Gut dysbiosis impairs recovery after spinal cord injury (original) (raw)
Kigerl et al. show that spinal cord injury causes profound changes in gut microbiota and that these changes in gut ecology are associated with activation of GALT immune cells. They show that feeding mice probiotics after SCI confers neuroprotection and improves functional recovery.
Abstract
The trillions of microbes that exist in the gastrointestinal tract have emerged as pivotal regulators of mammalian development and physiology. Disruption of this gut microbiome, a process known as dysbiosis, causes or exacerbates various diseases, but whether gut dysbiosis affects recovery of neurological function or lesion pathology after traumatic spinal cord injury (SCI) is unknown. Data in this study show that SCI increases intestinal permeability and bacterial translocation from the gut. These changes are associated with immune cell activation in gut-associated lymphoid tissues (GALTs) and significant changes in the composition of both major and minor gut bacterial taxa. Postinjury changes in gut microbiota persist for at least one month and predict the magnitude of locomotor impairment. Experimental induction of gut dysbiosis in naive mice before SCI (e.g., via oral delivery of broad-spectrum antibiotics) exacerbates neurological impairment and spinal cord pathology after SCI. Conversely, feeding SCI mice commercial probiotics (VSL#3) enriched with lactic acid–producing bacteria triggers a protective immune response in GALTs and confers neuroprotection with improved locomotor recovery. Our data reveal a previously unknown role for the gut microbiota in influencing recovery of neurological function and neuropathology after SCI.
Introduction
Commensal microbes, most of which reside in the gut, outnumber mammalian cells ∼10:1 and contain ∼100-fold more genes than the human genome (Savage, 1977; Bäckhed et al., 2005; Gill et al., 2006; Belkaid and Naik, 2013). This vast microbial network is critical for the development and maintenance of cellular metabolism, digestion and nutrient absorption, and immune system development (Round and Mazmanian, 2009; Hooper et al., 2012; Nicholson et al., 2012). Recent data indicate that gut microbes also regulate both normal development and disease pathogenesis in the central nervous system (CNS; Collins et al., 2012; Wang and Kasper, 2014; Benakis et al., 2016; Winek et al., 2016).
Gut microbiota “talk” to the CNS by directly interacting with immune cells or nerve fibers and indirectly by secreting metabolites that bypass the blood–brain barrier. Most (∼70–80%) immune cells in the body are located within gut-associated lymphoid tissues (GALTs). There, an ongoing dialogue between immune cells and gut bacteria produces cytokines that affect CNS function (Brandtzaeg, 1989; Castro and Arntzen, 1993). Gut microbes also produce neuroactive metabolites (i.e., short chain fatty acids [SCFAs] and choline) and neurotransmitters (γ-aminobutyric acid, serotonin, dopamine, and acetylcholine; Clarke et al., 2014; Tillisch, 2014) that act on both enteric neurons in the gut and also within the CNS (Wikoff et al., 2009; Clarke et al., 2014; Forsythe et al., 2014).
Dysbiosis develops when the composition of the gut microbiota is altered such that beneficial nonpathogenic gut bacteria (i.e., probionts) are depleted or become overwhelmed by pathogenic inflammatory bacteria (i.e., pathobionts). Autoimmune diseases (e.g., multiple sclerosis, type I diabetes, and rheumatoid arthritis), allergies, and metabolic disorders have been linked to gut dysbiosis (Berer et al., 2011; Kriegel et al., 2011; Lee et al., 2011; Tilg and Kaser, 2011; Murri et al., 2013; Cao et al., 2014; Soyucen et al., 2014). Similarly, dysbiosis has been implicated in the onset or progression of neurological diseases including autism, pain, depression, anxiety, and stroke (Ait-Belgnaoui et al., 2006; Rousseaux et al., 2007; Cryan and Dinan, 2012; Foster and McVey Neufeld, 2013; Hsiao et al., 2013; de Theije et al., 2014a,b; Benakis et al., 2016; Winek et al., 2016). Common causes of gut dysbiosis include antibiotic use, stress, and gastrointestinal dysfunction (Hawrelak and Myers, 2004; Round and Mazmanian, 2009; Bailey et al., 2010, 2011). Many of these dysbiosis triggers also cause bacterial translocation (BT) or “leaky gut,” a process whereby gut bacteria migrate from the intestinal lumen into extraintestinal sites (Liu et al., 2004; MacFie, 2004; Magnotti and Deitch, 2005; Gatt et al., 2007). A healthy gut microbiota is critical for preventing BT (Diehl et al., 2013).
Traumatic spinal cord injury (SCI) causes severe neurological and psychological complications and requires secondary care needs that likely predispose SCI individuals to gut dysbiosis. For example, acute and often chronic psychological stress is expected because of the sudden and dramatic life changes experienced by someone with an SCI (Boekamp et al., 1996; Elliott and Frank, 1996). Neurogenic bladder and bowel develop after SCI as a result of damage to spinal autonomic circuitry (Chung and Emmanuel, 2006; Enck et al., 2006; Karlsson, 2006; Riegger et al., 2007, 2009). SCI-induced dysautonomia also impairs immune function, increasing the need for repeat dosing with antibiotics to fight infections (Evans et al., 2011, 2013; Failli et al., 2012). Here, novel data show that SCI disrupts the gut microbiota and that SCI-induced gut dysbiosis is associated with profound changes in GALT immune cell activation. Dysbiosis also exacerbates intraspinal inflammation and impairs recovery of neurological function. Importantly, feeding mice a medical-grade probiotic (VSL#3) after SCI confers neuroprotection and improves functional recovery.
Results
SCI induces BT and persistent gut dysbiosis
Chronic immune suppression, intestinal obstruction, and impaired intestinal motility, all complications of experimental and clinical SCI, can independently cause BT (MacFie, 2004; Magnotti and Deitch, 2005; Gatt et al., 2007). To determine whether SCI causes BT, we first sought evidence that SCI causes indigenous bacteria to migrate from the gastrointestinal tract into sterile extraintestinal sites.
Using sterile techniques, mesenteric LNs (MLNs), livers, spleens, kidneys, and blood were collected from naive mice and mice that had received a midthoracic (T9) spinal contusion injury 1, 3, or 7 d earlier. Collected tissues were homogenized and then plated onto BBL CHROMagar plates. To control for environmental contamination, mice were housed in microisolator cages in the same laboratory environment. Importantly, bacteria were never found in cultures from tissues isolated from uninjured mice (Fig. 1 A). Conversely, bacteria were detected in all tissues from SCI mice by 7 d postinjury (dpi; Fig. 1 A). These data indicate that SCI causes commensal bacteria to translocate from the gut into peripheral tissues.
Figure 1.

SCI causes BT and gut dysbiosis. (A) Tissue samples collected from mice at various times after SCI were processed using sterile techniques and then were plated onto BBL CHROMagar plates to assess bacterial growth. No bacteria were found in tissues from naive mice or mice at 1 dpi. By 7 dpi, bacteria could be cultured from all tissues (χ2 contingency; n = 4 per time point). (B) Intestinal permeability was tested by oral gavage of FITC-dextran (4 kD) with subsequent analysis of FITC fluorescence in the blood. At 7 dpi, there was a significant increase in leakage of FITC-dextran from the gut in SCI mice (n = 7 per group). Horizontal bars represent the mean of the sample. (C) mRNA expression of genes encoding transcription factors or epithelial tight junction proteins that regulate paracellular permeability or the proliferation and differentiation of epithelia were increased at 7 dpi compared with control samples (one-way ANOVA; n = 8 per group). (D) Fecal cultures from SCI mice reveal the onset of gut dysbiosis by 7 dpi with persistent changes in composition of gut flora through 21 dpi (n = 6 per time point). *, P < 0.05; **, P < 0.01. Bars, 20 mm.
BT can occur as a result of an increase in paracellular transport through compromised epithelial tight junctions (Cruz et al., 1994; Han et al., 2004; Clark et al., 2005). To test whether intestinal barrier permeability increases after SCI, mice were gavaged with FITC-labeled dextran (4 kD) at a time when bacteria were detected in extraintestinal sites after SCI (7 dpi), and then FITC levels were measured in blood. On average, gut permeability increased 20% after SCI (vs. sham-injured mice; Fig. 1 B). In these mice, various genes encoding epithelial tight junction proteins that regulate paracellular permeability were dramatically affected by SCI (Fig. 1 C). Several transcription factors critical for the proliferation (Tcf7l2) and differentiation (Cdx1 and Cdx2) of intestinal epithelia increased significantly after SCI (James et al., 1994; Korinek et al., 1998; Subramanian et al., 1998; Beck et al., 1999; Silberg et al., 2000; van de Wetering et al., 2002). Expression of eight other genes that encode different tight junction proteins also increased in the gut of SCI mice (Fig. 1 C).
A healthy gut microbiota prevents BT (Diehl et al., 2013). Because BT and intestinal permeability increase after SCI (Fig. 1, A and B), we predicted that a microbial imbalance would also develop in the intestines of SCI mice. To test this hypothesis, bacterial cultures were grown from feces collected from SCI mice. Data in Fig. 1 D show that after SCI, the number and types of bacterial colonies that can be cultured from feces change; differences in gut microbe composition appear within the first week after SCI and persist for at least 21 dpi.
To unequivocally prove that SCI causes dysbiosis, fecal samples were collected and prepared for bacterial 16S ribosomal RNA (rRNA) gene sequencing. To prevent coprophagia and cross-colonization of gut microbiota between individual mice, all mice were individually housed. To reduce the potential effects of stress on the microbiota, fecal samples were not collected until mice acclimated in home cages for at least 1 wk. Dietary effects on the microbiota were controlled for by ensuring that daily food intake was equilibrated across all mice. Finally, no antibiotics or dietary supplements were given.
16S rRNA sequencing revealed SCI-dependent changes in the gut microbiome (Fig. 2 A). Hierarchical clustering of naive, sham-injured, and SCI samples showed a profound time-dependent effect of SCI on gut microbiome composition (Fig. 2 A). Specifically, Bacteroidales and Clostridiales, the two most prevalent bacterial taxa in mouse gut (Eckburg et al., 2005; Krych et al., 2013), were inversely regulated by SCI: Bacteroidales decreased while Clostridiales increased (Fig. 2, B and C). Significant changes in these taxa were accompanied by lesser but consistent changes in minor taxa, including Anaeroplasmatales, Turicibacterales, and Lactobacillales (Fig. 2 A). Open-field locomotor (Basso mouse scale [BMS]) scores were negatively correlated with the relative abundance of Clostridiales and Anaeroplasmatales, suggesting that the proportion of these bacteria can predict the magnitude of functional recovery and might be useful as a biomarker of injury severity (Fig. 2, D and E).
Figure 2.

16S rRNA gene sequencing reveals significant changes in the gut microbiome after SCI. (A) Unbiased hierarchical clustering techniques reveal significant changes in the relative abundance of major and minor bacteria taxa after SCI. The majority of the SCI samples cluster together on the left half of the heatmap, whereas the majority of the naive and laminectomy control samples cluster on the right half. (B and C) Bacteria from the two major orders of gut bacteria, Bacteriodales and Clostridiales, are reciprocally regulated after SCI. Relative abundance of Bacteroidales decreases as a function of time after SCI (B), whereas Clostridiales increases (C). Box and whisker plots show the minimum to maximum values. The horizontal line in each box represents the median of that group. n = 6–10 per time point. Trend lines show significant rates of change as functions of time after injury using the mixed effects model with repeated measures. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (D and E) Importantly, the relative abundance of Clostridiales and Anaeroplasmatales inversely correlate with locomotor recovery, as assessed by BMS (Pearson correlation; n = 6–10 per time point; each dot represents an individual mouse).
SCI triggers an immune response in GALT
SCI-induced gut dysbiosis could activate mucosal immune cells in GALT, which could subsequently affect systemic and intraspinal inflammation (Diehl et al., 2013). Two prominent areas of immune activation in GALT include Peyer’s patches (PPs) and MLNs. Immune surveillance of the intestinal lumen is provided by the PPs. Antigens or activated immune cells drain from the PPs into the MLNs, which act as a firewall between the gut and the rest of the body (Koboziev et al., 2010). Flow cytometry analyses of cells isolated from MLNs and PPs show that SCI-induced gut dysbiosis elicits inflammation in GALT (Table 1 and Fig. 3). By 3 dpi, B lymphocytes, CD8+ T lymphocytes, DCs, and macrophages increased in the MLNs relative to those in control mice (Fig. 3, A–E; and Table 1). TNF, IL-1β, TGF-β, and IL-10 mRNA expression also increased in MLNs by 7 dpi (Fig. 3 F). Conversely, CD4+ T lymphocytes decreased by 3 dpi in MLNs and remained below control levels throughout the duration of the study (Fig. 3 C). Similar cellular changes were noted in PPs, although the magnitude of change was smaller and the duration of effect was shorter when compared with postinjury changes in MLNs (Table 1). Cytokine changes in PPs were not measured.
Table 1. SCI alters the inflammatory cell phenotype in GALT.
| MLNs | B cells (B220+) | T cells (CD4+) | T cells (CD8+) | Macrophages (CD11b+) | DCs (CD11c+) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cells | MFI | Cells | MFI | Cells | MFI | Cells | MFI | Cells | MFI | |
| % | % | % | % | % | ||||||
| Naive | 26.23 | 1,486 | 58.20 | 6,287 | 18.33 | 5,619 | 1.42 | 499 | 3.43 | 167 |
| 3 dpi | 53.83a | 1,355 | 41.03a | 5,943 | 34.65a | 10,801 | 2.58a | 501 | 3.7 | 413.8a |
| 7 dpi | 40.45 | 1,859 | 37.88a | 6,117 | 34.98a | 7,016 | 2.15a | 461 | 4.65 | 216 |
| 28 dpi | 43.38 | 2,181a | 38.48a | 6,505 | 32.28a | 9,237 | 2.04a | 471 | 4.73 | 339.5a |
| PPs | ||||||||||
| Naive | 86.38 | 1,219 | 14.525 | 4,398 | 5.57 | 1,038 | 0.74 | 336 | 2.83 | 151.0 |
| 3 dpi | 74.33 | 1,692a | 21.23 | 4,721 | 14.95a | 5,048 | 0.71 | 348 | 3.91a | 214.5a |
| 7 dpi | 85.88 | 1,692a | 13.65 | 3,998 | 9.60 | 4,386 | 0.32 | 308 | 2.87 | 199.8a |
| 28 dpi | 87.43 | 1,992a | 13.48 | 3,556 | 9.04 | 3,238 | 0.24 | 362 | 2.90 | 192.8a |
Figure 3.
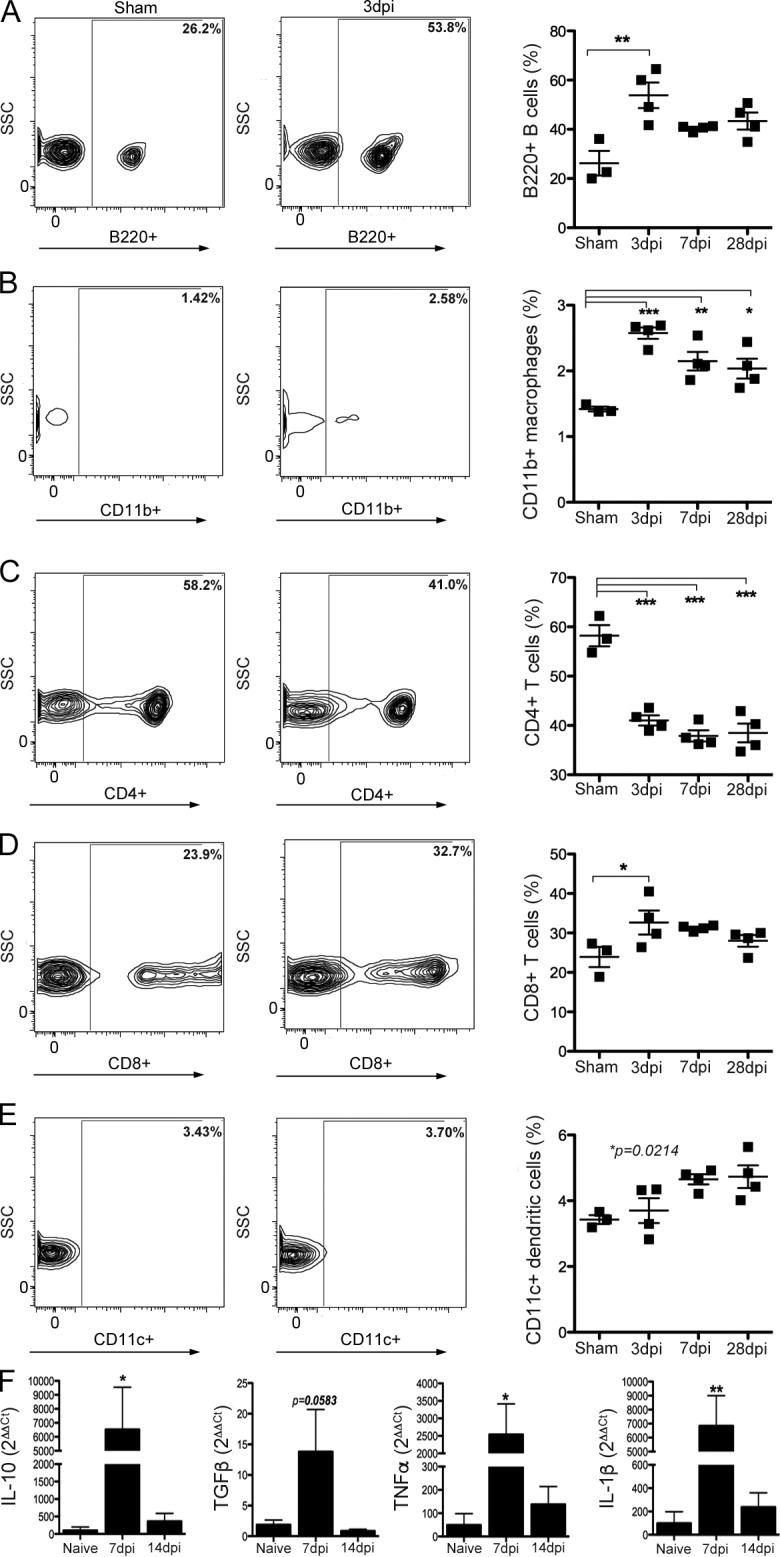
SCI activates an immune response in GALT. (A–E) Numbers of B lymphocytes (A), macrophages (B), CD8+ T lymphocytes (D), and CD11c+ DCs (E) increase in the MLNs after SCI. Conversely, CD4+ T lymphocytes (C) decrease and remain below preinjury levels indefinitely. Horizontal bars in scatter plots represent the mean of the sample. n = 3–4 per time point. (F) SCI also increases the expression of IL-10, TGF-β, TNF, and IL-1β mRNA. n = 3–5 per time point. One-way ANOVA; *, P < 0.05; **, P < 0.01; ***, P < 0.001 versus sham control.
Gut dysbiosis impairs locomotor recovery and exacerbates lesion pathology after SCI
Data in Figs. 1, 2, and 3 show that SCI causes BT, intestinal dysbiosis, and activation of mucosal immune cells. To determine the potential consequences of this break in gut–immune axis homeostasis, a gain-of-function study was performed in which mice with established gut dysbiosis received an SCI (Fig. 4 A). Dysbiosis was induced before SCI by mixing a cocktail of broad-spectrum antibiotics into mouse drinking water (Chen et al., 2008). To confirm depletion of gut bacteria and the onset of dysbiosis, fecal samples from individual mice were cultured before starting the antibiotic regimen and again 7 d after (but before SCI; Fig. 4, A and B). Bacteria were notably absent in feces from mice receiving antibiotics (Fig. 4 B). However, despite maintenance on antibiotics, 7–14 d after SCI in these same mice, there was an overgrowth in the gut of antibiotic-resistant bacteria (Fig. 4 B). This gut dysbiosis persisted for at least 21 dpi (Fig. 4 B). In a separate experiment, fecal samples from SCI mice with and without antibiotic-induced dysbiosis were analyzed using 16S rRNA sequencing (Table S3). In these samples, total sequence counts from mice given antibiotics were too low to generate reliable data, indicating that the antibiotic regimen induces bacterial depletion and dysbiosis (Table S3).
Figure 4.

Gut dysbiosis impairs recovery after SCI. (A) Fecal cultures from SCI mice with antibiotic treatment show altered gut microbiota. No culturable bacteria were found in preinjury samples from mice given antibiotics for 1 wk before SCI (0 dpi). (B) However, after SCI, antibiotic-resistant bacteria repopulate the gut and overgrow the culture (7 and 14 dpi). This effect remained even after antibiotic treatment was discontinued (21 dpi; n = 6 per time point). Bar, 20 mm. (C–E) Mice receiving antibiotics starting 1 wk before SCI until 14 dpi had impaired locomotor recovery as shown by a reduction in BMS scores (repeated measures two-way ANOVA; *, P < 0.05; n = 6 per group). (D and E) Horizontal bars represent the mean of the sample. (F–H) In addition to impaired functional recovery, antibiotic-induced dysbiosis caused larger lesions to form after SCI (two-way ANOVA; P < 0.0001; **, P < 0.01; n = 6 per group) with less white matter sparing (F and H, EC/neurofilament). (I–L) Mice receiving antibiotics had enhanced intraspinal inflammation after 14 dpi; the CD11b+ CNS macrophage response was enhanced at the lesion epicenter (I and J), as was the total number of infiltrating CD45R+ B cells (K) and CD3+ T cells (L). Student’s t test; *, P < 0.05; n = 5 per group. Error bars represent mean ± SEM. Bars, 200 µm.
When compared with standard recovery profiles in SCI mice, spontaneous locomotor recovery was significantly impaired in mice with antibiotic-induced dysbiosis (Fig. 4, C–E). SCI mice normally recover frequent or consistent plantar stepping and can coordinate use of fore and hind limbs (BMS score = 6). Conversely, SCI mice with antibiotic-induced dysbiosis achieved frequent or consistent plantar stepping but without coordination (BMS score = 5), and at the peak of recovery (28 and 35 dpi) locomotion was impaired by consistent paw rotation during placement and significant trunk instability (Fig. 4, C–E).
Antibiotic-induced dysbiosis also exacerbated lesion pathology and intraspinal inflammation (Fig. 4, F–L). When compared with standard spinal contusion lesions, total lesion volumes were ∼30% larger, with ∼25% less spared white matter at the injury epicenter in SCI mice with antibiotic-induced dysbiosis (Fig. 4, F–H). Also, in these same mice at 14 dpi, the peak of intraspinal inflammation after SCI (Kigerl et al., 2006), the magnitude of the microglia/macrophage reaction (CD11b+ cells) and the total numbers of infiltrating T (CD3+) and B (CD45R+) lymphocytes increased (Fig. 4, I–L).
The antibiotic cocktail used to induce dysbiosis includes aminoglycosides (gentamycin and streptomycin) and quinolones (ciprofloxacin). Both classes of antibiotics are used to treat pneumonia, urinary tract infections, and wound infections; however, they can also cause sickness-like behaviors and act directly on the nervous system to affect neurological or neuromuscular function (Grill and Maganti, 2011). As such, it is possible that the exacerbated pathology and impaired locomotor recovery that occurs in gain-of-function dysbiotic SCI mice were caused by direct neuroactive effects of the antibiotics. Because we delivered antibiotics for 18 d, beginning 1 wk before SCI, it is possible that the spinal cord or brain was damaged before delivering a spinal contusion injury. To explore this possibility, weight loss/gain and spontaneous motor function and exploratory behaviors were quantified. At 0 dpi, mice in the SCI + dysbiosis group had already been dosed with antibiotics for 7 d (starting 7 d preinjury). Preinjury weights in these mice and spontaneous movement (not depicted) were indistinguishable from control SCI mice (i.e., there were no signs of cachexia or sickness-like behaviors). Similarly, patterns of postinjury weight loss/gain tracked together with SCI control mice through 1 mo after injury (Fig. 5 B). Spontaneous movement and overall distance traveled at 7 and 14 dpi, times preceding and corresponding with the onset of antibiotic-induced dysbiosis, respectively, also were indistinguishable between groups (Fig. 5, A and B).
Figure 5.

Antibiotic-induced gut dysbiosis does not cause sickness behavior or increase blood–spinal cord barrier permeability. (A) Accuscan activity monitoring revealed no obvious behavioral changes in mice receiving antibiotics to trigger gut dysbiosis; no differences between groups were found for total distance traveled, movement time, or rest time over a 30-min testing period. P > 0.05; n = 5 per group per time point. Error bars represent mean ± SEM. (C) Delaying the induction of intestinal dysbiosis until 14 dpi, a time when functional recovery begins to plateau, does not impair recovery of function. P > 0.05; n = 6 per group. (B, D, and E) Antibiotics did not affect weight loss after SCI (B) or increase blood–spinal cord barrier permeability (D and E). cc, central canal; bv, blood vessel; AP, area postrema; 4V, fourth ventricle. Bars, 100 μm.
To determine whether antibiotics affect blood–spinal cord barrier permeability, a possible consequence of early antibiotic-induced gut dysbiosis or neurotoxicity, Evans blue dye was injected into intact mice dosed with saline or antibiotics for 7 d. As expected, Evans blue dye can be detected in CNS parenchyma adjacent to regions containing fenestrated capillaries (e.g., circumventricular organs; Fig. 5 D). However, Evans blue dye was never detected in the spinal cord, indicating that antibiotics do not compromise the blood–spinal cord barrier (Fig. 5 E).
Because antibiotics are used to treat bacteremia in SCI patients (Waites et al., 2001; Evans et al., 2009; Dinh et al., 2016), it was important to determine whether antibiotics and the subsequent onset of dysbiosis can cause delayed neurological impairment. To test this, mice received an SCI, and, 2 wk later after locomotor function plateaued, antibiotics were added to the drinking water of half the mice. Surprisingly, delayed induction of gut dysbiosis with antibiotics did not impair locomotor function (Fig. 5 C).
Together, these data indicate that antibiotics induce gut dysbiosis, which exacerbates intraspinal pathology and impairs neurological function. However, the effects of gut dysbiosis on spinal cord structure and function may be time dependent (i.e., limited to the acute postinjury phase). Alternatively, any delayed effects of antibiotic-induced dysbiosis may be overwhelmed by preexisting gut dysbiosis caused by SCI.
Postinjury probiotic treatment improves locomotor recovery after SCI and induces an antiinflammatory immune phenotype in GALT
The natural microbiota contains small amounts of probiotic lactic acid bacteria, and oral delivery of probiotics can reverse the pathological effects of gut dysbiosis. In SCI patients, probiotics have been used to treat urinary tract infections and gastrointestinal distress (Anukam et al., 2009; Wong et al., 2014). However, whether probiotics can confer neuroprotection or ameliorate neurological dysfunction caused by SCI has not been tested.
In an effort to develop a clinically feasible therapeutic protocol, SCI mice received VSL#3 probiotics (5 × 109 bacteria/dose via oral gavage) starting immediately after injury and then again daily until 35 dpi. In VSL#3-treated SCI mice, locomotor recovery was significantly improved relative to vehicle-treated SCI mice (Fig. 6, A and B). VSL#3 increased the frequency of plantar stepping and fore limb–hind limb coordination with concomitant improvements in paw position and trunk stability (Fig. 6, A and B). VSL#3 also reduced lesion volume and axon/myelin pathology at the injury epicenter (Fig. 6, C and D). Smaller lesion volumes in the VSL#3-treated mice correlated with improved behavioral recovery (*, P = 0.03; not depicted).
Figure 6.

Probiotics (VSL#3) confer neuroprotection and improve locomotor recovery after SCI. (A and B) Daily treatment (oral gavage) with VSL#3 improved locomotor recovery in SCI mice (BMS scores [A] and subscores [B]; repeated measure two-way ANOVA; n = 7–8 per group). (C and D) VSL#3 treatment reduced lesion size and spared myelinated axons at the lesion epicenter. (E–H) Probiotic treatment increased the number of CD4+CD25+FoxP3+ T reg cells and the CD11c+ DCs in the MLNs of SCI mice (G and H). (C–H) Student’s t test; n = 7–8 per group. (B, F, and H) Horizontal bars represent the mean of the sample. (I) An independent replicate experiment in a separate cohort of mice (and different batch of VSL#3) verified the ability of daily probiotic treatment to improve locomotor recovery after SCI (repeated measures two-way ANOVA; n = 8 per group). (J and K) Fecal samples collected from these mice show that the proportion of probionts, Lactobacilliales and Bifidobacteriales, increase only in the gut of VSL#3-treated SCI mice (two-way ANOVA; n = 8 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. Error bars represent mean ± SEM.
Probiotics influence mucosal homeostasis through various mechanisms, including regulation of intestinal microbial homeostasis, stabilizing gut epithelial barrier function, and modulation of local and systemic immune responses (Verna and Lucak, 2010; van Baarlen et al., 2013). Probiotics also can normalize or reverse pathological immune responses in GALT, primarily via activation of regulatory T lymphocytes (T reg lymphocytes; Lavasani et al., 2010; Kwon et al., 2013). To determine whether VSL#3 increased T reg lymphocytes, flow cytometry was used to quantify CD4+CD25+FoxP3+ T cells and CD11c+ DCs in MLNs. Both leukocyte subsets increased in MLNs from SCI mice treated with VSL#3 as compared with MLNs from vehicle-treated SCI mice (Fig. 6, E–H).
To increase rigor and document robustness of effect, an independent replicate experiment was completed using a separate cohort of mice and a new lot of VSL#3. These experiments were separated in time by >1 yr. Data in Fig. 6 I show that daily feeding of VSL#3 improves functional recovery after SCI. 16S rRNA sequencing of fecal pellets from SCI mice treated with VSL#3 (or vehicle) revealed transient but significant changes in gut microbe composition. Notably, Bifidobacteriales and Lactobacillales, which are enriched in VSL#3, increased only in VSL#3-treated mice (Fig. 6, J and K).
Together, these data indicate that probiotics may have therapeutic value after SCI. VSL#3, a medical-grade probiotic, can increase the relative abundance of lactic acid–producing bacteria in the gut, overcoming the pathogenic effects of SCI-induced gut dysbiosis on both intraspinal pathology and recovery of neurological function.
Discussion
The gut microflora create a complex ecosystem that is essential for maintenance of human health. Any factors that cause sustained changes in the composition of gut bacteria create a state of gut dysbiosis that can cause or exacerbate disease. Data in this study indicate that traumatic SCI causes profound gut dysbiosis that persists for at least 4 wk after injury, although even longer durations are expected in humans and animals because neurogenic bladder/bowel and gastrointestinal dysfunction are chronic complications of mammalian SCI. In mice with SCI-induced gut dysbiosis, gut permeability and BT are increased. These changes are associated with sustained activation of mucosal immune cells in GALT (e.g., MLNs and PPs). A causal role for gut dysbiosis in the secondary pathophysiology of SCI was proved using an experimental tool analogous to a genetic gain-of-function study. Specifically, an antibiotic cocktail was used to induce gut dysbiosis before SCI. In mice given antibiotics, intraspinal pathology and inflammation were exacerbated, and recovery of locomotor function was significantly impaired relative to normal SCI mice. Importantly, the effects of SCI dysbiosis were time dependent and could be blocked by treating mice with probiotics after SCI. Postinjury probiotics increased the presence of neuroprotective lactic acid–producing bacteria in the gut of SCI mice and activated immune T reg lymphocytes in GALT. Both changes were associated with neuroprotection and improved neurological recovery. Together, these data highlight a previously unappreciated role for the gut–CNS–immune axis in regulating recovery after SCI.
16S rRNA sequencing identified in the SCI mouse gut reciprocal changes in Bacteroidetes and Firmicutes, two major bacterial taxa in both mouse and human microbiota (Eckburg et al., 2005; Krych et al., 2013). The relative abundance of Bacteroidetes (order Bacteroidales) decreased significantly as a function of time after injury and corresponded with a time-dependent increase in Firmicutes (order Clostridiales). A similar reciprocal change in the Bacteroidetes/Firmicutes ratio occurs in obese humans and rodents, and obesity can be transmitted by microbiota transplantation (Ley et al., 2006; Turnbaugh et al., 2006, 2008). Thus, gut microbiota represent novel genetic determinants that, together with changes in diet and lifestyle, contribute to the pathophysiology of obesity (Turnbaugh and Gordon, 2009; Ley, 2010; Tilg and Kaser, 2011; Baothman et al., 2016). Precisely how obesity dysbiosis causes or exacerbates adiposity is not known, but a high Firmicutes/Bacteroidetes ratio was found to increase energy harvest from the diet. Indeed, obese microbiota are more efficient than nonobese microbiota at metabolizing and absorbing dietary substrates (Bäckhed et al., 2004, 2007; Turnbaugh et al., 2006). It is intriguing to speculate that SCI-induced changes in gut ecology contribute to the high incidence of insulin resistance, obesity, and metabolic disease in SCI humans (Manns et al., 2005; Nelson et al., 2007; Maruyama et al., 2008).
By reducing the availability of microbe-derived neuroactive metabolites, including serotonin precursors (Clarke et al., 2013; O’Mahony et al., 2015; Yano et al., 2015) and SCFAs (Cryan and Dinan, 2012; Erny et al., 2015), dysbiosis also could contribute to the onset and progression of intraspinal and systemic pathology after SCI. There is a large literature documenting the health benefits of SCFAs, including butyrate, and recent data show that post-SCI dysbiosis causes a preferential loss of butyrate-producing gut bacteria in humans (Ferrante et al., 2003; Sun et al., 2015; Butchbach et al., 2016; Gungor et al., 2016; Li et al., 2016). This may explain why daily dosing with VSL#3, a medical-grade probiotic consisting of eight distinct lactic acid bacteria, primarily Lactobacillus and Bifidobacterium, is therapeutic in SCI mice. Lactobacillus and Bifidobacterium produce butyrate, other SCFAs, and neurotransmitters (serotonin, dopamine, and γ-aminobutyric acid; Furusawa et al., 2013; El Aidy et al., 2015; O’Mahony et al., 2015). These neurometabolites, produced locally in the gut, can spill into the circulation, bypass the blood–brain barrier, and influence CNS structure and function (Wikoff et al., 2009; Furusawa et al., 2013; Yano et al., 2015; Rothhammer et al., 2016). Probiotics have been shown to reduce neurological dysfunction or pathology caused by experimental autoimmune disease (e.g., experimental autoimmune encephalomyelitis), depression/anxiety, traumatic brain injury, and autism spectrum disorders (Lavasani et al., 2010; Tan et al., 2011; Dinan et al., 2013; Hsiao et al., 2013; Kwon et al., 2013).
The therapeutic effects of VSL#3 may also be attributed to the induction of T reg lymphocytes. T reg lymphocytes, a population of CD4+CD25+ T cells that express the transcription factor FoxP3, play a crucial role in maintaining immune homeostasis. Loss of T reg lymphocyte function is implicated in the onset or progression of multiple sclerosis, rheumatoid arthritis, graft versus host disease, and inflammatory bowel disease. Probiotics, especially those containing Lactobacillus and Bifidobacterium, significantly enhance T reg lymphocyte activity and can reduce inflammatory pathology within and outside the CNS (Lavasani et al., 2010; Bilate and Lafaille, 2012; Kwon et al., 2013). We found that in the MLNs of SCI mice treated with VSL#3, numbers of CD4+CD25+FoxP3+ T reg lymphocytes increased significantly, signifying that the probiotics had the intended effect on activating these neuroprotective mucosal immune cells. Whether enhanced T reg lymphocyte activity was causal in reducing pathology or improving recovery in VSL#3-treated mice is not known. Indeed, any or all of the mechanisms described previously are plausible and could explain how postinjury dysbiosis contributes to pathology after SCI and, similarly, how VSL#3 blocks or reverses these effects.
Data in this study also identify enhanced gut epithelial permeability with BT as mechanisms that explain how systemic inflammation develops after SCI (Gris et al., 2008; Bao et al., 2011; Fleming et al., 2012). In healthy mice, immune responses to gut bacteria are compartmentalized within the mucosal immune system where MLNs act as lymphatic firewalls that block gut bacteria and commensal-loaded DCs from entering the circulation (Macpherson and Smith, 2006; Hill and Artis, 2010). When gut dysbiosis develops, the MLN firewall can fail, allowing gut bacteria to pass into the circulation and then distribute throughout the body (Hill and Artis, 2010; Diehl et al., 2013). After SCI, the early appearance of bacteria in the lung can be explained by the presence of a gut–lung axis (Fig. 1). Lymph draining from the MLNs enters the subclavian vein via the thoracic duct, which then empties directly into the heart and lungs (Deitch, 2010). Thus, the lung would be one of the first organs to come in contact with intestinal bacteria or inflammatory factors derived from a dysbiotic gut. This would explain why lung inflammation develops as early as 2 h after SCI (Gris et al., 2008) and why spontaneous lung infections also develop in experimental stroke models (Meisel et al., 2004).
Compromised liver function may exacerbate the magnitude and effects of BT after SCI. Signs of liver disease (e.g., nonalcoholic steatohepatitis [NASH]) develop soon after SCI, and hepatic inflammation has been implicated as a key mediator of intraspinal inflammation (Campbell et al., 2008; Fleming et al., 2012; Anthony and Couch, 2014; Sauerbeck et al., 2015). Like the MLNs, the liver acts as a firewall, filtering gut microbes that drain from the intestine into the hepatic portal vein (Jenne and Kubes, 2013; Balmer et al., 2014). In SCI animals and humans, liver damage or dysfunction can cause the hepatic filtration capacity to become overwhelmed, allowing gut microbes to bypass the liver and elicit systemic inflammation. NASH is a risk factor for metabolic disease and is associated with altered gut microbiota (Dumas et al., 2006; Abu-Shanab and Quigley, 2010). Thus, the gut microbiota may represent a novel therapeutic target for modulating systemic inflammation and metabolic diseases after SCI. In turn, ameliorating these postinjury sequelae could positively affect intraspinal pathology and functional recovery.
In parallel with the increase in gut permeability and BT, the expression of genes encoding transcription factors or epithelial tight junction proteins that regulate paracellular permeability increased in SCI mice. This could signify the onset of repair responses in damaged or dysfunctional gut epithelia after SCI. As an example, Tcf7l2 is essential for stem cell proliferation and intestinal self-renewal in mice (van Es et al., 2012), and Cdx2 is critical for maintaining the differentiation status of intestinal epithelia (Beck et al., 1999; Hryniuk et al., 2012). Changes in gut permeability, epithelial repair, and GALT immune cell activation coincide and may delineate a therapeutic window for manipulating the adverse effects of gut dysbiosis after SCI. Indeed, initiating antibiotic-induced dysbiosis 2 wk after injury (i.e., after the onset of maximal gut permeability and after mice reach a plateau in functional recovery) did not further impair locomotor function. Also, the lasting neuroprotective effects of VSL#3 do not correspond with chronic increases in gut lactic acid–producing bacteria (Fig. 6). Instead, Lactobacillus and Bifidobacterium increase in the gut early after SCI, but both taxa diminish even though SCI mice continue to gain neurological function. These data suggest that the therapeutic effects of probiotics are established early after SCI; however, many factors could affect the therapeutic window for modulating gut dysbiosis. For example, the magnitude and duration of SCI dysbiosis will vary as a function of an individual’s baseline gut microflora, changes in diet and lifestyle, and injury level. Future studies should consider whether dysbiosis contributes to, exacerbates, or causes neurological deterioration, systemic inflammation (e.g., lungs, kidneys, and liver), immune suppression, deficits in hematopoiesis, metabolic syndrome, cardiovascular disease, or nutritional deficiencies that are common after SCI. No longer should spinal-centric repair approaches dominate SCI research or standards of clinical care for affected individuals.
Materials and methods
Animals and SCI
All surgical and postoperative care procedures were performed in accordance with The Ohio State University Institutional Animal Care and Use Committee. All mice were anesthetized with an i.p. cocktail of 80 mg/kg ketamine/10 mg/kg xylazine, after which a partial laminectomy was performed at T9. All mice received a moderate 75-kilodyne spinal contusion injury using the Infinite Horizons injury device. Postoperatively, animals were hydrated with 2 ml Ringer’s solution (s.c.) for 5 d. Bladders were voided manually at least twice daily for the duration of the study. For all SCI studies, mice were randomly assigned to cages upon arrival at the housing facility. Because gut bacteria can cross-colonize mice living in the same cage, animals within a cage received the same treatment (i.e., vehicle, antibiotic, or probiotic; Figs. 4, 5, 6). Cages were randomly assigned to groups using an online random number calculator (QuickCalcs; GraphPad Software). Experimenters were blinded to treatment group throughout the duration of the study.
Bacterial culture
At 0 (naive), 1, 3, and 7 dpi, mice were euthanized, and blood, lung, liver, spleen, and MLNs were aseptically collected (n = 4 per time point × 2 independent replications). Tissue samples were homogenized in sterile PBS, and 100 µl was plated onto orientation plates (BBL CHROMagar; BD), incubated at 37°C for 48 h, and assessed for bacterial growth. From a separate set of animals, fecal samples were aseptically collected from mice at various times after injury (n = 6 per time point) and homogenized in 500 µl of sterile PBS. Mice were housed according to condition (i.e., vehicle vs. antibiotic treatment) to prevent cross-colonization of gut bacteria between groups. Samples were diluted to 104, and 100 µl of solution was plated onto BBL CHROMagar orientation plates and incubated at 37°C for 48 h.
Bacterial 16S rRNA gene sequencing
All mice used for microbiota sequencing studies (Fig. 2; Fig. 6, J and K; and Tables S1, S2, and S3) were individually housed to prevent cross-colonization of mice in the same cage. Female C57BL/6 mice were used for sample collection at the following time intervals: preinjury (naive mice), 1–3 dpi, 5–7 dpi, 12–14 dpi, 19–21 dpi, and 26–28 dpi. Fecal samples were collected daily over each time interval and then pooled across the time interval for each mouse (e.g., days 1, 2, and 3 after injury were pooled into a single 1–3-dpi sample for each mouse). After cleaning the walls and floor of a plexiglass open field with a chemical disinfectant (Roccal), mice were placed in the open field for 30 min. Fecal pellets were collected into sterile tubes at the end of the 30-min period and were immediately frozen and stored at −80°C.
DNA extraction and 16S rRNA gene sequencing
DNA was extracted from individual mouse fecal samples by the repeated bead beating plus column purification method (Yu and Morrison, 2004). DNA quality was checked by agarose gel and quantified by the Quant-iT dsDNA Assay kit (Thermo Fisher Scientific). DNA were subjected to Miseq sequencing (Illumina) following the 2 × 300 pair ended protocol. The V4–V5 hypervariable regions (primer sets 515F and 806R) of the 16S rRNA gene were amplified and sequenced using the manufacturer’s method/manual.
Sequencing data processing and analysis
Illumina sequencing data from each experiment were processed and analyzed using QIIME (v 1.9.0, scikit-bio; Caporaso et al., 2010). In brief, paired reads were trimmed to remove low-quality bases (Q < 25) and then merged to single contigs using fastq-join (Aronesty, 2011). The barcodes and primers were trimmed from the joined contigs and, after trimming, sequences <248 bp were filtered out. The reads were assigned to species-equivalent operational taxonomic units (OTUs) at 97% similarity by QIIME (pick_open reference_otus.py) using the uclust algorithm (Edgar, 2010) against the Silva_119 released reference sequences (http://www.arb-silva.de/download/archive/qiime/). Chimera sequences were identified using ChimeraSlayer (Haas et al., 2011) against the default Greengenes database (gg_13_08) of QIIME. If an OTU represented <0.005% of the total bacterial sequences (Bokulich et al., 2013) or didn’t have an abundance of at least 1% in at least one sample (Jervis-Bardy et al., 2015), it was filtered out.
Statistics
For the heatmap, the relative abundance of identified OTUs was first standardized by means and standard deviations across samples to obtain Z scores (Wilkinson and Friendly, 2009). The unbiased hierarchical clustering method was used to cluster genus and samples according to the Euclidean distance. The mixed effect model, incorporating repeated measures, was performed to test the difference in relative abundance between the time after injury and the rate of change as a function of time after injury (Verbeke and Molenberghs, 2000). The association between behavior scores (BMS) and relative OTU abundance was evaluated using the Pearson correlation method. The Microarray Software Suite MeV4.9 (http://mev.tm4.org) was used to generate the heatmap, and SAS 9.4 statistical software (SAS, Inc.) was used for data analysis.
Data availability
All sequence data are accessible in the SRA database (accession nos. SRP078936 [Fig. 2 and Table S1], SRP078937 [Fig. 6, J and K; and Table S2], and SRP078940 [Table S3]).
FITC-dextran assay
At 7 dpi, after a 75-kilodyne T9 Infinite Horizons contusion SCI (or T9 laminectomy), female C57BL/6 mice (n = 8 per group) were given 0.6 g/kg FITC-dextran (4 kD; Sigma-Aldrich) dissolved in sterile PBS via gastric gavage (food and water were removed from the cage the previous night). 4 h after FITC-dextran gavage, mice were anesthetized with ketamine/xylazine, and blood was collected via cardiac puncture. Blood was collected into Microtainer SST tubes (BD), and serum was isolated according to the manufacturer’s instructions. Blood was allowed to clot for 30 min, and then samples were centrifuged for 90 s at 6,000 g. Serum was diluted with an equal volume of sterile PBS, and 100 µl of this dilution was added to individual wells of a black 96-well plate. Samples were run in duplicate. Fluorescence was measured on a plate reader (SpectraMax; Molecular Devices) at an excitation of 481 nm and an emission of 524 nm. The concentration of FITC-dextran was calculated from standard curve measurements run on the same plate.
Tight junction PCR array
RNA was isolated from TRIzol homogenates according to the manufacturer’s instructions and was followed by treatment with 1 µg/µl DNase I to eliminate genomic DNA (Invitrogen). 1 µg DNase-treated RNA was primed with random hexamers (1 µM; Applied Biosystems) and reverse transcribed using SuperScript II reverse transcription (Applied Biosystems) in a 20-µl reaction. cDNA was diluted 1:10 with RNase-free sterile water and loaded onto a 384-well plate preloaded with primers for tight junction genes (PrimePCR Pathway Mouse Tight Junctions 384; Bio-Rad Laboratories). PCR reactions were performed with SYBR Green Master Mix (Applied Biosystems) in 10-µl reactions on a 7300 system (Applied Biosystems). Melting point analyses were performed for each reaction to confirm single amplified products. Expression was normalized to GAPDH for each sample.
Flow cytometry
MLNs and PPs were collected after PBS perfusion (n = 3–4 per time point; Fig. 2). Tissues were mashed through a Falcon 40-µm screen (BD) and washed with RPMI medium. Cells were counted, and 106 cells were used for flow cytometry. Cells were first incubated in blocking solution (anti-CD16/32 antibody; eBioscience) for 15 min at 4°C and then stained with the following antibodies from BD: V450-conjugated anti-CD11b, PerCP-Cy5.5–conjugated anti-CD4, PE-conjugated CD8, APC-conjugated CD11c, and FITC-conjugated B220 for 30 min at 4°C. T reg cells were labeled using a mouse T reg cell staining kit (88–8118; eBioscience) as per the manufacturer’s instructions (n = 7–8 per group; Fig. 5). Antibodies include CD4 FITC (RM4-5), CD25 PE (PC61.5), and FoxP3 APC (FJK-16S). After staining was completed, cells were washed and analyzed using a FACSCanto II flow cytometer or an LSRII flow cytometer with FACSDiva software (BD). Subsequent data analyses were completed using FlowJo software (version 9.6; Tree Star). Isotype control antibodies (BD) were matched for fluorochrome and used for cursor/gate placement. Data were normalized to control group (SCI only) samples analyzed on the same day, and data are expressed as a percentage of the SCI-only group. Data are presented as the percentage rather than the absolute cell number because the amount of starting material (MLNs or PPs) varied between animals (i.e., equivalent numbers of MLNs or PPs could not be found or isolated from each mouse).
Cytokine PCR
MLNs and PPs were isolated and homogenized into 500 µl TRIzol (n = 3–5 per time point; Invitrogen). RNA was isolated from the TRIzol homogenates according to the manufacturer’s instructions, followed by treatment with 1 µg/µl DNase I to eliminate genomic DNA (Invitrogen). 1 µg DNase-treated RNA was primed with random hexamers (1 µM; Applied Biosystems) and reverse transcribed using SuperScript II reverse transcription (Applied Biosystems) in a 20-µl reaction. cDNA was stored at −20°C until analysis by PCR. Reactions were performed in triplicate using 1 µl cDNA/reaction. RNA was analyzed using primers specific to the gene of interest and SYBR Green Master Mix in 10-µl reactions. All PCR reactions were performed using a 7300 system, and melting point analyses were performed for each reaction to confirm single amplified products. Expression was normalized to 18 s for each sample. Primer sequences were TNF, forward (5′-GTGATCGGTCCCCAAAGG-3′) and reverse (5′-GGTCTGGGCCATAGAACTGATG-3′); IL-1β, forward (5′-CAGGCTCCGAGATGAACAAC-3′) and reverse (5′-GGTGGAGAGCTTTCAGCTCATAT-3′); IL-10, forward (5′-CAGCCGGGAAGACAATAACTG-3′) and reverse (5′-CCGCAGCTCTAGGAGCATGT-3′); and TGF-β, forward (5′-TGAGTGGCTGTCTTTTGACGTC-3′) and reverse (5′-TTCATGTCATGGATGGTGCC-3′).
Tissue processing
At designated times after injury, mice were anesthetized and then perfused intracardially with 100 ml of 0.1 M PBS, pH 7.4, followed by 100 ml of 4% paraformaldehyde (PF). Perfused spinal cords were immersion fixed in 4% PF for 2 h. Fixed tissues were rinsed and stored overnight at 4°C in 0.2 M phosphate buffer and then cryoprotected in 30% sucrose for 48 h. Spinal cords were blocked into 1-cm segments centered on the impact site and then were embedded in optimum cutting temperature (VWR International). 10-µm serial cross sections were cut through each block using a Microm cryostat (HM 505 E) and then collected on SuperFrost Plus slides (Thermo Fisher Scientific) and stored at −20°C.
Experimentally induced dysbiosis (preinjury) and probiotic treatment
To induce intestinal dysbiosis, mice were started on antibiotic treatment 7 d before SCI. An antibiotic cocktail (2 g/L streptomycin, 0.17 g/L gentamicin, 0.125 mg/L ciprofloxacin, and 1 g/L bacitracin) was given via drinking water until 11 d after injury. Maple syrup was added to both treatment and vehicle (control) groups to make water palatable. After 11 dpi, all mice were returned to normal drinking water (n = 6 per treatment × 2 independent replications). To assess the effects of delayed dysbiosis on functional recovery (Fig. 5), mice were started on antibiotic treatment (2 g/L streptomycin, 0.17 g/L gentamicin, 0.125 mg/L ciprofloxacin, and 1 g/L bacitracin) 14 d after SCI and remained on antibiotics for 3 wk (until 35 dpi; n = 6 per group). Probiotic treatment with VSL#3 (Sigma-Tau Pharmaceuticals) was given daily, starting on the day of SCI and continuing for 35 dpi (n = 7–8 per group; Fig. 6). VSL#3 was reconstituted with sterile water, and a dose of 5 × 109 bacteria was delivered in 100 µl via oral gavage. Vehicle-treated mice also received a daily oral gavage of 100 µl of sterile water. VSL#3 contains eight probiotic strains: Lactobacillus casei, Lactobacillus plantarum, Lactobacillus acidophilus, Lactobacillus delbrueckii subsp_. bulgaricus_, Bifodobacterium longum, Bifidobacterium breve, Bifidobacterium infantis, and Streptococcus salivarius subsp. thermophiles.
Evans blue dye permeability assay
An antibiotic cocktail (2 g/L streptomycin, 0.17 g/L gentamicin, 0.125 mg/L ciprofloxacin, and 1 g/L bacitracin) was given to C57BL/6 mice via drinking water for 7 d. Maple syrup was added to both treatment (n = 3) and vehicle (control; n = 3) groups to make water palatable. On day 7, mice were injected i.p. with a 2% solution of Evans blue dye at 4 ml/kg. 2 h after injecting Evans blue dye, mice were anesthetized and then perfused intracardially with 100 ml of 0.1 M PBS, pH 7.4, followed by 100 ml of 4% PF. Perfused spinal cords were postfixed via immersion in 4% PF for 2 h. Fixed tissues were rinsed and stored overnight at 4°C in 0.2 M phosphate buffer and then cryoprotected in 30% sucrose for 48 h. 10-µm sections were assessed under a fluorescent microscope for the appearance of Evans blue dye, signifying leakage into the tissue parenchyma.
Quantitative analysis of lesion volume and immunohistochemistry
Immunohistochemistry was performed as previously described (Kigerl et al., 2006). Sections were rinsed (0.1 M PBS) and then overlaid with blocking serum for 1 h at room temperature. Primary antibody (CD11b, MCA-74G [AbD Serotec]; CD3, 555273 [BD]; B220/CD45R, MCA 1258G [AbD Serotec]; and NF-H [NFH Aves Labs]) was incubated overnight at 4°C in humidified chambers. Sections were rinsed with 0.1 M PBS and then overlaid with biotinylated conjugated secondary antibodies for 2 h at room temperature. Bound antibody was visualized using Elite-ABC reagent (Vector Laboratories) with diaminobenzidine as a substrate (Vector Laboratories). Immunoperoxidase-labeled sections were dehydrated through ascending alcohols, cleared in histoclear, and then coverslipped with Permount (Thermo Fisher Scientific). Eriochrome cyanine (EC) staining was used to visualize myelin. Frozen sections cut through the rostro-caudal extent of the lesion were incubated in EC for 30 min at 20°C, washed in distilled H2O, and then were differentiated in 5% iron alum and then borax-ferricyanide for 5–10 min. The injury epicenter was defined visually as the spinal cord section with the smallest visible rim of spared myelin/neurofilament. To calculate the lesion volume, digital images of equidistant EC-stained sections spanning the injury epicenter were captured using an Axioplan 2 Imaging microscope (ZEISS). A point grid of the known area was overlaid with random orientation onto printed digital images, and myelin sparing was calculated according to the Cavalieri method using the formula V = T · a/p · n∑p, where T equals the distance between sections, a/p equals the calculated area per point, and n∑p equals the sum of points counted across all sections (Kigerl et al., 2006). Lesion area is expressed as the percent lesion of the entire cross-sectional area. Microglia/macrophage activation was quantified using image analysis as described previously (Kigerl et al., 2006). Manual counts of T and B cells were done at 40× magnification throughout the entire cross section from the epicenter of the lesion site. To ensure that individual cells were not doubly counted on adjacent sections, profile counts were performed on sections separated by 200 µm.
Analysis of locomotor function
Open-field locomotor function was assessed using the BMS for locomotion (Basso et al., 2006) at 1, 3, 7, 10, 14, 21, 28, and 35 dpi. In addition, general indices of locomotion and activity were assessed with an activity monitor (AccuScan Intruments). Mice were recorded using the AccuScan system for 30 min at 7 and 14 dpi.
Online supplemental material
Tables S1, S2, and S3 include raw data obtained from 16S rRNA gene sequencing. Specifically, these tables provide raw sequence counts for each sample, the number of reads subjected to OTU picking, the reads assigned an OTU, and the final number of reads after filtering.
Supplementary Material
Supplemental Materials (PDF)
Acknowledgments
This work was funded by Craig H. Neilsen Foundation grant 164246, the Ray W. Poppleton Endowment, and National Institutes of Health grant R01NS083942.
The authors declare no competing financial interests.
Footnotes
Abbreviations used:
BMS
Basso mouse scale
BT
bacterial translocation
CNS
central nervous system
dpi
day postinjury
EC
eriochrome cyanine
GALT
gut-associated lymphoid tissue
MLN
mesenteric LN
OTU
operational taxonomic unit
PF
paraformaldehyde
PP
Peyer’s patch
rRNA
ribosomal RNA
SCFA
short chain fatty acid
SCI
spinal cord injury
References
- Abu-Shanab A., and Quigley E.M.. 2010. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 7:691–701. 10.1038/nrgastro.2010.172 [DOI] [PubMed] [Google Scholar]
- Ait-Belgnaoui A., Han W., Lamine F., Eutamene H., Fioramonti J., Bueno L., and Theodorou V.. 2006. Lactobacillus farciminis treatment suppresses stress induced visceral hypersensitivity: a possible action through interaction with epithelial cell cytoskeleton contraction. Gut. 55:1090–1094. 10.1136/gut.2005.084194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony D.C., and Couch Y.. 2014. The systemic response to CNS injury. Exp. Neurol. 258:105–111. 10.1016/j.expneurol.2014.03.013 [DOI] [PubMed] [Google Scholar]
- Anukam K.C., Hayes K., Summers K., and Reid G.. 2009. Probiotic Lactobacillus rhamnosus GR-1 and Lactobacillus reuteri RC-14 may help downregulate TNF-Alpha, IL-6, IL-8, IL-10 and IL-12 (p70) in the neurogenic bladder of spinal cord injured patient with urinary tract infections: a two-case study. Adv. Urol. 2009:680363 10.1155/2009/680363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronesty E. 2011. ea-utils: Command-line tools for processing biological sequencing data. Expression Analysis. Available at: https://github.com/ExpressionAnalysis/ea-utils
- Bäckhed F., Ding H., Wang T., Hooper L.V., Koh G.Y., Nagy A., Semenkovich C.F., and Gordon J.I.. 2004. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA. 101:15718–15723. 10.1073/pnas.0407076101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F., Ley R.E., Sonnenburg J.L., Peterson D.A., and Gordon J.I.. 2005. Host-bacterial mutualism in the human intestine. Science. 307:1915–1920. 10.1126/science.1104816 [DOI] [PubMed] [Google Scholar]
- Bäckhed F., Manchester J.K., Semenkovich C.F., and Gordon J.I.. 2007. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA. 104:979–984. 10.1073/pnas.0605374104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey M.T., Dowd S.E., Parry N.M., Galley J.D., Schauer D.B., and Lyte M.. 2010. Stressor exposure disrupts commensal microbial populations in the intestines and leads to increased colonization by Citrobacter rodentium. Infect. Immun. 78:1509–1519. 10.1128/IAI.00862-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey M.T., Dowd S.E., Galley J.D., Hufnagle A.R., Allen R.G., and Lyte M.. 2011. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav. Immun. 25:397–407. 10.1016/j.bbi.2010.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer M.L., Slack E., de Gottardi A., Lawson M.A., Hapfelmeier S., Miele L., Grieco A., Van Vlierberghe H., Fahrner R., Patuto N., et al. . 2014. The liver may act as a firewall mediating mutualism between the host and its gut commensal microbiota. Sci. Transl. Med. 6:237ra66 10.1126/scitranslmed.3008618 [DOI] [PubMed] [Google Scholar]
- Bao F., Brown A., Dekaban G.A., Omana V., and Weaver L.C.. 2011. CD11d integrin blockade reduces the systemic inflammatory response syndrome after spinal cord injury. Exp. Neurol. 231:272–283. 10.1016/j.expneurol.2011.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baothman O.A., Zamzami M.A., Taher I., Abubaker J., and Abu-Farha M.. 2016. The role of gut microbiota in the development of obesity and diabetes. Lipids Health Dis. 15 10.1186/s12944-016-0278-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso D.M., Fisher L.C., Anderson A.J., Jakeman L.B., McTigue D.M., and Popovich P.G.. 2006. Basso mouse scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J. Neurotrauma. 23:635–659. 10.1089/neu.2006.23.635 [DOI] [PubMed] [Google Scholar]
- Beck F., Chawengsaksophak K., Waring P., Playford R.J., and Furness J.B.. 1999. Reprogramming of intestinal differentiation and intercalary regeneration in Cdx2 mutant mice. Proc. Natl. Acad. Sci. USA. 96:7318–7323. 10.1073/pnas.96.13.7318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkaid Y., and Naik S.. 2013. Compartmentalized and systemic control of tissue immunity by commensals. Nat. Immunol. 14:646–653. 10.1038/ni.2604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benakis C., Brea D., Caballero S., Faraco G., Moore J., Murphy M., Sita G., Racchumi G., Ling L., Pamer E.G., et al. . 2016. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat. Med. 22:516–523. 10.1038/nm.4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berer K., Mues M., Koutrolos M., Rasbi Z.A., Boziki M., Johner C., Wekerle H., and Krishnamoorthy G.. 2011. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 479:538–541. 10.1038/nature10554 [DOI] [PubMed] [Google Scholar]
- Bilate A.M., and Lafaille J.J.. 2012. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu. Rev. Immunol. 30:733–758. 10.1146/annurev-immunol-020711-075043 [DOI] [PubMed] [Google Scholar]
- Boekamp J.R., Overholser J.C., and Schubert D.S.. 1996. Depression following a spinal cord injury. Int. J. Psychiatry Med. 26:329–349. 10.2190/CMU6-24AH-E4JG-8KBN [DOI] [PubMed] [Google Scholar]
- Bokulich N.A., Subramanian S., Faith J.J., Gevers D., Gordon J.I., Knight R., Mills D.A., and Caporaso J.G.. 2013. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods. 10:57–59. 10.1038/nmeth.2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandtzaeg P. 1989. Overview of the mucosal immune system. Curr. Top. Microbiol. Immunol. 146:13–25. [DOI] [PubMed] [Google Scholar]
- Butchbach M.E., Lumpkin C.J., Harris A.W., Saieva L., Edwards J.D., Workman E., Simard L.R., Pellizzoni L., and Burghes A.H.. 2016. Protective effects of butyrate-based compounds on a mouse model for spinal muscular atrophy. Exp. Neurol. 279:13–26. 10.1016/j.expneurol.2016.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S.J., Zahid I., Losey P., Law S., Jiang Y., Bilgen M., van Rooijen N., Morsali D., Davis A.E., and Anthony D.C.. 2008. Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology. 55:780–787. 10.1016/j.neuropharm.2008.06.074 [DOI] [PubMed] [Google Scholar]
- Cao S., Feehley T.J., and Nagler C.R.. 2014. The role of commensal bacteria in the regulation of sensitization to food allergens. FEBS Lett. 588:4258–4266. 10.1016/j.febslet.2014.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J.G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F.D., Costello E.K., Fierer N., Peña A.G., Goodrich J.K., Gordon J.I., et al. . 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro G.A., and Arntzen C.J.. 1993. Immunophysiology of the gut: a research frontier for integrative studies of the common mucosal immune system. Am. J. Physiol. 265:G599–G610. [DOI] [PubMed] [Google Scholar]
- Chen G.Y., Shaw M.H., Redondo G., and Núñez G.. 2008. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 68:10060–10067. 10.1158/0008-5472.CAN-08-2061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E.A., and Emmanuel A.V.. 2006. Gastrointestinal symptoms related to autonomic dysfunction following spinal cord injury. Prog. Brain Res. 152:317–333. 10.1016/S0079-6123(05)52021-1 [DOI] [PubMed] [Google Scholar]
- Clark E., Hoare C., Tanianis-Hughes J., Carlson G.L., and Warhurst G.. 2005. Interferon γinduces translocation of commensal Escherichia coli across gut epithelial cells via a lipid raft-mediated process. Gastroenterology. 128:1258–1267. 10.1053/j.gastro.2005.01.046 [DOI] [PubMed] [Google Scholar]
- Clarke G., Grenham S., Scully P., Fitzgerald P., Moloney R.D., Shanahan F., Dinan T.G., and Cryan J.F.. 2013. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry. 18:666–673. 10.1038/mp.2012.77 [DOI] [PubMed] [Google Scholar]
- Clarke G., Stilling R.M., Kennedy P.J., Stanton C., Cryan J.F., and Dinan T.G.. 2014. Minireview: Gut microbiota: the neglected endocrine organ. Mol. Endocrinol. 28:1221–1238. 10.1210/me.2014-1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S.M., Surette M., and Bercik P.. 2012. The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol. 10:735–742. 10.1038/nrmicro2876 [DOI] [PubMed] [Google Scholar]
- Cruz N., Alvarez X., Berg R.D., and Deitch E.A.. 1994. Bacterial translocation across enterocytes: results of a study of bacterial-enterocyte interactions utilizing Caco-2 cells. Shock. 1:67–72. 10.1097/00024382-199401000-00012 [DOI] [PubMed] [Google Scholar]
- Cryan J.F., and Dinan T.G.. 2012. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 13:701–712. 10.1038/nrn3346 [DOI] [PubMed] [Google Scholar]
- Deitch E.A. 2010. Gut lymph and lymphatics: a source of factors leading to organ injury and dysfunction. Ann. NY Acad. Sci. 1207(Suppl 1):E103–E111. 10.1111/j.1749-6632.2010.05713.x [DOI] [PubMed] [Google Scholar]
- de Theije C.G., Koelink P.J., Korte-Bouws G.A., Lopes da Silva S., Korte S.M., Olivier B., Garssen J., and Kraneveld A.D.. 2014a Intestinal inflammation in a murine model of autism spectrum disorders. Brain Behav. Immun. 37:240–247. 10.1016/j.bbi.2013.12.004 [DOI] [PubMed] [Google Scholar]
- de Theije C.G., Wopereis H., Ramadan M., van Eijndthoven T., Lambert J., Knol J., Garssen J., Kraneveld A.D., and Oozeer R.. 2014b Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain Behav. Immun. 37:197–206. 10.1016/j.bbi.2013.12.005 [DOI] [PubMed] [Google Scholar]
- Diehl G.E., Longman R.S., Zhang J.X., Breart B., Galan C., Cuesta A., Schwab S.R., and Littman D.R.. 2013. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX3CR1hi cells. Nature. 494:116–120. 10.1038/nature11809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinan T.G., Stanton C., and Cryan J.F.. 2013. Psychobiotics: a novel class of psychotropic. Biol. Psychiatry. 74:720–726. 10.1016/j.biopsych.2013.05.001 [DOI] [PubMed] [Google Scholar]
- Dinh A., Saliba M., Saadeh D., Bouchand F., Descatha A., Roux A.L., Davido B., Clair B., Denys P., Annane D., et al. . 2016. Blood stream infections due to multidrug-resistant organisms among spinal cord-injured patients, epidemiology over 16 years and associated risks: a comparative study. Spinal Cord. 54:720–725. 10.1038/sc.2015.234 [DOI] [PubMed] [Google Scholar]
- Dumas M.E., Barton R.H., Toye A., Cloarec O., Blancher C., Rothwell A., Fearnside J., Tatoud R., Blanc V., Lindon J.C., et al. . 2006. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA. 103:12511–12516. 10.1073/pnas.0601056103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg P.B., Bik E.M., Bernstein C.N., Purdom E., Dethlefsen L., Sargent M., Gill S.R., Nelson K.E., and Relman D.A.. 2005. Diversity of the human intestinal microbial flora. Science. 308:1635–1638. 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 26:2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- El Aidy S., Dinan T.G., and Cryan J.F.. 2015. Gut microbiota: The conductor in the orchestra of immune-neuroendocrine communication. Clin. Ther. 37:954–967. 10.1016/j.clinthera.2015.03.002 [DOI] [PubMed] [Google Scholar]
- Elliott T.R., and Frank R.G.. 1996. Depression following spinal cord injury. Arch. Phys. Med. Rehabil. 77:816–823. 10.1016/S0003-9993(96)90263-4 [DOI] [PubMed] [Google Scholar]
- Enck P., Greving I., Klosterhalfen S., and Wietek B.. 2006. Upper and lower gastrointestinal motor and sensory dysfunction after human spinal cord injury. Prog. Brain Res. 152:373–384. 10.1016/S0079-6123(05)52025-9 [DOI] [PubMed] [Google Scholar]
- Erny D., Hrabě de Angelis A.L., Jaitin D., Wieghofer P., Staszewski O., David E., Keren-Shaul H., Mahlakoiv T., Jakobshagen K., Buch T., et al. . 2015. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 18:965–977. 10.1038/nn.4030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans C.T., Burns S.P., Chin A., Weaver F.M., and Hershow R.C.. 2009. Predictors and outcomes of antibiotic adequacy for bloodstream infections in veterans with spinal cord injury. Arch. Phys. Med. Rehabil. 90:1364–1370. 10.1016/j.apmr.2009.02.012 [DOI] [PubMed] [Google Scholar]
- Evans C.T., Rogers T.J., Weaver F.M., and Burns S.P.. 2011. Providers’ beliefs and behaviors regarding antibiotic prescribing and antibiotic resistance in persons with spinal cord injury or disorder. J. Spinal Cord Med. 34:16–21. 10.1179/107902610X12886261091794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans C.T., Rogers T.J., Chin A., Johnson S., Smith B., Weaver F.M., and Burns S.P.. 2013. Antibiotic prescribing trends in the emergency department for veterans with spinal cord injury and disorder 2002-2007. J. Spinal Cord Med. 36:492–498. 10.1179/2045772312Y.0000000076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Failli V., Kopp M.A., Gericke C., Martus P., Klingbeil S., Brommer B., Laginha I., Chen Y., DeVivo M.J., Dirnagl U., and Schwab J.M.. 2012. Functional neurological recovery after spinal cord injury is impaired in patients with infections. Brain. 135:3238–3250. 10.1093/brain/aws267 [DOI] [PubMed] [Google Scholar]
- Ferrante R.J., Kubilus J.K., Lee J., Ryu H., Beesen A., Zucker B., Smith K., Kowall N.W., Ratan R.R., Luthi-Carter R., and Hersch S.M.. 2003. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 23:9418–9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming J.C., Bailey C.S., Hundt H., Gurr K.R., Bailey S.I., Cepinskas G., Lawendy A.R., and Badhwar A.. 2012. Remote inflammatory response in liver is dependent on the segmental level of spinal cord injury. J. Trauma Acute Care Surg. 72:1194–1201. 10.1097/TA.0b013e31824d68bd [DOI] [PubMed] [Google Scholar]
- Forsythe P., Bienenstock J., and Kunze W.A.. 2014. Vagal pathways for microbiome-brain-gut axis communication. Adv. Exp. Med. Biol. 817:115–133. 10.1007/978-1-4939-0897-4_5 [DOI] [PubMed] [Google Scholar]
- Foster J.A., and McVey Neufeld K.A.. 2013. Gut–brain axis: how the microbiome influences anxiety and depression. Trends Neurosci. 36:305–312. 10.1016/j.tins.2013.01.005 [DOI] [PubMed] [Google Scholar]
- Furusawa Y., Obata Y., Fukuda S., Endo T.A., Nakato G., Takahashi D., Nakanishi Y., Uetake C., Kato K., Kato T., et al. . 2013. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 504:446–450. 10.1038/nature12721 [DOI] [PubMed] [Google Scholar]
- Gatt M., Reddy B.S., and MacFie J.. 2007. Review article: bacterial translocation in the critically ill - evidence and methods of prevention. Aliment. Pharmacol. Ther. 25:741–757. 10.1111/j.1365-2036.2006.03174.x [DOI] [PubMed] [Google Scholar]
- Gill S.R., Pop M., Deboy R.T., Eckburg P.B., Turnbaugh P.J., Samuel B.S., Gordon J.I., Relman D.A., Fraser-Liggett C.M., and Nelson K.E.. 2006. Metagenomic analysis of the human distal gut microbiome. Science. 312:1355–1359. 10.1126/science.1124234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill M.F., and Maganti R.K.. 2011. Neurotoxic effects associated with antibiotic use: management considerations. Br. J. Clin. Pharmacol. 72:381–393. 10.1111/j.1365-2125.2011.03991.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gris D., Hamilton E.F., and Weaver L.C.. 2008. The systemic inflammatory response after spinal cord injury damages lungs and kidneys. Exp. Neurol. 211:259–270. 10.1016/j.expneurol.2008.01.033 [DOI] [PubMed] [Google Scholar]
- Gungor B., Adiguzel E., Gursel I., Yilmaz B., and Gursel M.. 2016. Intestinal microbiota in patients with spinal cord injury. PLoS One. 11:e0145878 10.1371/journal.pone.0145878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas B.J., Gevers D., Earl A.M., Feldgarden M., Ward D.V., Giannoukos G., Ciulla D., Tabbaa D., Highlander S.K., Sodergren E., et al. Human Microbiome Consortium . 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21:494–504. 10.1101/gr.112730.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X., Fink M.P., Yang R., and Delude R.L.. 2004. Increased iNOS activity is essential for intestinal epithelial tight junction dysfunction in endotoxemic mice. Shock. 21:261–270. 10.1097/01.shk.0000112346.38599.10 [DOI] [PubMed] [Google Scholar]
- Hawrelak J.A., and Myers S.P.. 2004. The causes of intestinal dysbiosis: a review. Altern. Med. Rev. 9:180–197. [PubMed] [Google Scholar]
- Hill D.A., and Artis D.. 2010. Intestinal bacteria and the regulation of immune cell homeostasis. Annu. Rev. Immunol. 28:623–667. 10.1146/annurev-immunol-030409-101330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper L.V., Littman D.R., and Macpherson A.J.. 2012. Interactions between the microbiota and the immune system. Science. 336:1268–1273. 10.1126/science.1223490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryniuk A., Grainger S., Savory J.G., and Lohnes D.. 2012. Cdx function is required for maintenance of intestinal identity in the adult. Dev. Biol. 363:426–437. 10.1016/j.ydbio.2012.01.010 [DOI] [PubMed] [Google Scholar]
- Hsiao E.Y., McBride S.W., Hsien S., Sharon G., Hyde E.R., McCue T., Codelli J.A., Chow J., Reisman S.E., Petrosino J.F., et al. . 2013. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 155:1451–1463. 10.1016/j.cell.2013.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James R., Erler T., and Kazenwadel J.. 1994. Structure of the murine homeobox gene cdx-2. Expression in embryonic and adult intestinal epithelium. J. Biol. Chem. 269:15229–15237. [PubMed] [Google Scholar]
- Jenne C.N., and Kubes P.. 2013. Immune surveillance by the liver. Nat. Immunol. 14:996–1006. 10.1038/ni.2691 [DOI] [PubMed] [Google Scholar]
- Jervis-Bardy J., Leong L.E., Marri S., Smith R.J., Choo J.M., Smith-Vaughan H.C., Nosworthy E., Morris P.S., O’Leary S., Rogers G.B., and Marsh R.L.. 2015. Deriving accurate microbiota profiles from human samples with low bacterial content through post-sequencing processing of Illumina MiSeq data. Microbiome. 3:19 10.1186/s40168-015-0083-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson A.K. 2006. Autonomic dysfunction in spinal cord injury: clinical presentation of symptoms and signs. Prog. Brain Res. 152:1–8. 10.1016/S0079-6123(05)52034-X [DOI] [PubMed] [Google Scholar]
- Kigerl K.A., McGaughy V.M., and Popovich P.G.. 2006. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J. Comp. Neurol. 494:578–594. 10.1002/cne.20827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboziev I., Karlsson F., and Grisham M.B.. 2010. Gut-associated lymphoid tissue, T cell trafficking, and chronic intestinal inflammation. Ann. NY Acad. Sci. 1207(Suppl 1):E86–E93. 10.1111/j.1749-6632.2010.05711.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V., Barker N., Moerer P., van Donselaar E., Huls G., Peters P.J., and Clevers H.. 1998. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat. Genet. 19:379–383. 10.1038/1270 [DOI] [PubMed] [Google Scholar]
- Kriegel M.A., Sefik E., Hill J.A., Wu H.J., Benoist C., and Mathis D.. 2011. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc. Natl. Acad. Sci. USA. 108:11548–11553. 10.1073/pnas.1108924108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krych L., Hansen C.H., Hansen A.K., van den Berg F.W., and Nielsen D.S.. 2013. Quantitatively different, yet qualitatively alike: a meta-analysis of the mouse core gut microbiome with a view towards the human gut microbiome. PLoS One. 8:e62578 10.1371/journal.pone.0062578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.K., Kim G.C., Kim Y., Hwang W., Jash A., Sahoo A., Kim J.E., Nam J.H., and Im S.H.. 2013. Amelioration of experimental autoimmune encephalomyelitis by probiotic mixture is mediated by a shift in T helper cell immune response. Clin. Immunol. 146:217–227. 10.1016/j.clim.2013.01.001 [DOI] [PubMed] [Google Scholar]
- Lavasani S., Dzhambazov B., Nouri M., Fåk F., Buske S., Molin G., Thorlacius H., Alenfall J., Jeppsson B., and Weström B.. 2010. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS One. 5:e9009 10.1371/journal.pone.0009009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.K., Menezes J.S., Umesaki Y., and Mazmanian S.K.. 2011. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA. 108(Suppl 1):4615–4622. 10.1073/pnas.1000082107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R.E. 2010. Obesity and the human microbiome. Curr. Opin. Gastroenterol. 26:5–11. 10.1097/MOG.0b013e328333d751 [DOI] [PubMed] [Google Scholar]
- Ley R.E., Turnbaugh P.J., Klein S., and Gordon J.I.. 2006. Microbial ecology: human gut microbes associated with obesity. Nature. 444:1022–1023. 10.1038/4441022a [DOI] [PubMed] [Google Scholar]
- Li H., Sun J., Wang F., Ding G., Chen W., Fang R., Yao Y., Pang M., Lu Z.Q., and Liu J.. 2016. Sodium butyrate exerts neuroprotective effects by restoring the blood-brain barrier in traumatic brain injury mice. Brain Res. 1642:70–78. 10.1016/j.brainres.2016.03.031 [DOI] [PubMed] [Google Scholar]
- Liu J., An H., Jiang D., Huang W., Zou H., Meng C., and Li H.. 2004. Study of bacterial translocation from gut after paraplegia caused by spinal cord injury in rats. Spine. 29:164–169. 10.1097/01.BRS.0000107234.74249.CD [DOI] [PubMed] [Google Scholar]
- MacFie J. 2004. Current status of bacterial translocation as a cause of surgical sepsis. Br. Med. Bull. 71:1–11. 10.1093/bmb/ldh029 [DOI] [PubMed] [Google Scholar]
- Macpherson A.J., and Smith K.. 2006. Mesenteric lymph nodes at the center of immune anatomy. J. Exp. Med. 203:497–500. 10.1084/jem.20060227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnotti L.J., and Deitch E.A.. 2005. Burns, bacterial translocation, gut barrier function, and failure. J. Burn Care Rehabil. 26:383–391. 10.1097/01.bcr.0000176878.79267.e8 [DOI] [PubMed] [Google Scholar]
- Manns P.J., McCubbin J.A., and Williams D.P.. 2005. Fitness, inflammation, and the metabolic syndrome in men with paraplegia. Arch. Phys. Med. Rehabil. 86:1176–1181. 10.1016/j.apmr.2004.11.020 [DOI] [PubMed] [Google Scholar]
- Maruyama Y., Mizuguchi M., Yaginuma T., Kusaka M., Yoshida H., Yokoyama K., Kasahara Y., and Hosoya T.. 2008. Serum leptin, abdominal obesity and the metabolic syndrome in individuals with chronic spinal cord injury. Spinal Cord. 46:494–499. 10.1038/sj.sc.3102171 [DOI] [PubMed] [Google Scholar]
- Meisel C., Prass K., Braun J., Victorov I., Wolf T., Megow D., Halle E., Volk H.D., Dirnagl U., and Meisel A.. 2004. Preventive antibacterial treatment improves the general medical and neurological outcome in a mouse model of stroke. Stroke. 35:2–6. 10.1161/01.STR.0000109041.89959.4C [DOI] [PubMed] [Google Scholar]
- Murri M., Leiva I., Gomez-Zumaquero J.M., Tinahones F.J., Cardona F., Soriguer F., and Queipo-Ortuño M.I.. 2013. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 11:46 10.1186/1741-7015-11-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M.D., Widman L.M., Abresch R.T., Stanhope K., Havel P.J., Styne D.M., and McDonald C.M.. 2007. Metabolic syndrome in adolescents with spinal cord dysfunction. J. Spinal Cord Med. 30:S127–S139. 10.1080/10790268.2007.11754591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson J.K., Holmes E., Kinross J., Burcelin R., Gibson G., Jia W., and Pettersson S.. 2012. Host-gut microbiota metabolic interactions. Science. 336:1262–1267. 10.1126/science.1223813 [DOI] [PubMed] [Google Scholar]
- O’Mahony S.M., Clarke G., Borre Y.E., Dinan T.G., and Cryan J.F.. 2015. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav. Brain Res. 277:32–48. 10.1016/j.bbr.2014.07.027 [DOI] [PubMed] [Google Scholar]
- Riegger T., Conrad S., Liu K., Schluesener H.J., Adibzahdeh M., and Schwab J.M.. 2007. Spinal cord injury-induced immune depression syndrome (SCI-IDS). Eur. J. Neurosci. 25:1743–1747. 10.1111/j.1460-9568.2007.05447.x [DOI] [PubMed] [Google Scholar]
- Riegger T., Conrad S., Schluesener H.J., Kaps H.P., Badke A., Baron C., Gerstein J., Dietz K., Abdizahdeh M., and Schwab J.M.. 2009. Immune depression syndrome following human spinal cord injury (SCI): a pilot study. Neuroscience. 158:1194–1199. 10.1016/j.neuroscience.2008.08.021 [DOI] [PubMed] [Google Scholar]
- Rothhammer V., Mascanfroni I.D., Bunse L., Takenaka M.C., Kenison J.E., Mayo L., Chao C.C., Patel B., Yan R., Blain M., et al. . 2016. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 22:586–597. 10.1038/nm.4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round J.L., and Mazmanian S.K.. 2009. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9:313–323. 10.1038/nri2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseaux C., Thuru X., Gelot A., Barnich N., Neut C., Dubuquoy L., Dubuquoy C., Merour E., Geboes K., Chamaillard M., et al. . 2007. Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat. Med. 13:35–37. 10.1038/nm1521 [DOI] [PubMed] [Google Scholar]
- Sauerbeck A.D., Laws J.L., Bandaru V.V., Popovich P.G., Haughey N.J., and McTigue D.M.. 2015. Spinal cord injury causes chronic liver pathology in rats. J. Neurotrauma. 32:159–169. 10.1089/neu.2014.3497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage D.C. 1977. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 31:107–133. 10.1146/annurev.mi.31.100177.000543 [DOI] [PubMed] [Google Scholar]
- Silberg D.G., Swain G.P., Suh E.R., and Traber P.G.. 2000. Cdx1 and Cdx2 expression during intestinal development. Gastroenterology. 119:961–971. 10.1053/gast.2000.18142 [DOI] [PubMed] [Google Scholar]
- Soyucen E., Gulcan A., Aktuglu-Zeybek A.C., Onal H., Kiykim E., and Aydin A.. 2014. Differences in the gut microbiota of healthy children and those with type 1 diabetes. Pediatr. Int. 56:336–343. 10.1111/ped.12243 [DOI] [PubMed] [Google Scholar]
- Subramanian V., Meyer B., and Evans G.S.. 1998. The murine Cdx1 gene product localises to the proliferative compartment in the developing and regenerating intestinal epithelium. Differentiation. 64:11–18. 10.1046/j.1432-0436.1998.6410011.x [DOI] [PubMed] [Google Scholar]
- Sun J., Wang F., Li H., Zhang H., Jin J., Chen W., Pang M., Yu J., He Y., Liu J., and Liu C.. 2015. Neuroprotective effect of sodium butyrate against cerebral ischemia/reperfusion injury in mice. BioMed Res. Int. 2015 10.1155/2015/395895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M., Zhu J.C., Du J., Zhang L.M., and Yin H.H.. 2011. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit. Care. 15:R290 10.1186/cc10579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H., and Kaser A.. 2011. Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Invest. 121:2126–2132. 10.1172/JCI58109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillisch K. 2014. The effects of gut microbiota on CNS function in humans. Gut Microbes. 5:404–410. 10.4161/gmic.29232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P.J., and Gordon J.I.. 2009. The core gut microbiome, energy balance and obesity. J. Physiol. 587:4153–4158. 10.1113/jphysiol.2009.174136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P.J., Ley R.E., Mahowald M.A., Magrini V., Mardis E.R., and Gordon J.I.. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 444:1027–1031. 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- Turnbaugh P.J., Bäckhed F., Fulton L., and Gordon J.I.. 2008. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 3:213–223. 10.1016/j.chom.2008.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Baarlen P., Wells J.M., and Kleerebezem M.. 2013. Regulation of intestinal homeostasis and immunity with probiotic lactobacilli. Trends Immunol. 34:208–215. 10.1016/j.it.2013.01.005 [DOI] [PubMed] [Google Scholar]
- van de Wetering M., Sancho E., Verweij C., de Lau W., Oving I., Hurlstone A., van der Horn K., Batlle E., Coudreuse D., Haramis A.P., et al. . 2002. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 111:241–250. 10.1016/S0092-8674(02)01014-0 [DOI] [PubMed] [Google Scholar]
- van Es J.H., Haegebarth A., Kujala P., Itzkovitz S., Koo B.K., Boj S.F., Korving J., van den Born M., van Oudenaarden A., Robine S., and Clevers H.. 2012. A critical role for the Wnt effector Tcf4 in adult intestinal homeostatic self-renewal. Mol. Cell. Biol. 32:1918–1927. 10.1128/MCB.06288-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke G., and Molenberghs G.. 2000. Linear Mixed Models for Longitudinal Data. Springer-Verlag, New York. 570 pp. [Google Scholar]
- Verna E.C., and Lucak S.. 2010. Use of probiotics in gastrointestinal disorders: what to recommend? Therap. Adv. Gastroenterol. 3:307–319. 10.1177/1756283X10373814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waites K.B., Canupp K.C., Chen Y., DeVivo M.J., and Moser S.A.. 2001. Bacteremia after spinal cord injury in initial versus subsequent hospitalizations. J. Spinal Cord Med. 24:96–100. 10.1080/10790268.2001.11753562 [DOI] [PubMed] [Google Scholar]
- Wang Y., and Kasper L.H.. 2014. The role of microbiome in central nervous system disorders. Brain Behav. Immun. 38:1–12. 10.1016/j.bbi.2013.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff W.R., Anfora A.T., Liu J., Schultz P.G., Lesley S.A., Peters E.C., and Siuzdak G.. 2009. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA. 106:3698–3703. 10.1073/pnas.0812874106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson L., and Friendly M.. 2009. The history of the cluster heatmap. Am. Stat. 63:179–184. 10.1198/tas.2009.0033 [DOI] [Google Scholar]
- Winek K., Engel O., Koduah P., Heimesaat M.M., Fischer A., Bereswill S., Dames C., Kershaw O., Gruber A.D., Curato C., et al. . 2016. Depletion of cultivatable gut microbiota by broad-spectrum antibiotic pretreatment worsens outcome after murine stroke. Stroke. 47:1354–1363. 10.1161/STROKEAHA.115.011800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S., Jamous A., O’Driscoll J., Sekhar R., Weldon M., Yau C.Y., Hirani S.P., Grimble G., and Forbes A.. 2014. A Lactobacillus casei Shirota probiotic drink reduces antibiotic-associated diarrhoea in patients with spinal cord injuries: a randomised controlled trial. Br. J. Nutr. 111:672–678. 10.1017/S0007114513002973 [DOI] [PubMed] [Google Scholar]
- Yano J.M., Yu K., Donaldson G.P., Shastri G.G., Ann P., Ma L., Nagler C.R., Ismagilov R.F., Mazmanian S.K., and Hsiao E.Y.. 2015. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 161:264–276. (published erratum appears in Cell. 2015. 163:258) 10.1016/j.cell.2015.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z., and Morrison M.. 2004. Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques. 36:808–812. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials (PDF)
Data Availability Statement
All sequence data are accessible in the SRA database (accession nos. SRP078936 [Fig. 2 and Table S1], SRP078937 [Fig. 6, J and K; and Table S2], and SRP078940 [Table S3]).