RAGE potentiates Aβ-induced perturbation of neuronal function in transgenic mice (original) (raw)
Abstract
Receptor for Advanced Glycation Endproducts (RAGE), a multiligand receptor in the immunoglobulin superfamily, functions as a signal-transducing cell surface acceptor for amyloid-beta peptide (Aβ). In view of increased neuronal expression of RAGE in Alzheimer's disease, a murine model was developed to assess the impact of RAGE in an Aβ-rich environment, employing transgenics (Tgs) with targeted neuronal overexpression of RAGE and mutant amyloid precursor protein (APP). Double Tgs (mutant APP (mAPP)/RAGE) displayed early abnormalities in spatial learning/memory, accompanied by altered activation of markers of synaptic plasticity and exaggerated neuropathologic findings, before such changes were found in mAPP mice. In contrast, Tg mice bearing a dominant-negative RAGE construct targeted to neurons crossed with mAPP animals displayed preservation of spatial learning/memory and diminished neuropathologic changes. These data indicate that RAGE is a cofactor for Aβ-induced neuronal perturbation in a model of Alzheimer's-type pathology, and suggest its potential as a therapeutic target to ameliorate cellular dysfunction.
Keywords: neuroinflammation, neuronal degeneration, NF-κB activation, synaptic plasticity
Introduction
Recent studies have focused attention on amyloid-β peptide (Aβ) as a key pathogenic entity in neuronal dysfunction and neuropathologic changes of Alzheimer's disease (AD). The steady accumulation of Aβ over years in AD may result in progressive displacement of essential neuronal structures. However, early in the disease process, when levels of deposited Aβ are low, mechanisms amplifying and focusing the effects of soluble neurotoxic Aβ assemblies on vulnerable cellular targets could contribute importantly to cellular dysfunction. Several mechanisms could potentially target Aβ to cellular elements. In this regard, cell surface-binding sites are logical to consider for multiple reasons: their capacity to concentrate Aβ at the plasma membrane, where it could directly damage membranes; the possibility that they could function as receptors which engage in intracellular signaling mechanisms; and, their ability to trigger endocytosis, potentially concentrating toxic species in the endolysosomal pathway where disruption of lysosomal integrity could induce severe cellular damage (Yang et al, 1999). As might be expected for a pleiotropic peptide such as Aβ, many cell surface interaction sites have been reported (Snow et al, 1995; Paresce et al, 1996; Yan et al, 1996; Wang et al, 2000). We have focused our attention on Receptor for Advanced Glycation Endproducts (RAGE) for several reasons; its enhanced expression in an Aβ-rich environment (Yan et al, 1995; Lue et al, 2001; Deane et al, 2003); the effectiveness with which it recruits and activates signal transduction mechanisms; and its presence on critical cells (neurons, microglia, and cells of the vessel wall) (Schmidt et al, 2001).
A key test of the possible involvement of RAGE (as well as other candidate cell surface binding sites/receptors) in cellular dysfunction associated with Aβ involves analysis of transgenic (Tg) models in which mutant human amyloid precursor protein (mAPP) and/or presenilins are overexpressed (Duff et al, 1996; Hsiao et al, 1996; Holcomb et al, 1998; Mucke et al, 2000). Since many of these Tg models display only subtle changes in neuronal function and neuropathology, especially at early time points, we began our analysis by developing Tg mice with neuronal overexpression of wild-type (wt) RAGE. Our results demonstrate that double Tg mice, with targeted neuronal expression of transgenes for mAPP and RAGE, display functional and pathologic evidence of neuronal perturbation at early times (3–4 months of age), before the steep rise in cerebral Aβ and plaque formation occur. At later times (14–18 months), activation of microglia/astrocytes was significantly increased in double Tgs, compared with single Tg mAPP mice. Consistent with these data, experiments in which Tg mice bearing a dominant-negative form of RAGE (DN-RAGE) targeted to neurons were crossed with Tg mAPP animals (to generate Tg mAPP/DN-RAGE mice) showed preservation of spatial learning/memory and diminished neuropathologic changes compared with Tg mAPP mice. Our data support the possibility that blockade of neuronal RAGE might have cytoprotective effects, especially with respect to preserving neuronal function early in the disease process.
Results
Generation of Tg RAGE and Tg mAPP/RAGE mice
RAGE is a multiligand receptor which binds Aβ in the nanomolar range (apparent _K_d's of 50–100 nM; Yan et al, 1996). In vitro, interaction of ligands with RAGE-bearing neuronal-like cell lines induces cell stress, characterized by nuclear translocation of NF-κB, and, eventually, loss of cell viability. In view of increased levels of RAGE in AD brain, including neurons (Yan et al, 1996; Lue et al, 2001), we sought to develop a model system in which neuronal expression of RAGE would be exaggerated so that consequences of receptor–ligand engagement could be assessed in an Aβ-rich environment. A transgene bearing full-length wt human RAGE driven by the platelet-derived growth factor (PDGF) B-chain promoter was constructed and used to generate Tg mice, termed Tg RAGE (or Tg wtRAGE), which were backcrossed into the C57BL/6 strain eight times. Southern blotting from two offspring of the same founder demonstrated the presence of the transgene (Supplementary Figure S1a, lanes 1 and 2). This founder was used to establish a line of Tg animals, which has shown normal gross development, reproductive fitness and lifespan (two other Tg RAGE founders were generated, and one of these was used to develop a second line of Tg RAGE mice). Further characterization of the line shown in panel S1a demonstrated increased expression of RAGE transcripts in total RNA harvested from the cerebral cortex of Tg animals compared with nonTg littermates (Supplementary Figure S1b, lanes 1–4). Western blotting of cortical homogenates confirmed the increase in RAGE expression observed in Tg mice, compared with nonTg littermates (Supplementary Figure S1c, lanes 1 and 2). Immunostaining of neocortex from a Tg mouse demonstrated enhanced expression of RAGE antigen, compared with nonTg littermates (Supplementary Figure S1d).
In order to test the effect of neuronal RAGE in an Aβ-rich environment, Tg RAGE mice were crossed with animals expressing a mutant form of human APP (Tg mAPP) also driven by PDGF B-chain promoter, and in the C57BL/6 background (Mucke et al, 2000). The Tg mAPP model was well suited to our strategy of determining whether overexpression of RAGE might enhance/accelerate pathologic changes in the brain, since these animals have been characterized with respect to neuropathologic, biochemical, and physiologic end points (Hsia et al, 1999; Mucke et al, 2000). The cross of Tg RAGE with Tg mAPP mice produced four genotypes, in the expected Mendelian ratios, single Tgs (Tg RAGE, Tg mAPP), double Tgs (Tg mAPP/RAGE), and nonTg littermate controls (Figure 1A).
Figure 1.
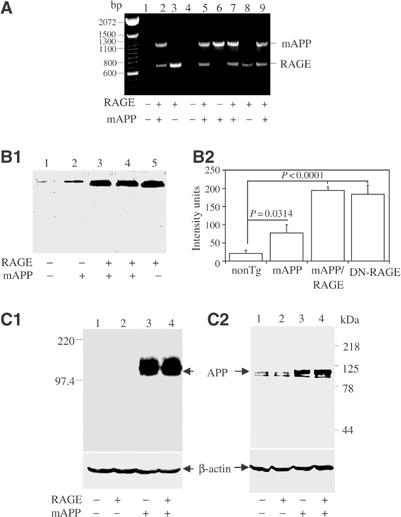
Identification and characterization of Tg mAPP/RAGE mice. (A) PCR analysis of tail DNA from one litter (+, RAGE or mAPP transgene present; −, transgene absent). (B) Western analysis of RAGE expression in the cerebral cortex of the four genotypes of mice. Homogenates of cerebral cortex (100 μg) were subjected to SDS–PAGE (reduced; 10%), followed by immunoblotting with anti-RAGE IgG (6.5 μg/ml) (B1). Densitometric analysis of gels is shown in (B2) (nonTg, _n_=7; Tg RAGE, _n_=5; Tg mAPP, _n_=7; Tg mAPP/RAGE, _n_=4). Results are means±s.e. (C) Western analysis of APP expression in the cerebral cortex of the four genotypes of mice. Homogenates of cortical protein were prepared as above and subjected to SDS–PAGE (reduced; 7.5%), followed by immunoblotting with either antibody 6E10 (C1; 1 μg/ml) or 369W (C2; 1:2000 dilution).
Characterization of transgene expression in Tg mAPP/RAGE mice
Expression of RAGE in each of the four genotypes was studied by immunoblotting of homogenates of cerebral cortex with an antibody that recognized both transgene-derived human and endogenous murine RAGE (Figure 1B). Lowest levels of RAGE were observed in nonTg littermates (lane 1), whereas Tg mAPP animals displayed an increase in RAGE antigen (lane 2). This is consistent with enhanced RAGE expression observed in other situations in which the cellular microenvironment is enriched for RAGE ligands, such as AD brain and diabetic vasculature (Lue et al, 2001; Schmidt et al, 2001; Deane et al, 2003). Tg RAGE mice had higher levels of RAGE antigen (Figure 1B1, lane 5) than Tg mAPP animals (Figure 1B1, lane 2), and double Tgs had RAGE levels comparable to Tg RAGE animals (Figure 1B1, lane 4). Levels of RAGE antigen were ∼3-fold greater in Tg mAPP/RAGE mice than in Tg mAPP mice. These results were obtained with animals at 3–4 months of age, and similar differences in RAGE expression were observed at 14–18 months (not shown).
Expression of the mAPP transgene was also studied in homogenates of cerebral cortex. Immunoblotting with an antibody selective for human APP revealed comparable expression of the transgene in Tg mAPP and Tg mAPP/RAGE (Figure 1C1, lanes 3–4). As expected, no material immunoreactive with murine monoclonal antibody 6E10 (targeted to human Aβ, residues 1–16) was observed in brain extracts from Tg RAGE or nonTg littermates (Figure 1C1, lanes 1–2). In order to assess the changes in total APP, both endogenous murine and Tg human APP, studies were performed with antibody 369W. Immunoblotting demonstrated similar lower levels of APP in nonTg littermates and Tg RAGE mice (Figure 1C2, lanes 1 and 2), and similar higher levels in Tg mAPP and Tg mAPP/RAGE (Figure 1C, lanes 3 and 4). These results were obtained using animals at 3–4 months of age, and levels of APP remained similar up to the oldest age group analyzed (14–18 months).
Cellular stress in Tg mAPP/RAGE mice: activation of NF-κB and inflammation
NF-κB activation has been identified in AD brain (Yan et al, 1996; Kaltschmidt et al, 1997). Based on the hypothesis that a receptor for Aβ should magnify cell stress at early stages of Aβ accumulation, nuclear binding activity for NF-κB was studied by gel shift analysis using nuclear extracts from cerebral cortex with a consensus 32P-labeled NF-κB probe. At 3–4 months of age, Tg mAPP/RAGE mice displayed a strong gel shift band compared with a virtually undetectable signal in nonTg littermates and single Tgs (Figure 2A1). The data shown in Figure 2A2 are representative of results obtained in mice and densitometry of autoradiograms from this larger group confirmed the striking increase in nuclear translocation of NF-κB in double Tgs.
Figure 2.
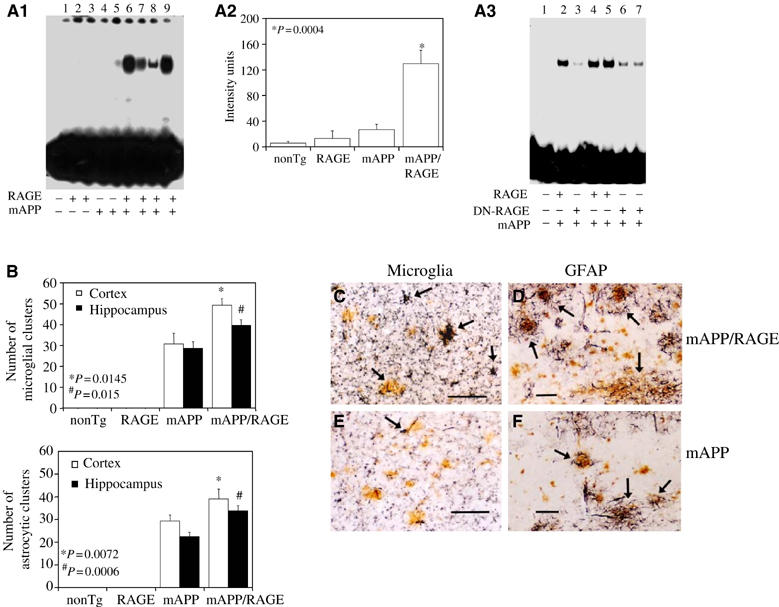
Activation of NF-κB and inflammation in Tg mAPP/RAGE mice. (A) Nuclear translocation of NF-κB was assessed in nuclear extracts from cerebral cortex (10 μg total protein/lane) of mice (age 3–4 months) with 32P-labeled consensus NF-κB probe (A1). The adjacent panel displays image analysis of results from a larger group of mice (4–6 mice/genotype) (A2). Panel A3 displays results of NF-κB gel shift experiments with Tg mAPP/RAGE, Tg mAPP/DN-RAGE, and nonTg littermates. Lane 1 shows migration of free probe. (B) Microgliosis and astrocytosis in brains of Tg mAPP/RAGE mice compared with the other groups. Results of image analysis are shown at 14–18 months in the cerebral cortex and hippocampus (4–6 mice/genotype). (C–F) Representative sections from Tg mAPP/RAGE (C, D) and Tg mAPP (E, F) mice at age 14–18 months to demonstrate microglia (C, E) and astrocytes (D, F). The arrows denote cells reactive with the indicated markers. Scale bar=10 μm. Multiple images similar to those in (C–F) were used to construct the histogram shown in panel (B).
In view of the association of NF-κB activation with inflammation, we considered the possibility that neurons in double Tg mice might incite an inflammatory response, even as early as 3–4 months. Thus, we compared microgliosis and astrocytosis in Tg mAPP/RAGE and Tg mAPP animals. Microglia were visualized immunohistochemically with antibodies to CD11b (complement receptor 3) or phosphotyrosine, and astrocytes were visualized with antibody to glial fibrillary acidic protein (GFAP). The results of image analysis of these sections are shown in the histogram in Figure 2, panel B, and representative micrographs are shown in panels c–f (animals were 14–18 months). Analysis of Tg mAPP/RAGE mice at 3–4 and 7–8 months of age did not reveal significantly increased microgliosis or astrocytosis compared with Tg mAPP, Tg RAGE or nonTg littermates (not shown). However, at 14–18 months, Tg mAPP/RAGE mice had significantly more plaque-associated reactive microglia and astrocytes (_P_⩽0.015; Figure 2B). It is possible that the latter reflect an exaggerated secondary inflammatory response to deposited Aβ.
Perturbation of neuronal function in Tg mAPP/RAGE mice
In view of accentuated cell stress in brains of Tg mAPP/RAGE mice, as reflected by NF-κB activation at early times, we sought to determine whether this would be reflected in impaired spatial learning/memory in the radial arm water maze (Diamond et al, 1999). At 3–4 months of age, nonTg littermates showed strong learning and memory capacity (Figure 3A). Similar results were obtained with Tg RAGE, as well as Tg mAPP mice (Figure 3A). In contrast, Tg mAPP/RAGE mice averaged about four errors by trials 4 or 5, indicative of impaired spatial memory for platform location between trials, as well as during the 30 min delay before trial 5 (Figure 3A). To determine whether combined overexpression of RAGE and mAPP impaired vision, motor coordination or motivation, we also tested mice with a visible platform task. The four genotypes showed no difference in their speed of swimming (Supplementary Figure S2a1) or in the time required to reach the platform in this version of the task (Supplementary Figure S2a2). At 5–6 months, the deficit in learning/memory in Tg mAPP/RAGE mice was worse (≈8 errors by trial 4 or 5) than in younger double Tg animals (Figure 3B). A deficit, though less severe, was also observed in age-matched Tg mAPP mice (≈6 errors by trials 4 or 5), whereas single Tg RAGE mice and nonTg littermates showed intact spatial memory (Figure 3B). Spatial memory impairment persisted in older double Tg animals (12–13 months; Supplementary Figure S3); their performance was worse than single Tg mAPP mice, Tg RAGE mice, and nonTg littermates.
Figure 3.
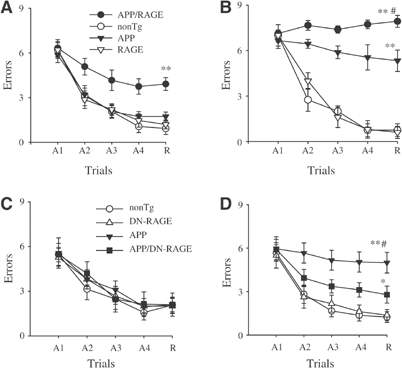
Functional neuronal deficits: spatial learning and memory in Tg mAPP/RAGE and Tg mAPP/DN-RAGE mice. (A, B) Spatial learning and memory was tested in the radial arm water maze at 3–4 (A) and 5–6 months of age (B) in mice of the indicated genotypes (in (A), _n_=7–8 male mice/genotype; in (B), _n_=5 male mice/genotype): Tg mAPP/RAGE (APP/RAGE), nonTg littermate (nonTg), Tg mAPP (APP), and Tg RAGE (RAGE). (A1–A4) denote the acquisition trials, and R denotes the retention trial. In panel A, **P<0.01 Tg mAPP/RAGE compared with nonTg mice (by repeated-measure ANOVA followed by Fisher's protected least significant difference for _post hoc_ comparisons in this and the following graphs). In panel B, **_P_<0.01 Tg mAPP/RAGE and Tg mAPP mice compared with nonTg mice; #_P_<0.01 Tg mAPP/RAGE compared with Tg mAPP mice. ANOVA revealed a main age effect in Tg mAPP/RAGE mice and Tg mAPP mice (_P_<0.05 for both), but not in nonTg and DN-RAGE mice (_P_>0.05 for both). (C, D) Effect of DN-RAGE transgene on spatial learning and memory in Tg mice at 3–4 months (_n_=5–8 male mice/genotype (C)) and 5–6 months (_n_=6–9 male mice/genotype (D)). The following genotypes were tested: nonTg littermate (nonTg), Tg DN-RAGE (DN-RAGE), Tg mAPP (APP), and Tg mAPP/DN-RAGE (APP/DN-RAGE). In panel D, **P<0.01 Tg mAPP mice compared with nonTg mice; #_P_<0.01 Tg mAPP mice compared with Tg mAPP/DN-RAGE mice, and *_P_<0.05 Tg mAPP/DN-RAGE mice compared with nonTg littermates. ANOVA revealed a main age effect in Tg mAPP mice (_P_<0.05) but not in Tg mAPP/DN-RAGE, DN-RAGE, and nonTg mice (_P_>0.05).
These results indicate an early perturbation of learning and memory, consequent to overexpression of RAGE and mAPP. Since cognitive abnormalities in AD are thought to be linked to synaptic dysfunction (Selkoe, 2002), we examined synaptic transmission under basal conditions and during long-term potentiation (LTP). Consistent with previous observations (Hsia et al, 1999), field-excitatory post-synaptic potential (fEPSPs) in the CA1 stratum radiatum revealed impaired basal synaptic transmission (BST) in young (3–5 months) double Tg mice (≈24% decrease in slope of the input–output curve) and Tg mAPP littermates (≈26% decrease) (Figure 4A). BST was further reduced in aged (8–10 months) Tg mAPP/RAGE (64% decrease) and Tg mAPP animals (≈58% decrease) (Figure 4A). Both younger and older Tg mice overexpressing RAGE alone did not show any significant difference in BST, compared with nonTg littermates (Figure 4A). CA1 LTP was normal in young (3–5 months) Tg mice (all genotypes) and nonTg littermates (Figure 4B). However, LTP was most severely affected in 8–10-month-old double Tg mice compared with the other groups (Figure 4C). At 60 min after LTP induction, we observed a 140±9% increase in potentiation in Tg mAPP/RAGE mice, whereas nonTg littermates displayed a 202±13% increase. Single Tg RAGE and Tg mAPP showed levels of LTP comparable to nonTg animals (188±20 and 203±17%, respectively). These findings are consistent with the hypothesis that RAGE-Aβ interaction alters synaptic plasticity. In addition, since BST is affected both in single Tg mAPP and double Tg mice, but LTP is reduced only in the double Tgs, we infer that overexpression of RAGE in an Aβ-rich environment further perturbs synaptic function in the older mice, impairing plasticity.
Figure 4.
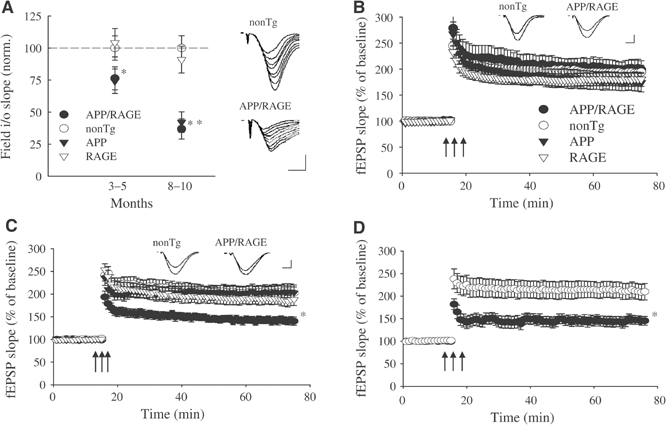
Functional neuronal deficits: decreased LTP in Tg mAPP/RAGE mice. (A) BST was reduced in the CA1 region of slices from 3–5-month-old Tg mAPP/RAGE (APP/RAGE) mice (*P<0.05) and Tg mAPP (APP) littermates compared with nonTg littermates (male animals were used throughout in electrophysiologic studies). A greater reduction in BST was observed in 10-month-old Tg mAPP/RAGE mice and Tg mAPP littermates (**P<0.01). Single Tg RAGE (RAGE) mice showed normal values of BST at both ages. The responsiveness of CA1 cells to increasing afferent fiber stimulation (slope of input–output (i/o) relation) was measured to assess BST strength. Results from 3–5-month-old animals were based on recordings in 16 slices from five male Tg mAPP/RAGE mice, 19 slices from six Tg mAPP mice, 16 slices from six Tg RAGE mice, and 19 slices from seven nonTg mice. Results from 8–10-month-old mice were based on recordings in 12 slices from five Tg mAPP/RAGE mice; 10 slices from four Tg mAPP mice; 15 slices from six Tg RAGE; and 12 slices from six nonTg mice. Insets show representative fEPSP for a nonTg mouse and a Tg mAPP/RAGE mouse, illustrating that far higher stimulation strengths are required to elicit synaptic responses in 10-month-old Tg mAPP/RAGE mice. Scale: 0.2 mV; 5 ms. (B) LTP was normal in the CA1 region of slices from 3–5-month-old Tg mice (all genotypes) and their nonTg littermates. Theta-burst stimulation indicated by the three arrows was delivered after recording a 15-min baseline (_n_=16 slices from five Tg mAPP/RAGE mice, _n_=19 slices from six male Tg mAPP mice, _n_=16 slices from six male Tg RAGE mice, and _n_=19 slices from seven male nonTg mice). Insets show traces taken 1 min before and 60 min after LTP induction on 3–5-month-old nonTg and Tg mAPP/RAGE mice. Scale: 0.1 mV (nonTg), 0.05 mV (Tg mAPP/RAGE mice); 2.5 ms. (C) LTP was reduced in the CA1 region of slices from 8–10-month-old double Tg mAPP/RAGE mice, whereas Tg mAPP, Tg RAGE, and nonTg mice showed normal potentiation (_n_=12 slices from five Tg mAPP/RAGE mice; _n_=10 slices from four Tg mAPP mice; _n_=15 slices from six Tg RAGE mice; and _n_=12 slices from six nonTg mice; *P<0.05). Insets show traces taken 1 min before and 60 min after LTP induction on 8–10-month-old nonTg and Tg mAPP/RAGE mice. Scale: 0.1 mV (nonTg), 0.03 mV (Tg mAPP/RAGE mice); 2.5 ms. (D) LTP was still reduced in double Tg mAPP/RAGE mice after enhancement of the theta-burst strength (_n_=7 slices from four Tg mAPP/RAGE mice; and _n_=8 slices from four nonTg mice; *P<0.05).
To further evaluate the cellular basis of the apparent learning/memory deficit, we tested the concept that impaired plasticity in older (14–18 months) Tg mAPP/RAGE mice might be due to insufficient release and/or entry of Ca2+ during the tetanus. CA1-LTP appears to be due to Ca2+ entrance through postsynaptic NMDA receptors following glutamate release during high-frequency stimulation of the presynaptic cell. Thus, an increase of the tetanus strength should release more neurotransmitter, leading to an increase in the amount of Ca2+ conveyed through NMDA receptors producing normal potentiation. Similar manipulations have previously been used to address this issue (Usdin et al, 1999). Slices from 8–10-month-old Tg mAPP/RAGE mice were subjected to enhanced tetanus consisting of a double number of bursts (see Materials and methods). Under these conditions, the amount of LTP was not increased (Tg mAPP/RAGE, 145±9%; nonTg, 209±18%; Figure 4D). These results suggest that impaired LTP in Tg mAPP/RAGE mice was not likely due to insufficient release and/or entry of Ca2+, but, rather, due to dysfunction at steps occurring after Ca2+ entrance.
Neuropathologic changes in Tg mAPP/RAGE mice
Diminished density of cholinergic fibers and synapses is associated with AD-like pathology (Masliah et al, 1989; Geula, 1998). We sought to determine if such changes might occur in Tg mAPP/RAGE mice by semiquantitatively analyzing acetylcholinesterase (AChE) activity-positive neurites (which predominately identifies cholinergic neurites), using a histochemical method, and synaptophysin (a marker of presynaptic terminals) by immunostaining. Based on the results of the above behavioral studies, we predicted that changes in these neuropathologic properties should be detectable by 3–4 months of age in the double Tgs, and should be progressive. AChE activity was determined histochemically and reported as the area occupied by positively staining neurites in the subiculum (Sb), CA1 and entorhinal cortex (EC). At the 3–4-month time point, the area occupied by AChE-positive neurites in the subiculum was decreased in Tg mAPP/RAGE mice (P<0.001) compared with single Tgs and nonTg littermates (Figure 5A1). This decrease in AChE-positive fibers in double Tgs appeared to be progressive at the 14–18-month time point (Figure 5A2). At this older age, there was also a statistically significant decrease in AChE-positive fibers in Tg mAPP mice, though not in Tg RAGE or nonTg littermates, indicating that loss of cholinergic fibers was not simply a function of age. A representative example of AChE histochemical-stained sections of subiculum from each genotype of mice used for image analysis is shown in Figure 5B. Similar results, with respect to the density of AChE neurites, were obtained in the EC and CA1 (not shown).
Figure 5.
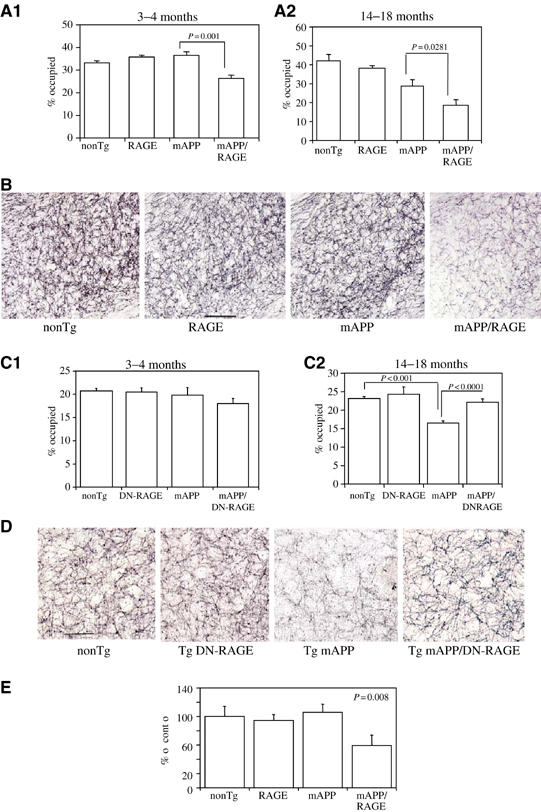
Characterization of Tg mAPP/RAGE and Tg mAPP/DN-RAGE mice: effect on AChE activity. (A1, A2) AChE activity was determined histochemically in the subiculum of mice of the indicated genotype at 3–4 (A1; _n_=4–9/group) and 14–18 months (A2; _n_=5–12/group) of age: nonTg littermate (nonTg), Tg RAGE (RAGE), Tg mAPP (mAPP), and Tg mAPP/RAGE (mAPP/RAGE). The bar graph shows the results of image analysis. (B) Representative images from AChE histochemical staining from the subiculum of mice of the indicated genotype from the experiment in A1. Scale bar=10 μm. (C1, C2) AChE activity was determined as above in the indicated mice (nonTg, Tg DN-RAGE (DN-RAGE), Tg mAPP (mAPP), and Tg mAPP/DN-RAGE (mAPP/DN-RAGE)) at 3–4 months (C1; _n_=7–12/group) and 14–18 months of age (C2; _n_=7–12/group). (D) Representative images of AChE staining for the indicated mice from the experiment in C2. Scale bar=10 μm. (E) Area occupied by synaptophysin immunoreactivity in the stratum lacunosum moleculare of the hippocampus in mice of the indicated genotype at 3–4 months of age (mean±s.e. are shown).
We also assessed the density of presynaptic terminals by determining synaptophysin immunoreactivity. Analysis of presynaptic terminals of defined signal intensity, which reacted with anti-synaptophysin antibody in the stratum lacunosum moleculare of the hippocampus, demonstrated a decrease in the area occupied by these structures in samples from Tg mAPP/RAGE mice, compared with single Tgs (Tg mAPP, Tg RAGE) and nonTg littermates, at 3–4 months of age (Figure 5E). Taken together, these data indicate that neuropathologic changes accompany altered neuronal function at both early and later time points, and appear to be accelerated in Tg mAPP/RAGE mice.
Analysis of Tg mAPP/DN-RAGE mice
In view of subtle behavioral and neuropathologic changes in Tg mAPP mice, we favored starting our analysis of the role of RAGE in Aβ-mediated neuronal stress by overexpressing the receptor and determining if pathologic changes would become more exaggerated. Next, we sought to determine whether RAGE was functioning solely as a cell surface-binding site for Aβ, possibly increasing the local levels of Aβ to interact with other sites, or whether it had a more direct role in transducing a signal important for Aβ-induced cellular perturbation. For these studies, we employed Tg mice overexpressing a DN-RAGE also under control of the PDGF B-chain promoter (Tg DN-RAGE) backcrossed seven times into the C57BL/6 strain. DN-RAGE comprises a truncated form of the receptor with intact extracellular and membrane-spanning portions, but a deleted cytosolic tail. In previous studies, we have found that expression of DN-RAGE, even in cells expressing wt RAGE, blocks RAGE-dependent cellular activation (Hofmann et al, 1999). Tg DN-RAGE animals did not manifest abnormalities with respect to reproductive fitness, development, basic neurologic function or longevity. Southern analysis of tail DNA from two offspring derived from one founder demonstrated the presence of the transgene (Supplementary Figure S1a, lanes 3 and 4). The level of expression of DN-RAGE, at the mRNA (Supplementary Figure S1b, lanes 6 and 8) and antigen levels (Supplementary Figure S1c (lane 3) and d (DN-RAGE)), appeared to be similar to that of the wtRAGE transgene in the line of Tg RAGE mice used in the above studies (see Supplementary Figure S1). DN-RAGE antigen is seen to migrate more rapidly on SDS–PAGE than wtRAGE (Supplementary Figure S1c), in view of deletion of the cytosolic tail in the dominant-negative variant. Tg DN-RAGE mice were crossed with Tg mAPP animals to produce Tg mAPP/DN-RAGE animals, as well as single Tgs (Tg mAPP, Tg DN-RAGE) and nonTg littermates.
First, activation of NF-κB was analyzed in nuclear extracts of cerebral cortex of animals expressing the DN-RAGE transgene at 3–4 months of age (Figure 2A3). In the electrophoretic mobility shift assay for NF-κB, Tg mAPP/DN-RAGE animals (Figure 2A3, lanes 3, 6, and 7) displayed a striking reduction in intensity of the gel shift band compared with Tg mAPP/RAGE mice (Figure 2A3, lanes 2, 4, and 5). These data were consistent with decreased evidence of cell stress in brains of Tg mAPP/DN-RAGE mice compared with Tg mAPP/RAGE mice, and led us to analyze the effect of introducing the DN-RAGE transgene on spatial memory/learning (Figure 3C and D). At 3–4 months of age, Tg mAPP/DN-RAGE, Tg DN-RAGE, Tg mAPP, and nonTg littermates all showed strong learning and memory capacity (they averaged about two errors by trials 4 or 5; Figure 3C). However, at 5–6 months of age, when Tg mAPP showed a significant deficit in spatial memory (about five errors by trials 4 and 5), Tg mAPP/DN-RAGE animals made fewer errors in the same trials (about three errors by trials 4 and 5) (Figure 3D). All groups performed similarly during the visible platform trials with respect to swim speed and time required to reach platform (not shown). Thus, compared with Tg mAPP/RAGE animals, there was preservation of spatial learning/memory at the 3–4-month time point in Tg mAPP/DN-RAGE mice. Furthermore, at 5–6 months of age, neurologic function in Tg mAPP/DN-RAGE mice was better maintained compared with Tg mAPP. Neuropathologic changes, as evaluated by the area occupied by AChE-positive fibers in the subiculum (Figure 5C and D), were virtually completely prevented in Tg mAPP/DN-RAGE mice, compared with Tg mAPP animals, at both 3–4 (Figure 6C1) and 14–18 months of age (Figure 5C2). Representative micrographs of AChE histochemical-stained sections are shown in Figure 5D. In contrast, there was accelerated loss of AChE-positive fibers in Tg mAPP/RAGE mice compared with Tg mAPP animals at these same time points (Figure 5A1–2). Taken together, these data are consistent with the concept that RAGE functions as a signal transduction receptor mediating neuronal dysfunction in the Aβ-rich environment provided by the mAPP transgene.
Figure 6.
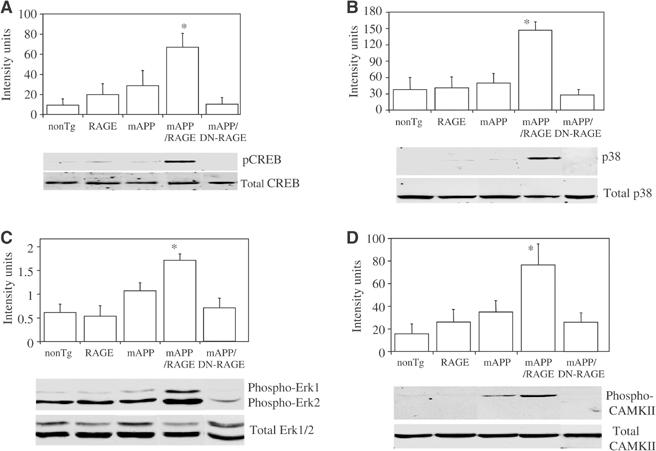
Phosphorylation/activation of CREB (A), MAP kinases (B, C), and CAMKII (D) in Tg mice. (A) Protein extracts were obtained from hippocampii of mice of the indicated genotypes (Tg mAPP, Tg (wt)RAGE, Tg mAPP/RAGE, Tg mAPP/DN-RAGE, nonTg) at 3–4 months of age and subjected to immunoblotting with antibody to total CREB or phospho (p)-CREB. The upper panel displays quantitation of the results with phospho-CREB by image analysis (_n_=5–8/group) and the lower panel shows representative immunoblots. (B, C) The experiment was performed as in (A), except that antibodies for immunoblotting were anti-total p38 and anti-phospho-p38 IgG (B), and anti-total Erk1/2 and anti-phospho-Erk1/2 IgG (C). The upper panel displays image analysis of results with the phospho antibodies and _n_=5–8/group (in the case of Erk, image analysis was performed on p44). In panel D, the experiment was performed with antibodies to total and phospho-CAMKII (CAMKII) and the animals were 14–18 months of age (image analysis shows results of phospho-CAMKII from 5–13 animals/group).
Markers of synaptic plasticity in Tg mAPP/RAGE mice
In view of these findings, we explored the possible mechanisms underlying accelerated neuronal dysfunction in Tg mAPP/RAGE mice and apparent protection in Tg mAPP/DN-RAGE animals, compared with findings in Tg mAPP mice, at the level of markers of synaptic plasticity. We therefore analyzed phosphorylation of CREB and activation of mitogen-activated protein kinases (MAP kinases) and calcium/calmodulin-dependent protein kinase II (CAMKII) in hippocampal extracts. Immunoblotting with antibody to total CREB showed no differences in 3–4-month-old mice from each of the genotypes (Figure 6A, lower panel). However, labeling of the same blot with anti-phospho-CREB (Ser-133) antibody revealed significantly greater levels of phospho-CREB in Tg mAPP/RAGE mice, than in single Tgs (Tg RAGE, Tg mAPP) and nonTg littermates (Figure 6A, lower panel). Furthermore, intensity of the phospho-CREB band was reduced to the background in Tg mAPP/DN-RAGE mice (Figure 6A, lower panel). Densitometric analysis of data from a larger group of animals (_n_=5–8/group) demonstrated the reproducibility of these observations (Figure 6A, upper panel).
Activation of MAP kinases is closely associated with synaptic plasticity (especially extracellular signal-regulated kinases, Erk1/2) and cell stress (p38 and jnk) (Kyriakis and Avruch, 2001; Sweatt, 2001). Furthermore, in previous studies, we have found that activation of MAP kinases occurs in RAGE-bearing tumor cells exposed to ligands of the receptor in vitro (Taguchi et al, 2000). Here, we focused our attention on animals at 3–4 months of age, because of impaired spatial learning/memory at this time, using antibodies to phosphorylated forms of MAP kinases and hippocampal extracts to estimate activation of the related kinases. Phosphorylation of p38 occurred selectively in Tg mAPP/RAGE mice (Figure 6B, lower panel; there was no change in the level of total p38), compared with the other groups, with ≈3.5-fold increased intensity of the immunoreactive band (_P_=0.0016; Figure 6B, upper panel). As in the experiments analyzing phospho-CREB (above), levels of phospho-p38 were reduced to the background in Tg mAPP/DN-RAGE mice, compared with Tg mAPP/RAGE animals (Figure 6B). In terms of Erk1/2, the phosphorylated forms of Erk1/2 (p44/42) were also increased in Tg mAPP/RAGE mice, compared with other groups (and diminished in Tg mAPP/DN-RAGE mice) (Figure 6C, lower panel). Densitometric analysis of gels (focusing on phospho-Erk1) confirmed this impression and indicated that it achieved statistical significance based on analysis of samples from 5–8 animals/group (Figure 6C, upper panel). However, p-Erk1/2 was also increased in Tg mAPP (though this did not achieve statistical significance), and there was a somewhat more variable inter-animal baseline level of p-Erk in nonTg controls. In contrast, similar studies with anti-phospho-JNK IgG showed no change in p-JNK immunoreactive material in samples from the Tg mAPP/RAGE animals, compared with Tg RAGE, Tg mAPP, and nonTg littermates (data not shown).
CAMKII is an important contributor to synaptic plasticity, and, shortly after its activation, becomes autophosphorylated at threonine 286 (Miller and Kennedy, 1986). Using an antibody specific for phospho-CAMKII, levels of the phosphorylated form were found to be increased in hippocampal extracts from Tg mAPP/RAGE mice compared with Tg mAPP animals at 14–18 months of age (Figure 6D, lower panel), whereas they were unchanged at the earlier time point, 3–4 months (not shown). Densitometric analysis (Figure 6D, upper panel) of autoradiograms from a larger group of mice (_n_=5–13 animals/group) confirmed this impression, and demonstrated that the p-CAMKII band was ≈2–3-fold more intense in the double Tg mAPP/RAGE mice than in Tg mAPP animals (_P_=0.014). In contrast, p-CAMKII remained at background levels in Tg mAPP/DN-RAGE animals. Total CAMK II antigen was similar in each of the groups of mice (Figure 6D, lower panel).
Discussion
Previous studies have linked RAGE to evolving immune/inflammatory responses, ranging from colitis to atherosclerosis (Schmidt et al, 2001). In vitro, interaction of Aβ with RAGE-bearing neuronal-like cells leads to activation of NF-κB and expression of macrophage-colony stimulating factor and interleukin-6, suggesting a possible role for neuronal RAGE in inflammation (Yan et al, 1997; Wang et al, 2000). However, despite strong activation of NF-κB in Tg mAPP/RAGE mice by 3–4 months of age, evidence of neuroinflammation, based on microgliosis and astrocytosis, was not significantly increased until much later, 14–18 months. In contrast, more subtle indications of neuronal dysfunction (impaired spatial learning/memory) were the earliest detectable functional defects in Tg mAPP/RAGE mice, and occurred at early times (3–4 months), when overall Aβ levels are low and there are few plaques (Mucke et al, 2000).
There were several findings temporally associated with altered spatial learning/memory in young Tg mAPP/RAGE mice. These findings included decreased area occupied by neurites positive for AChE activity in the subiculum, as well as EC and CA1; decreased area occupied by synaptophysin-positive presynaptic terminals in the striatum lacunosum moleculare of the hippocampus; and altered activation of markers of synaptic plasticity. In each case, our data are consistent with the concept that increased cellular RAGE renders neurons more vulnerable to Aβ-induced dysfunction. Furthermore, decreased density of presynaptic terminals (synaptophysin immunoreactivity) and cholinergic fibers (the predominant population of AChE-positive neurites) are consistent with pathologic findings in AD. Increased phosphorylation of CREB, CAMKII and the MAP kinases (p38, Erk1/2) might not have been expected to be associated with decreased hippocampal function (spatial learning/memory). However, strongly increased basal activation of these kinases and CREB in Tg mAPP/RAGE mice probably has very different functional consequences than transient activation in wt animals associated with stimuli that accompany physiologic processes (such as LTP or fear conditioning) (Kyriakis and Avruch, 2001; Sweatt, 2001). The capacity of RAGE to activate multiple signaling pathways (in addition to those analyzed in the current experiments), including recruitment of NADPH oxidase, p21ras (Wautier et al, 2001), phosphatidylinositol 3-kinase, Rho GTPases (cdc42 and rac), and the Jak/stat pathway (Miller and Kennedy, 1986; Lander et al, 1997; Deora et al, 1998; Huttunen et al, 1999; Taguchi et al, 2000; Huang et al, 2001), indicates that receptor-mediated neuronal stimulation could exert effects on diverse cellular functions.
The role of RAGE as a signal transduction receptor mediating the effects of an Aβ-rich environment is highlighted by our experiments in Tg mAPP/DN-RAGE mice. Although the latter animals expressed about the same levels of RAGE as Tg mAPP/(wt)RAGE animals and the receptor encoded by the DN-RAGE transgene is fully competent with respect to ligand binding, signal transduction from the truncated DN-RAGE variant is blocked (Yan et al, 2000). Tg mAPP/DN-RAGE mice displayed decreased activation of NF-κB and apparent preservation of spatial learning/memory at early times. In addition, there was apparent sparing of AChE-positive neuronal fibers in the subiculum of Tg mAPP/DN-RAGE mice, compared with Tg mAPP animals. These data provide strong support for the concept that RAGE functions as a signaling receptor, rather than solely as a site tethering ligands to the cell surface. Detailed mechanisms linking occupancy of RAGE to NF-κB activation, diminished neuronal/synaptic function, and neuropathologic changes remain to be elucidated. However, in previous studies, we have found that ligand engagement of RAGE results in activation of MAP kinases and NF-κB. RAGE-dependent activation of MAP kinases appears to proceed via an oxidant-senstive mechanism involving p21ras (Lander et al, 1997). Links between IkB phosphorylation and activation of protein kinase A suggest a further mechanism potentially joining the NF-κB and the cAMP pathways, the latter closely linked to synaptic plasticity (Zhong et al, 1997).
The results of our studies add to a growing body of evidence in murine models, suggesting that even before the amyloid deposition in the brain Aβ can disrupt neuronal integrity and functions, including synaptic plasticity (Hsia et al, 1999; Buttini et al, 2002). The current experiments demonstrate that RAGE can act as a cofactor critically magnifying the effects of Aβ on neuronal function.
Materials and methods
Generation of Tg mice and characterization of transgene expression
Selective overexpression of human RAGE in neurons was achieved using the PDGF B chain promoter, in view of its previous success in driving overexpression of mAPP (Mucke et al, 2000). A transgene driving expression of human RAGE or DN-RAGE was similarly placed under control of PDGF B chain promoter. Further description of these Tg mice is provided in Supplementary data.
Immunocytochemical, histochemical and immunoblotting analyses are described in Supplementary data.
NF-κB activation in Tg mice
For gel shift analysis, nuclear extracts (total protein, 10 μg/lane) prepared from mouse cerebral cortex were incubated with 32P-labeled consensus NF-κB probe and subjected to PAGE (5%) as described (Yan et al, 2000).
Behavioral and electrophysiological studies
Behavioral testing and electrophysiological studies were performed as described (Petrone et al, 2003; Trinchese et al, 2004). Details of these experimental methods are described in Supplementary data.
Statistical analysis
Statistical analyses using STATVIEW were performed with a two-way ANOVA for repeated measure, followed by Fisher's protected least significant difference for post hoc comparisons. Results were expressed as mean±standard error mean (s.e.m.).
Supplementary Material
Supplementary Figures
Acknowledgments
This work was supported by grants from the USPHS (AG60901, AG16223, AG17490, 1KO7AG00959, IIRG-00-2076, AG18345), NYS and Surgical Research Fund. We thank Dr R Bourtchouladze for helpful discussion in performing the behavioral experiments and Dr F Battaglia for assistance with the statistical analysis.
References
- Buttini M, Yu GQ, Shockley K, Huang Y, Jones B, Masliah E, Mallory M, Yeo T, Longo FM, Mucke L (2002) Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid β peptides but not plaque formation. J Neurosci 22: 10539–10548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Yan SD, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B (2003) RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat Med 9: 907–913 [DOI] [PubMed] [Google Scholar]
- Deora A, Win T, Vanhaesebroeck B, Lander H (1998) A redox-triggered Ras-effector interaction: recruitment of phosphatidylinositol 3′-kinase to Ras by redox stress. J Biol Chem 273: 29923–29928 [DOI] [PubMed] [Google Scholar]
- Diamond D, Park C, Herman K, Rose G (1999) Exposing rats to a predator impairs spatial working memory in the radial arm water maze. Hippocampus 9: 542–552 [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon M, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S (1996) Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 83: 710–713 [DOI] [PubMed] [Google Scholar]
- Geula C (1998) Abnormalities of neuronal circuitry in Alzheimer's disease: hippocampus and cortical cholinergic innervation. Neurology 51: S18–S29 [DOI] [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97: 889–901 [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon J, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada C-M, Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97–100 [DOI] [PubMed] [Google Scholar]
- Hsia A, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka R, Nicoll R, Mucke L (1999) Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 96: 3228–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274: 99–102 [DOI] [PubMed] [Google Scholar]
- Huang J, Guh J, Chen H, Hung W, Lai Y, Chuang L (2001) Role of RAGE and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J Cell Biochem 81: 102–113 [DOI] [PubMed] [Google Scholar]
- Huttunen H, Fages C, Rauvala H (1999) RAGE-mediated neurite outgrowth and activation of NF-κB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem 274: 19919–19924 [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Volk B, Baeuerle P, Kaltschmidt C (1997) Transcription factor NF-κB is activated in primary neurons by Aβ and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA 94: 2642–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis J, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81: 807–869 [DOI] [PubMed] [Google Scholar]
- Lander H, Tauras J, Ogiste J, Moss R, Schmidt AM (1997) Activation of RAGE triggers a MAP kinase pathway regulated by oxidant stress. J Biol Chem 272: 17810–17814 [DOI] [PubMed] [Google Scholar]
- Lue LF, Walker D, Brachova L, Rogers J, Shen Y, Schmidt AM, Stern DM, Yan SD (2001) Involvement of RAGE–microglial interactions in Alzheimer's disease: in vivo and in vitro studies. Exp Neurol 171: 29–45 [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry R, DeTeresa R, Hansen L (1989) Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett 103: 234–239 [DOI] [PubMed] [Google Scholar]
- Miller S, Kennedy M (1986) Regulation of brain type II calcium calmodulin-dependent protein kinase by autophosphorylation by calcium-triggered molecular switch. Cell 44: 861–870 [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L (2000) High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050–4058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paresce D, Ghosh R, Maxfield F (1996) Microglial cells internalize aggregates of the Alzheimer's disease amyloid β-protein via a scavenger receptor. Neuron 17: 553–565 [DOI] [PubMed] [Google Scholar]
- Petrone A, Battaglia F, Wang C, Dusa A, Su J, Zagzag D, Bianchi R, Casaccia-Bonnefil P, Arancio O, Sap J (2003) Receptor protein tyrosine phosphatase alpha (RPTPα) is essential for hippocampal neuronal migration, long-term potentiation, and working memory. EMBO J 22: 4121–4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt AM, Yan SD, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108: 949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298: 789–791 [DOI] [PubMed] [Google Scholar]
- Snow A, Kinsella M, Parks E, Sekiguchi R, Miller J, Kimata K, Wight T (1995) Differential binding of vascular cell-derived proteoglycans (perlecan, biglycan, decorin, and versican) to the beta-amyloid protein of Alzheimer's disease. Arch Biochem Biophys 320: 84–95 [DOI] [PubMed] [Google Scholar]
- Sweatt J (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76: 1–10 [DOI] [PubMed] [Google Scholar]
- Taguchi A, DelToro G, Canet A, Lee D, Tanji N, Lu Y, Ingram M, Lalla E, Hofmann M, Fu J, Kislinger T, Lu A, Stern DM, Schmidt A-M (2000) Blockade of RAGE/amphoterin suppresses tumor growth and metastases. Nature 405: 354–360 [DOI] [PubMed] [Google Scholar]
- Trinchese F, Liu S, Battaglia F, Walter S, Mathews P, Arancio O (2004) Progressive age-related development of Alzheimer-like pathology in APP/PS1 mice. Ann Neurol 55: 801–814 [DOI] [PubMed] [Google Scholar]
- Usdin M, Shelbourne P, Myers R, Madison D (1999) Impaired synaptic plasticity in mice carrying the Huntington's disease mutation. Hum Mol Genet 8: 839–846 [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee D, D'Andrea M, Peterson P, Shank R, Reitz A (2000) Aβ(1–42) binds to α7 nicotinic acetylcholine receptor with high affinity. J Biol Chem 275: 5626–5632 [DOI] [PubMed] [Google Scholar]
- Wautier MP, Chappey O, Corda S, Stern D, Schmidt A-M, Wautier JL (2001) Activation of NADPH oxidase by AGEs links oxidant stress to altered gene expression via RAGE. Am J Physiol 280: E685–E694 [DOI] [PubMed] [Google Scholar]
- Yan SD, Chen X, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern DM, Schmidt A-M (1996) RAGE and Aβ peptide neurotoxicity in Alzheimer's disease. Nature 382: 685–691 [DOI] [PubMed] [Google Scholar]
- Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith M, Perry G, Godman G, Nawroth P, Zweier J, Stern DM (1995) Nonenzymatically glycated tau in Alzheimer's disease induces neuronal oxidant stress resulting in cytokine gene expression and release of Aβ. Nat Med 1: 693–699 [DOI] [PubMed] [Google Scholar]
- Yan SD, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, Rajavashisth T, Chen X, Godman GC, Stern D, Schmidt AM (1997) RAGE-Aβ interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci USA 94: 5296–5301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Zhu H, Zhu A, Golabek A, Roher A, Yu J, Soto C, Schmidt AM, Stern DM, Kindy M (2000) Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med 6: 643–651 [DOI] [PubMed] [Google Scholar]
- Yang A, Chandswangbhuvana D, Shu T, Henschen A, Glabe C (1999) Intracellular accumulation of insoluble, newly synthesized Aβ1–42 in amyloid precursor protein-transfected cells that have been treated with Aβ1–42. J Biol Chem 274: 20650–20656 [DOI] [PubMed] [Google Scholar]
- Zhong H, SuYang H, Bromage H, Tempst P, Ghosh S (1997) The transcriptional activity of NF-κB is regulated by IkB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 89: 413–424 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures