Scaffolding by ERK3 regulates MK5 in development (original) (raw)
Abstract
Extracellular-regulated kinase 3 (ERK3, MAPK6) is an atypical member of the ERKs, lacking the threonine and tyrosine residues in the activation loop, carrying a unique C-terminal extension and being mainly regulated by its own protein stability and/or by autophosphorylation. Here we show that ERK3 specifically interacts with the MAPK-activated protein kinase 5 (MK5 or PRAK) in vitro and in vivo. Expression of ERK3 in mammalian cells leads to nuclear-cytoplasmic translocation and activation of MK5 and to phosphorylation of both ERK3 and MK5. Remarkably, activation of MK5 is independent of ERK3 enzymatic activity, but depends on its own catalytic activity as well as on a region in the C-terminal extension of ERK3. In mouse embryonic development, mRNA expression patterns of ERK3 and MK5 suggest spatiotemporal coexpression of both kinases. Deletion of MK5 leads to strong reduction of ERK3 protein levels and embryonic lethality at about stage E11, where ERK3 expression in wild-type mice is maximum, indicating a role of this signalling module in development.
Keywords: MAP kinases, MAPKAP kinases, nucleocytoplasmic translocation, protein phosphorylation
Introduction
Besides the well-known members of the extracellular-regulated mitogen-activated protein kinases (MAPKs), ERK1 and ERK2, which are central members of this MAPK pathway (Boulton et al, 1991; Johnson and Lapadat, 2002), several other ERK-related genes and corresponding proteins were identified such as ERK3 (MAPK6) (Zhu et al, 1994; Meloche et al, 1996), ERK4 (ERK3-related, ERK3β, p63 MAPK, MAPK4) (Gonzalez et al, 1992), ERK5 (BMK) (Zhou et al, 1995), ERK7 (Abe et al, 1999) and ERK8 (Abe et al, 2002). Of these, only ERK3 and ERK4 lack the characteristic activation loop signature TEY and instead display a SEG motif. The serine residue within this motif (S189 in ERK3) can be phosphorylated by a partially purified and characterised ERK3 kinase (Cheng et al, 1996) and ERK3 itself displays kinase activity against in vitro substrates such as myelin basic protein or histone H1 (Zhu et al, 1994). A unique feature of ERK3 is its C-terminal domain of about 400 amino acids with no homology to other proteins, which is only partially present in ERK4 (170 amino acids). So far, relevant stimuli, activators and in vivo substrates for ERK3 and ERK4 have not been identified.
Recently, it became clear that ERK3, unlike other ERKs, is an unstable protein containing two destabilisation regions in the N-terminal kinase lobe, which is constitutively degraded by the proteasome pathway in proliferating cells (Coulombe et al, 2003). During differentiation, ERK3 is stabilised by an unknown mechanism and its intracellular accumulation is paralleled by cell cycle arrest in G1 (Coulombe et al, 2003). Interestingly, ERK3 carries a nuclear export signal (NES), which interacts with exportin 1, and nucleocytoplasmic shuttling of ERK3 is required for its negative regulatory effect on cell cycle progression (Julien et al, 2003). During mouse embryogenesis, ERK3 mRNA shows a sharp peak of strong expression at embryonic day (E)11, while only weak expression can be detected at E13 and E15 (Turgeon et al, 2000).
Downstream to MAPKs, there exists a family of MAPK-activated protein kinases (MKs; for a recent review, see Roux and Blenis, 2004). Based on their sequence homologies, the MKs can be classified into five subgroups. Besides the RSK, MSK, MNK and MK(MAPKAPK)2/3 subfamilies, the kinase MK5 is regarded the only member of the fifth subgroup (Roux and Blenis, 2004). MK5 displays about 40% amino-acid sequence identity with the p38 MAPK-activated kinases MK2 and MK3 (New et al, 1998; Ni et al, 1998; Underwood et al, 2003). Similar to MK2 and MK3, MK5 carries a nuclear localisation signal (NLS) C-terminal to its kinase domain, which causes nuclear accumulation of the kinase in resting cells (Seternes et al, 2002; New et al, 2003). Besides the regulatory phosphorylation site at the activation loop, MK2 and MK3 possess another regulatory phosphorylation site in the hinge region between the catalytic domain and the C-terminus (Stokoe et al, 1992; Ben-Levy et al, 1995; Engel et al, 1995). Phosphorylation of this site regulates activity of a C-terminal NES and triggers nuclear-cytoplasmic translocation of MK2 and MK3 (Ben-Levy et al, 1998; Engel et al, 1998; Neininger et al, 2001). Since such regulatory phosphorylation site is not present in the C-terminus of MK5, this kind of coupling phosphorylation-dependent regulation of activity and localisation of MK5 is unlikely. Although MK5 was first described as p38-regulated/activated protein kinase (PRAK) (New et al, 1998), recent data challenged this finding, because endogenous MK5 activity is not significantly increased by stimulation of the p38 MAPK cascade (Shi et al, 2003). In addition, MK5 shows only weak interaction and no stabilisation of endogenous p38 MAPK, as MK2 did (Shi et al, 2003). MK5 displays in vitro activity against the small heat shock proteins Hsp25 (mouse) and Hsp27 (human), but in MK5-deficient cells no reduction of Hsp25 phosphorylation in response to stress could be detected, indicating that other protein kinases such as MK2 and MK3 are responsible for stress-induced phosphorylation of these proteins in vivo (Shi et al, 2003). Hence, as for ERK3, stimuli, activators and physiological relevant substrates for MK5 remain to be identified.
Results
Specific interaction between MK5 and ERK3
Since the mechanism of activation of MK5 is unclear, we were interested in identification of MK5 interacting partners. A two-hybrid screen using two different prey libraries, mouse 11-day-old embryo and adult mouse brain, was applied. Mouse MK5 and the structurally related kinase MK2 (Engel et al, 1993) were used as baits and analysed in more than 107 mating events. The MK2 screen led to the identification of p38 MAPKα as prey in nine out of about 200 positive clones, but none of the 52 positive clones of the MK5 screen overlapped with the positive clones from the MK2 experiment. Interestingly, three independent clones contained ERK3-Gal 4 fusion proteins as interacting molecules for MK5. The specific interaction of MK5 with ERK3 but not with p38 MAPK was confirmed by selection of yeast growth on medium lacking leucine, tryptophan, histidine and adenine. After several days, colony growth at the selection medium was observed only for MK2–p38 and MK5–ERK3 matings. However, after 2 weeks of incubation at 30°C, we could also detect colonies for the MK2–ERK3 mating (Figure 1A). To compare semiquantitatively p38 and ERK3 interactions with MK5, we used a luminometric β-galactosidase assay for quantification of yeast two-hybrid interactions (Figure 1B). While p38–MK5 interaction leads to about a two-fold increase in β-galactosidase activity compared with the negative control, ERK3–MK5 interaction is monitored by a more than 10-fold increase in enzyme activity, indicating a significantly higher affinity of ERK3 for MK5. By in vitro pull-down of GST-MK5, GST-MK2 and, as control, GST alone using nickel–agarose bound with recombinant hexahistidine-tagged ERK3 (Figure 1C) as well as by probing interaction in HEK293 cells cotransfected with GST-ERK3 or His-ERK3 and MK2- or MK5- tandem affinity purification constructs (Shi et al, 2003) (Figure 1D), we could further demonstrate specific interaction between MK5 and ERK3. For analysing whether endogenous MK5 interacts with ERK3, we transfected mouse embryonic fibroblasts (MEFs) and, as a negative control, MEFs derived from MK5-deficient mice with a biotinylatable tag-fused ERK3 protein. After expression and in vivo biotinylation, cells were lysed, the ERK3 fusion protein together with proteins bound was purified using streptavidin beads and endogenous MK5 protein could be detected by Western blot (Figure 1E). Finally, we analysed whether endogenous ERK3 can be co-immunoprecipitated from MEF lysates together with endogenous MK5 (Figure 1F). For wild-type (WT) and MK2-deficient cells, ERK3 is detectable in Western blot of the MK5 immunoprecipitate, while in the negative control, MK5-deficient cells, no ERK3 could be detected, indicating a complex of endogenous MK5 and ERK3 in vivo. Interestingly, in MK5-deficient MEFs, a significant reduction in ERK3 level could also be detected (see below).
Figure 1.
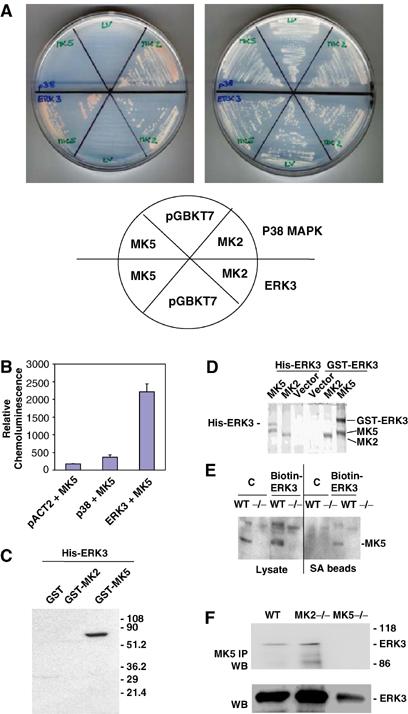
Detection of specific MK5–ERK3 interaction. (A) pACT2-p38 or -ERK3 in strain Y187 (MATα) was mated with pGBKT7-, pGBKT7-MK5- or -MK2- transformed AH109 (MATa) and plated on an SDΔAHLT plate (left) and an SDΔLT plate (right). The positions of the different matings are schematically shown. (B) Quantification of β-galactosidase activity in yeast two-hybrid system. (C) Pull-down of GST-MK5, GST-MK2 and GST alone from the Escherichia coli lysate using recombinant hexahistidine-tagged ERK3 bound to nickel–agarose. Co-purification of GST or GST fusion proteins is detected by Western blot using anti-GST antibodies. (D) MK5–ERK3 interaction in 293 cells transfected with plasmids coding for expression of two differentially tagged forms of ERK3, GST-ERK3 and His-ERK3, and cotransfected with MK2 and MK5 tandem affinity purification constructs (Shi et al, 2003). Coomassie protein stain of tandem affinity-purified proteins demonstrates GST-ERK3 and His-ERK3 as specific binding partners for MK5. (E) Binding of endogenous MK5 to a biotinylated ERK3 fusion protein in MEFs (WT). MK5, which can be identified by its absence in the MK5 knockout cells (−/−), can be purified bound to the _in vivo_-biotinylated ERK fusion protein but not to the control where the biotinylated 72-amino-acid peptide (derived from the C-terminus amino acids 524–595) of the Klebsiella pneumoniae oxalacetate decarboxylase was fused to β-galactosidase (Schwarz et al, 1988). A similar experiment was carried out also for WT and MK2-deficient MEFs, but no endogenous MK2 can be detected bound to the biotinylated ERK3 fusion protein (data not shown). (F) Co-immunoprecipitation of endogenous ERK3 with endogenous MK5 from MEFs. MK5 was immunoprecipitated from cell lysates and IP was applied to SDS–PAGE and Western blot using ERK3 antibodies (upper panel). Whole cell lysates were analysed in lower panel.
Coexpression of ERK3 causes cytoplasmic translocation of nuclear MK5
ERK3 contains a functional NES (Julien et al, 2003), while MK5 carries an NLS and displays nuclear localisation in resting cells (Seternes et al, 2002). To test whether physiologically relevant interaction between MK5 and ERK3 is prevented by different localisation, we coexpressed tagged versions of both proteins and analysed their subcellular localisation in HEK293 cells, which do not express endogenous ERK3 mRNA or protein to a detectable level. GFP-ERK3 localisation is almost exclusively cytoplasmic and not changed by coexpression of MK5 or MK2 (Figure 2A). Remarkably, coexpression of hexahistidine-tagged ERK3 completely changes GFP-MK5 nuclear localisation to cytoplasmic, while localisation of MK2 remains nuclear (Figure 2A). As a control, expression of p38 MAPKα could change GFP-MK2's localisation to cytoplasmic but leaves MK5 in the nucleus (Figure 2A). Similarly, when yellow fluorescent protein (YFP)-ERK3 and cyan fluorescent protein (CFP)-MK5 or CFP-MK2 were cotransfected, a specific translocation of MK5 and not of MK2 to the cytoplasm in ERK3-expressing cells is detected (Figure 2B). In cells cotransfected with lower amounts of plasmids (Supplementary Figure 1), the unstable ERK3 protein could hardly be detected after 24 h, while MK5 is still detectable and, as expected for stoichiometric deficiency of ERK3, mainly in the nucleus. Subcellular localisation of both ERK3 and MK5 in HEK293 cells is fusion tag- and cell type-independent, since an HA-tagged version of MK5 shows the same translocation as GFP-MK5 and similar localisation was observed also in HeLa cells (Supplementary Figure 2). While the p38 MAPKα,β inhibitor SB203580 (Lee et al, 1994) used in a 10 μM concentration and the export inhibitor leptomycin B (200 nM) inhibit MK2's translocation (Engel et al, 1998)), these agents are not able to inhibit ERK3-dependent translocation of MK5 (Supplementary Figure 3). Obviously, a specific interaction between MK5 and ERK3 in vivo leads to translocation of MK5 by cytoplasmic anchoring of MK5 by ERK3.
Figure 2.
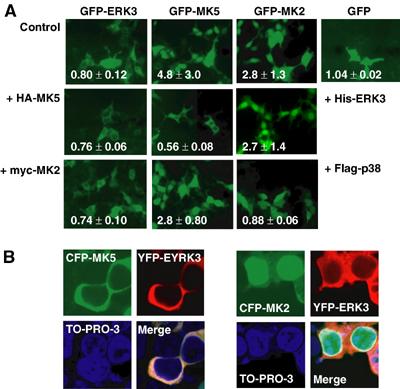
Coexpression of ERK3 changes subcellular localisation of MK5. (A) Localisation of GFP-tagged ERK3 and MK5 analysed by fluorescence microscopy and characterised quantitatively by the nuclear/cytoplasmic localisation index i (lower left in each image; i<1 stands for predominantly cytoplasmic localisation, while _i_>1 indicates nuclear accumulation). Cytoplasmic localisation of GFP-ERK3 is not significantly changed by coexpression of epitope-tagged versions of MK2 (myc-MK2) or MK5 (HA-MK5) in HEK293 cells. Nuclear localisation of GFP-MK5 but not of GFP-MK2 is changed to cytoplasmic by coexpression of His-tagged ERK3. Flag-tagged p38 MAPK completely changes localisation of GFP-MK2 but only slightly shifts localisation of GFP-MK5 when coexpressed. GFP is equally distributed in the cells. (B) YFP-ERK3 and CFP-MK5 or CFP-MK2 were cotransfected and detected in parallel. Nuclei are stained using TO-PRO-3 (Molecular Probes, Invitrogen).
Coexpression of ERK3 leads to phosphorylation and activation of MK5
We were then interested in whether ERK3 regulates enzymatic activity of MK5. So far, no stimulus that activates ERK3 has been described and it is assumed that ERK3 activity is mainly regulated by degradation-dependent changes of its level of expression during development (Coulombe et al, 2003). Furthermore, no MK5-specific cellular substrate has been identified so far, while small heat shock protein Hsp25 is a suitable substrate for MK5 in vitro (Shi et al, 2003). We expressed His-ERK3 together with a GFP-tagged MK5 in HEK293 cells and analysed the activity of MK5 by immunoprecipitation (IP) kinase assay using anti-GFP antibodies and Hsp25 as substrate. As controls, we analysed coexpression of Flag-tagged p38 MAPKα and GFP-MK5, and treated the cells with sodium arsenite, a strong activator of the p38 MAPK cascade (Rouse et al, 1994). Coexpression of ERK3 leads to more than 10-fold increase of GFP-MK5 activity, which is not further stimulated by arsenite treatment and could not be inhibited by 10 μM SB203580 (Figure 3A and B). In contrast, there is no significant stimulation of MK5 activity by coexpression of p38 MAPK alone, and only weaker (about three-fold) stimulation of this activity as a result of coexpression of p38 MAPK and arsenite stimulation, which is completely SB203580-dependent. There is also no significant arsenite stimulation of MK5 in cells expressing only the endogenous p38 MAPK (control in Figure 3A and B; Shi et al, 2003). Phosphorylation of GFP-MK5 parallels its activity towards Hsp25 and, in cells coexpressing ERK3, also phosphorylation of ERK3. Since overexpression of GFP-MK5 in HEK293 cells could provide nonphysiological results by titrating out other signalling components, we decided to analyse endogenous MK5 activity in MEFs dependent on ERK3 expression. WT and, as a negative control, MK5-deficient MEFs were transfected with His-tagged ERK3 and endogenous MK5 activity was determined by IP kinase assay. In this experiment, MK5 activity can only be detected in WT MEFs transfected with ERK3 (Figure 3C), indicating that ERK3 is able to specifically activate endogenous MK5.
Figure 3.
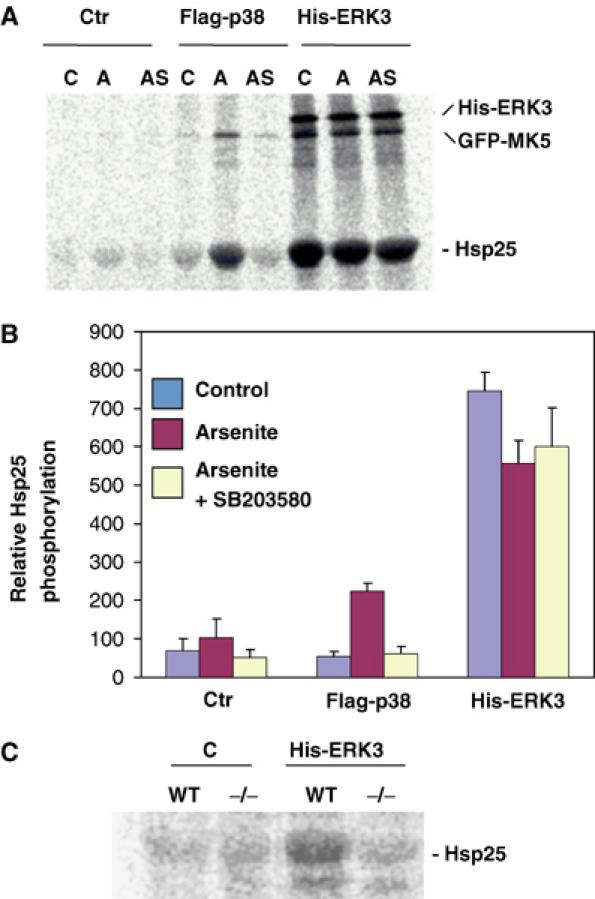
Expression of ERK3 stimulates activity of MK5. (A, B) Coexpression of MK5 and ERK3 in HEK293 cells. (A) Autoradiograph of MK5 IP kinase assay. GFP-MK5 was precipitated by anti-GFP antibodies from unstimulated cells (C), HEK293 cells stimulated by 150 μM arsenite treatment (A) and from arsenite-stimulated cells pretreated for 1 h with 10 μM SB203580 (AS) expressing GFP-MK5 alone (Ctr), or coexpressing Flag-p38 or His-ERK3. MK5 kinase activity in the IP was determined by incorporation of phosphate from [γ-33P]ATP into the in vitro substrate Hsp25. In the IP, ERK3 and GFP-MK5 were also phosphorylated. (B) Quantification of Hsp25 phosphorylation by phospho-imaging of two independent experiments, each with double determinations. (C) Autoradiograph of IP kinase assay using MK5 antibodies for WT and MK5-deficient (−/−) MEFs transfected with control plasmid (C) or expressing His-ERK3.
Catalytic activity of ERK3 is not required for MK5 translocation and activation
An obvious mechanism for MK5 activation could be its regulatory phosphorylation in the activation loop at T182 directly by ERK3. To prove this, we investigated whether catalytic activity of ERK3 is necessary in the signalling module. Two ATP-binding pocket mutants and an activation loop catalytic dead mutant of ERK3, ERK3-K49,50R, -K49,50A and -S189A, which were tested to be catalytically inactive in a myelin basic protein in gel kinase assay (not shown), were analysed for their ability to translocate and activate MK5 when coexpressed in HEK293 cells (Figure 4). Unexpectedly, all mutants are able to translocate (Figure 4A and not shown) and activate MK5 (Figure 4B). Coexpression of all mutants leads to significant phosphorylation of MK5 and its in vitro substrate Hsp25. Furthermore, a significant phosphorylation of WT ERK3 and all mutants could be observed, suggesting that ERK3 itself might be a direct substrate for MK5. Since the activation loop mutant ERK3-S189A shows comparable phosphorylation to WT ERK3, the putative phosphorylation site(s) must be distinct from S189, a site that is a target of a previously characterised ERK3 kinase (Cheng et al, 1996).
Figure 4.
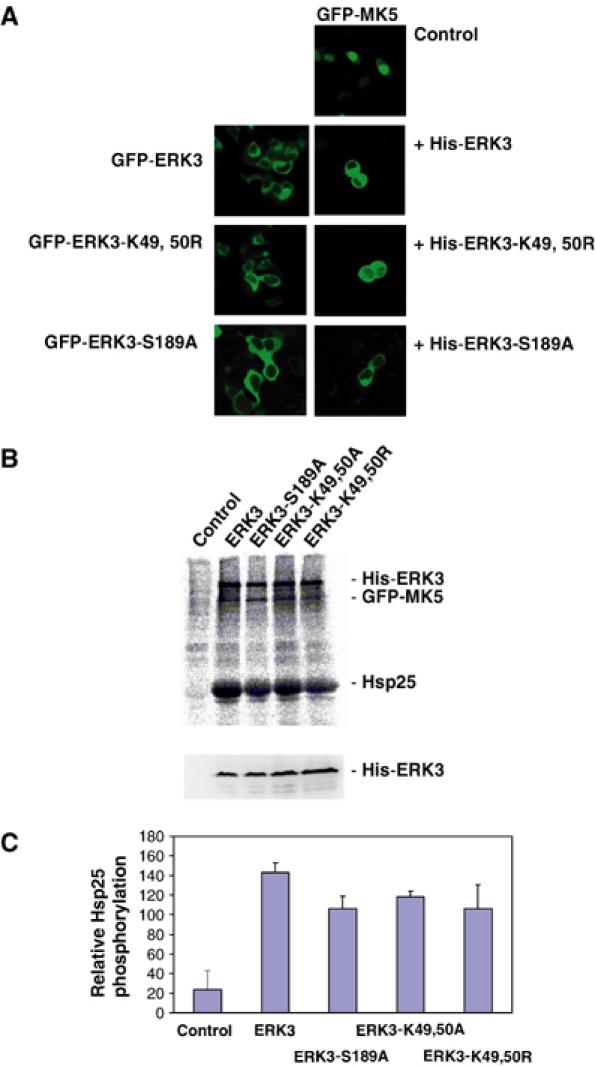
Catalytic activity of ERK3 is not necessary for translocation and activation of MK5. (A) GFP-ERK3 and its catalytic dead mutants, GFP-ERK3-K49,50R and GFP-ERK3-S189A, are mainly in the cytoplasmic compartment of HEK293 cells. When coexpressed as His-tagged proteins, ERK3 and its mutants change MK5 localisation from nuclear (control) to cytoplasmic. (B) Upper panel: Phospho-image of IP kinase assay using anti-GFP antibodies and HEK293 cells transfected with GFP-MK5 (control) and cotransfected with ERK3 and its mutants. The positions of His-ERK3 and its mutants, GFP-MK5 and the MK5 in vitro substrate Hsp25 are indicated. Lower panel: Western blot using anti-ERK3 antibody (Santa Cruz, sc156) for the HEK293 lysates used for the IP kinase assay as expression control for His-ERK3 protein and its mutants. (C) Quantification of MK5 and ERK3 activity by phospho-imaging in two independent experiments, each with double determinations.
Identification of C-terminal regions in ERK3 necessary for MK5 binding, translocation and activation
Since enzymatic activity of ERK3 is dispensable for MK5 activation, we were interested in whether domains outside the catalytic region of ERK3 are involved. By stepwise deletion of the C-terminal extension of ERK3 (Figure 5A), regions necessary for MK5 translocation and activation were identified. Deletion of the complete C-terminus of ERK3 (amino acids 301–720, ERK3-ΔC3), which leaves only the catalytic domain of ERK3 intact, leads to loss of MK5–ERK3 interaction (Figure 5B) as well as of MK5 translocation and activation (Figure 5C and D). In contrast, the mutant ERK3-ΔC2 (amino acids 358–720 deleted) is sufficient for MK5 binding and translocation but not for its activation. Finally, the mutant ERK3-ΔC1 (amino acids 472–720 deleted) is able to bind, translocate and activate MK5. For effective activation of MK5, the binding region of ERK3 and the resulting translocation are not sufficient, but the region between amino acids 358 and 472 is needed. As for the kinase dead mutants of ERK3, activation of MK5 is paralleled by phosphorylation of ERK3 and of ERK3-ΔC1 (Figures 4B and 5D), indicating that after activation MK5 might act as a direct ERK3 kinase.
Figure 5.
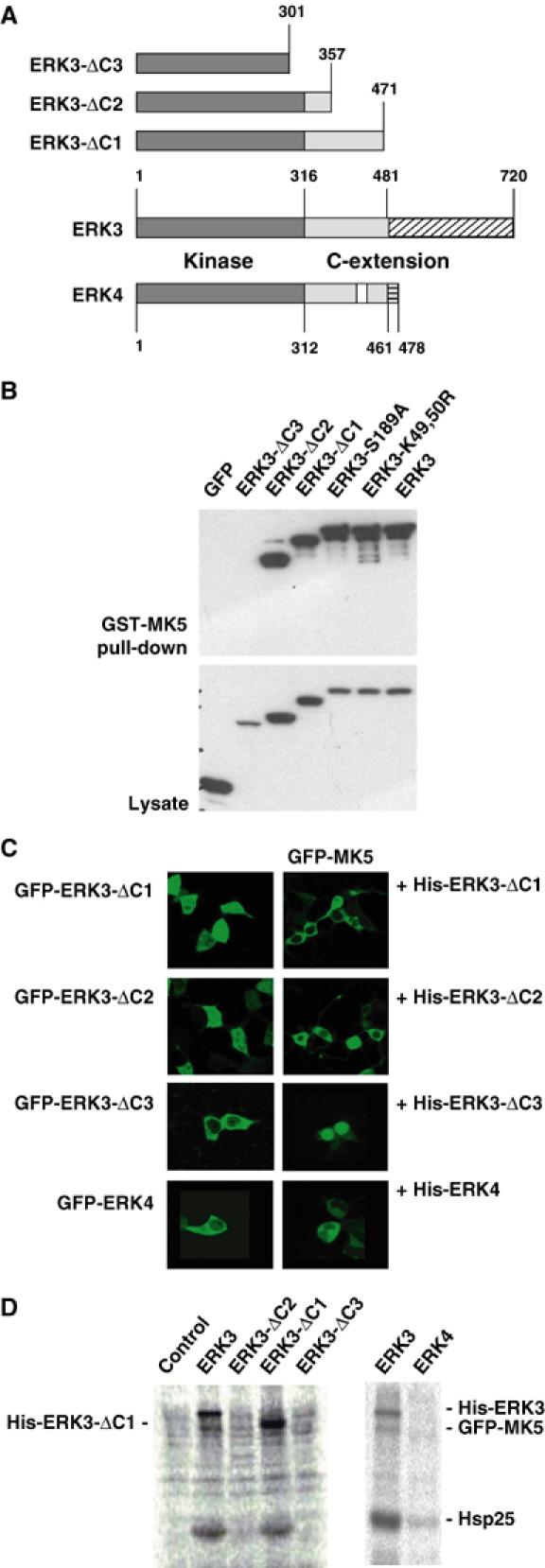
Deletion analysis of ERK3–MK5 interaction and ERK3-dependent activation of MK5. (A) Schematic representation of the C-terminal deletions of ERK3 used and of the amino-acid sequence similarity between mouse ERK3 (gi 31560797) and mouse ERK4 (gi 27369906). Dark grey: catalytic domain; light grey: parts of C-terminal extention similar between ERK3 and ERK4; white: similarity gap in C-terminus ERK4; hatched: C-terminal regions of ERK3 and ERK4 without sequence similarity. (B) GST-MK5 pull-down of GFP-tagged ERK3 and its mutants from lysates of transfected HEK293 cells. GFP-ERK3 was detected in pull-down and whole lysate (as control) by Western blot against GFP. (C) Subcellular localisation of GFP-ERK3 deletion mutants and of GFP-ERK4 is mainly cytoplasmic. GFP-MK5 is only translocated by the MK5-binding ERK3 mutants ΔC1 and ΔC2 and by ERK4, which also binds MK5 (not shown). (D) The C-terminal extension of ERK3 (amino acids 358–471) missing in ΔC2 and ΔC3 is necessary for activation of MK5 and for subsequent phosphorylation of ERK3. ERK4, which displays a homology gap (see A) in the region between amino acids 358 and 471, is not able to activate MK5. GFP-MK5 activity was monitored by IP kinase assay as shown in Figure 3A. His-tagged versions of ERK3 and ERK4 were coexpressed with GFP-MK5 or, as a negative control, with GFP-MK2, and activity of MK5 was determined by α-GFP IP kinase assay.
The ERK3-related kinase ERK4 (also designated ERK3β or p63 MAPK) (Gonzalez et al, 1992) shares homology with ERK3 within the catalytic domain and, in part, also within the C-terminal extension (Figure 5A). ERK4 also interacts with MK5 in a pull-down assay (not shown) and translocates MK5 to the cytoplasm (Figure 5C). However, ERK4 cannot activate MK5 (Figure 5D), supporting the notion towards a specific contribution of the region between amino acids 357 and 471 of ERK3, which shows a lower degree of homology to ERK4 (cf. Figure 5A).
The C-terminus but not the D-domain in MK5 is necessary for ERK3 binding
In the C-terminal region of ERK3 necessary for MK5 binding (amino acids 301–357), two common docking (CD)-like motifs (Tanoue and Nishida, 2003), which could interact with basic D-domains of MAPK substrates such as MKs, can be identified. However, these CD-like motifs show some differences from the CD motifs present in ERK1,2, JNKs and p38 MAPKs. Furthermore, mutation of conserved residues, for example D339, does not prevent interaction with MK5 (not shown). Deletion of the D-domain in MK5 (cf. Figure 6A), which overlaps with the NLS of this kinase (Seternes et al, 2002), from RKRK to GTGT (amino acids 361–364, MK5-GTGT) leads to cytoplasmic localisation of MK5 independent of ERK3 (Figure 6D). Interestingly, this mutation does neither block MK5's interaction with ERK3 (Figure 6B) nor its ERK3-dependent activation (Figure 6C). In contrast, deletion of the C-terminus of MK5 starting from amino acid 369 (MK5-1-368) leads to a mutant with intact D-domain, which can no longer bind to ERK3 and which is not translocated and activated (Figure 6). This indicates that ERK3–MK5 interaction proceeds between the region of amino acids 301–358 in ERK3 and 369–473 in MK5 and is different from the CD interaction of other MAPKs with their MKs.
Figure 6.
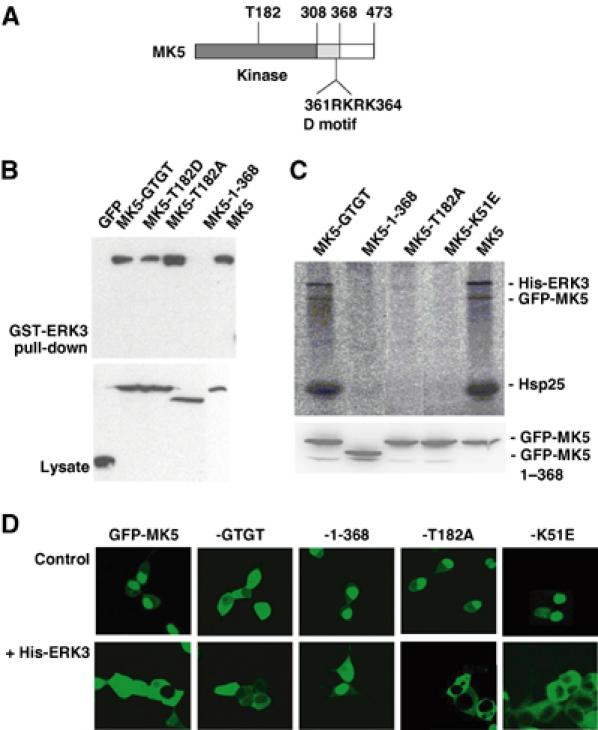
Mutational analysis of MK5 properties necessary for ERK3 binding and its activation. (A) Schematic structure of MK5. (B) GST-ERK3 pull-down of GFP-tagged MK5 and its mutants (MK5-GTGT—D-domain mutant; MK5-1-368—C-terminal deletion of amino acids 369–473; GFP-MK5-T182A (activation loop)) from lysates of transfected HEK293 cells. GFP-MK5 was detected in pull-down and whole lysate (as control) by Western blot against GFP. (C) Upper panel: Phospho-image of IP kinase assay as in Figure 3A of cells coexpressing His-ERK3 and GFP-MK5 or -MK5 mutants (including also the kinase-dead ATP-binding pocket mutant MK5-K51E). Lower panel: Expression control for GFP-MK5 and its mutants in the cell lysates used for IP kinase assay. (D) GFP-MK5 and its mutants were transiently cotransfected with His-ERK3 or, as a control, transfected alone, and localisation of the GFP fusion proteins was analysed by fluorescence microscopy as described.
Catalytic activity and phosphorylation of T182 in the activation loop of MK5 are necessary for its activation but not for its translocation to the cytoplasm
To decide whether MK5 catalytic activity is necessary for ERK3-regulated activation of MK5, we analysed the GFP fusion protein of the activation loop mutant T182A and of the ATP-binding site mutant K51E (Seternes et al, 2002) in the IP kinase assay (Figure 6C). Although it binds to ERK3 (Figure 6B), GFP-MK5-T182A is not phosphorylated as a result of coexpression of His-ERK3 and, as expected, no kinase activity against the substrates Hsp25 and ERK3 can be detected (Figure 6C). This indicates that ERK3-mediated activation of MK5 depends on phosphorylation of its activation loop at T182 and that both Hsp25 and ERK3 phosphorylations are due to MK5 activity. More interestingly, the ATP-binding mutant MK5-K51E was not phosphorylated on existing T182, indicating that, in complex with ERK3, MK5 activity is necessary for its own phosphorylation and activation, suggesting ERK3-initiated autophosphorylation of MK5. This idea is supported by the observation that no other proline-directed kinases of the ERK, JNK and p38 MAPK family could be detected in the immunoprecipitated ERK3–MK5 complex by Western blot using pan-ERK, pan-JNK and pan-p38 MAPK antibodies (not shown).
It is known that the MK5-related enzyme MK2 is translocated to the cytoplasm as a result of phosphorylation at T317 in the C-terminal hinge region independent of phosphorylation of T205 in the activation loop (Ben-Levy et al, 1998; Engel et al, 1998). Similar to MK2, the activation loop mutant MK5-T182A shows also ERK3-dependent translocation to the cytoplasm (Figure 6D). However, in contrast to MK2, phosphorylation of MK5 as well as phosphorylation of ERK3 is not necessary for MK5's ERK3-driven translocation at all (cf. Figure 6C and D).
Coexpression of ERK3 and MK5 in mouse embryogenesis
It has been recently shown that ERK3 markedly accumulates during differentiation and increased ERK3 level inhibits proliferation by a G1 arrest blocking S-phase entry (Coulombe et al, 2003; Julien et al, 2003). In mouse embryonic development, ERK3 mRNA expression peaks at day E11, while at days E13 and E15 there is only weak expression and at days E9, E17 and P1 no ERK3 mRNA is detectable (Turgeon et al, 2000). We analysed ERK3 and MK5 expression by in situ hybridisation at E11 and E14.5 (Figure 7). ERK3 mRNA is widely expressed in E11 embryos (Figure 7A), while in E14.5 embryos its expression appears to be reduced and signal is restricted to some tissues including lung and subregions of the brain (arrows in Figure 7B). Similar to ERK3, MK5 mRNA seems to be widely expressed at E11 (Figure 7C), but restricted to low levels and few specific sites at E14.5 (arrows in Figure 7D). Background levels of control hybridisation using sense MK5 riboprobe were higher than in the control experiments for ERK3 (not shown);therefore, further studies will be required to define the specificity and extent of overlaps in MK5 and ERK3 expression in more detail. These results do, however, support the notion towards spatiotemporal coexpression of MK5 and ERK3 during mouse embryogenesis.
Figure 7.
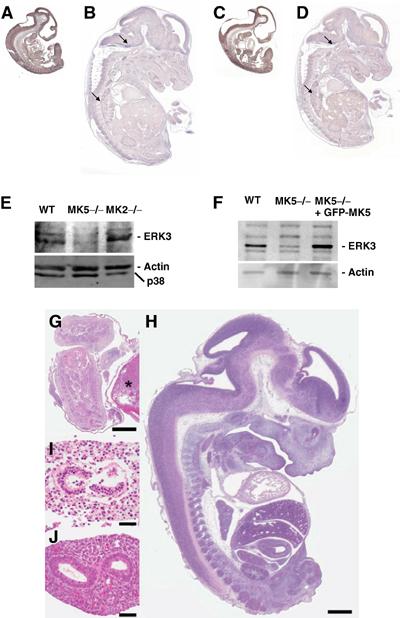
Expression of ERK3 and MK5 in mouse embryos. Spatiotemporal coexpression of ERK3 (A, B) and MK5 mRNA (C, D) detected by in situ hybridisation in embryos of stages E11 (A, C) and E14.5 (B, D). (E) Western blot detection of ERK3 in WT, MK5-deficient (MK5−/−) and MK2-deficient (−/−) primary MEFs obtained from day 12.5 embryos. As a control, the stripped blot was developed with anti-p38 MAPK and anti-β-actin antibodies. (F) Rescue of ERK3 level in MK5-deficient MEFs by transient transfection of GFP-MK5. As control, ERK3 expression in WT MEFs (WT) was analysed. (G) Dead and autolytic MK5−/− embryo at day E13.5 with an overall length of approximately 5 mm (asterisk denotes placenta) when WT littermates were approximately 14.5 mm long (H, WT littermate), suggesting premature death of the MK5−/− embryo around day E11.5. All organs of the MK5−/− embryos were disintegrated and autolytic at day E13.5 (intestine is shown in I) when compared to the organs of WT littermates (WT intestine shown in J). Bars=1 mm (G, H) and 50 μm (I, J). Photographs were generated from formalin-fixed, paraffin-embedded tissue sections, stained with haematoxylin and eosin.
Reduced ERK3 levels in MK5-deficient cells
Recently, we generated MK5-deficient mice and could show that these animals do not exhibit a significant phenotype in the mixed 129 × C57/B6 genetic background (Shi et al, 2003). Meanwhile, these mice were backcrossed to the C57/B6 genetic background. We decided to analyse ERK3 expression in embryonic cells derived from these mice. Primary embryonic fibroblasts were derived from E12.5 stage of WT, MK5-deficient and, as another control, MK2-deficient animals and subjected to Western blot detection of ERK3 and p38 MAPK (Figure 7E). Since MK2 is a major interaction partner of p38 MAPKα, its absence in MK2-deficient cells leads to reduced levels of p38 MAPKα as seen in the control. Interestingly, in MK5-deficient cells, the relatively high level of expression of ERK3 in WT embryonic fibroblasts is significantly reduced, indicating that endogenous MK5 is a major stabilising interaction partner of endogenous ERK3 in these cells. Conversely, ERK3 downregulation by siRNA technique or absence of ERK3 due to deletion by homologous recombination also leads to significantly reduced MK5 activity (Seternes and Keyse, personal communication). In SV40 large T immortalised MK5-deficient embryonic fibroblasts (Shi et al, 2003), a significantly reduced expression of ERK3 is also detected (Figure 7F). Since these cells can be efficiently transfected using standard methods, we ask whether reintroduction of MK5 by expression of GFP-MK5 can rescue the ERK3 level. Overexpression of GFP-MK5 leads to strongly increased ERK3 levels in these cells (Figure 7F), further supporting the ERK3-stabilising interaction between both protein kinases.
MK5 deficiency causes embryonic lethality around E11
Interestingly, in the C57/B6 genetic background, MK5-deficient mice showed embryonic lethality with incomplete penetrance. Homozygous mutants were under-represented at least after E12, where major deviation from the expected Mendelian ratios was already observed with only about 50% of the expected number of MK5−/− embryos detectable (Table I). In addition, we observed an increased number of MK5-deficient autolytic pups from E13 (Figure 7G–J), which may have resulted from developmental defects at earlier times. It is to be noted that autolytic embryos were 5 mm long, a body length that is usually seen around day E11.5, strongly suggesting that embryonic death must have occurred around day E11.5 where ERK3 mRNA expression in wild mice is maximum (Turgeon et al, 2000). These observations support the notion that ERK3 and MK5 cooperate in regulation of mouse development and differentiation at a stage close to E11.
Table 1.
Embryonic lethality of MK5-deficient mice displayed by genetic under-representation of homozygous MK5 mutant mice after embryonic day 11
| Intercross | Day | +/+ | +/− | −/− |
|---|---|---|---|---|
| MK5+/− | E11–12 (_n_=14) | 1.0 (4) | 2.0 (8) | 0.5 (2) |
| × MK5+/− | ||||
| E13–15 (_n_=16) | 1.0 (5) | 1.8 (9) | 0.4 (2) | |
| P17 (_n_=155) | 1.0 (42) | 2.2 (91) | 0.5 (22) | |
| MK5+/− | E11–12 (_n_=14) | — | 1.0 (9) | 0.55 (5) |
| × MK5−/− | ||||
| E13–15 (_n_=31) | — | 1.0 (21) | 0.48 (10) | |
| Values represent the relative ratio of genotype frequency compared to WT and MK5+/−, respectively. The total number of embryos and newborn animals analysed for a specific cross in a specific stage, n, and the number of organisms representing the different MK5 genotypes are given in parentheses. Genotyping was carried out by PCR as described previously (Shi et al, 2003). |
Discussion
Specific molecular interaction between the C-terminus of MK5 and amino acids 301–358 in the C-terminal domain of ERK3 results in translocation of MK5 from the nucleus to the cytoplasmic compartment of the cell. Obviously, ERK3 binding and translocation is not sufficient for MK5 activation, which requires a further C-terminal region between amino acids 358 and 471 of ERK3 as well as MK5 catalytic activity itself. The finding that the C-terminal region of ERK3 but not its catalytic activity is necessary for MK5 activation indicates a scaffolding and translocator function of ERK3 for MK5. Since MK5 catalytic activity is required for its own activation and since ERK3 undergoes MK5-dependent phosphorylation, a scaffolding for MK5 by cytoplasmic ERK3 followed by an ERK3-mediated autophosphorylation and autoactivation of MK5 and subsequent phosphorylation of ERK3 by activated MK5 is supposed (cf. Figure 8). In most aspects, this is different from the well-known mechanisms by which a catalytically active kinase stimulates a downstream kinase via phosphorylation of its regulatory sites. It is known that other catalytically active protein kinases, such as the yeast protein kinase Pbs2p, which binds Hog1 and Ste11p, can also act as scaffold (Posas and Saito, 1997). In the case of the ERK3/MK5 module, no catalytic activity of the scaffolding ‘kinase' is required for phosphorylation and activation of the downstream element MK5 although ERK3 is known to display some catalytic activity (Julien et al, 2003). Hence, our finding is more similar to the unfolded protein response receptor Ire1, where a conformational change in the catalytically active kinase domain, triggered by occupancy of its active site with a ligand, stimulates all known downstream functions without the need for catalytic activity (Papa et al, 2003), or to protein kinases carrying both catalytic and pseudokinase domains, such as Janus kinases where the pseudokinase domain is necessary for efficient activation of the enzyme (Saharinen et al, 2000).
Figure 8.
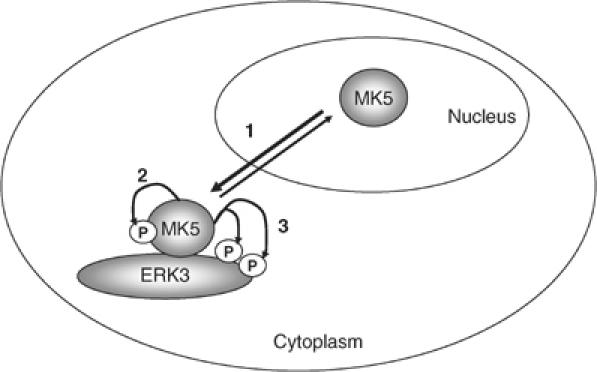
Schematic representation of our model of MK5 activation by ERK3. Increased level of cytoplasmic ERK3 leads to increased cytoplasmic anchoring of MK5, which shuttles between nucleus and cytoplasm (1). Cytoplasmic scaffolding of MK5 by ERK3 facilitates activation of MK5 by autophosphorylation (2) and activated MK5 phosphorylates ERK3 (3).
Recently, activation of the protein kinase LKB1, a gene mutated in Peutz–Jeghers cancer syndrome and involved in polarisation of epithelial cells, by the pseudokinase STRAD has been reported (Baas et al, 2003; 2004). In this case, binding between LKB1 and STRAD also induces nuclear-cytoplasmic translocation of LKB1, phosphorylation of both LKB1 and STRAD and activation of LKB1 (Baas et al, 2003). It was shown that LKB1 exhibits STRAD-mediated autophosphorylation and that other components in the complex, such as MO25 (Boudeau et al, 2003), may stimulate this process.
ERK3 is a protein kinase containing an N-terminal catalytically active domain. The C-terminal extension between amino acids 442 and 720 displays at least weak homology to a protein kinase catalytic domain, since it has been described as a member of an MAPK cluster in euKaryotic Orthologous Groups (KOG0660, NCBI; Marchler-Bauer et al, 2003), which contains also other catalytic kinase domains. Similar to STRAD, essential conserved kinase subdomains are lacking in the C-terminus of ERK3. By the fact that parts of the C-terminal ‘pseudokinase' domain of ERK3 are essential for MK5 translocation and activation, the role of STRAD for LKB1 is resembled. Furthermore, our observation that a functional ATP-binding pocket of MK5 is necessary for its activation suggests ERK3-mediated autophosphorylation of MK5 similar to STRAD-mediated autophosphorylation of LKB1. Also, both STRAD and ERK3 translocate the target protein to the cytoplasm and are phosphorylated after activation of their target. Finally, nuclear-cytoplasmic shuttling of both STRAD and ERK3 has been reported to be necessary for cell cycle arrest (Baas et al, 2003; Julien et al, 2003). Hence, this activation mechanism could be of general importance in growth regulation and development and could assign a new role to other pseudokinases lacking residues essential for catalysis. One may speculate that an activation mechanism that does not require catalytic activity of a phosphorylation-regulated activator kinase but only its expression and binding to the target kinase may be well suited for more sustained activation of the target kinase during development and differentiation. Apart from transient phosphorylation-dependent signalling, which often occurs in response to extracellular signals and which can be rapidly reverted by dephosphorylation of the activator kinase, changes in expression and stability of pseudokinase-like translocators and activators may add another regulatory level in the orchestration of signalling. With regard to this idea, one should be aware that in the human kinome, 50 protein kinases were identified that lack residues essential for catalysis and are predicted to act as catalytically inactive scaffolding proteins or pseudokinases (Manning et al, 2002).
The reason for incomplete penetrance of the embryonic lethal phenotype of homozygous MK5 mutant mice is still enigmatic. The remaining viable homozygous MK5 mutant mice are smaller after birth (e.g. body mass P17: MK5+/−: 7.6±0.26 g; MK5−/−: 6.7±0.28 g; P<0.001), but do not display morphological or histological abnormalities when analysed after 3, 6 and 24 weeks. We also inspected the maternal placenta of hemizygous MK5 mutants used for the intercrossing, but could not detect abnormalities that explain the incomplete penetrance of the offspring lethality. Hence, the detailed developmental effect of the ERK3/MK5 signalling module in mouse embryogenesis and the MK5 targets involved remain to be identified and knowing the phenotype of the ERK3 knockout mouse will be certainly helpful. Apart from this, understanding of the detailed molecular mechanism leading to ERK-dependent autoactivation of MK5 within this signalling module and of the role of MK5-dependent ERK3 phosphorylation in its regulation, and identification of additional components of the ERK3/MK5 complex require further investigation.
Materials and methods
Yeast two-hybrid screen
A pretransformed mouse 11-day embryo MATCHMAKER pACT2-cDNA library (Clontech MY4012AH) or a mouse brain library (MY4008AH) in strain Y187 (MATα) was mated with pGBKT7-MK5-transformed AH109 (MATa) and plated on 20 SDΔHLT plates with 15 mM aminotriazol and on 20 SDΔAHLT plates. The plates were incubated for 3–21 days at 30°C. For semiquantitative luminometric analysis of protein–protein interactions in yeast, the Galacto-Light plus system (Applied Biosystems) was used.
Cloning and site-directed mutagenesis
For cloning into pENTR/D-TOPO (Invitrogen), mouse ERK3 cDNA was amplified from the identified two-hybrid clone pACT2-cDNA-ERK3 by PCR using the primer pair 5′-CCA CAT GGC AGA GAA ATT CGA AAG TCT C-3′ (forward) and 5′-TTA GTT CAG ATG TTT CAG AAT GCT GC-3′ (reverse). For cloning into pEGFP-C1 and pEYFP-C1, pACT2-cDNA-ERK3 was digested by _Eco_RI, refilled with Klenow enzyme and redigested by _Xho_I and inserted into _Bsp_EI-cleaved, Klenow-filled and _Xho_I-cut dephosphorylated vector. Site-directed mutagenesis was performed in pENTR/D-ERK3-WT and pEGFP-C1-ERK3-WT using the Quik-change XL Site-directed Mutagenesis Kit (Stratagene). The recombination reaction between the entry clone and the pDEST26 vector for His-tagged ERK3 expression was achieved with the LR Clonase Kit (Invitrogen). C-terminal deletion mutants of ERK3, ERK3-ΔC1, -ΔC2, -ΔC3, were generated from both pENTR/D-ERK3 and pEGFP-C1-ERK3.
Most MK5 constructs were a kind gift from Dr Ole Morten Seternes and are described elsewhere (Seternes et al, 2002). An _Eco_R1/_Kpn_I MK5-coding fragment of pEGFP-C1-MK5 was ligated into _Eco_R1/_Kpn_I-cut pECFP-C1 to give pECFP-C1-MK5.
Expression of fusion proteins in HEK293 and detection of subcellular localisation of GFP fusions
A total of 2.5 × 106 HEK293 cells were transfected using 500 μl Optimem (Gibco) and Lipofectamine PLUS reagent (Invitrogen). A 0.5 μg portion of expression plasmid for GFP-MK5 and 5 μg portion of expression plasmid for His-tagged ERK3 were used in cotransfection experiments for subcellular localisation studies; otherwise, equimolar amounts of plasmids were used. For subcellular localisation of GFP-tagged protein, the transfected cells were replated in Chambered Coverglass (Labtek, Nunc) and analysed using a Leica DM IRBE microscope with the Leica TCS confocal systems program.
Nuclear and cytoplasmic fluorescence intensity was determined using MetaMorph software (Universal Imaging Corporation) and the measure pixel option for at least seven randomly chosen cells (_n_⩾7). Subcellular localisation index i was calculated for each cell as _i_=(_I_n−_I_b)/(_I_c−_I_b), where _I_n is the nuclear fluorescence intensity, _I_b is the background intensity and _I_c is the cytoplasmic intensity. Hence i<1 stands for predominantly cytoplasmic localisation, while _i_>1 indicates nuclear accumulation.
In vitro pull-down assay
A total of 5 × 106 transfected HEK293 cells expressing different GST-tagged forms of MK5 or ERK3 were washed with ice-cold PBS and lysed in 1 ml lysis buffer containing 1% (v/v) Triton X-100, 10% (v/v) glycerine, 150 mM NaCl, 50 mM Hepes pH 7.5, 1,5 mM MgCl2 and 1 mM EGTA for 30 min on ice. After centrifugation (16 000 g, 4°C), the supernatant was transferred to a new tube and incubated with 25 μl glutathione–Sepharose 4B beads (Amersham Biosciences) overnight tumbling at 4°C. Proteins bound were analysed by Western blot against GFP (anti-GFP B2, Santa Cruz).
Alternatively, purified GST-tagged protein was incubated with recombinant hexahistidine-tagged ERK3 bound to nickel–agarose (Qiagen). Binding of GST or GST fusion proteins was detected by Western blot using anti-GST antibodies (B14, Santa Cruz).
MK2 and MK5 tandem affinity constructs used are described by Shi et al (2003).
Purification and detection of biotin-ERK3 binding proteins from MEFs
A total of 7 × 106 immortalised MEFs (Shi et al, 2003) were transfected with 5 μg pcDNA6/BioEase-ERK3 (generated from pcDNA6/BioEase-DEST (Invitrogen) and pENTR/D-ERK3) together with 1 μg pEGFP-C1. After washing and centrifugation in ice-cold PBS, cells were resuspended in lysis buffer containing 50 mM Tris pH 7.8, 150 mM NaCl, 1% NP-40 and 1 mM PMSF and incubated for 15 min on ice. The cleared lysate was incubated with 30 μl streptavidin–agarose suspension (1:1) tumbling overnight at 4°C. After six washes, proteins bound to beads were analysed by Western blot using sheep anti-MK5 antibodies (kind gift from Dr P Cohen, Dundee).
IP kinase assays
IP kinase assay was performed as described (Shi et al, 2003) using anti-GFP (B2, Santa Cruz) and 25 μl of 50% Protein G–Sepharose suspension (Amersham Biosciences) or sheep anti-MK5 antibodies and the substrate Hsp25. Radioactivity incorporated into Hsp25 was quantified by phospho-imaging using a Fuji Bas-1500.
In situ hybridisation
Embryos were dissected from timed-pregnant mice at E11 and E14.5. E11 embryos were fixed in 4% paraformaldehyde for 2 h, transferred through a dilution series into Tris–HCl saline buffer containing 0.5 M sucrose for cryoprotection and embedded in 4% gelatin. E14.5 embryos were embedded in TissueTek medium (Sakura) immediately after dissection. All samples were quick-frozen at −60°C and cryosectioned at 25 μm thickness.
Templates for riboprobes were generated by PCR using gene-specific primers with attached SP6- and T7-RNA polymerase recognition sites (capitals in primer sequences). Primers for MK5 were T7-FW (5′-AAG GTA ATA CGA CTC ACT ATA GGG aga gct att tca cag aat cag cc-3′) and SP6-RV (5′-AGA GAT TTA GGT GAC ACT ATA Gaa aga gca tcc ctc agg agc ttg cat tcg-3′), covering nucleotide positions 1027–2013 of GenBank entry NM_010765. Primers for ERK3 were T7-FW (5′-AAG GTA ATA CGA CTC ACT ATA GGG aga ccg aga gaa gta tct aga gg-3′) and SP6-RV (5′-AGA GAT TTA GGT GAC ACT ATA Gaa gag aaa tgt ctg ctg agg ttt ag-3′), covering nucleotide positions 1484–2458 of GenBank entry NM_015806. Templates were tested for correct size and absence of by-products by agarose gel electrophoresis and sequenced to confirm their identity with expected sequence. Digoxigenin-labelled antisense and sense riboprobes were generated by standard methods with SP6- and T7-RNA polymerase, respectively.
In situ hybridisation on cryosections was performed using an automated liquid handling system essentially as described (Herzig et al, 2001).
Miscellaneous
ERK3 Western blot was performed using anti-ERK3 (I-15) from Santa Cruz. Pathological inspection of mouse embryos was carried out as described (Shi et al, 2003).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We thank Dr Ole-Morten Seternes (University Tromsoe, Norway) for several MK5 constructs, Dr Maria Schubert for the pECFP-MK2 construct, Dr Sir Philip Cohen (University Dundee, Scotland) for the MK5 antibodies, Tatiana Iakovleva for help with mice breeding and genotyping, Polina Spies and Kornelia Maslo for help with in situ hybridisation and Drs Helmut Holtmann and Michael Kracht for critical reading of the manuscript. We also thank Drs Ole-Morten Seternes and Steve Keyse (Cancer Research, Ninewells Hospital, Dundee, Scotland) for communicating results prior to publication. This work was supported by the Research Training Network Programme of the European Community (HPRN-CT-2002-00255), by the DFG and by the German Ministry of Research (01 KW9965).
References
- Abe MK, Kuo WL, Hershenson MB, Rosner MR (1999) Extracellular signal-regulated kinase 7 (ERK7), a novel ERK with a C-terminal domain that regulates its activity, its cellular localization, and cell growth. Mol Cell Biol 19: 1301–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe MK, Saelzler MP, Espinosa R III, Kahle KT, Hershenson MB, Le Beau MM, Rosner MR (2002) ERK8, a new member of the mitogen-activated protein kinase family. J Biol Chem 277: 16733–16743 [DOI] [PubMed] [Google Scholar]
- Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, Clevers HC (2003) Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J 22: 3062–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas AF, Kuipers J, van der Wel NN, Batlle E, Koerten HK, Peters PJ, Clevers HC (2004) Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell 116: 457–466 [DOI] [PubMed] [Google Scholar]
- Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ (1998) Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr Biol 8: 1049–1057 [DOI] [PubMed] [Google Scholar]
- Ben-Levy R, Leighton IA, Doza YN, Attwood P, Morrice N, Marshall CJ, Cohen P (1995) Identification of novel phosphorylation sites required for activation of MAPKAP kinase-2. EMBO J 14: 5920–5930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, Alessi DR (2003) MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J 22: 5102–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD (1991) ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65: 663–675 [DOI] [PubMed] [Google Scholar]
- Cheng M, Zhen E, Robinson MJ, Ebert D, Goldsmith E, Cobb MH (1996) Characterization of a protein kinase that phosphorylates serine 189 of the mitogen-activated protein kinase homolog ERK3. J Biol Chem 271: 12057–12062 [DOI] [PubMed] [Google Scholar]
- Coulombe P, Rodier G, Pelletier S, Pellerin J, Meloche S (2003) Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin–proteasome pathway defines a novel paradigm of mitogen-activated protein kinase regulation during cellular differentiation. Mol Cell Biol 23: 4542–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel K, Kotlyarov A, Gaestel M (1998) Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J 17: 3363–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel K, Plath K, Gaestel M (1993) The MAP kinase-activated protein kinase 2 contains a proline-rich SH3-binding domain. FEBS Lett 336: 143–147 [DOI] [PubMed] [Google Scholar]
- Engel K, Schultz H, Martin F, Kotlyarov A, Plath K, Hahn M, Heinemann U, Gaestel M (1995) Constitutive activation of mitogen-activated protein kinase-activated protein kinase 2 by mutation of phosphorylation sites and an A-helix motif. J Biol Chem 270: 27213–27221 [DOI] [PubMed] [Google Scholar]
- Gonzalez FA, Raden DL, Rigby MR, Davis RJ (1992) Heterogeneous expression of four MAP kinase isoforms in human tissues. FEBS Lett 304: 170–178 [DOI] [PubMed] [Google Scholar]
- Herzig U, Cadenas C, Sieckmann F, Sierralta W, Thaller C, Visel A, Eichele G (2001) Development of high-throughput tools to unravel the complexity of gene expression patterns in the mammalian brain. Novartis Found Symp 239: 129–146 [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298: 1911–1912 [DOI] [PubMed] [Google Scholar]
- Julien C, Coulombe P, Meloche S, Rodier G, Pelletier S, Pellerin J, Turgeon B, Lang BF, Robinson MJ, Xu Be BE, Stippec S, Cobb MH, Zimmermann J, Lamerant N, Grossenbacher R, Furst P (2003) Nuclear export of ERK3 by a CRM1-dependent mechanism regulates its inhibitory action on cell cycle progression. J Biol Chem 278: 42615–42624 [DOI] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372: 739–746 [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298: 1912–1934 [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, DeWeese-Scott C, Fedorova ND, Geer LY, He S, Hurwitz DI, Jackson JD, Jacobs AR, Lanczycki CJ, Liebert CA, Liu C, Madej T, Marchler GH, Mazumder R, Nikolskaya AN, Panchenko AR, Rao BS, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Vasudevan S, Wang Y, Yamashita RA, Yin JJ, Bryant SH (2003) CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res 31: 383–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloche S, Beatty BG, Pellerin J (1996) Primary structure, expression and chromosomal locus of a human homolog of rat ERK3. Oncogene 13: 1575–1579 [PubMed] [Google Scholar]
- Neininger A, Thielemann H, Gaestel M (2001) FRET-based detection of different conformations of MK2. EMBO Rep 2: 703–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- New L, Jiang Y, Han J (2003) Regulation of PRAK subcellular location by p38 MAP kinases. Mol Biol Cell 14: 2603–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- New L, Jiang Y, Zhao M, Liu K, Zhu W, Flood LJ, Kato Y, Parry GC, Han J (1998) PRAK, a novel protein kinase regulated by the p38 MAP kinase. EMBO J 17: 3372–3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H, Wang XS, Diener K, Yao Z (1998) MAPKAPK5, a novel mitogen-activated protein kinase (MAPK)-activated protein kinase, is a substrate of the extracellular-regulated kinase (ERK) and p38 kinase. Biochem Biophys Res Commun 243: 492–496 [DOI] [PubMed] [Google Scholar]
- Papa FR, Zhang C, Shokat K, Walter P (2003) Bypassing a kinase activity with an ATP-competitive drug. Science 302: 1533–1537 [DOI] [PubMed] [Google Scholar]
- Posas F, Saito H (1997) Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science 276: 1702–1705 [DOI] [PubMed] [Google Scholar]
- Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR (1994) A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell 78: 1027–1037 [DOI] [PubMed] [Google Scholar]
- Roux PP, Blenis J (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68: 320–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen P, Takaluoma K, Silvennoinen O (2000) Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol 20: 3387–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz E, Oesterhelt D, Reinke H, Beyreuther K, Dimroth P (1988) The sodium ion translocating oxalacetate decarboxylase of Klebsiella pneumoniae. Sequence of the biotin-containing alpha-subunit and relationship to other biotin-containing enzymes. J Biol Chem 263: 9640–9645 [PubMed] [Google Scholar]
- Seternes OM, Johansen B, Hegge B, Johannessen M, Keyse SM, Moens U (2002) Both binding and activation of p38 mitogen-activated protein kinase (MAPK) play essential roles in regulation of the nucleocytoplasmic distribution of MAPK-activated protein kinase 5 by cellular stress. Mol Cell Biol 22: 6931–6945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Kotlyarov A, Laaß K, Gruber AD, Butt E, Marcus K, Meyer HE, Friedrich A, Volk HD, Gaestel M (2003) Elimination of protein kinase MK5/PRAK activity by targeted homologous recombination. Mol Cell Biol 23: 7732–7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M (1992) Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett 313: 307–313 [DOI] [PubMed] [Google Scholar]
- Tanoue T, Nishida E (2003) Molecular recognitions in the MAP kinase cascades. Cell Signal 15: 455–462 [DOI] [PubMed] [Google Scholar]
- Turgeon B, Saba-El-Leil MK, Meloche S (2000) Cloning and characterization of mouse extracellular-signal-regulated protein kinase 3 as a unique gene product of 100 kDa. Biochem J 346: 169–175 [PMC free article] [PubMed] [Google Scholar]
- Underwood KW, Parris KD, Federico E, Mosyak L, Czerwinski RM, Shane T, Taylor M, Svenson K, Liu Y, Hsiao CL, Wolfrom S, Maguire M, Malakian K, Telliez JB, Lin LL, Kriz RW, Seehra J, Somers WS, Stahl ML (2003) Catalytically active MAP KAP kinase 2 structures in complex with staurosporine and ADP reveal differences with the autoinhibited enzyme. Structure (Camb) 11: 627–636 [DOI] [PubMed] [Google Scholar]
- Zhou G, Bao ZQ, Dixon JE (1995) Components of a new human protein kinase signal transduction pathway. J Biol Chem 270: 12665–12669 [DOI] [PubMed] [Google Scholar]
- Zhu AX, Zhao Y, Moller DE, Flier JS (1994) Cloning and characterization of p97MAPK, a novel human homolog of rat ERK-3. Mol Cell Biol 14: 8202–8211 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3