Anti-VEGF therapy induces ECM remodeling and mechanical barriers to therapy in colorectal cancer liver metastases (original) (raw)
. Author manuscript; available in PMC: 2017 Oct 12.
Published in final edited form as: Sci Transl Med. 2016 Oct 12;8(360):360ra135. doi: 10.1126/scitranslmed.aaf5219
Abstract
The survival benefit of anti–vascular endothelial growth factor (VEGF) therapy in metastatic colorectal cancer (mCRC) patients is limited to a few months because of acquired resistance. We show that anti-VEGF therapy induced remodeling of the extracellular matrix with subsequent alteration of the physical properties of colorectal liver metastases. Preoperative treatment with bevacizumab in patients with colorectal liver metastases increased hyaluronic acid (HA) deposition within the tumors. Moreover, in two syngeneic mouse models of CRC metastasis in the liver, we show that anti-VEGF therapy markedly increased the expression of HA and sulfated glycosaminoglycans (sGAGs), without significantly changing collagen deposition. The density of these matrix components correlated with increased tumor stiffness after anti-VEGF therapy. Treatment-induced tumor hypoxia appeared to be the driving force for the remodeling of the extracellular matrix. In preclinical models, we show that enzymatic depletion of HA partially rescued the compromised perfusion in liver mCRCs after anti-VEGF therapy and prolonged survival in combination with anti-VEGF therapy and chemotherapy. These findings suggest that extracellular matrix components such as HA could be a potential therapeutic target for reducing physical barriers to systemic treatments in patients with mCRC who receive anti-VEGF therapy.
INTRODUCTION
Systemic chemotherapy is the main treatment option for patients with inoperable metastatic colorectal cancer (mCRC). The effectiveness of chemotherapy depends on the delivery of the drugs into the tumor, which in turn is dependent on tumor blood perfusion (1–4). There is increasing evidence that the solid stress generated by proliferating cells in a growing tumor mass can cause compression of blood vessels and reduced perfusion (5). Components of the extracellular matrix (ECM) play an important role in the solid stress–induced blood vessel collapse because they transmit the mechanical stress created by proliferating cells within the confined space of a tumor (6). Targeting the ECM components has therefore been suggested as a strategy to improve perfusion, drug delivery, and, ultimately, outcomes in patients with solid malignancies (3, 7).
The anti–vascular endothelial growth factor (VEGF) antibody bevacizumab in combination with chemotherapy is the current standard of care for mCRC, based on an overall survival improvement (8). This survival benefit, however, is modest, and the disease ultimately progresses (9). The underlying mechanisms of acquired resistance to antiangiogenic therapy remain unclear (7). In part, this is a result of the limited understanding of the effects of anti-VEGF therapy on the microenvironment of metastatic lesions. Recent preclinical studies have shown that antiangiogenic therapy increases collagen expression in primary tumors, as a consequence of increasing hypoxia (10, 11). The effect of antiangiogenic therapy on the expression of noncollagenous matrix components such as hyaluronic acid [also known as hyaluronan (HA)] or sulfated glycosaminoglycans (sGAGs) in metastatic lesions is not well studied. As an abundant and highly hydrated matrix molecule with negatively charged chains that resist compression, HA has gathered increasing attention as a biologically relevant and potentially targetable cause of vessel compression and poor drug delivery in desmoplastic tumors (12–15). Recently, liver metastases from pancreatic cancer have been reported to be desmoplastic with high concentrations of HA and collagen that correlated with patients’ survival (16). Here, we investigated the effects of antiangiogenic therapy on the composition of the ECM, both collagenous and noncollagenous, and blood perfusion as mechanisms of acquired resistance to antiangiogenic therapy in liver mCRC.
RESULTS
Bevacizumab increases HA expression in human CRC liver metastases
Given the lack of data on expression of HA in human liver mCRC, we first performed immunohistochemical analyses of surgical specimens from mCRC patients who underwent metastasectomy. We found higher expression of HA in the metastases compared to that of the uninvolved liver parenchyma, where HA expression was restricted to the periportal fields (fig. S1). Next, we examined the impact of preoperative treatment on HA deposition in 49 liver metastases resected from 43 patients (table S1). Although there was no difference in HA between patients with and without preoperative chemotherapy, we found significantly increased HA expression in liver mCRC tissues from patients treated with preoperative bevacizumab and chemotherapy (P < 0.001) (Fig. 1, A and B). To further confirm this potential effect of bevacizumab, we analyzed serial samples from patients who underwent multiple liver resections for CRC liver metastases and carried out intraindividual comparisons of HA expression. These analyses confirmed the significant increase in HA expression after preoperative treatment including bevacizumab (P = 0.024, paired t test), with relatively low intensity of staining in samples from the same patients who were resected at different time points without previous exposure to anti-VEGF therapy (Fig. 1, C and D).
Fig. 1. Treatment with bevacizumab increases HA expression in human CRC liver metastases.
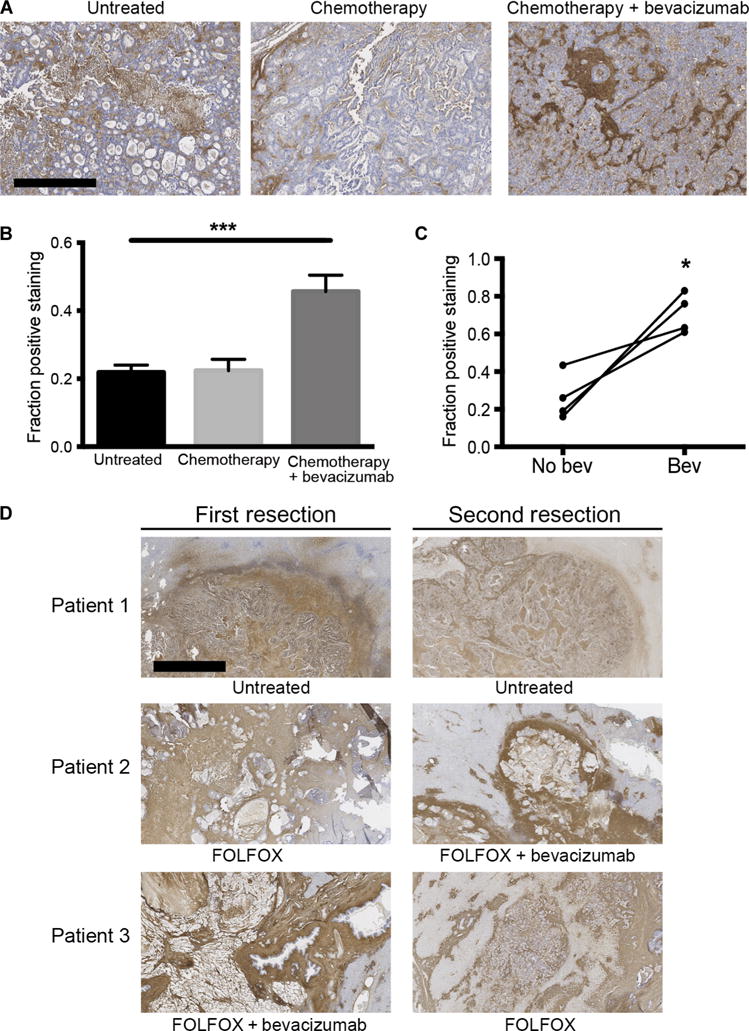
(A) Representative images showing HA expression in liver metastases from CRC patients: left, no treatment; middle, preoperative chemotherapy alone; right, preoperative chemotherapy in combination with the anti-VEGF antibody bevacizumab. Scale bar, 200 μm. (B) Immunohistochemical analysis of HA concentration in human CRC liver metastases [***P < 0.001, analysis of variance (ANOVA); n = 8 per group; mean ± SEM]. (C) Intraindividual comparisons of HA expression in paired samples from patients who underwent repeated resections for CRC liver metastases with and without preoperative bevacizumab treatment (*P = 0.024, paired t test; n = 4 per group; mean ± SEM). (D) Intraindividual comparison of HA expression in colorectal liver metastases of patients who underwent repeated liver resections after different preoperative treatments or no treatment. FOLFOX, folinic acid, 5-fluorouracil (5-FU), and oxaliplatin; FOLFIRI, folinic acid, 5-FU, and irinotecan. Scale bar, 200 μm. *P < 0.05 and ***P < 0.001 as compared to corresponding controls.
Anti-VEGF therapy alters mechanical properties of murine CRC liver metastasis
Next, to investigate the mechanisms and consequences of increased HA expression after VEGF-targeted therapy, we used two syngeneic mouse models of liver mCRC (SL4 and CT26) (17, 18). The effectiveness of anti-VEGF therapy was different in these models. Whereas treatment with B20.4-1.1 [a VEGF-blocking antibody abbreviated as B20 from here onward (19)] significantly prolonged survival of mice bearing liver metastatic SL4 tumors (median survival, 14.25 versus 10.4 days; P < 0.001, log-rank test), the survival advantage in the CT26 model did not reach statistical significance (median survival, 13.5 versus 12 days; _P_ = 0.072, log-rank test) (Fig. 2A). The macroscopic appearance of the metastatic lesions in the anti-VEGF treatment groups was different from that seen in the control groups and was characterized by a stiff texture, suggesting changes in the amount and/or composition of the ECM. We also found that the DNA content, normalized to wet weight, was significantly lower in SL4 (_P_ = 0.011) and CT26 (_P_ = 0.023) liver mCRCs of mice treated with B20, suggesting a shift in the cell-to-matrix ratio (fig. S2, A and B). To quantify the changes in the mechanical properties of liver mCRCs after anti-VEGF therapy, we performed stiffness measurements using unconfined compression tests of metastases harvested from animals treated with B20. These experiments confirmed a dose-dependent increase in tumor stiffness in SL4 (_P_ = 0.002) and CT26 (_P_ < 0.0001) liver mCRC models (Fig. 2B). In addition, a recently developed method tomeasure growth-induced solid stress (6) revealed significantly higher solid stress in anti-VEGF–treated mCRCs. The normalized mean tumor opening—a measure of solid stress—increased from 0.690 to 1.506 for SL4 tumors (_P_ = 0.018) and from 1.830 to 5.884 for CT26 tumors (_P_ = 0.019) (Fig. 2C). Anti-VEGF therapy caused >80% reduction in tumor perfusion (Fig. 2D). Together, these data indicate that anti-VEGF therapy (at 5 mg/kg) alters the mechanical properties of liver metastases by inducing tumor desmoplasia.
Fig. 2. Anti-VEGF therapy increases tumor desmoplasia, stiffness, and solid stress in colorectal liver metastases.
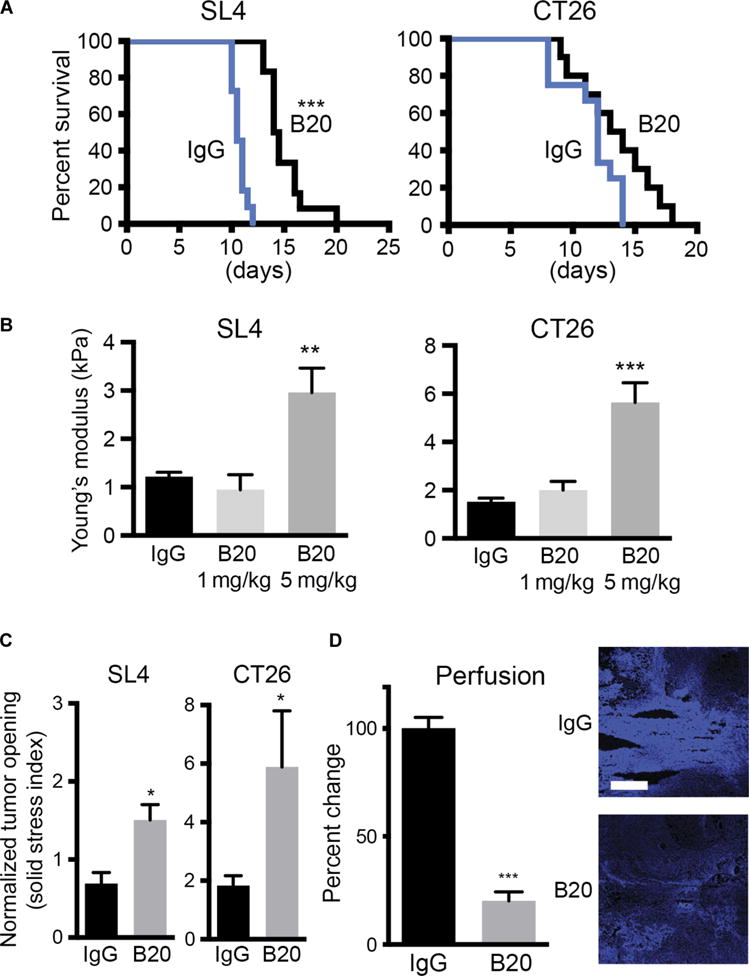
(A) Survival of C57BL/6 wild-type (WT) mice bearing SL4 liver metastases (***P < 0.001, log-rank test) and BALB/c WT mice bearing CT26 liver metastases (P = 0.072, log-rank test) treated with either B20 or control immunoglobulin G (IgG) (n = 10 to 12 per group). (B) Stiffness of SL4 (**P = 0.002, ANOVA) and CT26 (***P < 0.0001, ANOVA) liver metastases treated with IgG, a low dose of B20 (1 mg/kg; twice a week), or a high dose of B20 (5 mg/kg; twice a week) and harvested at the end of the survival study (n = 9 to 12 per group). (C) Solid stress measured as normalized tumor opening in SL4 (*P = 0.018, Student’s t test) and CT26 (*P = 0.01, Student’s t test) liver metastases treated with IgG or high dose of B20 (5 mg/kg; twice a week) and harvested at the terminal endpoint (n = 3 to 5 per group). (D) Effects of B20 therapy on perfusion of SL4 liver metastases in C57BL/6 WT mice after injection of Hoechst 33342 (***P = 0.0001, Student’s t test) (n = 4 to 7 per group). Data presented as percent change compared to controls. Scale bar, 200 μm. Data are shown as means ± SEM; *P <0.05, **P < 0.01, and ***P < 0.001 as compared to corresponding controls.
HA and sGAG in colorectal liver metastases increase after anti-VEGF treatment
To evaluate the effect of anti-VEGF therapy on the ECM, we first measured the relative abundance of the major ECM components. In line with the clinical findings of increased HA expression in human liver mCRCs after bevacizumab treatment, we found a threefold increase of HA in SL4 (0.503 ± 0.035 versus 1.766 ± 0.233 μg HA/mg tissue; P < 0.0001) and CT26 (0.055 ± 0.0108 versus 0.143 ± 0.014 μg HA/mg tissue; P < 0.0001) liver mCRCs in mice (Fig. 3A). In addition, the expression of CD44, the HA receptor, was increased in metastatic lesions of SL4 (P = 0.008) and CT26 (P = 0.005) tumors after anti-VEGF therapy (fig. S3, A and B). Moreover, we found a twofold increase in sGAG expression after anti-VEGF therapy in both mouse models (SL4: 0.231 ± 0.022 versus 0.43 ± 0.044 μg sGAG/mg tissue; P = 0.0005; CT26: 0.149 ± 0.021 versus 0.277 ± 0.028 μg sGAG/mg tissue; P = 0.001) (Fig. 3B). The increased expression of sGAG was then confirmed in liver mCRCs in patients treated with preoperative chemotherapy with and without bevacizumab (fig. S4). The increase in collagen deposition was less pronounced and reached statistical significance only in the SL4 model (P = 0.01) (Fig. 3C), a finding consistent with the data from patients with liver mCRCs after preoperative chemotherapy with and without bevacizumab (20). To rule out the possibility that the changes in the ECM are off-target effects related to anti-VEGF therapy, we treated non–tumor-bearing mice with B20 for 3 weeks. Histological analysis did not reveal obvious parenchymal changes in the liver (fig. S5A). Moreover, biochemical analysis of the liver tissue did not show major differences between the groups with respect to HA and sGAG abundance (fig. S5, B and C). We then sought to correlate the stiffness of the liver mCRCs with the presence of specific ECM components. We found a strong correlation between stiffness and tumor concentration of HA (SL4: r = 0.72; P < 0.0001; CT26: r = 0.554; P < 0.0001) or sGAG (SL4: r = 0. 665; P < 0.0001; CT26: r = 0.524; P = 0.0002) but no association with collagen content (Fig. 3D).
Fig. 3. Anti-VEGF treatment increases noncollagenous ECM in CRC liver metastases.
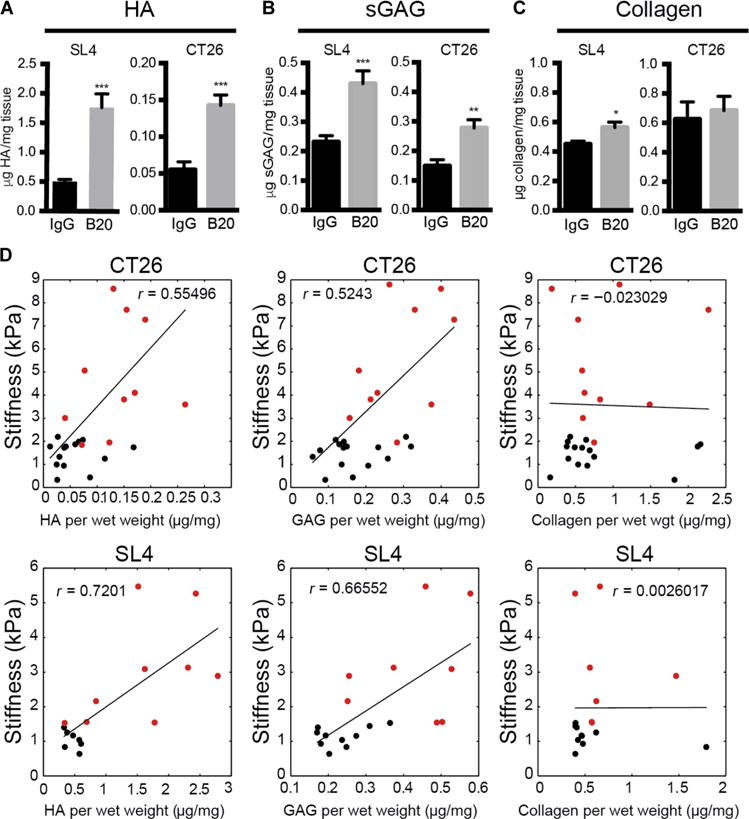
(A to C) Quantification of HA concentration by enzyme-linked immunosorbent assay (ELISA) (SL4, ***P < 0.0001; CT26, ***P < 0.0001, Student’s t test) (A), sGAG concentration by dimethylmethylene blue (DMMB) assay (SL4, ***P = 0.0005; CT26, **P = 0.001, Student’s t test) (B), and collagen by hydroxyproline assay (SL4, *P = 0.01; CT26, P = 0.69, Student’s t test) (C) in SL4 and CT26 liver metastases after treatment with IgG or B20 (5 mg/kg; twice a week) until moribund (n = 6 to 15 per group). (D) Correlation of matrix components versus stiffness of SL4 (HA, P < 0.0001; sGAG, P < 0.0001; collagen, P = 0.98, Pearson’s correlation) and CT26 (HA, P < 0.0001; sGAG, P = 0.0002; collagen, P = 0.88, Pearson’s correlation) liver metastases grown in C57BL/6 WT and BALB/c WT mice, respectively, and treated with IgG or B20 (5 mg/kg; twice a week) until moribund. Black dots indicate controls and red dots indicate animals treated with B20. Data are shown as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to corresponding controls.
Inflammatory cell depletion does not alter HA production after anti-VEGF therapy
Immune cells, specifically CD11b+Gr1+ myeloid-derived suppressor cell (MDSC) populations such as Ly6Chi inflammatory monocytes (IMs), play a role in liver fibrosis, tumor progression, and resistance to antiangiogenic therapy, including primary liver tumors (11, 21–24). To evaluate the potential role of these cell populations in ECM deposition after anti-VEGF therapy, we performed flow cytometry analysis of tumor tissues from mice treated with B20 or IgG. The results demonstrate a significant influx of Ly6Chi IMs as well as metastasis-associated neutrophils (MANs) in SL4 liver mCRCs, at 3 days (Ly6Chi IMs: P = 0.016; MANs: P = 0.024, Student’s t test) and 6 days (Ly6Chi IM: P = 0.0001; MAN: P = 0.005, Student’s t test) after initiation of anti-VEGF therapy (Fig. 4, A and B). These results were confirmed in the CT26 liver mCRCs (fig. S6). Flow cytometric analysis of the spleen tissue—a “reservoir” for tumor-associated monocytes and neutrophils (25, 26)—and peripheral blood cells showed that increased counts of these cells in metastases and in peripheral blood associated with lower counts in the spleen. These data suggested a release of these cell populations from the spleen into the circulation and the mCRC tissues (Fig. 4, B and C). There were no significant differences in the cell numbers in the bone marrow at 3 days (Ly6Chi IM: P = 0.85; MAN: P = 0.81, Student’s t test) and 6 days (Ly6Chi IM: P = 0.68; MAN: P = 0.22) after initiation of anti-VEGF therapy (fig. S7).
Fig. 4. MDSCs do not increase noncollagenous matrix deposition after anti-VEGF therapy.
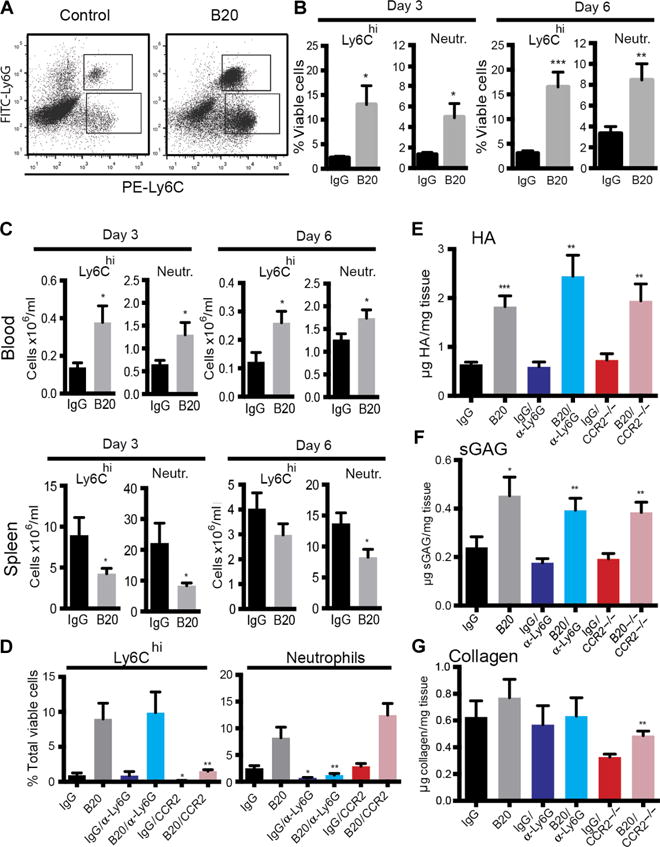
(A) Representative flow cytometry plot presenting the influx of CD45+CD11b+Ly6Chi monocytes and CD45+CD11b+Ly6G+ neutrophils into SL4 liver metastases after anti-VEGF therapy. FITC, fluorescein isothiocyanate. (B) Fluorescence-activated cell sorting (FACS) analysis of Ly6Chi IMs and MANs in SL4 liver metastases grown in C57BL/6 WT mice after 3 days (Ly6Chi IMs, *P = 0.016; neutrophils, *P = 0.024, Student’s t test) and 6 days (Ly6Chi IMs, *P = 0.0001; neutrophils, *P = 0.005, Student’s t test) of treatment with IgG or B20 (n = 7 to 10 per group). (C) FACS analyses of the blood (day 3: Ly6Chi IMs, *P = 0.017; neutrophils, *P = 0.03; day 6: Ly6Chi IMs, *P = 0.018; neutrophils, *P = 0.045, Student’s t test) and spleen (day 3: Ly6Chi IMs, *P = 0.037; neutrophils, P = 0.03; day 6: Ly6Chi IMs, P = 0.025; neutrophils, P = 0.21, Student’s t test) compartments of C57BL/6 WT mice bearing SL4 liver metastases after 3 and 6 days of treatment with IgG or B20 (n = 5 to 9 per group). (D) FACS analysis of Ly6Chi IMs and neutrophils in SL4 liver metastases grown in C57BL/6 WT mice after 7 days of treatment with IgG or B20 with or without anti-Ly6G therapy (5 mg/kg every 2 days), in _Ccr2_−/− mice after 7 days of treatment with IgG or B20 (Ly6Chi IMs, *P = 0.015; neutrophils, *P = 0.013, one-way ANOVA) with or without anti-Ly6G therapy (5 mg/kg every 2 days) (Ly6Chi IMs, *P = 0.014; neutrophils, *P = 0.98, one-way ANOVA), or in _Ccr2_−/− mice after 7 days of treatment with IgG or B20 (Ly6Chi IMs, *P = 0.93; neutrophils, *P < 0.0001, one-way ANOVA) (n = 4 to 9 per group). The number of Ly6Chi IMs in liver mCRCs was significantly decreased in _Ccr2_−/− mice as compared with WT mice under IgG (*P = 0.016) or B20 treatment (**P = 0.009). On the other hand, anti-Ly6G antibody significantly decreased MANs under IgG (*P = 0.016) or B20 (**P = 0.002) treatment. (E to G) Quantification of HA concentration by ELISA (IgG versus B20, **P = 0.003; IgG versus B20 in _Ccr2_−/− mice, **P = 0.005; IgG versus B20/anti-Ly6G, ***P < 0.0001, one-way ANOVA) (E), sGAG concentration by DMMB assay (IgG versus B20, **P = 0.005; IgG versus B20 in _Ccr2_−/− mice, *P = 0.015; IgG versus B20/anti-Ly6G, *P = 0.013, one-way ANOVA) (F), and collagen concentration by hydroxyproline assay (IgG versus B20, P = 0.67; IgG versus B20 in _Ccr2_−/− mice, P = 0.68; IgG versus B20/nti-Ly6G, P = 0.97; one-way ANOVA) (G) in SL4 liver metastases treated with IgG or B20 after depletion of Ly6Chi IMs (_Ccr2_−/− mice) or MANs (anti-Ly6G antibody) (n = 7 to 10 per group). B20 treatments (5 mg/kg): on day 0 for 3-day experiments and on day 0 and day 3 for 6- and 7-day experiments. Data are shown as means ± SEM; *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to corresponding controls.
To further evaluate the biological relevance of these immune cell populations in liver mCRC response to anti-VEGF therapy, we selectively depleted these cells by using a genetically engineered mouse model (_Ccr2_−/− mice) for depletion of Ly6Chi IM and a monoclonal antibody against Ly6G for depletion of MANs. Liver metastases from _Ccr2_−/− mice had an 83% reduction in Ly6Chi IM compared to WT mice receiving VEGF-targeted therapy (P = 0.009, Student’s t test), whereas combined treatment with B20 and the anti-Ly6G antibody reduced MAN by 85% compared to B20 alone (P = 0.002, Student’s t test). Furthermore, depletion of one MDSC population did not result in compensatory recruitment of the other cell type (Fig. 4D). Biochemical analysis of liver mCRCs collected from these mice showed persistent enrichment of HA in mice treated with B20 despite depletion of Ly6Chi IMs (0.731 ± 0.132 versus 1.941 ± 0.347 μg HA/mg tissue; P = 0.004, Student’s t test) or MANs (0.592 ± 0.099 versus 2.445 ± 0.432 μg HA/mg tissue; P = 0.002, Student’s t test) (Fig. 4E). Similarly, depletion of Ly6Chi IMs (0.192 ± 0.022 versus 0.384 ± 0.042 μg sGAG/mg tissue; P = 0.001, Student’s t test) or MANs (0.176 ± 0.018 versus 0.393 ± 0.049 μg sGAG/mg tissue; P = 0.002, Student’s t test) did not abrogate the increased deposition of sGAG after anti-VEGF therapy (Fig. 4F). We did observe a decrease in collagen in _Ccr2_−/− mice (Fig. 4G), a finding consistent with the role of Ly6Chi IMs in the pathogenesis of liver fibrosis (24). Collectively, our results show that the accumulation of HA or sGAG in liver mCRCs after anti-VEGF therapy is independent of the increased tumor infiltration by MDSC populations.
Angiotensin II receptor 1 deletion does not affect HA expression in mCRC after anti-VEGF therapy
The angiotensin II receptor 1 (AT1) signaling pathway is a relevant mediator of ECM deposition through transforming growth factor–β (TGF-β) activation and a potential target to decrease tumor desmoplasia (27, 28). Thus, we next investigated the role of AT1 in tumor stiffness and increased deposition of noncollagenous matrix after anti-VEGF therapy of liver mCRCs. To this end, we generated SL4 liver metastases in angiotensin II type 1a receptor knockout (_Agtr1a_−/−) mice, measured tumor stiffness, and analyzed the ECM composition after treatment with B20 or IgG. We found a significant increase in tumor stiffness after anti-VEGF therapy in liver mCRCs in _Agtr1a_−/− mice (1.11 ± 0.23 versus 5.93 ± 0.93 kPa; P = 0.0002, Student’s t test) (fig. S8A). Similarly, we observed a significant increase in HA (0.49 ± 0.036 versus 2.14 ± 0.57 μg HA/mg tissue; P = 0.008, Student’s t test) and sGAG (0.23 ± 0.018 versus 0.402 ± 0.044 μg sGAG/mg tissue; P = 0.005, Student’s t test) (fig. S8, B and C) and no significant change in collagen expression (fig. S8D). In line with these data, the concentration of TGF-β was either decreased (in SL4 tumors, P = 0.028, Student’s t test) or not changed (in CT26 tumors, P = 0.15, Student’s t test) after anti-VEGF therapy (fig. S9, A and B). Collectively, these data indicate that the abnormal deposition of HA and GAGs after anti-VEGF therapy in liver mCRCs is independent of the AT1 pathway.
Hypoxia drives HA expression in colorectal liver metastases after anti-VEGF therapy
Previous studies have shown that increased collagen deposition after anti-VEGF therapy is related to tumor hypoxia (10, 11). To explore the mechanisms by which HA accumulates in liver mCRCs after anti-VEGF therapy and to understand the temporal relationship between the treatment-induced increase in hypoxia and the abnormal ECM deposition, we performed time course experiments. These studies revealed a significant decrease of microvessel density after 3 days of anti-VEGF therapy (SL4: 66% reduction; P < 0.0001; CT26: 83% reduction; P = 0.01, Student’s t test; n = 4 to 6 per group). This effect persisted after 6 days in both mCRC models (SL4: 77% reduction; P = 0.0001; CT26: 93% reduction; P < 0.0001, Student’s t test; n = 4 to 6 per group) (Fig. 5, A and B, and fig. S10A) and was accompanied by an increased hypoxic area fraction in both models (Fig. 5, C and D, and fig. S10B). We then measured the ECM components and found only modest changes after 3 days of therapy in both models. However, analyses of samples obtained 6 days after initiation of B20 therapy showed a marked increase of HA deposition (SL4: 0.537 ± 0.088 versus 1.04 ± 0.143 μg HA/mg tissue; P = 0.009; CT26: 0.025 ± 0.01 versus 0.102 ± 0.021 μg HA/mg tissue; P = 0.006, Student’s t test; n = 6 to 8 per group) and sGAG (SL4: 0.283 ± 0.031 versus 0.597 ± 0.108 μg sGAG/mg tissue; P = 0.014; CT26: 0.197 ± 0.029 versus 0.402 ± 0.054 μg sGAG/mg tissue; P = 0.008, Student’s t test; n = 5 to 8 per group) in liver mCRCs (Fig. 5E). To further investigate the link between hypoxia and HA synthesis, we then performed a detailed image analysis of the localization of HA expression. HA expression was significantly colocalized in hypoxic regions (81.54 ± 9.23% versus 18.46 ± 9.232%; P = 0.0003, Student’s t test; n = 8 per group) (Fig. 5F), indicating that hypoxia is associated with HA up-regulation after anti-VEGF therapy.
Fig. 5. Increased HA deposition after anti-VEGF therapy in colorectal liver metastases is mediated by hypoxia.
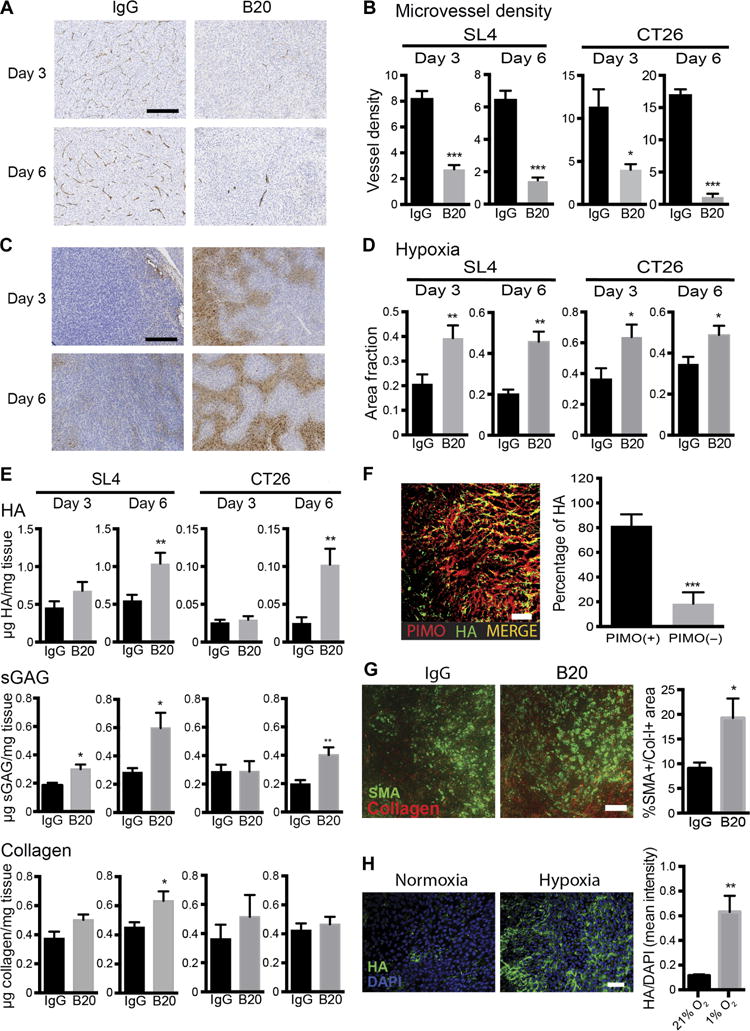
(A and B) Representative immunohistochemistry images of SL4 liver metastases (A) and quantification (B) of microvessel density (CD34 staining) in SL4 and CT26 liver metastases grown in C57BL/6 and BALB/c WT mice after 3 days (SL4, ***P < 0.0001; CT26, *P = 0.01, Student’s t test) and 6 days (SL4, ***P = 0.0001; CT26, ***P < 0.0001, Student’s t test) of treatment with IgG or B20. Scale bar, 100 μm (n = 6 to 8 per group). (C and D) Representative immunohistochemistry images of hypoxic area (staining for pimonidazole adducts) of SL4 liver metastases (C) and quantification of hypoxic area fraction in SL4 and CT26 liver metastases grown in C57BL/6 and BALB/c WT mice after 3 days (SL4, **P = 0.009; CT26, *P = 0.026, Student’s t test) and 6 days (SL4, **P = 0.005; CT26, *P = 0.037, Student’s t test) (n = 6 to 8 per group) (D). (E) Quantification of HA concentration by ELISA after 3 days (SL4, P = 0.153; CT26, P = 0.542, Student’s t test) and 6 days (SL4, **P = 0.009; CT26, **P = 0.006, Student’s t test) of treatment (top), sGAG concentration by DMMB assay after 3 days (SL4, *P = 0.011; CT26, P = 0.984, Student’s t test) and 6 days (SL4, *P = 0.014; CT26, **P = 0.008, Student’s t test) of treatment (middle), and collagen concentration by hydroxyproline assay after 3 days (SL4, P = 0.069; CT26, P = 0.412) and 6 days (SL4, *P = 0.028; CT26, P = 0.575, Student’s t test) of treatment (bottom) with IgG or B20 in SL4 and CT26 liver metastases. (F) Immunofluorescence staining demonstrating colocalization of HA expression with hypoxic areas in SL4 liver metastases treated with B20 for 7 days (***P = 0.0003, Student’s t test) (n = 8 per group). Scale bar, 100 μm. (G) Immunofluorescence staining demonstrating an increase in matrix-producing α-SMA+/collagen-I+ cells in SL4 liver metastases after anti-VEGF therapy (*P = 0.043, Student’s t test) (n = 6 to 8 per group). Scale bar, 100 μm. (H) Hypoxia increases HA expression in human HSCs. Cells were cultured in normoxic (21% O2) versus hypoxic conditions (1% O2) for 48 hours (**P = 0.003, Student’s t test). Scale bar, 100 μm. B20 treatments (5 mg/kg): on day 0 for 3-day experiments and on day 0 and day 3 for 6- and 7-day experiments. All data are shown as means ± SEM; *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to corresponding controls. DAPI, 4′,6-diamidino-2-phenylindole.
Hepatic stellate cells (HSCs) are the primary source of matrix synthesis in the liver (29). However, the impact of anti-VEGF therapy on HSC activation in liver mCRCs remains unclear. Also, the role of hypoxia in HA synthesis by HSCs is unknown. We found an increased amount of α-smooth muscle actin (α-SMA)+/collagen-I+–activated, matrix-producing HSCs in SL4 liver mCRCs after treatment with B20 (P = 0.043, Student’s t test) (Fig. 5G). To confirm that hypoxia mediates HA synthesis in HSC, we carried out in vitro studies using human HSCs cultured in normoxic versus hypoxic conditions for 48 hours. Hypoxia caused a 4.5-fold increase in HA expression in human HSCs (P = 0.003, Student’s t test) (Fig. 5H). Collectively, these results indicate that increased hypoxia is the likely driving stimulus for HA production by activated HSCs after anti-VEGF therapy. On the basis of the spatiotemporal analyses, hypoxia induced by anti-VEGF therapy appears to be the initiating event in this context. Then, resultant aggravation of desmoplasia compromises tumor perfusion further and induces a vicious cycle of hypoxia, desmoplasia, and immunosuppression.
Targeting HA increases perfusion and improves efficacy of chemotherapy in liver metastases after anti-VEGF therapy
Enzymatic targeting of HA improves vascular function and perfusion in pancreatic cancer (12). To investigate whether depletion of HA can prevent the changes induced by anti-VEGF therapy, we treated mice with B20 with or without intravenous administration of a polyethylene glycol conjugated (PEGylated) hyaluronidase (PEG-HAse). Treatment with PEG-HAse in combination with B20 resulted in a 74% reduction of HA in SL4 liver mCRCs compared to B20 alone (P < 0.0001) (Fig. 6A), which is consistent with previous observations in preclinical tumor models (30). To investigate the effects of HA depletion on blood perfusion of liver mCRCs after anti-VEGF therapy, we measured tissue distribution of Hoechst 33342 dye as a blood perfusion marker (31). Combined treatment of B20 with PEG-HAse significantly increased tumor perfusion compared to B20 monotherapy (perfused area fraction, 0.211 ± 0.044 versus 0.436 ± 0.0915; P = 0.036) (Fig. 6B). Because anti-VEGF therapy is commonly administered together with systemic chemotherapy in mCRC patients (32), and given the effects of HA depletion on perfusion of liver mCRCs treated with anti-VEGF therapy in mice, we next examined the effect of adding chemotherapy to anti-VEGF therapy and PEG-HAse. As expected, PEG-HAse treatment significantly lowered HA concentrations in SL4 liver mCRCs (P = 0.001, Student’s t test) (fig. S11). Furthermore, combination of PEG-HAse with B20 and chemotherapy significantly prolonged survival compared to B20 and chemotherapy only (median survival, 19.06 versus 17.13 days; P = 0.008, log-rank test) (Fig. 6C). Thus, enzymatic targeting of HA could potentiate the efficacy of anti-VEGF therapy with chemotherapy in liver mCRC.
Fig. 6. Targeting HA in liver metastases increases tumor perfusion after anti-VEGF therapy.
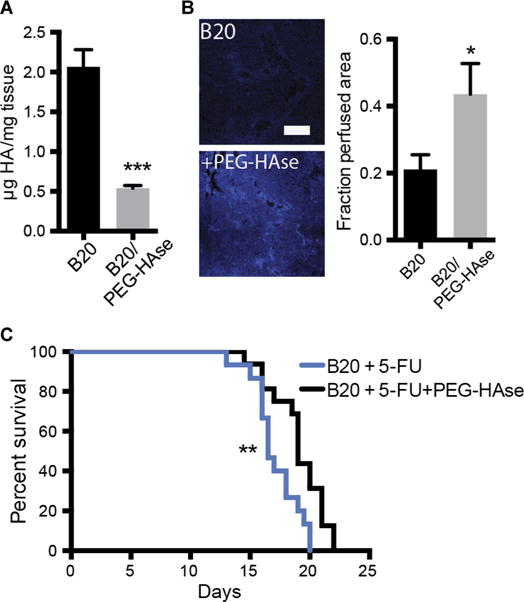
(A) Quantification of HA concentration in SL4 liver metastases after treatment with B20 (5 mg/kg; twice a week for 7 days) with and without PEG-HAse (4.5 mg/kg) (***P < 0.0001, Student’s t test). (B) Effects of PEG-HAse on perfusion of SL4 liver metastases treated with B20 (5 mg/kg; twice a week for 7 days) after intraportal injection of Hoechst 33342 (*P = 0.036, Student’s t test). Scale bar, 200 μm. (C) Survival of C57BL/6 WT mice bearing SL4 liver metastases treated with either B20 (5 mg/kg) and 5-FU (50 mg/kg) or B20 (5 mg/kg), 5-FU (50 mg/kg), and PEG-HAse (4.5 mg/kg) (**P = 0.008, log-rank test) (n = 15 to 16 per group). All data are shown as means ± SEM; *P < 0.05 and ***P < 0.001 as compared to corresponding controls.
DISCUSSION
We report a mechanism of acquired resistance to anti-VEGF therapy in liver mCRC. As shown in patient samples and orthotopic mouse models of liver mCRC, anti-VEGF therapy results in abnormal deposition of ECM components and, in particular, HA and sGAG. Our data suggest that increased HA expression after anti-VEGF therapy is a consequence of treatment-induced hypoxia and increases solid stress in liver mCRCs. Finally, we show that depleting HA can result in improved tumor perfusion and treatment efficacy in mouse models of liver mCRC.
Several mechanisms of resistance to antiangiogenic therapy have been proposed thus far (9, 33). In particular, treatment-induced hypoxia and acidosis have multiple adverse effects on tumor cells and the tumor microenvironment that fuel progression and mediate treatment resistance (7). Besides induction of the epithelial-to-mesenchymal program that favors an invasive and metastatic tumor cell phenotype (34), hypoxia is thought to select for tumor cells with cancer stem cell properties that might further mediate resistance to cytotoxics, as well as favoring an immunosuppressive microenvironment by reducing the activity of cytotoxic T cells and antigen-presenting cells and by skewing the polarization of tumor-associated macrophages toward the protumorigenic and immunosuppressive M2 phenotype (35–39). Increased fibrosis is another effect of hypoxia on the microenvironment of tissues and is dependent on hypoxia-inducible factor 1α (40). In line with these data, we also found an increased recruitment of immunosuppressive MDSCs after anti-VEGF therapy (41, 42). Although the increased infiltration of MDSCs and HA production are both driven by hypoxia in liver mCRC, these mechanisms appear to be independent.
We have previously demonstrated that desmoplasia affects tumor perfusion and effectiveness of chemotherapy by impairing vascular function (6, 27). We have also shown that ECM, specifically HA, in primary tumors contributes to growth-induced solid stress, and thus, reducing solid stress may reduce compression of tumor vessels (5, 6). Our recent studies showed that HA impairs vascular function and drug delivery into the tumor in animal models of pancreatic and prostate cancers (12, 15, 43, 44). Here, we show that increased HA deposition in liver metastatic lesions is an important mechanism of acquired resistance to systemic treatment in this setting.
Because of the uniformly elevated interstitial fluid pressure, diffusion represents the primary mechanism of transport within tumors, which in turn depends on the size, charge, and configuration of the substance being transported. In addition, the physicochemical properties of the ECM—a complex network built up by collagenous and noncollagenous components—are critical determinants of interstitial transport (45, 46). Apart from their role in creation of intratumor solid stress, HA and sGAGs, with their negative charge and high hydration, function as interstitial barriers to the delivery of cancer therapeutics by forming aggregates and increasing viscoelasticity of the interstitial matrix (2, 47–49).
Moreover, HA and sGAG affect various intracellular signaling pathways that can promote tumor cell proliferation, motility, and invasiveness as well as angiogenesis and stromal cell recruitment (50–53). In a recent study, Ropponen et al. reported the expression of HA in the CRC stroma as well as in the cytoplasm and/or pericellular region of cancer cells, which carried a poor prognosis (54). These authors confirmed these findings in other cancer types (55–58). There are several mechanisms of how HA can directly affect tumor cell viability and/or phenotype that ultimately promote disease progression and resistance to therapy. Interaction of HA with its specific cell surface receptors CD44 and RHAMM (receptor for HA-mediated motility) mediates evasion of apoptosis, particularly in anchorage-independent conditions (59–61), which might be explained by activation of several signaling cascades such as the phosphatidylinositol 3-kinase (PI3K)/AKT and focal adhesion kinase (FAK) pathways (62, 63). In addition, HA promotes a migratory and invasive tumor phenotype, in part, through the production and cell surface presentation of matrix metalloproteinases and cytoskeletal rearrangement (64–66). These effects may collectively explain the more aggressive phenotype associated with increased HA in our preclinical and clinical studies of liver mCRC (54–56, 67–69).
Our study has several limitations. Depletion of HA in addition to chemotherapy and anti-VEGF therapy modestly prolonged survival. Although this confirmed this study’s hypothesis, the results of this animal experiment should be validated in a controlled clinical trial designed to evaluate whether HA depletion can translate into a survival benefit in liver mCRC patients when using clinically relevant doses from phase 1b trials (70). On the basis of recent reports, HA depletion appeared more effective in patients with tumors with high baseline amounts of HA (70). We found a substantial variability in the increase in HA expression in human liver mCRC samples after anti-VEGF therapy. Although our study serves as a proof of principle that preventing anti-VEGF treatment–induced HA deposition could improve the efficacy of systemic therapy, the importance of this mechanism versus other mechanisms of acquired resistance remains to be determined in patients. It also remains to be seen whether alternative targets within the ECM can further improve the effects of systemic therapy in these patients. For example, direct targeting of HSCs (or metastasis-associated fibroblasts), the main source of ECM components within liver mCRCs, may offer a therapeutic strategy. Treatment with vitamin D receptor ligands might be another strategy to reduce or prevent the activation of HSCs (71, 72). Finally, as shown in Fig. 2B, the increase in stiffness and hence mechanical forces could be abrogated by using a lower dose of anti-VEGF antibody. This lower “vascular normalizing” dose may actually increase oxygenation, as demonstrated in preclinical models of breast cancer (7, 31).
In conclusion, our results show that anti-VEGF therapy at the dose used here and in the clinic up-regulates the expression of HA in liver mCRC both in mouse models and in patient samples. This may be a mechanism of acquired resistance in this setting because depletion of HA improved perfusion of liver mCRC in mice and their response to systemic chemotherapy. Thus, targeting the noncollagenous ECM is a potential strategy to enhance the efficacy of systemic treatments for mCRC patients.
MATERIALS AND METHODS
Study design
The objective of the present study was to evaluate the remodeling of the ECM and its clinical relevance in colorectal liver metastases after a VEGF-targeted therapy. The sample sizes of the experiments were selected on the basis of previous experience. Data collection was stopped at a priori defined time points. Animal experiments were performed in a confirmatory fashion with an a priori hypothesis without repetition. In vitro experiments were carried out in triplicate. Experiments were carried out in an unblinded fashion except for analyses of immunohistochemistry images.
Patient samples
Patient samples were obtained from the Heidelberg University Hospital. Surgical specimens of patients who underwent liver resection for colorectal liver metastases without preoperative chemotherapy, after preoperative chemotherapy without bevacizumab, or after preoperative chemotherapy with bevacizumab were analyzed. Analysis of human samples was in line with the Declaration of Helsinki. Collection of patient samples for scientific purposes was approved by the local ethics committee (323/2004), and written informed consent was obtained.
Cell and animal models
The SL4 mouse CRC cells were a gift from T. Irimura (17). The mouse CRC cell line CT26 was purchased from the American Type Culture Collection (catalog #CRL-2638). HSCs were purchased from ScienCell (catalog #5300). To model liver metastasis, the spleen was split into two sectors, and 1 × 105 cells were injected into the distal caudal sector, which was then resected. The other hemispleen remained in place. Tumor burden was assessed by measuring blood concentration of Gaussia luciferase (Gluc) from Gluc-transduced tumors (73, 74) or using a high-frequency ultrasound imaging system (VisualSonics). Mice were randomly assigned to the treatment groups, and treatments were initiated after the development of macroscopic liver metastases (~8 to 12 days after injection). See the Supplementary Materials and Methods for more details.
Treatments
The anti-VEGF monoclonal antibody B20.4-1.1 (Genentech) was administered by intraperitoneal injection twice a week at 5 or 1 mg/kg. For neutrophil depletion, an anti-Ly6G antibody (clone RB6-8C5) was administered intraperitoneally every 2 days at a dose of 5 mg/kg. PEG-HAse (see the Supplementary Materials and Methods) was administered intravenously twice a week or 24 hours before administration of chemotherapy at a dose of 4.5 mg/kg (43). 5-FU chemotherapy was administered intravenously twice a week at a dose of 50 mg/kg.
Histochemistry
Frozen blocks were cut at a thickness of 20 μm for immunofluorescence staining for α-SMA, collagen-I, HA, and hypoxia. Whole-tumor mosaic images were obtained from each sample with an Olympus (FV1000) confocal laser-scanning microscope. Paraffin blocks were cut at a thickness of 5 μm and stained with hematoxylin and eosin, CD34, CD44, and HA. For hypoxia staining, pimonidazole hydrochloride (60 mg/kg) (Hypoxyprobe) was injected intraperitoneally 60 min before tissue collection. For perfusion measurement, Hoechst 33342 (Sigma) was administered by portal infusion 5 min before tumor tissue collection (31). Image analyses were carried out in an automated fashion using a custom algorithm in MATLAB (MathWorks). See the Supplementary Materials and Methods for more details.
Cytometry
Tissue samples from tumors, spleen, and bone marrow were digested and filtered through 70- and 40-μm cell strainers. Single cells were stained with CD45, CD11b, Ly6G, and Ly6C and analyzed using an LSR II flow cytometer (Becton Dickinson). See the Supplementary Materials and Methods for more details.
Biochemical assays
sGAG content in finely dispersed tissues was determined by the Di-methylmethylene Blue Assay (DMMB) (75). Total collagen content was determined by measuring hydroxyproline content of the hydrolysate samples (76). Mouse HA and TGF-β1 were quantified using ELISA kits (R&D Systems). See the Supplementary Materials and Methods for more details.
Physical parameters
The stiffness (Young’s modulus) of tumors was determined by unconfined compression tests (77). Solid stress was determined using a recently proposed method (6). In line with this method, the normalized tumor opening was used as a readout for growth-induced stress. See the Supplementary Materials and Methods for more details.
Statistical analysis
Data are presented as means ± SEM and compared using Student’s t test, one-way ANOVA, or two-way ANOVA when appropriate. Survival was estimated by Kaplan-Meier curves and compared using the log-rank test. All tests were two-sided. Statistical analyses were carried out using GraphPad Prism software. A P value of less than 0.05 was considered statistically significant.
Supplementary Material
Supplement
Fig. S1. Expression of HA is increased in CRC liver metastases compared to liver parenchyma.
Fig. S2. DNA content in murine CRC liver metastases is decreased after anti-VEGF therapy.
Fig. S3. CD44 expression is increased in liver metastases after anti-VEGF therapy.
Fig. S4. Bevacizumab increases sGAG expression in human CRC liver metastases.
Fig. S5. Anti-VEGF therapy does not alter ECM expression in healthy liver parenchyma.
Fig. S6. Anti-VEGF therapy results in recruitment of MDSCs into CT26 liver metastases.
Fig. S7. Anti-VEGF therapy does not alter IM and neutrophil counts in the bone marrow.
Fig. S8. Agtr1a knockout does not alter anti-VEGF–induced tumor stiffness and expression of noncollagenous matrix.
Fig. S9. Anti-VEGF therapy does not increase TGF-β1 expression in CRC liver metastases.
Fig. S10. Microvessel density is decreased, and hypoxia is increased in CT26 liver metastases after anti-VEGF therapy.
Fig. S11. Treatment with PEG-HAse lowers HA content in SL4 liver metastases.
Table S1. Clinicopathologic data of patients with CRC liver metastases.
Acknowledgments
We thank C. Smith, S. Roberge, and A. Khachatryan for expert assistance with immunohistochemistry and ELISA, T. Irimura for providing the SL4 mouse CRC cells, P. Huang for providing mice, O. Iliopoulos for the use of a hypoxia incubator, L. Fisher for providing the collagen-I (LF-68) antibody, B. A. Tannous for providing a lentivirus vector encoding green fluorescent protein and secreted Gluc, E. Herpel for providing paired samples of patients with colorectal liver metastases from the tissue bank of the National Center for Tumor Diseases (Heidelberg, Germany) in accordance with the regulations of the tissue bank and the approval of the ethics committee of the University of Heidelberg, and R. Sasisekharan for helpful discussions.
Funding: This study was supported, in part, by the NIH (CA080124 to R.K.J., D.F., D.G.D., and Y.B., T32DK007191 to D.K., CA126642 and CA197743 to R.K.J., CA096915 to D.F., CA139168 and CA159258 to D.G.D., and CA098706 and CA173518 to Y.B.), the American Cancer Society (grant 120733-RSG-11-073-01-TBG to D.G.D.), fellowship from the German Research Foundation (DFG) (to N.N.R.), fellowship from the Foundation for Science and Technology (FCT, Portugal), and the Portuguese Human Potential Operational Program/European Social Fund (POPH/FSE) funding program (to J.I.).
Footnotes
Author contributions: N.N.R., D.K., J.I., W.W.H., H.T.N., C.M.E., K.J., J.D., I.C., T.H., J.D.M., and Y.H. performed experiments. N.N.R., D.K., J.I., H.T.N., C.R., Y.B., J.W., A.J.G., D.G.D., R.K.J., J.W.C., N.M., and D.F. designed the experiments and analyzed the data. N.N.R., D.K., R.K.J., and D.F. wrote the paper. All authors read and agreed on the final version of the manuscript.
Competing interests: R.K.J. received consultant fees from Dyax, XTuit Pharmaceuticals, Enlight Biosciences, Ophthotech, SPARC, and SynDevRx. R.K.J. owns equity in Enlight Biosciences, Ophthotech, SynDevRx, and XTuit Pharmaceuticals and serves on the Board of Directors of XTuit Pharmaceuticals and the Boards of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, and the Tekla Healthcare Opportunities Fund. No reagents or funding from these companies was used in these studies. Y.B. received consultant fees from XTuit Pharmaceuticals. D.G.D. received grants from Merrimack, Bayer, and HealthCare Pharmaceuticals and served as a consultant for Hexal.
REFERENCES AND NOTES
- 1.Jain RK. The next frontier of molecular medicine: Delivery of therapeutics. Nat Med. 1998;4:655–657. doi: 10.1038/nm0698-655. [DOI] [PubMed] [Google Scholar]
- 2.Chauhan VP, Stylianopoulos T, Boucher Y, Jain RK. Delivery of molecular and nanoscale medicine to tumors: Transport barriers and strategies. Annu Rev Chem Biomol Eng. 2011;2:281–298. doi: 10.1146/annurev-chembioeng-061010-114300. [DOI] [PubMed] [Google Scholar]
- 3.Jain RK. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205–2218. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nat Mater. 2013;12:958–962. doi: 10.1038/nmat3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stylianopoulos T, Martin JD, Snuderl M, Mpekris F, Jain SR, Jain RK. Coevolution of solid stress and interstitial fluid pressure in tumors during progression: Implications for vascular collapse. Cancer Res. 2013;73:3833–3841. doi: 10.1158/0008-5472.CAN-12-4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, Smith BL, Ferrone CR, Hornicek FJ, Boucher Y, Munn LL, Jain RK. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci USA. 2012;109:15101–15108. doi: 10.1073/pnas.1213353109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain RK. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605–622. doi: 10.1016/j.ccell.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 9.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguilera KY, Rivera LB, Hur H, Carbon JG, Toombs JE, Goldstein CD, Dellinger MT, Castrillon DH, Brekken RA. Collagen signaling enhances tumor progression after anti-VEGF therapy in a murine model of pancreatic ductal adenocarcinoma. Cancer Res. 2014;74:1032–1044. doi: 10.1158/0008-5472.CAN-13-2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y, Chen Y, Huang Y, Reiberger T, Duyverman AM, Huang P, Samuel R, Hiddingh L, Roberge S, Koppel C, Lauwers GY, Zhu AX, Jain RK, Duda DG. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/C-X-C receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59:1435–1447. doi: 10.1002/hep.26790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kultti A, Li X, Jiang P, Thompson CB, Frost GI, Shepard HM. Therapeutic targeting of hyaluronan in the tumor stroma. Cancers. 2012;4:873–903. doi: 10.3390/cancers4030873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whatcott CJ, Han H, Posner RG, Hostetter G, Von Hoff DD. Targeting the tumor microenvironment in cancer: Why hyaluronidase deserves a second look. Cancer Discov. 2011;1:291–296. doi: 10.1158/2159-8290.CD-11-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chauhan VP, Boucher Y, Ferrone CR, Roberge S, Martin JD, Stylianopoulos T, Bardeesy N, DePinho RA, Padera TP, Munn LL, Jain RK. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell. 2014;26:14–15. doi: 10.1016/j.ccr.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, Hostetter G, Shepard HM, Von Hoff DD, Han H. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res. 2015;21:3561–3568. doi: 10.1158/1078-0432.CCR-14-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morimoto-Tomita M, Ohashi Y, Matsubara A, Tsuiji M, Irimura T. Mouse colon carcinoma cells established for high incidence of experimental hepatic metastasis exhibit accelerated and anchorage-independent growth. Clin Exp Metastasis. 2005;22:513–521. doi: 10.1007/s10585-005-3585-0. [DOI] [PubMed] [Google Scholar]
- 18.Griswold DP, Corbett TH. A colon tumor model for anticancer agent evaluation. Cancer. 1975;36:2441–2444. doi: 10.1002/1097-0142(197512)36:6<2441::aid-cncr2820360627>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 19.Liang WC, Wu X, Peale FV, Lee CV, Meng YG, Gutierrez J, Fu L, Malik AK, Gerber HP, Ferrara N, Fuh G. Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J Biol Chem. 2006;281:951–961. doi: 10.1074/jbc.M508199200. [DOI] [PubMed] [Google Scholar]
- 20.Wicherts DA, de Haas RJ, Sebagh M, Corrales ES, Gorden DL, Lévi F, Paule B, Azoulay D, Castaing D, Adam R. Impact of bevacizumab on functional recovery and histology of the liver after resection of colorectal metastases. Br J Surg. 2011;98:399–407. doi: 10.1002/bjs.7368. [DOI] [PubMed] [Google Scholar]
- 21.Cotechini T, Medler TR, Coussens LM. Myeloid cells as targets for therapy in solid tumors. Cancer J. 2015;21:343–350. doi: 10.1097/PPO.0000000000000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27:462–472. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanahan D, Coussens LM. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 24.Karlmark KR, Weiskirchen R, Zimmerman HW, Gassler N, Ginhoux F, Weber C, Merad M, Luedde T, Trautwein C. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50:261–274. doi: 10.1002/hep.22950. [DOI] [PubMed] [Google Scholar]
- 25.Cortez-Retamozo V, Etzrodt M, Newton I, Rauch PJ, Chudnovskiy A, Berger C, Ryan RJH, Iwamoto Y, Marinelli B, Gorbatov R, Forghani R, Novobrantsev TI, Koteliansky V, Figueireod JL, Chen JW, Anderson DG, Nahrendorf M, Swirski FK, Weissleder R, Pittet MJ. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci USA. 2012;109:2491–2496. doi: 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, Popović Z, Huang P, Bawendi MG, Boucher Y, Jain RK. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516. doi: 10.1038/ncomms3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci USA. 2011;108:2909–2914. doi: 10.1073/pnas.1018892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Friedman SL. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singha NC, Nekoroski T, Zhao C, Symons R, Jiang P, Frost GI, Huang Z, Shepard HM. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol Cancer Ther. 2015;14:523–532. doi: 10.1158/1535-7163.MCT-14-0580. [DOI] [PubMed] [Google Scholar]
- 31.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F, Leblan P, Munn LL, Huang P, Duda DG, Fukumura D, Jain RK, Poznansky MC. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci USA. 2012;109:17561–17566. doi: 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strickler JH, Hurwitz HI. Bevacizumab-based therapies in the first-line treatment of metastatic colorectal cancer. Oncologist. 2012;17:513–524. doi: 10.1634/theoncologist.2012-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–3200. doi: 10.1172/JCI70212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47–71. doi: 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- 35.Casazza A, Di Conza G, Wenes M, Finisguerra V, Deschoemaeker S, Mazzone M. Tumor stroma: A complexity dictated by the hypoxic tumor microenvironment. Oncogene. 2014;33:1743–1754. doi: 10.1038/onc.2013.121. [DOI] [PubMed] [Google Scholar]
- 36.Motz GT, Coukos G. Deciphering and reversing tumor immune suppression. Immunity. 2013;39:61–73. doi: 10.1016/j.immuni.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noy R, Pollard JW. Tumor-associated macrophages: From mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 39.Palazón A, Aragonés J, Morales-Kastresana A, de Landázuri MO, Melero I. Molecular pathways: Hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res. 2012;18:1207–1213. doi: 10.1158/1078-0432.CCR-11-1591. [DOI] [PubMed] [Google Scholar]
- 40.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, Ochiai H, Kitahara S, Unan EC, Reddy TP, Fan C, Huang P, Bardeesy N, Zhu AX, Jain RK, Duda DG. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61:1591–1602. doi: 10.1002/hep.27665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, Lolkema MP, Jiang P, Kultti A, Thompson CB, Maneval DC, Jodrell DI, Frost GI, Shepard HM, Skepper JN, Tuveson DA. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson CB, Shepard HM, O’Connor PM, Kadhim S, Jiang P, Osgood RJ, Bookbinder LH, Li X, Sugarman BJ, Connor RJ, Nadjsombati S, Frost GI. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther. 2010;9:3052–3064. doi: 10.1158/1535-7163.MCT-10-0470. [DOI] [PubMed] [Google Scholar]
- 45.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jain RK. Transport of molecules in the tumor interstitium: A review. Cancer Res. 1987;47:3039–3051. [PubMed] [Google Scholar]
- 47.Dvorak HF. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 48.Alexandrakis G, Brown EB, Tong RT, McKee TD, Campbell RB, Boucher Y, Jain RK. Two-photon fluorescence correlation microscopy reveals the two-phase nature of transport in tumors. Nat Med. 2004;10:203–207. doi: 10.1038/nm981. [DOI] [PubMed] [Google Scholar]
- 49.Thorne RG, Lakkaraju A, Rodriguez-Boulan E, Nicholson C. In vivo diffusion of lactoferrin in brain extracellular space is regulated by interactions with heparan sulfate. Proc Natl Acad Sci USA. 2008;105:8416–8421. doi: 10.1073/pnas.0711345105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sironen RK, Tammi M, Tammi R, Auvinen PK, Antilla M, Kosma VM. Hyaluronan in human malignancies. Exp Cell Res. 2011;317:383–391. doi: 10.1016/j.yexcr.2010.11.017. [DOI] [PubMed] [Google Scholar]
- 51.Toole BP. Hyaluronan: From extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528–539. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- 52.Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U. Roles of heparan-sulphate glycosaminoglycans in cancer. Nat Rev Cancer. 2002;2:521–528. doi: 10.1038/nrc842. [DOI] [PubMed] [Google Scholar]
- 53.Kozlowski EO, Pavao MS. Effect of sulfated glycosaminoglycans on tumor invasion and metastasis. Front Biosci. 2011;3:1541–1551. doi: 10.2741/244. [DOI] [PubMed] [Google Scholar]
- 54.Ropponen K, Tammi M, Parkkinen J, Eskelinen M, Tammi R, Lipponen P, Ågren U, Alhava E, Kosma V-M. Tumor cell-associated hyaluronan as an unfavorable prognostic factor in colorectal cancer. Cancer Res. 1998;58:342–347. [PubMed] [Google Scholar]
- 55.Auvinen P, Tammi R, Parkkinen J, Tammi M, Ågren U, Johansson R, Hirvikoski P, Eskelinen M, Kosma V-M. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am J Pathol. 2000;156:529–536. doi: 10.1016/S0002-9440(10)64757-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pirinen R, Tammi R, Tammi M, Hirvikoski P, Parkkinen JJ, Johansson R, Böhm J, Hollmén S, Kosma V-M. Prognostic value of hyaluronan expression in non-small-cell lung cancer: Increased stromal expression indicates unfavorable outcome in patients with adenocarcinoma. Int J Cancer. 2001;95:12–17. doi: 10.1002/1097-0215(20010120)95:1<12::aid-ijc1002>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 57.Tammi RH, Kultti A, Kosma VM, Pirinen R, Auvinen P, Tammi MI. Hyaluronan in human tumors: Pathobiological and prognostic messages from cell-associated and stromal hyaluronan. Semin Cancer Biol. 2008;18:288–295. doi: 10.1016/j.semcancer.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 58.Wang C, Tammi M, Guo H, Tammi R. Hyaluronan distribution in the normal epithelium of esophagus, stomach, and colon and their cancers. Am J Pathol. 1996;148:1861–1869. [PMC free article] [PubMed] [Google Scholar]
- 59.Ghatak S, Misra S, Toole BP. Hyaluronan oligosaccharides inhibit anchorage-independent growth of tumor cells by suppressing the phosphoinositide 3-kinase/Akt cell survival pathway. J Biol Chem. 2002;277:38013–38020. doi: 10.1074/jbc.M202404200. [DOI] [PubMed] [Google Scholar]
- 60.Kosaki R, Watanabe K, Yamaguchi Y. Overproduction of hyaluronan by expression of the hyaluronan synthase Has2 enhances anchorage-independent growth and tumorigenicity. Cancer Res. 1999;59:1141–1145. [PubMed] [Google Scholar]
- 61.Zoltan-Jones A, Huang L, Ghatak S, Toole BP. Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J Biol Chem. 2003;278:45801–45810. doi: 10.1074/jbc.M308168200. [DOI] [PubMed] [Google Scholar]
- 62.Fujita Y, Kitagawa M, Nakamura S, Azuma K, Ishii G, Higashi M, Kishi H, Hiwasa T, Koda K, Nakajima N, Harigaya K. CD44 signaling through focal adhesion kinase and its anti-apoptotic effect. FEBS Lett. 2002;528:101–108. doi: 10.1016/s0014-5793(02)03262-3. [DOI] [PubMed] [Google Scholar]
- 63.Hall CL, Wang C, Lange LA, Turley EA. Hyaluronan and the hyaluronan receptor RHAMM promote focal adhesion turnover and transient tyrosine kinase activity. J Cell Biol. 1994;126:575–588. doi: 10.1083/jcb.126.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999;13:35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bourguignon LY, Gunja-Smith Z, Iida N, Zhu HB, Young LJ, Muller WJ, Cardiff RD. CD44v3,8–10 is involved in cytoskeleton-mediated tumor cell migration and matrix metalloproteinase (MMP-9) association in metastatic breast cancer cells. J Cell Physiol. 1998;176:206–215. doi: 10.1002/(SICI)1097-4652(199807)176:1<206::AID-JCP22>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 66.Koochekpour S, Pilkington GJ, Merzak A. Hyaluronic acid/CD44H interaction induces cell detachment and stimulates migration and invasion of human glioma cells in vitro. Int J Cancer. 1995;63:450–454. doi: 10.1002/ijc.2910630325. [DOI] [PubMed] [Google Scholar]
- 67.Itano N, Sawai T, Miyaishi O, Kimata K. Relationship between hyaluronan production and metastatic potential of mouse mammary carcinoma cells. Cancer Res. 1999;59:2499–2504. [PubMed] [Google Scholar]
- 68.Yu Q, Toole BP, Stamenkovic I. Induction of apoptosis of metastatic mammary carcinoma cells in vivo by disruption of tumor cell surface CD44 function. J Exp Med. 1997;186:1985–1996. doi: 10.1084/jem.186.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lipponen P, Aaltomaa S, Tammi R, Tammi M, Ågren U, Kosma V-M. High stromal hyaluronan level is associated with poor differentiation and metastasis in prostate cancer. Eur J Cancer. 2001;37:849–856. doi: 10.1016/s0959-8049(00)00448-2. [DOI] [PubMed] [Google Scholar]
- 70.Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, Tjulandin SA, Gladkov OA, Holcombe RF, Korn R, Raghunand N, Dychter S, Jiang P, Shepard HM, Devoe CE. Phase Ib study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clin Cancer Res. 2016;22:2848–2854. doi: 10.1158/1078-0432.CCR-15-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ding N, Yu RT, Subramaniam N, Sherman MH, Wilson C, Rao R, Leblanc M, Coulter S, He M, Scott C, Lau SL, Atkins AR, Barish GD, Gunton JE, Liddle C, Downes M, Evans RM. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153:601–613. doi: 10.1016/j.cell.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lynn Lau S, Gunton JE, Wahl GM, Hunter T, Drebin JA, O’Dwyer PJ, Liddle C, Tuveson DA, Downes M, Evans RM. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tannous BA. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat Protoc. 2009;4:582–591. doi: 10.1038/nprot.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chung E, Yamashita H, Au P, Tannous BA, Fukumura D, Jain RK. Secreted Gaussia luciferase as a biomarker for monitoring tumor progression and treatment response of systemic metastases. PLOS ONE. 2009;4:e8316. doi: 10.1371/journal.pone.0008316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sah RLY, Kim YJ, Doong JY, Grodzinsky AJ, Plaas AHK, Sandy JD. Biosynthetic response of cartilage explants to dynamic compression. J Orthop Res. 1989;7:619–636. doi: 10.1002/jor.1100070502. [DOI] [PubMed] [Google Scholar]
- 76.Woessner J., Jr The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 77.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 78.Sugaya T, Nishimatsu S-i, Tanimoto K, Takimoto E, Yamagishi T, Imamura K, Goto S, Imaizumi K, Hisada Y, Otsuka A, Uchida H, Sugiura M, Fukuta K, Fukamizu A, Murakami K. Angiotensin II type 1a receptor-deficient mice with hypotension and hyperreninemia. J Biol Chem. 1995;270:18719–18722. doi: 10.1074/jbc.270.32.18719. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement
Fig. S1. Expression of HA is increased in CRC liver metastases compared to liver parenchyma.
Fig. S2. DNA content in murine CRC liver metastases is decreased after anti-VEGF therapy.
Fig. S3. CD44 expression is increased in liver metastases after anti-VEGF therapy.
Fig. S4. Bevacizumab increases sGAG expression in human CRC liver metastases.
Fig. S5. Anti-VEGF therapy does not alter ECM expression in healthy liver parenchyma.
Fig. S6. Anti-VEGF therapy results in recruitment of MDSCs into CT26 liver metastases.
Fig. S7. Anti-VEGF therapy does not alter IM and neutrophil counts in the bone marrow.
Fig. S8. Agtr1a knockout does not alter anti-VEGF–induced tumor stiffness and expression of noncollagenous matrix.
Fig. S9. Anti-VEGF therapy does not increase TGF-β1 expression in CRC liver metastases.
Fig. S10. Microvessel density is decreased, and hypoxia is increased in CT26 liver metastases after anti-VEGF therapy.
Fig. S11. Treatment with PEG-HAse lowers HA content in SL4 liver metastases.
Table S1. Clinicopathologic data of patients with CRC liver metastases.