoma.47 When a low or high dose of JX‐594 was infused, OS was significantly longer in the high dose arm compared with the low dose arm (n = 14 vs 16, median OS 14.1 vs 6.7 months, respectively). A phase III trial in patients with advanced stage hepatocellular carcinoma began enrolling patients in late 2015 (PHOCUS, NCT02562755). In this trial, JX‐594 (109 pfu) is administered intralesionally three times bi‐weekly at days 1, 15 and 29, followed by sorafenib at day 43, whereas, in the control arm, sorafenib begins on Day 1 at 400 mg twice daily.
CG0070
CG0070 is an oncolytic adenovirus developed by Ramesh et al.48 Ad5 adenovirus was engineered so that the human E2F‐1 promoter drives the E1A gene, and the human GM‐CSF gene is inserted. E2F‐1 is regulated by the retinoblastoma tumor suppressor protein (Rb), which is commonly mutated in bladder cancer, and a loss of Rb binding results in a transcriptionally active E2F‐1.49
A phase I trial of CG0070 was conducted in patients with non‐muscle‐invasive bladder cancer who did not respond to BCG therapy.50 Single or multiple (every 28 days × 3 and/or weekly six times) dose(s) of up to 3 × 1013 virus particles (vp) were administered intravesically. No clinically significant serious adverse events related to treatment were reported, and the most common adverse events observed were grade 1–2 bladder toxicities, such as dysuria, bladder pain and frequency.50 The overall response rate was 48.6% (17 of 35), which increased to 63.6% (14 of 22) in the multi‐dose cohort. In the following randomized phase II/III trial in patients with non‐muscle‐invasive bladder cancer, 15 patients received CG0070 and 7 control patients received other standard intravesical therapies (BOND, NCT01438112). Although there was no apparent difference in the initial CR (8 patients of CG0070 [53%] vs 4 of control group [57%]), CG0070 treatment demonstrated a better durable response in a subset of high‐risk patients.51 In a single arm phase III trial that is underway, patients with BCG‐refractory non‐muscle‐invasive bladder cancer are given CG0070 intravesically at a dose of 1012 vp weekly for 6 weeks. Patients who achieved a partial or complete response at 6 months after the first intervention are maintained with the same induction cycle every 6 months (BOND2, NCT02365818).
Naturally occurring oncolytic viruses
The idea of using naturally occurring viruses for the treatment of cancer was almost abandoned after vigorous attempts during the 1960s and 1970s because of the lack of means to control viral pathogenicity at the time. However, the idea was revived along with the emerging development of genetically engineered viruses, and newly developed naturally occurring viruses are typically those that are not pathogenic in humans.
Reolysin
Reoviruses are double‐stranded RNA viruses that replicate preferentially in transformed cell lines but not in normal cells.52, 53, 54 In theory, oncolytic properties of reovirus depend on activated Ras signaling.55, 56 Reolysin is the T3D strain of reovirus, which has been most extensively studied among several serotypes as an anticancer agent, and is currently the only therapeutic wild‐type reovirus in clinical development.57
The first phase I trial involved intralesional administration of Reolysin in patients with advanced solid tumors.58 The most common treatment‐related adverse events were nausea (79%), vomiting (58%), erythema at the injection site (42%), fevers/chills (37%) and transient flu‐like symptoms (32%).58 Further phase I studies demonstrated the safety and broad anticancer activity of Reolysin in prostate cancer,59 malignant glioma,60 metastatic colorectal cancer,61, 62 multiple myeloma63 and solid cancers.64, 65 Multiple phase II studies have investigated intralesional injection of Reolysin together with local irradiation for the treatment of refractory or metastatic solid tumors,66 intravenous administration of Reolysin for metastatic melanoma67 and intravenous administration of Reolysin in combination with chemotherapy for head and neck cancer or lung squamous cell carcinoma.68, 69
A randomized double‐blinded phase III trial has been performed, comparing intravenous Reolysin in combination with paclitaxel and carboplatin versus chemotherapy alone, in patients with metastatic and/or recurrent head and neck cancer (NCT01166542). Patients were treated with intravenous administration of 3 × 1010 tissue culture infectious dose‐50 (TCID50) of Reolysin on days 1–5 with standard doses of intravenous paclitaxel and carboplatin on day 1 only every 21 days, versus standard doses of intravenous paclitaxel and carboplatin alone. According to a report by the company developing Reolysin, of 165 patients analyzed, 118 patients had regional head and neck cancer with/without distant metastases and 47 patients had distant metastases only. In patients with regional cancer, a significant improvement in OS was observed for the Reolysin group versus the control group (P = 0.0146).57 The FDA in the USA granted Reolysin an orphan drug designation for malignant glioma, ovarian cancer and pancreatic cancer in 2015.
Limitations of oncolytic virus therapy
A wide variety of oncolytic viruses are currently under clinical development worldwide, and, as described in this review, each oncolytic virus carries the characteristics of the parental wild‐type virus, not only the advantages but also the disadvantages. For example, in regards to oncolytic HSV‐1, such as T‐Vec and G47∆, because HSV‐1 spreads from cell to cell and does not naturally cause viremia, oncolytic HSV‐1 is best administered intralesionally and may not be well suited for intravenous delivery. However, as proven by the phase III study of T‐Vec in melanoma patients at advanced stages,13 local intralesional injections with oncolytic HSV‐1 can act on remote lesions via induction of systemic antitumor immunity and prolong survival. It has been shown that expression of GM‐CSF does not augment the efficacy of oncolytic HSV‐1, while IL‐12 expression does, in immunocompetent mouse tumor models.31 Therefore, it is likely that the systemic effect via antitumor immunity was due to the characteristics of HSV‐1 itself rather than the effect by GM‐CSF.
One major concern of oncolytic virus therapy has been that the efficacy may be diminished by the presence of circulating antibodies.57 Viruses that naturally cause viremia are likely vulnerable to neutralizing antibodies; therefore, for such viruses, the antitumor effect of intravenous administration may be limited in patients who have had previous treatment or vaccination. An unfavorable effect of circulating antibodies was well documented in a clinical trial using oncolytic measles virus (MV‐NIS) in patients with multiple myeloma.70 In this dose escalation study, it was only after the dosing level reached a very high dose of 1011 TCID50 that intravenous infusion with MV‐NIS showed efficacy. In a preclinical study using tumor‐bearing immunocompetent mice, intravenous treatment with reovirus resulted in regrowth of tumors 3 weeks after initial tumor growth inhibition, which coincided with the rise in serum anti‐reovirus antibody titers.71 Phase I data showed that the maximum neutralizing anti‐reovirus antibody titers were reached by day 7 in 12 (36%) of 33 patients and at day 14 in 20 patients (61%).72 It was, therefore, recommended that, for systemic treatment, reovirus should be administered in rapid, repeated, high doses within the first week of treatment before the rise of serum neutralizing antibodies, and that it should be used in combination with other anticancer therapies.57
Oncolytic virus as immunotherapy
All genetically engineered oncolytic viruses described in this review were designed to enhance the induction of antitumor immunity that accompanies the oncolytic activity. Both T‐Vec and G47∆ have a deletion in the α_47_ gene, the product of which inhibits the transporter associated with antigen presentation; therefore, cancer cells subjected to the oncolytic activities of these viruses are vulnerable to immune surveillance, and the processing by antigen presenting cells is likely facilitated.21, 22 A combination with systemic administration of immune checkpoint inhibitor is a reasonable strategy to enhance the efficacy of oncolytic viruses. In a preclinical study, intralesional Reolysin treatment in combination with intravenous anti‐PD‐1 antibody administration was significantly more efficacious than Reolysin or anti‐PD‐1 alone in mice with subcutaneous melanoma.73 A phase Ib/II clinical trial of T‐Vec in combination with ipilimumab (anti–CTLA4) is currently ongoing in patients with stage IIIb‐IV melanoma (NCT01740297). Preliminary results from the first 18 patients showed that the median time to response was 5.3 months, and the 18‐month PFS and OS rates were 50% and 67%, respectively, with a median follow‐up of 17 months.74. An open‐label Phase Ib/III study in patients with previously untreated, unresected stage IIIb–IVM1c melanoma will further evaluate the safety and efficacy of the combination of T‐Vec and pembrolizumab (anti–PD‐1) compared with pembrolizumab alone (NCT02263508).75 A phase I study of T‐Vec in combination with pembrolizumab has also started for head and neck cancer in late 2015 (Masterkey232, NCT02626000). For all oncolytic virus therapy, long‐term side effects from the induction of systemic antitumor immunity, including development of autoimmune diseases, should be closely investigated.
Like T‐Vec, JX‐594 and CG0070 that have the GM‐CSF gene inserted in the viral genome, “arming” oncolytic viruses with transgene(s) is a useful strategy to add certain antitumor functions to oncolytic viruses. According to preclinical studies with oncolytic HSV‐1, however, GM‐CSF is not exactly an ideal transgene for “arming”; rather, interleukin 12, interleukin 18 or soluble B7‐1 would significantly enhance the antitumor efficacy via augmenting the antitumor immunity induction.31, 32, 76 Besides immunostimulatory genes, various transgenes of other antitumor functions, including antiangiogenesis, have been utilized to arm oncolytic viruses.77, 78, 79
Conclusion
It would not be too early to say that oncolytic virus therapy is now established as an approach to treat cancer. Because an induction of specific antitumor immunity in the course of oncolytic activities is the common feature that plays an important role in presenting antitumor effects, the efficacy of oncolytic virus therapy is expected to improve further when combined with immunotherapy. By arming oncolytic viruses with functional transgenes, a whole panel of oncolytic viruses with a variety of antitumor functions would be available in the future, from which a combination of appropriate viruses can be chosen according to the type and stage of cancer. A new era of cancer treatment seems at dawn, where cancer patients can freely choose oncolytic virus therapy as a treatment option.
Disclosure Statement
Tomoki Todo owns the patent right for G47∆ in multiple countries including Japan. Tomoki Todo is the principal investigator of the ongoing phase II clinical trial of G47∆ in glioblastoma patients in Japan, which is funded by research grants from the MHLW of Japan, the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, and the Japan Agency of Medical Research and Development (AMED).
Acknowledgments
The clinical development of G47Δ is supported in part by the Translational Research Network Program of the MEXT of Japan, research grants from the MHLW of Japan and the AMED. G47∆ clinical trials are supported in part by the Research Hospital, the Institution of Medical Science, The University of Tokyo, and The University of Tokyo Hospital.
Cancer Sci 107 (2016) 1373–1379
Funding Information
Translational Research Network Program of the MEXT of Japan; Research Grants from MHLW of Japan and the AMED.
References
- 1.Larson C, Oronsky B, Scicinski J_et al_Going viral: a review of replication‐selective oncolytic adenoviruses. Oncotarget 2015; 6: 19976–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith RR, Huebner RJ, Rowe WP, Schatten WE, Thomas LB. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer 1956; 9: 1211–8. [DOI] [PubMed] [Google Scholar]
- 3.Hoster H, Zanes R, von Haam E. The association of “viral” hepatitis and Hodgkin's disease. Cancer Res 1949; 9: 473–80. [PubMed] [Google Scholar]
- 4.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther 2007; 15: 651–9. [DOI] [PubMed] [Google Scholar]
- 5.Platanias LC. Mechanisms of type‐I‐ and type‐II‐interferon‐mediated signaling. Nat Rev Immunol 2005; 5: 375–86. [DOI] [PubMed] [Google Scholar]
- 6.Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991; 252: 854–6. [DOI] [PubMed] [Google Scholar]
- 7.Todo T, Rabkin SD, Sundaresan P_et al_Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication‐competent herpes simplex virus. Hum Gene Ther 1999; 10: 2741–55. [DOI] [PubMed] [Google Scholar]
- 8.Todo T, Rabkin SD, Chahlavi A_et al_Corticosteroid administration does not affect viral oncolytic activity, but inhibits antitumor immunity in replication‐competent herpes simplex virus tumor therapy. Hum Gene Ther 1999; 10: 2869–78. [DOI] [PubMed] [Google Scholar]
- 9.Heise C, Sampson‐Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX‐015, an E1B gene‐attenuated adenovirus, causes tumor‐specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med 1997; 3: 639–45. [DOI] [PubMed] [Google Scholar]
- 10.Xia ZJ, Chang JH, Zhang L_et al_Phase III randomized clinical trial of intratumoral injection of E1B gene‐deleted adenovirus (H101) combined with cisplatin‐based chemotherapy in treating squamous cell cancer of head and neck or esophagus. Ai Zheng 2004; 23: 1666–70. [PubMed] [Google Scholar]
- 11.Garber K. China approves world's first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst 2006; 98: 298–300. [DOI] [PubMed] [Google Scholar]
- 12.Coffin R. Interview with Robert Coffin, inventor of T‐VEC: the first oncolytic immunotherapy approved for the treatment of cancer. Immunotherapy 2016; 8: 103–6. [DOI] [PubMed] [Google Scholar]
- 13.Andtbacka RH, Kaufman HL, Collichio F_et al_Talimogene laherparepvec Improves durable response rate in patients with advanced melanoma. J Clin Oncol 2015; 33: 2780–8. [DOI] [PubMed] [Google Scholar]
- 14.Hu JCC, Coffin RS, Davis CJ. A phase I study of OncoVEXGM‐CSF, a second‐generation oncolytic herpes simplex virus expressing granulocyte macrophage colony‐stimulating factor. Clin Cancer Res 2006; 12: 6737–47. [DOI] [PubMed] [Google Scholar]
- 15.Chou J, Roizman B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc Natl Acad Sci USA 1992; 89: 3266–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He B, Chou J, Brandimarti R_et al_Suppression of the phenotype of gamma(1)34.5‐herpes simplex virus 1: failure of activated RNA‐dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J Virol 1997; 71: 6049–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu BL, Robinson M, Han ZQ_et al_ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti‐tumour properties. Gene Ther 2003; 10: 292–303. [DOI] [PubMed] [Google Scholar]
- 18.Markert JM, Parker JN, Buchsbaum DJ_et al_Oncolytic HSV‐1 for the treatment of brain tumours. Herpes 2006; 13: 66–71. [PubMed] [Google Scholar]
- 19.Agarwalla PK, Aghi MK. Oncolytic herpes simplex virus engineering and preparation. Methods Mol Biol 2012; 797: 1–19. [DOI] [PubMed] [Google Scholar]
- 20.Goldsmith K, Chen W, Johnson DC_et al_Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med 1998; 187: 341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Früh K, Ahn K, Djaballah H_et al_A viral inhibitor of peptide transporters for antigen presentation. Nature 1995; 375: 415–8. [DOI] [PubMed] [Google Scholar]
- 22.York IA, Roop C, Andrews DW_et al_A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 1994; 77: 525–35. [DOI] [PubMed] [Google Scholar]
- 23.Poppers J, Mulvey M, Khoo D_et al_Inhibition of PKR activation by the proline‐rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J Virol 2000; 74: 11215–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Senzer NN, Kaufman HL, Amatruda T_et al_Phase II clinical trial of a granulocyte‐macrophage colony‐stimulating factor‐encoding, second‐generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 2009; 27: 5763–71. [DOI] [PubMed] [Google Scholar]
- 25.Kaufman HL, Kim DW, DeRaffele G_et al_Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM‐CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol 2010; 17: 718–30. [DOI] [PubMed] [Google Scholar]
- 26.Mineta T, Rabkin SD, Yazaki T_et al_Attenuated multi‐mutated herpes simplex virus‐1 for the treatment of malignant gliomas. Nat Med 1995; 1: 938–43. [DOI] [PubMed] [Google Scholar]
- 27.Todo T, Martuza RL, Rabkin SD_et al_Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc Natl Acad Sci USA 2001; 98: 6396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aghi M, Visted T, Depinho RA_et al_Oncolytic herpes virus with defective ICP6 specifically replicates in quiescent cells with homozygous genetic mutations in p16. Oncogene 2008; 27: 4249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaur B, Chiocca EA, Cripe TP. Oncolytic HSV‐1 virotherapy: clinical experience and opportunities for progress. Curr Pharm Biotechnol 2012; 13: 1842–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu R, Martuza RL, Rabkin SD. Intracarotid delivery of oncolytic HSV vector G47Delta to metastatic breast cancer in the brain. Gene Ther 2005; 12: 647–54. [DOI] [PubMed] [Google Scholar]
- 31.Varghese S, Rabkin SD, Liu R_et al_Enhanced therapeutic efficacy of IL‐12, but not GM‐CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther 2006; 13: 253–65. [DOI] [PubMed] [Google Scholar]
- 32.Fukuhara H, Ino Y, Kuroda T_et al_Triple gene‐deleted oncolytic herpes simplex virus vector double‐armed with interleukin 18 and soluble B7‐1 constructed by bacterial artificial chromosome‐mediated system. Cancer Res 2005; 65: 10663–8. [DOI] [PubMed] [Google Scholar]
- 33.Fukuhara H, Martuza RL, Rabkin SD_et al_Oncolytic herpes simplex virus vector g47delta in combination with androgen ablation for the treatment of human prostate adenocarcinoma. Clin Cancer Res 2005; 11: 7886–90. [DOI] [PubMed] [Google Scholar]
- 34.Prabhakar S, Messerli SM, Stemmer‐Rachamimov AO_et al_Treatment of implantable NF2 schwannoma tumor models with oncolytic herpes simplex virus G47Delta. Cancer Gene Ther 2007; 14: 460–7. [DOI] [PubMed] [Google Scholar]
- 35.Wang JN, Hu P, Zeng MS_et al_Anti‐tumor effect of oncolytic herpes simplex virus G47delta on human nasopharyngeal carcinoma. Chin J Cancer 2011; 30: 831–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J, Xu L, Zeng W_et al_Treatment of human hepatocellular carcinoma by the oncolytic herpes simplex virus G47delta. Cancer Cell Int 2014; 14: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffmann D, Bangen JM, Bayer W_et al_Synergy between expression of fusogenic membrane proteins, chemotherapy and facultative virotherapy in colorectal cancer. Gene Ther 2006; 13: 1534–44. [DOI] [PubMed] [Google Scholar]
- 38.Antoszczyk S, Spyra M, Mautner VF_et al_Treatment of orthotopic malignant peripheral nerve sheath tumors with oncolytic herpes simplex virus. Neuro Oncol 2014; 16: 1057–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang JN, Xu LH, Zeng WG_et al_Treatment of human thyroid carcinoma cells with the g47delta oncolytic herpes simplex virus. Asian Pac J Cancer Prev 2015; 16: 1241–5. [DOI] [PubMed] [Google Scholar]
- 40.Cheema TA, Wakimoto H, Fecci PE_et al_Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci USA 2013; 110: 12006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ino Y, Todo T. Clinical development of a third‐generation HSV‐1 (G47∆) for malignant glioma. Gene Ther Regul 2010; 5: 101–11. [Google Scholar]
- 42.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi‐mechanistic therapeutic class for cancer. Nat Rev Cancer 2009; 9: 64–71. [DOI] [PubMed] [Google Scholar]
- 43.Kim JH, Oh JY, Park BH_et al_Systemic armed oncolytic and immunologic therapy for cancer with JX‐594, a targeted poxvirus expressing GM‐CSF. Mol Ther 2006; 14: 361–70. [DOI] [PubMed] [Google Scholar]
- 44.Parato KA, Breitbach CJ, Le Boeuf F_et al_The oncolytic poxvirus JX‐594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther 2012; 20: 749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park BH, Hwang T, Liu TC_et al_Use of a targeted oncolytic poxvirus, JX‐594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol 2008; 9: 533–42. [DOI] [PubMed] [Google Scholar]
- 46.Breitbach CJ, Bruke J, Jonker D_et al_Intravenous delivery of a multi‐mechanistic cancer‐targeted oncolytic poxvirus in humans. Nature 2011; 477: 99–102. [DOI] [PubMed] [Google Scholar]
- 47.Heo J, Reid T, Ruo L_et al_Randomized dose‐finding clinical trial of oncolytic immunotherapeutic vaccinia JX‐594 in liver cancer. Nat Med 2013; 19: 329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramesh N, Ge Y, Ennist DL_et al_CG0070, a conditionally replicating granulocyte macrophage colony‐stimulating factor–armed oncolytic adenovirus for the treatment of bladder cancer. Clin Cancer Res 2006; 12: 305–13. [DOI] [PubMed] [Google Scholar]
- 49.Zwicker J, Müller R. Cell cycle‐regulated transcription in mammalian cells. Prog Cell Cycle Res 1995; 1: 91–9. [DOI] [PubMed] [Google Scholar]
- 50.Burke JM, Lamm DL, Meng MV_et al_A first in human phase 1 study of CG0070, a GM‐CSF expressing oncolytic adenovirus, for the treatment of nonmuscle invasive bladder cancer. J Urol 2012; 188: 2391–7. [DOI] [PubMed] [Google Scholar]
- 51.Packiam VT, Campanile AN, Barocas DA_et al_A phase II/III Trial of CG0070, an oncolytic adenovirus, for BCG‐refractory non‐muscle‐invasive bladder cancer (NMIBC). J Urol 2016; 195: e142. [Google Scholar]
- 52.Vidal L, Yap TA, White CL_et al_Reovirus and other oncolytic viruses for the targeted treatment of cancer. Target Oncol 2006; 1: 130–50. [Google Scholar]
- 53.Hashiro G, Loh PC, Yau JT. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch Virol 1977; 54: 307–15. [DOI] [PubMed] [Google Scholar]
- 54.Duncan MR, Stanish SM, Cox DC. Differential sensitivity of normal and transformed human cells to reovirus infection. J Virol 1978; 28: 444–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Norman KL, Hirasawa K, Yang AD, Shields MA, Lee PW. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl Acad Sci USA 2004; 101: 11099–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res 1989; 49: 4682–9. [PubMed] [Google Scholar]
- 57.Gong J, Sachdev E, Mita AC, Mita MM. Clinical development of reovirus for cancer therapy: an oncolytic virus with immune‐mediated antitumor activity. World J Methodol 2016; 6: 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gollamudi R, Ghalib MH, Desai KK_et al_REO‐001: intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients in advanced solid tumors. Invest New Drugs 2010; 28: 641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thirukkumaran CM, Nodwell MJ, Hirasawa K_et al_Oncolytic viral therapy for prostate cancer: efficacy of reovirus as a biological therapeutic. Cancer Res 2010; 70: 2435–44. [DOI] [PubMed] [Google Scholar]
- 60.Kicielinski KP, Chiocca EA, Yu JS, Gill GM, Coffey M, Markert JM. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol Ther 2014; 22: 1056–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Adair RA, Roulstone V, Scott KJ_et al_Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med 2012; 4: 138ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ocean AJ, Bekaii‐Saab TS, Chaudhary I_et al_A multicenter phase I study of intravenous administration of reolysin in combination with irinotecan/fluorouracil/leucovorin (FOLFIRI) in patients (pts) with oxaliplatin‐refractory/intolerant KRAS‐mutant metastatic colorectal cancer (mCRC). J Clin Oncol 2013; 31: 2318. [Google Scholar]
- 63.Sborov DW, Nuovo GJ, Stiff A_et al_A phase I trial of single‐agent reolysin in patients with relapsed multiple myeloma. Clin Cancer Res 2014; 20: 5946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karapanagiotou EM, Roulstone V, Twigger K_et al_Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res 2012; 18: 2080–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Phelps M, Cohn DE, O'Malley DM_et al_Reovirus replication in ovarian and peritoneal tumors after intravenous administration. Cancer Res 2010; 70: 2594. [Google Scholar]
- 66.Saunders M, Anthoney A, Coffey M_et al_Results of a phase II study to evaluate the biological effects of intratumoral (ITu) reolysin in combination with low dose radiotherapy (RT) in patients (Pts) with advanced cancers. J Clin Oncol 2009; 27: e14514. [Google Scholar]
- 67.Galanis E, Markovic SN, Suman VJ_et al_Phase II trial of intravenous administration of Reolysin(®) (Reovirus Serotype‐3‐dearing Strain) in patients with metastatic melanoma. Mol Ther 2012; 20: 1998–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karnad A, Haigentz M, Miley T, Coffey M, Gill G, Mita M. A phase II study of intravenous wild‐type reovirus (Reolysin) in combination with paclitaxel plus carboplatin in patients with platinum refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. Mol Cancer Ther 2011; 10: C22. [Google Scholar]
- 69.Mita AC, Argiris A, Coffey M, Gill GM, Mita M. A Phase 2 study of intravenous administration of Reolysin (R) (Reovirus Type 3 Dearing) in combination with paclitaxel (P) and carboplatin (C) in patients with squamous cell carcinoma of the lung. J Thorac Oncol 2013; 8: S617–8. [Google Scholar]
- 70.Russell SJ, Federspiel MJ, Peng KW_et al_Remission of disseminated cancer after systemic oncolytic virotherapy. Mayo Clin Proc 2014; 89: 926–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hirasawa K, Nishikawa SG, Norman KL_et al_Systemic reovirus therapy of metastatic cancer in immune‐competent mice. Cancer Res 2003; 63: 348–53. [PubMed] [Google Scholar]
- 72.White CL, Twigger KR, Vidal L_et al_Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther 2008; 15: 911–20. [DOI] [PubMed] [Google Scholar]
- 73.Rajani K, Parrish C, Kottke T_et al_Combination therapy with reovirus and anti‐PD‐1 blockade controls tumor growth through innate and adaptive immune Responses. Mol Ther 2016; 24: 166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Puzanov IMM, Andtbacka RH, Minor DR_et al_Survival, safety, and response patterns in a phase 1b multicenter trial of talimogene laherparepvec (T‐VEC) and ipilimumab (ipi) in previously untreated, unresected stage IIIB‐IV melanoma. J Clin Oncol 2015; 33(Suppl): Abstract 9063. [Google Scholar]
- 75.Harrington KJ, Puzanov I, Hecht JR_et al_Clinical development of talimogene laherparepvec (T‐VEC): a modified herpes simplex virus type‐1‐derived oncolytic immunotherapy. Expert Rev Anticancer Ther 2015; 15: 1389–403. [DOI] [PubMed] [Google Scholar]
- 76.Ino Y, Saeki Y, Fukuhara H_et al_Triple combination of oncolytic herpes simplex virus‐1 vectors armed with interleukin‐12, interleukin‐18, or soluble B7‐1 results in enhanced antitumor efficacy. Clin Cancer Res 2006; 12: 643–52. [DOI] [PubMed] [Google Scholar]
- 77.Liu TC, Zhang T, Fukuhara H. Oncolytic HSV armed with platelet factor 4, an antiangiogenic agent, shows enhanced efficacy. Mol Ther 2006; 14: 789–97. [DOI] [PubMed] [Google Scholar]
- 78.Liu TC, Zhang T, Fukuhara H_et al_Dominant‐negative fibroblast growth factor receptor expression enhances antitumoral potency of oncolytic herpes simplex virus in neural tumors. Clin Cancer Res 2006; 12: 6791–9. [DOI] [PubMed] [Google Scholar]
- 79.Tsuji T, Nakamori M, Iwahashi M_et al_An armed oncolytic herpes simplex virus expressing thrombospondin‐1 has an enhanced in vivo antitumor effect against human gastric cancer. Int J Cancer 2013; 132: 485–94. [DOI] [PubMed] [Google Scholar]