An Acetylation Switch Modulates the Transcriptional Activity of Estrogen-Related Receptor α (original) (raw)
Abstract
Posttranslational modifications are instrumental to achieve gene- and tissue-specific regulatory outcomes by transcription factors. Nuclear receptors are dynamically modulated by several types of posttranslational modifications including phosphorylation, methylation, acetylation, ubiquitination, and sumoylation. The estrogen-related receptor α (ERRα, NR3B1) is phosphorylated on multiple sites, and sumoylated in the amino-terminal region in a phosphorylation-dependent manner. Here we demonstrate that ERRα interacts with and is acetylated by p300 coactivator associated factor (PCAF) in vitro and in mouse liver. Purified PCAF acetylated the DNA-binding domain of ERRα on four highly-conserved lysines. In addition, coexpression of PCAF reduced the transcriptional activity of ERRα and, reciprocally, a deacetylase screen identified histone deacetylase 8 (HDAC8) and sirtuin 1 homolog (Sirt1) as independent enhancers of ERRα transcriptional function. HDAC8 and Sirt1 were also demonstrated to interact directly with ERRα in vivo and to deacetylate and increase the DNA binding affinity of ERRα in vitro. The removal of PCAF increases the DNA binding of ERRα in vivo, whereas the removal of Sirt1 and HDAC8 decreases it as assessed by chromatin immunoprecipitation assay. Altogether, our results show that ERRα is an acetylated protein and imply the existence of a dynamic acetylation/deacetylation switch involved in the control of ERRα transcriptional activity.
This study reveals the existence of a dynamic acetylation/deacetylation switch involved in the control of ERRα transcriptional activity.
The estrogen-related receptor α (ERRα, NR3B1) is an orphan nuclear receptor involved in metabolic control ( 1, 2). In association with coregulatory proteins such as peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) and prospero-related homeobox protein 1 (Prox1), ERRα regulates genes involved in glycolysis, oxidative phosphorylation, and the tricarboxylic acid cycle ( 1, 2, 3). ERRα is involved in cardiac energy homeostasis and fuel sensing ( 4), and its metabolic role in the heart is supported by the observation that ERRα is required for the bioenergetic and functional adaptation to cardiac pressure overload ( 5). In addition, the ERRα-deficient mice are resistant to high-fat diet-induced obesity ( 6) and are more sensitive to exposure to cold than their wild-type littermates ( 7). The expression of ERRα in mouse liver is induced by fasting and caloric restriction in a manner that parallels the expression of PGC-1α ( 8).
Posttranslational modifications (PTMs) of transcription factors are crucial for the regulation of their function. Evidence is accumulating to support the notion that multiple PTMs interact together and constitute a code dictating the activity of transcriptional regulators ( 9). To understand how this PTM code is affecting gene expression patterns, it is imperative to identify the multiple PTMs present on and affecting the activity of transcription factors. We and others ( 10, 11, 12, 13) have previously identified ERRα as a phosphoprotein that is modified on multiple sites within its amino-terminal region and DNA-binding domain (DBD). Recently, an interplay between phosphorylation and sumoylation on the N-terminal domain of ERRα has been uncovered by our group, a finding that strengthened the hypothesis that a PTM code is involved in the control of ERRα transcriptional activity ( 14). Although the identification of the complete set of PTMs modifying ERRα is a prerequisite for understanding the role played by the PTM code in modulating ERRα physiological functions, modification of ERRα activity via other PTMs such as acetylation, methylation, or ubiquitination is still largely understudied.
PGC-1α is acetylated on 13 different lysines via interaction with the nonredundant p300/cAMP response element-binding protein-binding protein-associated factor (PCAF) homolog GCN5 (or PCAF-B) and deacetylation by the metabolic regulator sirtuin 1 homolog (Sirt1) was shown to increase the expression of PGC-1α target genes ( 15, 16), several of these having also been identified as ERRα-regulated genes ( 4, 17, 18). Furthermore, it has been demonstrated that pharmacological activation of Sirt1 in mouse liver enhanced expression of ERRα target genes including Acadm, Pdk4, and Ndufb5, whereas hepatic deletion of Sirt1 reduced the levels of ERRα targets including Acadm, Cpt1a, and Cycs ( 19, 20). ERRα mRNA levels are lower in _Sirt1_−/− mouse embryonic fibroblasts (MEFs) ( 21), and both Sirt1 and ERRα have been demonstrated as activators of the AMP-activated protein kinase pathway ( 4, 22, 23). In the liver, Sirt1 activity is modulated by fasting, caloric restriction, high-fat diet, and synthetic activators ( 15, 19, 24, 25). Taken together, the observations linking the activity of ERRα, PGC-1α, and the Sirt1 deacetylase prompted us to investigate whether the activity of ERRα itself was modulated by acetylation. In this report, we demonstrate that ERRα is indeed acetylated within its DBD on four different lysines and that this modification represses the function of ERRα by altering its DNA-binding activity. Moreover, a deacetylase screen identified HDAC8 and Sirt1 as two independent activators of ERRα, both capable of deacetylating ERRα and increasing its DNA binding activity. Altogether, we present a dynamic model whereby an acetylation/deacetylation switch involving PCAF, Sirt1, and HDAC8 regulates the transcriptional activity of ERRα via modulation of its DNA binding properties.
Results
ERRα is an acetylated protein that interacts with and is acetylated by PCAF
We first probed whether acetylated ERRα can be detected in a cellular context. To this end, we expressed ERRα in 293 cells, immunoprecipitated ERRα, and performed Western blots using an ERRα antibody or a pan-acetylated lysine antibody. As shown in Fig. 1A, ERRα expressed in 293 cells is recognized by the pan-acetylated-lysine antibody. Because the PCAF acetyltransferase has been shown to influence the activity of several nuclear receptors ( 26), we next tested whether ERRα interacts directly with PCAF. As shown in Fig. 1B (right panel), transfected ERRα in 293 cells associates with endogenous PCAF protein as determined by coimmunoprecipitation. We then explored whether endogenous ERRα exists in the acetylated state in vivo. Using a mouse liver extract, endogenous ERRα was immunoprecipitated with the ERRα-specific antibody and blotted using the pan-acetylated-lysine antibody, with ERRα blotted as a positive control (Fig. 1C, top panel). As can be observed in Fig. 1C (middle panel), ERRα is acetylated in vivo in the liver. To further address whether endogenous PCAF and ERRα interacted with each other in this physiological context, we stripped the acetylated-lysine blot and reprobed using a PCAF antibody. As shown in Fig. 1C (lower panel), PCAF can be coimmunoprecipitated specifically with ERRα. These results thus demonstrate cellular acetylation of ERRα and validate the physiological relevance of the PCAF-ERRα interaction.
Fig. 1.

ERRα is an acetylated protein that interacts with and is acetylated by PCAF on the DBD. Panel A, ERRα was transiently transfected into 293 cells, immunoprecipitated with either preimmune serum (P) or anti-ERRα serum [antibody (Ab)], and blotted with an antibody directed against ERRα or a pan-antiacetylated lysine antibody. Arrow points to ERRα. Panel B, In a similar experiment the immunoprecipitated proteins were blotted with antibodies for ERRα or PCAF. Arrow points to PCAF; i, input. Panel C, ERRα interacts with PCAF and is acetylated in vivo. Lysate from mouse liver was immunoprecipitated with either preimmune serum (P) or anti-ERRα serum (Ab) and blotted for ERRα (top panel), acetylated lysine (middle panel), or PCAF (lower panel). Arrows point to ERRα (top and middle panels) and PCAF (lower panel). Panel D, In vitro acetylation assays were performed on bacterially expressed GST-tagged proteins encoding different domains of ERRα using a recombinant PCAF and 14C-radiolabeled acetyl-CoA substrate. N, Amino-terminal domain (amino acids 1–74; 34.7 kDa), DBD (amino acids 68–173; 37.6 kDa but migrates slightly faster due to the covalent zinc fingers structure), C, Carboxy-terminal domain (amino acids 165–423; 54.5 kDa). GST and histones (H) were used, respectively, as negative and positive controls. Upper panel is the acetylation assay, and lower is Coomassie staining to show equal protein loading. Panel E, A similar in vitro acetylation assay was performed with GST-DBD fusion proteins of the three members of the ERR family (ERRα/β/γ). H, histones. Bottom panel is Coomassie staining to show equal protein loading. Panel F, GST-tagged full-length ERRα was transiently expressed in immortalized wild-type (WT) or PCAF-null [knockout (KO)] MEFs. Equal amounts of saturated GST agarose beads were immunoblotted with an ERRα or pan-antiacetyl-lysine antibody. WB, Western blot.
To assess which domains of the ERRα protein were acetylated, we cloned and expressed ERRα as three overlapping domains (amino terminus, DBD, carboxy terminus) as glutathione-S-transferase (GST)-tagged proteins in bacteria and performed in vitro acetylation assays with the purified proteins and recombinant PCAF acetyltransferase domain in the presence of the radiolabeled [14C]acetyl-coenzyme A (CoA) cofactor. We observed multiple acetylated bands for the DBD region of ERRα and no acetylation for either amino or carboxy ends of the protein or the GST-alone control (Fig. 1D). Because the lysine residues present in the DBD of ERRα are conserved between species and between the three members of the subfamily (Supplemental Fig. 1A published on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org), we sought to investigate whether the DBD of ERRβ and ERRγ were also acetylated by PCAF. As expected, the DBD of all three ERR isoforms were acetylated in vitro with a similar pattern indicating that the entire ERR family is a target of PCAF (Fig. 1E).
Having established that ERRα interacts with PCAF in cells and is acetylated in vitro by PCAF, we asked whether the absence of PCAF would be sufficient to abrogate acetylation of ERRα. We obtained transformed fibroblasts originating from wild-type and PCAF-null mice ( 27) and transiently transfected these cells to express a full-length GST-tagged ERRα. Equal amounts of purified GST-ERRα extracted from both cell lines were blotted for acetyl-lysine. As can be observed in Fig. 1F, acetylated ERRα can only be detected in wild-type MEFs. Taken together, these data indicate that ERRα is an acetylated protein, and that PCAF acts as the main acetyltransferase responsible for this PTM in this context.
PCAF antagonizes the transcriptional activity of ERRα
As we recently reported, ERRα acts in concert with PGC-1α to directly activate the Got1 promoter ( 4). We thus used this promoter to assess whether coexpression of PCAF would affect ERRα activity in this system. Coexpression of PCAF led to a significant reduction in the ability of the ERRα-PGC-1α complex to activate the Got1 promoter (Fig. 2). This was an unexpected result given that PCAF has been described mainly as a transcriptional coactivator. To rule out the possibility that introduction of PCAF would affect the activity of this promoter in a nonspecific manner, we repeated the experiment with the transcription factor NKX2.5, which can also activate Got1. In contrast, PCAF acted to enhance NKX2.5-induced activity of Got1 (Fig. 2). These results demonstrate that the repressive effect of PCAF is specific to the ERRα-PGC-1α complex.
Fig. 2.
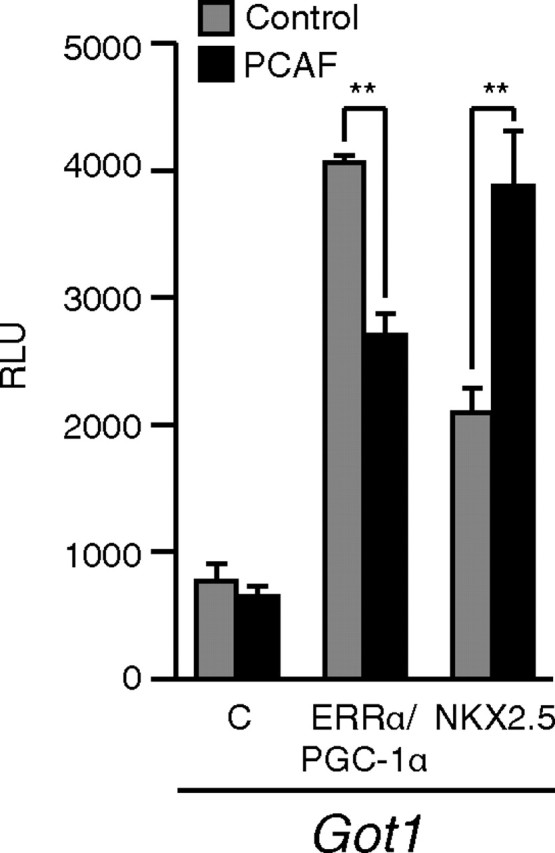
PCAF acts to reduce the transcriptional activation by ERRα. ERRα plus PGC-1α coactivator or NKX2.5 was transiently transfected in COS-1 cells with either vector alone (gray bars) or PCAF (black bars) with a luciferase reporter construct driven by the Got1 promoter. **, P < 0.01; Student’s t test. C, Control.
Four lysine residues within the DBD of ERRα are targets of PCAF acetylation
Having determined that the acetylation of ERRα occurred within the DBD, we next proceeded to map which specific lysine residue(s) were acetylated in ERRα. For this analysis, we first used in vitro PCAF-acetylated GST-ERRα DBD protein (to enrich for DBD peptides) for mass spectrometry analysis. Although we only achieved partial peptide coverage of the DBD, we observed acetylation of K138 using this technique (Fig. 3A). However, this left five lysine residues within the DBD that remained unaccounted for regarding their acetylation status. To address whether these five residues were also acetylated, we generated two intra-DBD deletion constructs, both of which excluded K138. We then mutated various lysine residues within these constructs to arginine to conserve the basic charge but exclude the possibility of acetylation. Equal amounts of all constructs were then tested in an in vitro acetylation assay with PCAF. This exclusion mapping identified K129, K160, and K162 as being acetylated by PCAF because mutations of these residues singly or in combination abrogated the acetylation observed with the wild-type protein (Fig. 3, B and C). Together, these techniques revealed that PCAF can acetylate the DBD of ERRα on multiple residues. We next generated a mutant GST-tagged full-length ERRα in which lysines 129/138/160/162 were mutated to arginines (referred to as 4KR). The two GST-ERRα expression constructs were cotransfected with PCAF in 293 cells, and the purified proteins were blotted using either the acetyl-lysine antibody or the ERRα antibody as a loading control. We observed that mutating the four lysine residues within the context of full-length ERRα was enough to completely abrogate acetylation by PCAF in cells, even with PCAF overexpression to ensure that the absence of acetylation of the mutant is not due to an increased ratio of ERRα to PCAF (Fig. 3D). Our analysis thus revealed that acetylation occurs on four lysines located within the second zinc finger and C-terminal extension (CTE) of the ERRα DBD (Fig. 3E).
Fig. 3.

The DBD of ERRα is acetylated by PCAF on four different lysine residues. A, GST-ERRα DBD was acetylated in vitro, tryptic digested, and subject to tandem mass spectrometry revealing ERRα peptides and K138 as a site of acetylation. B and C, In vitro acetylation assays were repeated with two different intra-DBD GST constructs [amino acid residues 101–137 (30 kDa; in panel C) and 143–170 (29 kDa; in panel D)] to further map sites of acetylation by PCAF. K129, K160, and K162 were identified as acetylation sites for PCAF. D, 293 cells were transfected to express either GST-ERRα full-length wild type (WT) or a mutant (4KR) in which lysine residues 129/138/160/162 were mutated to a nonacetylatable arginine. These constructs were both cotransfected with identical amounts of PCAF to enrich for PCAF-mediated acetylation. GST agarose was then used to purify equal amount of proteins that were then blotted for ERRα or acetylated lysine. E, Schema of the ERRα DBD region with the four acetylated lysines identified by our analysis. WB, Western blot.
A deacetylase screen identifies HDAC8 and Sirt1 as enhancers of ERRα function
Having identified ERRα acetylation by PCAF as a possible repressive PTM, we hypothesized that deacetylation of ERRα would restore/enhance its transcriptional activation function. To this end, we screened various deacetylation factors and assessed their impact on the ERRα/PGC-1α activity on two well-described ERRα target promoters (Got1 and Cycs). We restricted our focus to the HDAC family and chose representative members from class I (HDAC1, -3, -8), class II (HDAC4, -5, -10), and class III (Sirt1). PGC-1α is known to be acetylated and, consequently, was used as basal condition to identify the HDACs affecting ERRα specifically. As expected, introduction of most HDACs affected both basal and ERRα-dependent PGC-1α activity in a similar manner suggesting a contribution of PGC-1α (Fig. 4). However, HDAC8 led to a significant increase in ERRα-dependent activity on the Got1 promoter as compared with other class I (Fig. 4A) or class II HDACs which affected the ERRα-PGC-1α combination in the exact same manner as PGC-1α alone (Fig. 4B). Also, the introduction of Sirt1, which is a known PGC-1α deacetylase ( 14), produced an increase in the transcriptional activation of the Got1 promoter in the presence of ERRα as compared with PGC1α alone (Fig. 4C). The same effect of HDACs overexpression was observed on the Cycs promoter (Fig. 4, D–F). In all cases, HDAC8 and Sirt1 produced coactivation in the presence of ERRα compared with PGC-1α alone. Our results demonstrate that at least two distinct proteins with deacetylase activity, HDAC8 and Sirt1, preferentially act to enhance the activity of ERRα in reporter gene assays.
Fig. 4.

A deacetylase screen identifies HDAC8 and Sirt1 as activators of ERRα. Panel A, Using the Got1 reporter, ERRα and PGC-1α were cotransfected with an empty vector [control (C)] or a series of class I HDACs in COS-1 cells. Panel B, Same as in Panel A, but with class II HDACs. Panel C, ERRα and PGC-1α were cotransfected with an empty vector (C) or the Sirt1 deacetylase together with the Got1 reporter. Panels D–F, Same as panels A–C but with Cycs reporter. ***, P < 0.001; **, P < 0.01; *, P < 0.05; Student’s t test.
HDAC8 and Sirt1 interact with and deacetylate ERRα and increases ERRα DNA-binding activity
We next investigated whether HDAC8 and Sirt1 could directly interact with and deacetylate ERRα. First, we used in vitro translated and [35S]-radiolabeled HDAC8 or Sirt1 and subjected these mixes to a pull-down assay using GST alone as a control protein, or GST-ERRα-DBD. As shown in Fig. 5, A and B, we observed a direct interaction between the DBD of ERRα and Sirt1 or HDAC8, respectively. Furthermore, in vitro acetylated GST-ERRα DBD was significantly deacetylated in vitro by recombinant Sirt1 and HDAC8 (Fig. 5C). We next assessed the effect of in vitro deacetylation directly on DNA binding using highly purified acetylated Flag-ERRα produced from mouse fibroblasts, in which the acetylation state of ERRα is confirmed (Fig. 1F). The purification was done in stringent acetylation-preserving radioimmune precipitation assay buffer, and the beads were washed extensively to prevent the coimmunoprecipitation of other proteins. Equal amounts of the purified Flag-ERRα protein were incubated with recombinant HDAC8, Sirt1, or buffer control in a deacetylation assay and used in a DNA-binding assay with a radiolabeled probe encoding the Got1 ERR response element (ERRE). An increase in DNA binding was observed after deacetylation by both Sirt1 and HDAC8 (Fig. 5D), indicating that the ERRα acetylation/deacetylation status directly modulates the DNA-binding affinity of ERRα in vitro. We then investigated the functional consequence of the interaction between ERRα and Sirt1 or HDAC8 in cells. The deacetylase and the receptor were cotransfected, and the cell lysate was used in a DNA-binding assay with the Got1 ERRE. In a manner similar to the in vitro deacetylated Flag-ERRα, an increase in DNA-binding activity of ERRα was observed in the presence of either Sirt1 or HDAC8 (Fig. 5E).
Fig. 5.
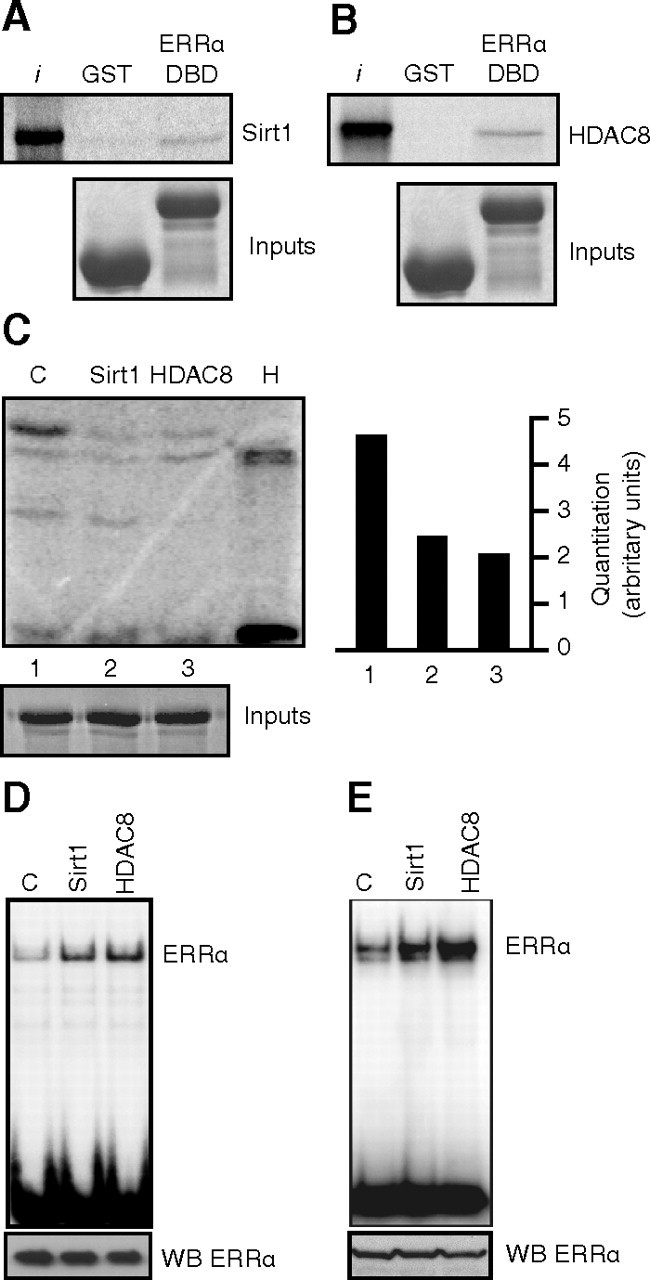
HDAC8 and Sirt1 interact with and deacetylate ERRα. Panel A, GST pull-down experiment demonstrates direct interactions between the ERRα DBD and Sirt1. Equal amounts of GST vector or GST-ERRα-DBD proteins were incubated with 35S-radiolabeled in vitro translated Sirt1. Bottom panels show equal loading of GST proteins by Coomassie. Panel B, Same as in panel A but with HDAC8. C, In vitro deacetylation assay. GST-ERRα-DBD was incubated with recombinant Sirt1 (with NAD+ cofactor), HDAC8, or buffer control in an in vitro acetylation assay, and levels of acetylation were quantified by phosphorimaging (right panel). Bottom panel is Coomassie staining showing equal loading of GST-ERRα-DBD. Panel D, DNA-binding assay using a 32P-labeled probe encoding the Got1 ERRE was performed using highly purified acetylated Flag-ERRα subsequently subjected to in vitro deacetylation by Sirt1 or HDAC8 (as in panel C, but without recombinant PCAF). The bottom panel displays an immunoblot for ERRα from the same lysates to show equal loading. Panel E, Same as in panel D but using 1 μg total cell lysates from 293 cells transfected with ERRα and either vector control, Sirt1, or HDAC8. The bottom panel displays an immunoblot for ERRα from the same lysates to show equal loading. WB, Western blot; I, input.
Next, we used mouse liver extracts and by coimmunoprecipitation, we observed that ERRα interacted with Sirt1 or HDAC8 in vivo (Fig. 6, A and B, respectively). To further confirm the role of Sirt1 and HDAC8 as bona fide ERRα deacetylases, we next assessed the effect of the removal of Sirt1 or HDAC8 by small interfering RNA (siRNA) on the acetylation states of ERRα. We observed that the knockdown of Sirt1 (Fig. 6C) and HDAC8 (Fig. 6D) increased the acetylation levels of ERRα. To determine the functional consequence of this increase in ERRα acetylation, we performed an ERRα chromatin immunoprecipitation (ChIP) assay in the presence of the siRNAs targeting Sirt1 or HDAC8. In both cases, and consistent with our in vitro observations, the knockdown of Sirt1 or HDAC8 (Fig. 6, E and F, respectively) led to a significant reduction of ERRα binding on the Got1 promoter (Fig. 6, E and F). The knockdown efficiencies were assessed by real-time qPCR and Western blotting (Supplemental Fig. 2).
Fig. 6.
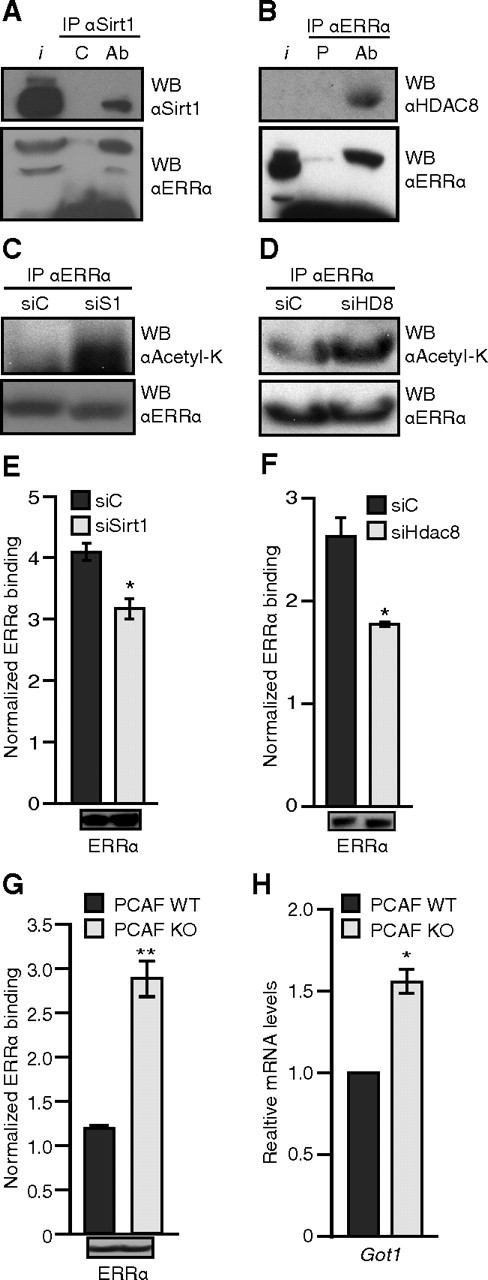
ERRα acetylation state correlates inversely with DNA-binding activity and Got1 expression. Panel A, ERRα interacts with Sirt1. Lysate from mouse liver was immunoprecipitated with either control antihemagglutinin antibody (C) or anti-Sirt1 antibody (Ab) and blotted for Sirt1 (top panel) and ERRα (bottom panel). Panel B, ERRα interacts with HDAC8. Lysate from mouse liver was immunoprecipitated with either preimmune (P) or anti-ERRα serum (Ab) and blotted for HDAC8 (top panel) and ERRα (bottom panel). Panel C, Endogenous ERRα was immunoprecipitated as in panel B but from Hepa1-6 mouse hepatocytes treated with Sirt1 siRNA (siS1) for 48 h and treated with TSA for 4 h before harvest and blotted for antiacetyl-lysine (top panel) and anti-ERRα (bottom panel). Panel D, Endogenous ERRα was immunoprecipitated as in panel B but from 293 cells treated with HDAC8 siRNA (siHD8) for 48 h and treated with TSA for 4 h before harvest and blotted for antiacetyl-lysine (top panel) and anti-ERRα (bottom panel). Panel E, ChiP assay for ERRα binding on Got1 promoter was performed from Hepa1–6 mouse hepatocytes treated with Sirt1 siRNA, as in panel C. The bottom panel shows the ERRα levels in the ChIP input. Panel F, Same as in panel E for HDAC8 in 293 cells. The bottom panel shows the ERRα levels in the ChIP input. Panel G, ChiP assay for ERRα binding on Got1 promoter was performed from wild-type (WT) and PCAF-null (KO) immortalized fibroblasts treated with TSA for 7 h before harvest. The bottom panel shows the ERRα levels in the ChIP input. Panel H, Relative expression of Got1 in the wild-type (WT) and PCAF null (KO) immortalized fibroblasts treated with TSA for 7 h before harvest. IP, Immunoprecipitation; WB, Western blot; I, input. *, P < 0.05; **, P < 0.01; Student’s t test vs. control.
To further confirm the mechanism and assess the effect of the absence of PCAF directly on ERRα DNA binding in vivo, we next performed an ERRα ChIP assay in the wild-type vs. PCAF-null fibroblasts. Whereas similar ERRα expression was observed between the two cell lines (Fig. 6G, bottom inset), the normalized DNA binding ratio of ERRα to the Got1 promoter was increased 2.4-fold in comparison with the wild-type fibroblasts (Fig. 6G). This result correlates with the acetylation levels of ERRα observed in these cells (Fig. 1F) and corresponds to an elevated level of Got1 mRNA in the PCAF-null cells (Fig. 6H). Taken together, our data support a dynamic acetylation/deacetylation switch model whereby acetylation of the ERRα DBD by PCAF weakens its DNA-binding activity whereas, conversely, the deacetylation of the ERRα DBD by Sirt1 or HDAC8 potentiates its DNA-binding activity.
Discussion
Our in vivo and in vitro studies identified acetylation as a PTM regulating the activity of ERRα and three new interacting coregulatory partners of ERRα. Specifically, our results show that PCAF acetylates ERRα on four lysines located in the DBD and that Sirt1 and HDAC8 deacetylate ERRα. The acetylation status of ERRα correlates with changes in DNA-binding and transcriptional activity. Acetylation can thus be added to the increasingly complex PTM code regulating the activity of ERRα.
A growing body of evidence links acetylation to nuclear receptor regulation and metabolic control, mainly through Sirt1 ( 28). However, the outcomes and mechanisms related to nuclear receptor acetylation reported to date display variability among members of the superfamily. In most cases, acetylation of nuclear receptors is associated with activation via various mechanisms. For example, the ligand-induced acetylation of the androgen receptor hinge region by the p300 coactivator was shown to enhance its activity by decreasing its interaction with the nuclear receptor corepressor ( 29, 30, 31). Consistent with this observation, Sirt1 was shown to deacetylate the androgen receptor and reduce its activity ( 32). As observed for ERRα, acetylation of the estrogen receptor α (ERα) was shown to repress its activity ( 33). However, the modified sites involved in this effect were mapped to the hinge region and acetylated by p300. Additionally, p300 was also shown to acetylate the DBD region of ERα on K266/K268 and to increase its DNA-binding capacity, whereas Sirt1 was shown to deacetylate ERα ( 34). In contrast, our findings show that acetylation of ERRα decreases its DBD activity and that deacetylation by Sirt1 positively regulates its transcriptional activity. Activation of nuclear receptor function by Sirt1 has been previously demonstrated for the liver X receptor factor, but in this case, the effect was mapped to a lysine adjacent to the domain encoding activation function 2 located within the ligand-binding domain ( 35), suggesting that the effect on liver X receptor involves changes in coregulator interactions rather than an effect on DNA binding. In ERRα, the acetylated lysines are located within the second zinc finger and in the carboxy-terminal extension (CTE) region of the DBD. These lysines are also conserved within ERRβ and -γ (Supplemental Fig. 1). Although the tertiary structure of DNA-bound ERRα is still unresolved, the elucidated solution structure of the DNA-bound ERRβ DBD ( 36) can provide insights into the mechanism described herein, owing to the high level of sequence identity of the ERRs DBDs (Supplemental Fig. 1). Indeed, based on the sequence conservation between the ERRs, acetylated ERRα K129 would sit directly adjacent to residues that contact the phosphate backbone of DNA, and ERRα K160/K162 would sit directly adjacent to the part of the AT-hook motif buried in the DNA minor groove, potentially influencing these interactions. The negative charge brought in by the acetyl group could produce an effect of charge neutralization, potentially decreasing the affinity of the receptor for the negatively charged DNA backbone. A similar effect of an adjacent PTM has been reported to affect DNA-binding affinity: a phosphorylation event on the DBD of NGFI-B (NR4A1), also bringing a negative charge, occurring three residues upstream of the AT-hook minor groove-binding motif leads to a significantly decreased DNA-binding affinity of the receptor ( 37). The acetylated lysines in ERRα CTE (K160/162) are also conserved within ERα (Supplemental Fig. 1).
Interestingly, although ERα and ERRα share two acetylated lysines within their respective DBDs, acetylation activates ERα but represses ERRα. This difference in the effect of acetylation on DNA-binding properties of ERα and ERRα may be important for target gene discrimination by the two related receptors. Our laboratory has recently demonstrated that despite ERRα’s ability to bind to the estrogen response element in vitro, few binding sites are recognized by both ERα and ERRα in vivo in intact chromatin ( 38). In fact, common target sites bound by both receptors are those that harbor a sequence that incorporates both an ERRα and ERα response element, on which the two receptors can compete for binding. It would be of interest to determine whether acetylation of ERRα and ERα is a determinant of binding site selectivity by the receptors on such elements.
Another point of interest is the physiological link between ERRα and Sirt1 on metabolism. Our finding that the activity of ERRα is modulated by an acetylation/deacetylation switch involving Sirt1 is consistent with the critical roles played by these proteins in metabolic control. A strong association exists between caloric restriction and metabolic health, which has been linked to the activity of sirtuins. The expression levels of both Sirt1 and ERRα have both been shown to be affected by fasting and long-term caloric restriction ( 8, 15, 24). In addition, both Sirt1 and ERRα have been implicated in the control of glucose and fat metabolism and as key regulators of energy homeostasis ( 1, 3, 19, 21, 39). Thus, the functional interaction between ERRα and Sirt1 and the effect of ERRα acetylation on its activity described in this study confer a cogent rationale for more in-depth investigations of the role of ERRα in aging-related diseases, an aspect of ERRα biology not yet explored.
Whereas knowledge on the function played by Sirt1 in physiology is increasing rapidly, HDAC8, on the other hand, is a poorly studied factor with little known beyond basic biochemical and tissue-expression studies. Our finding that HDAC8 is a regulator of ERRα activity thus opens a novel avenue for future investigation of HDAC8 in metabolic control and related diseases. Our study, although showing that HDAC8 and Sirt1 act as deacetylases for the DBD of ERRα, precludes neither further testing of the remaining members of the HDAC family in different experimental or physiological contexts nor the occurrence of alternative acetylation-dependent mechanisms involved in ERRα regulation. Finally, acetylation of ERRα is not an off/on switch but, as previously observed for the interaction between phosphorylation and sumoylation ( 14), is likely to act in combination with other PTMs to more precisely control the activity of ERRα and its transcriptional outcomes on specific genes and biochemical pathways. Only when all of these modifications are mapped and considered together will we be able to define the entire ERRα PTM code and predict with confidence its effects on metabolic control. Drugs targeting specific ERRα forms can then be generated to shift metabolism toward specific ERRα-regulated pathways in response to physiological and metabolic cues. Acetylation of ERRα may ultimately provide an additional means to modulate ERRα function in several disease states such as cancer, diabetes, or the metabolic syndrome via targeted therapies.
Materials and Methods
Cell lines and transfection assays
COS-1 and 293 cells, as well as Hepa1–6 mouse hepatocytes and wild-type and PCAF-null immortalized fibroblasts, were all maintained in DMEM supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin, in a 5% CO2 environment. For transfection cells were routinely seeded the night before and transfected using Fugene (Roche Canada, Laval, QC, Canada), according to manufacturer’s guidelines. For siRNA transfection, the cells were plated the night before transfection of the indicated siRNA (Smartpools On target plus; Dharmacon, Lafayette, CO) using Hiperfect (QIAGEN, Mississauga, ON, Canada), according to the manufacturer’s instructions.
Plasmids
Human ERRα amino-terminal domain (expressing amino acid residues 1–74), DBD (amino acid residues 68–173), and carboxy-terminal domain (amino acid residues 165–end) were cloned into the vector pGEX-2T as _Bam_HI/_EcoR_I fragments. Equivalent DBDs for human ERRβ and ERRγ were also cloned as _Bam_HI/_Eco_RI fragments into the same vector. GST-ERRα was inserted into the vector pcDNA3 as a _Kpn_I/_Eco_RI fragment for expression in mammalian cells. HDAC plasmids were a kind gift from Dr. X. J. Yang (McGill University). PCAF and Sirt1 plasmids were obtained from Addgene, Inc. (Cambridge, MA). The ERRα K129/138/160/162R mutant was engineered by PCR mutagenesis and verified by sequencing. pGL3-Got1 and pGL3-Cycs as well as CMX-myc-ERRα have been described previously ( 4).
In vitro acetylation/deacetylation assays
Recombinant PCAF acetylation-domain, HDAC8, and Sirt1 proteins were obtained from Biomol International (Enzo Life Sciences International, Plymouth Meeting, PA). Bacterially expressed GST-purified construct (1 μg) was incubated with recombinant PCAF protein and 14C-labeled acetyl-CoA at 30 C for 45 min in histone acetyltransferase buffer [50 mm Tris-HCl (pH 8), 10% glycerol, 0.1 mm EDTA, 1 mm dithiothreitol, 1 mm phenylmethylsulfonylfluoride; 300 nm Trichostatin A (TSA)], with agitation to resuspend the beads every 5 min. Samples were then boiled in protein loading buffer to stop the reaction and run on a 12% SDS-PAGE gel, fixed in MeOH-acetic acid and washed with amplify solution for 30 min (GE Healthcare, Piscataway, NJ) before being dried down and exposed to a phosphorimaging screen. Histones were used as a positive control for acetylation, and GST alone was the negative control. For the deacetylation assay, the acetylation assay described above also included HDAC8 or Sirt1 (plus 10 μm NAD+ cofactor) or buffer control, and TSA was excluded from all buffers. In all cases, protein levels were monitored using a separate gel for Coomassie staining.
Immunoprecipitations and immunoblots
CMX-myc-ERRα-transfected 293 cells were harvested in buffer B [150 mm NaCl, 20 mm Tris (pH 8), 0.1% Nonidet P-40 (NP-40), 5 mm MgCl2, 10% glycerol, 300 nm TSA, 10 mm nicotinamide, 1× complete protease inhibitors], immunoprecipitated at 4 C for 1 h with equal amounts of anti-ERRα or preimmune serum, and blotted for ERRα ( 40), PCAF (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), or acetyl-lysine (Cell Signaling Technology, Inc., Beverly, MA).
Alternatively, pcDNA3-GST-ERRα or the acetylation-deficient mutant was transfected into 15-cm plates of 293 cells (or PCAF-null fibroblasts). For wild type, 10 μg was used in fibroblast cells, and 2 μg was used in 293 cells (+8 μg pcDNA3 vector) vs. 10 μg pcDNA3-GST-ERRα K129/138/160/162R mutant. PCAF expression vector (5 μg) was also cotransfected into 293 cells. Cells were harvested 48 h later, by scraping in buffer B+ (500 mm NaCl; 20 mm Tris, pH 8; 1% NP-40; 10% glycerol; 5 mm MgCl2; 0.1% Triton X-100; 300 nm TSA; 10 mm nicotinamide; 1× complete protease inhibitor cocktail; 500 μl per plate), having been incubated in a mix of 10 mm nicotinamide and 300 nm TSA (by addition to cell culture medium) for 16 h before harvesting. After harvesting, cells were briefly sonicated to break apart precipitation, and the debris was pelleted by centriguation at 4 C. Remaining lysate was incubated with 50 μl of 50% GST-bead slurry, rotating at 4 C for 2 h. Saturated beads were then centrifuged, transferred to Eppendorf tubes and washed seven times in buffer B+ before being boiled in 40 μl sample loading buffer (30 μl used for acetyl-lysine immunoblot, and 10 μl for the ERRα immunoblot). Samples were then run on an 8% SDS-PAGE gel. Acetyllysine monoclonal (9681) and polyclonal (9441) antibodies were obtained from Cell Signaling Technology and used according to manufacturer’s guidelines. PCAF antibody (H-369) was obtained from Santa Cruz Biotechnology. Visualization was performed by chemiluminescence using Lumi-Light, or Lumi-LightPLUS (Roche).
For in vivo immunoprecipitations, 2- to 3-month-old male C57/Bl6HSD mouse liver was prepared in modified buffer K (20 mm phosphate buffer, pH 7; 250 mm NaCl; 0.1% NP-40; 0.1% Triton X-100; 5 mm EDTA, pH 8.3; 1 mm phenylmethylsulfonylfluoride; 100 mm anacardic acid; 300 nm TSA; 10 mm nicotinamide; and 1× complete miniprotease inhibitor tablet). Briefly, mouse livers were homogenized on ice in modified buffer K for 3 × 15 sec using a polytron-type homogenizer (IKA T-10 basic ultra-turrax). After a 1-h incubation period at 4 C under constant rotation, the crude homogenate was centrifuged at 4 C for 10 min at 10,000 rpm. The concentration of the total liver extract (supernatant) was determined by the Bradford method, and 2 mg of protein were subjected to a 20-min preclearing step with 50 μl of 50% protein G agarose slurry followed by immunoprecipitation in modified buffer K using 6 μl of preimmune serum or 4 μl of anti-ERRα antiserum for 1 h at 4 C under constant rotation. The ERRα immunocomplexes were then immobilized on protein G agarose resin for 45 min at 4 C under constant rotation, pelleted by centrifugation, washed three times with 500 μl of modified buffer K, and solubilized by boiling in reducing sodium dodecyl sulfate sample buffer for separation by SDS-PAGE and detection by autoradiography. The anti-ERRα antiserum was used at 1:10,000 and the anti-acetylated-lysine mouse monoclonal antibody (Cell Signaling Technology, catalog no. 9681) was used according to the manufacturer’s instruction for subsequent Western blot analysis. Blotting for PCAF (Santa Cruz Biotechnology, catalog no. SC8999) involved stripping the acetylation blot, washing in TBS, blocking in 3% BSA for 1 h at room temperature, and then blotting overnight at 4 C with anti-PCAF antibody. The Sirt1 and HDAC8 antibodies were obtained from Santa Cruz Biotechnology (catalog nos. SC74465 and SC17778, respectively).
In vitro GST pull-down assay
HDAC8 or Sirt1 was in vitro translated with [35S]methionine and incubated with GST-ERRα-DBD for 2 h in GST-pull-down NET-N buffer (150 mm NaCl; 1 mm EDTA; 50 mm Tris, pH 8; 1% Triton X-100; 1× complete protease inhibitors). Samples were washed seven times, then run on an SDS-PAGE gel, fixed in methanol-acetic acid, and exposed overnight to a phosphorimaging screen.
Reporter gene assays
Cloning and characterization of the luciferase reporter genes have previously been reported ( 4). Additionally, COS-1 cells were transfected with 200 ng of either PCAF, HDAC, or Sirt1 expression vectors where indicated.
Mass spectrometry
In vitro acetylation assay was performed as above, on the GST-ERRα-DBD recombinant protein, except using nonradioactive acetyl-CoA as the substrate. Five different acetylation assays were then recombined and run on a SDS-PAGE gel. Proteins were stained with Coomassie G250 and cut from the SDS-PAGE gel for analysis. Liquid chromatography-mass spectrometry was performed after tryptic digestion, using a LC-QToF micro machine (Genome Quebec, Quebec, Canada).
EMSA
EMSA using the Got1 ERRE probe derived from the murine promoter has been described previously ( 4).
ChIP assays
Chromatin was prepared from wild-type and PCAF-null fibroblasts (treated with 300 nm TSA for 7 h), Hepa 1–6 cells, and from 293 cells (transfected with the indicated siRNA for 48 h and treated with TSA 4 h before harvest). ChIP was performed as described previously ( 41). Quantification of ChIP enrichment by real-time Q-PCR was carried out using the LightCycler 480 instrument (Roche).
Acknowledgments
We thank Dr. X. J. Yang (McGill University, Montréal, Québec, Canada) for HDAC expression plasmids, Dr. A. Kralli (Scripps, La Jolla, CA) for pcDNA3-HA-PGC1α, and Dr. J. DeCaprio (Harvard University, Cambridge, MA) for wild-type and PCAF-null immortalized fibroblasts.
NURSA Molecule Pages:
- Coregulators: P/CAF | Sirt1;
- Nuclear Receptors: ERR-α.
Footnotes
This work was supported by an operating grant (MOP-77763) from the Canadian Institutes for Health Research (to V.G.) and an infrastructure grant by the Fonds de le Recherche en Santé du Québec. A.M.T. was supported by a Canada Graduate Scholarship doctoral award from Canadian Institutes for Health Research. B.J.W. was supported by a postdoctoral fellowship from the Research Institute of the McGill University Health Centre; and G.D. was supported by a doctoral studentship award from Fonds de le Recherche en Santé du Québec.
Disclosure Summary: The authors have nothing to disclose.
First Published Online May 19, 2010
1
B.J.W. and A.M.T. contributed equally to this work.
Abbreviations: ChIP, Chromatin immunoprecipitation; CoA, coenzyme A; CTE, C-terminal extension; DBD, DNA-binding domain; ERα, estrogen receptor-α; ERR, estrogen-related receptor; ERRE, ERR response element; GST, glutathione-_S_-transferase; HDAC, histone deacetylase; 4KR, mutant GST-tagged full-length ERRα in which lysines 129/138/160/162 were mutated to arginines; MEFs, mouse embryonic fibroblasts; NP-40, Nonidet P-40; PCAF, p300/CBP associated factor; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α; PTM, posttranslational modification; siRNA, small interfering RNA; Sirt1, sirtuin 1 homolog; TSA, Trichostatin A.
References
- 1.Giguère V2008. Transcriptional control of energy homeostasis by the estrogen-related receptors. Endocr Rev 29:677–696 [DOI] [PubMed] [Google Scholar]
- 2.Villena JA, Kralli A2008. ERRα: a metabolic function for the oldest orphan. Trends Endocrinol Metab 19:269–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Charest-Marcotte A, Dufour CR, Wilson BJ, Tremblay AM, Eichner LJ, Arlow DH, Mootha VK, Giguère V2010. The homeobox protein Prox1 is a negative modulator of ERRα/PGC-1α bioenergetic functions. Genes Dev 24:537–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguère V2007. Genome-wide orchestration of cardiac functions by orphan nuclear receptors ERRα and γ. Cell Metab 5:345–356 [DOI] [PubMed] [Google Scholar]
- 5.Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguère V, Murphy E, Kelly DP2007. The nuclear receptor ERRα is required for the bioenergetic and functional adaption to cardiac pressure overload. Cell Metab 6:25–37 [DOI] [PubMed] [Google Scholar]
- 6.Luo J, Sladek R, Carrier J, Bader JA, Richard D, Giguère V2003. Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor α. Mol Cell Biol 23:7947–7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Villena JA, Hock MB, Chang WY, Barcas JE, Giguère V, Kralli A2007. Orphan nuclear receptor ERRα is essential for adaptive thermogenesis. Proc Natl Acad Sci USA 104:1418–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranhotra HS2009. Up-regulation of orphan nuclear estrogen-related receptor α expression during long-term caloric restriction in mice. Mol Cell Biochem 332:59–65 [DOI] [PubMed] [Google Scholar]
- 9.Yang XJ, Seto E2008. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 31:449–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barry JB, Giguère V2005. Epidermal growth factor-induced signaling in breast cancer cells results in selective target gene activation by orphan nuclear receptor estrogen-related receptor α. Cancer Res 65:6120–6129 [DOI] [PubMed] [Google Scholar]
- 11.Sladek R, Bader JA, Giguère V1997. The orphan nuclear receptor estrogen-related receptor α is a transcriptional regulator of the human medium-chain acyl coenzyme A dehydrogenase gene. Mol Cell Biol 17:5400–5409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villèn J, Li J, Cohn MA, Cantley LC, Gygi SP2004. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci USA 101:12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villèn J, Beausoleil SA, Gerber SA, Gygi SP2007. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci USA 104:1488–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tremblay AM, Wilson BJ, Yang XJ, Giguère V2008. Phosphorylation-dependent sumoylation regulates ERRα and γ transcriptional activity through a synergy control motif. Mol Endocrinol 22:570–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P2005. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434:113–118 [DOI] [PubMed] [Google Scholar]
- 16.Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P2006. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1α. Cell Metab 3:429–438 [DOI] [PubMed] [Google Scholar]
- 17.Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A2004. The estrogen-related receptor α (ERRα) functions in PPARγ coactivator 1α (PGC-1α)-induced mitochondrial biogenesis. Proc Natl Acad Sci USA 101:6472–6477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sonoda J, Laganière J, Mehl IR, Barish GD, Chong LW, Li X, Scheffler IE, Mock DC, Bataille AR, Robert F, Lee CH, Giguère V, Evans RM2007. Nuclear receptor ERRα and coactivator PGC-1β are effectors of IFN-γ induced host defense. Genes Dev 21:1909–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J2008. Specific SIRT1 activation mimics low energy levels and protects against diet- induced metabolic disorders by enhancing fat oxidation. Cell Metab 8:347–358 [DOI] [PubMed] [Google Scholar]
- 20.Purushotham A, Schug TT, Xu Q, Surapureddi S, Guo X, Li X2009. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab 9:327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P2007. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J 26:1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hou X, Xu S, Maitland-Toolan KA, Sato K, Jiang B, Ido Y, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, Cohen RA, Zang M2008. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem 283:20015–20026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lan F, Cacicedo JM, Ruderman N, Ido Y2008. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem 283:27628–27635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L2008. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev 22:1753–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith JJ, Kenney RD, Gagne DJ, Frushour BP, Ladd W, Galonek HL, Israelian K, Song J, Razvadauskaite G, Lynch AV, Carney DP, Johnson RJ, Lavu S, Iffland A, Elliott PJ, Lambert PD, Elliston KO, Jirousek MR, Milne JC, Boss O2009. Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst Biol 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blanco JC, Minucci S, Lu J, Yang XJ, Walker KK, Chen H, Evans RM, Nakatani Y, Ozato K1998. The histone acetylase PCAF is a nuclear receptor coactivator. Genes Dev 12:1638–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poulin DL, Kung AL, DeCaprio JA2004. p53 targets simian virus 40 large T antigen for acetylation by CBP. J Virol 78:8245–8253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C, Powell MJ, Popov VM, Pestell RG2008. Acetylation in nuclear receptor signaling and the role of sirtuins. Mol Endocrinol 22:539–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu M, Rao M, Wang C, Sakamaki T, Wang J, Di Vizio D, Zhang X, Albanese C, Balk S, Chang C, Fan S, Rosen E, Palvimo JJ, Jänne OA, Muratoglu S, Avantaggiati ML, Pestell RG2003. Acetylation of androgen receptor enhances coactivator binding and promotes prostate cancer cell growth. Mol Cell Biol 23:8563–8575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu M, Wang C, Wang J, Zhang X, Sakamaki T, Yeung YG, Chang C, Hopp T, Fuqua SA, Jaffray E, Hay RT, Palvimo JJ, Jänne OA, Pestell RG2002. Androgen receptor acetylation governs trans activation and MEKK1-induced apoptosis without affecting in vitro sumoylation and trans-repression function. Mol Cell Biol 22:3373–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gong J, Zhu J, Goodman Jr OB, Pestell RG, Schlegel PN, Nanus DM, Shen R2006. Activation of p300 histone acetyltransferase activity and acetylation of the androgen receptor by bombesin in prostate cancer cells. Oncogene 25:2011–2021 [DOI] [PubMed] [Google Scholar]
- 32.Fu M, Liu M, Sauve AA, Jiao X, Zhang X, Wu X, Powell MJ, Yang T, Gu W, Avantaggiati ML, Pattabiraman N, Pestell TG, Wang F, Quong AA, Wang C, Pestell RG2006. Hormonal control of androgen receptor function through SIRT1. Mol Cell Biol 26:8122–8135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang C, Fu M, Angeletti RH, Siconolfi-Baez L, Reutens AT, Albanese C, Lisanti MP, Katzenellenbogen BS, Kato S, Hopp T, Fuqua SA, Lopez GN, Kushner PJ, Pestell RG2001. Direct acetylation of the estrogen receptor α hinge region by p300 regulates transactivation and hormone sensitivity. J Biol Chem 276:18375–18383 [DOI] [PubMed] [Google Scholar]
- 34.Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL2006. Acetylation of estrogen receptor α by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol 20:1479–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L2007. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell 28:91–106 [DOI] [PubMed] [Google Scholar]
- 36.Gearhart MD, Holmbeck SM, Evans RM, Dyson HJ, Wright PE2003. Monomeric complex of human orphan estrogen related receptor-2 with DNA: a pseudo-dimer interface mediates extended half-site recognition. J Mol Biol 327:819–832 [DOI] [PubMed] [Google Scholar]
- 37.Hirata Y, Kiuchi K, Chen HC, Milbrandt J, Guroff G1993. The phosphorylation and DNA-binding of the DNA-binding domain of the orphan nuclear receptor NGFI-B. J Biol Chem 268:24808–24812 [PubMed] [Google Scholar]
- 38.Deblois G, Hall JA, Perry MC, Laganière J, Ghahremani M, Park M, Hallett M, Giguère V2009. Genome-wide identification of direct target genes implicates estrogen-related receptor α as a determinant of breast cancer heterogeneity. Cancer Res 69:6149–6157 [DOI] [PubMed] [Google Scholar]
- 39.Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J2006. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 127:1109–1122 [DOI] [PubMed] [Google Scholar]
- 40.Laganière J, Tremblay GB, Dufour CR, Giroux S, Rousseau F, Giguère V2004. A polymorphic autoregulatory hormone response element in the human estrogen related receptor α (ERRα) promoter dictates PGC-1α control of ERRα expression. J Biol Chem 279:18504–18510 [DOI] [PubMed] [Google Scholar]
- 41.Laganière J, Deblois G, Giguère V2005. Functional genomics identifies a mechanism for estrogen activation of the retinoic acid receptor α1 gene in breast cancer cells. Mol Endocrinol 19:1584–1592 [DOI] [PubMed] [Google Scholar]