ProbCons: Probabilistic consistency-based multiple sequence alignment (original) (raw)
Abstract
To study gene evolution across a wide range of organisms, biologists need accurate tools for multiple sequence alignment of protein families. Obtaining accurate alignments, however, is a difficult computational problem because of not only the high computational cost but also the lack of proper objective functions for measuring alignment quality. In this paper, we introduce probabilistic consistency, a novel scoring function for multiple sequence comparisons. We present ProbCons, a practical tool for progressive protein multiple sequence alignment based on probabilistic consistency, and evaluate its performance on several standard alignment benchmark data sets. On the BAliBASE, SABmark, and PREFAB benchmark alignment databases, ProbCons achieves statistically significant improvement over other leading methods while maintaining practical speed. ProbCons is publicly available as a Web resource.
Given a set of biological sequences, a multiple alignment provides a way of identifying and visualizing patterns of sequence conservation by organizing homologous positions across different sequences in columns. As sequence similarity often implies divergence from a common ancestor or functional similarity, sequence comparisons facilitate evolutionary and phylogenetic studies (Phillips et al. 2000; Castillo-Davis et al. 2004) and isolation of the most relevant regions (Attwood 2002) for a variety of biological analyses. In particular, conserved amino acid stretches in proteins are strong indicators of preserved three-dimensional structural domains, so protein alignments have been widely used in aiding structure prediction (Rost and Sander 1994; Jones 1999) and characterization of protein families (Sonnhammer et al. 1998; Johnson and Church 1999; Bateman et al. 2004). However, when sequence identity falls below 30%, called the “twilight zone” of protein alignments, the accuracies of most automatic sequence alignment methods drop considerably (Rost 1999; Thompson et al. 1999b). As a result, alignment quality is often the limiting factor in biological analyses of amino acid sequences (Jaroszewski et al. 2002).
The problem of alignment construction consists of defining either explicitly or implicitly an objective function for assessing alignment quality and employing an efficient algorithm to find the optimal, or a near optimal, alignment according to the objective function. Two-sequence alignments are usually evaluated by addition of match/mismatch scores for aligned pairs of positions and affine gap penalties for unaligned amino acids (Needleman and Wunsch 1970; Smith and Waterman 1981). Quantitatively, scores for aligned residues are given by log-odds (Altschul 1991) substitution matrices such as PAM (Dayhoff et al. 1978), GONNET (Gonnet et al. 1992), or BLOSUM (Henikoff and Henikoff 1992). Estimation of appropriate gap penalties, however, is often regarded as a “black art” based on trial and error (Vingron and Waterman 1994). For two sequences of length L, an optimal alignment according to this metric may be computed in O(_L_2) time (Gotoh 1982) and O(L) space (Myers and Miller 1988) via dynamic programming.
Pair-hidden Markov models (HMMs) provide an alternative formulation of the sequence alignment problem in which alignment generation is directly modeled as a first-order Markov process involving state emissions and transitions. In this approach, model parameters obtain an intuitive probabilistic interpretation and can be trained on real data using standard supervised or unsupervised likelihood-based methods. The Viterbi (1967) algorithm computes the highest probability alignment of two input sequences according to an alignment pair-HMM. In the standard three-state pair-HMM for alignment, the Viterbi algorithm may be viewed as an instantiation of the Needleman-Wunsch algorithm in which alignment parameters are determined by a log-odds transformation of the HMM scoring scheme (Durbin et al. 1998).
Since they specify a conditional probability distribution over the space of all suboptimal alignments, pair-HMMs also allow the computation of the posterior probability, P(xi ∼ yj ∈ a*| x, y), that particular positions xi and yj of two sequences x and y, respectively, will be matched in an alignment a* generated by the model. Running the Needleman-Wunsch algorithm with these posterior probabilities as substitution scores and no gap penalties gives rise to the maximum expected accuracy alignment method (see Methods), also known as optimal accuracy alignment (Holmes and Durbin 1998).
In the general case of multiple sequence comparisons, theoretically sound and biologically motivated scoring methods are not straightforward to devise. In practice, ad hoc sum-of-pairs schemes (Carrillo and Lipman 1988), which combine the projected pairwise log-odds scores for all pairs of sequences in the alignment, and their weighted variants (Altschul et al. 1989) are commonly used. Unfortunately, direct application of dynamic programming is too inefficient for alignment of more than a few sequences. Instead, a variety of heuristic strategies have been proposed, including genetic algorithms (Notredame and Higgins 1996), simulated annealing (Kim et al. 1994), alignment to a profile HMM (Krogh et al. 1994; Eddy 1995), or greedy assemblage of multiple segment-to-segment comparisons (Morgenstern et al. 1996). By far, the most popular heuristic strategies involve tree-based progressive alignment (Feng and Doolittle 1987) in which groups of sequences are assembled into a complete multiple alignment via several pairwise alignment steps. As with any hierarchical approach, however, errors at early stages in the alignment not only propagate to the final alignment but also may increase the likelihood of misalignment due to incorrect conservation signals. Post-processing steps such as iterative refinement (Gotoh 1996) alleviate some of the errors made during progressive alignment.
Consistency-based schemes take the alternative view that “prevention is the best medicine.” Note that for any multiple alignment, the induced pairwise alignments are necessarily _consistent_—that is, given a multiple alignment containing three sequences x, y, and z, if position xi aligns with position zk and position zk aligns with yj in the projected _x_-z and _z_-y alignments, then xi must align with yj in the projected _x_-y alignment. Consistency-based techniques apply this principle in reverse, using evidence from intermediate sequences to guide the pairwise alignment of x and y, such as needed during the steps of a progressive alignment. By adjusting the score for an xi ∼ yj residue pairing according to support from some position zk that aligns to both xi and yj in the respective _x_-z and _y_-z pairwise comparisons, consistency-based objective functions incorporate multiple sequence information in scoring pairwise alignments.
Gotoh (1990) first introduced consistency to identify anchor points for reducing the search space of a multiple alignment. A mathematically elegant reformulation of consistency in terms of boolean matrix multiplication was later given by Vingron and Argos (1991) and implemented in the program MALI, which builds multiple alignments from dot matrices (Vingron and Argos 1989). An alternative formulation of consistency was employed in the DIALIGN tool, which finds ungapped local alignments via segment-to-segment comparisons, determines new weights for these alignments using consistency, and assembles them into a multiple alignment by a greedy selection procedure (Morgenstern et al. 1996).
More recently, Notredame et al. (1998) introduced COFFEE, a new consistency-based objective function for scoring residue pairs in a pairwise alignment. In this approach, an alignment library is computed by merging consistent CLUSTALW (Thompson et al. 1994) global and LALIGN (Huang and Miller 1991) local pairwise alignments to form three-way alignments, which are assigned percent identity weights. Then, the score for aligning xi to yj is defined to be the sum of the weights of all alignments in the library containing that aligned residue pair. The program T-Coffee (Notredame et al. 2000), which implements multiple sequence alignment under this objective function using progressive maximum weight trace computations (Kececioglu 1993), has demonstrated superior accuracy on the BAliBASE test suite (Thompson et al. 1999a) over competing methods, including CLUSTALW, DIALIGN, and PRRP (Gotoh 1996).
In this article, we introduce probabilistic consistency, a novel modification of the traditional sum-of-pairs scoring system that incorporates HMM-derived posterior probabilities and three-way alignment consistency. We discuss the theoretical motivations behind the probabilistic consistency scoring system and demonstrate its applicability with ProbCons, a protein progressive multiple alignment tool based on this technique. To assess the utility of our methods, we compared ProbCons to several current leading alignment tools including Align-m (Van Walle et al. 2004), CLUSTALW, DIALIGN, MAFFT (Katoh et al. 2002), MUSCLE (Edgar 2004), and T-Coffee on the BAliBASE, SABmark (Van Walle et al. 2004), and PREFAB (Edgar 2004) benchmark alignment databases, using commonly accepted accuracy measures for validating alignment quality. In this comparison, ProbCons shows a clear statistically significant improvement in accuracy over all other alignment tools in every benchmark test, while maintaining practical running times. Moreover, all parameters for the program are derived through unsupervised training methods without making any manual adjustments. ProbCons is publicly available as a Web resource. Source code and executables are available as public domain software at http://probcons.stanford.edu.
Results
Algorithm overview
Fundamentally, ProbCons is a pair-hidden Markov model-based progressive alignment algorithm that primarily differs from most typical approaches in its use of maximum expected accuracy rather than Viterbi alignment, and of the probabilistic consistency transformation to incorporate multiple sequence conservation information during pairwise alignment. ProbCons uses the HMM shown in Figure 1 to specify the probability distribution over all alignments between a pair of sequences. Emission probabilities, which correspond to traditional substitution scores, are based on the BLOSUM62 matrix (Henikoff and Henikoff 1992). Transition probabilities, which correspond to gap penalties, are trained with unsupervised expectation maximization (EM).
Figure 1.

Basic pair-HMM for sequence alignment between two sequences, x and y. State M emits two letters, one from each sequence, and corresponds to the two letters being aligned together. State Ix emits a letter in sequence x that is aligned to a gap, and similarly state Iy emits a letter in sequence y that is aligned to a gap. Finding the most likely alignment according to this model by using the Viterbi algorithm corresponds to applying Needleman-Wunsch with appropriate parameters. The logarithm of the emission probability function p(.,.) at M corresponds to a substitution scoring matrix, while affine gap penalty parameters can be derived from the transition probabilities δ and ε (Durbin et al. 1998).
ProbCons algorithm
Given m sequences, S ={s(1),..., s(m)}:
Step 1: Computation of posterior-probability matrices
For every pair of sequences x, y ∈ S and all i ∈ {1,...,|x|}, j ∈ {1,...,|y|}, compute the matrix Pxy, where Pxy(i, j)= P(xi ∼ yj ∈ a*| x, y) is the probability that letters xi and yj are paired in a*, an alignment of x and y generated by the model.
Step 2: Computation of expected accuracies
Define the expected accuracy of a pairwise alignment a between x and y to be the expected number of correctly aligned pairs of letters, divided by the length of the shorter sequence:
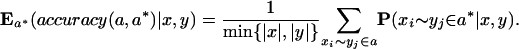
For each pair of sequences x, y ∈ S, compute the alignment a that maximizes expected accuracy by dynamic programming, and set E(x, y)= Ea*(accuracy(a, a*)| x, y).
Step 3: Probabilistic consistency transformation
Reestimate the match quality scores P(xi ∼ yj ∈ a*| x, y) by applying the probabilistic consistency transformation, which incorporates similarity of x and y to other sequences from S into the _x_-y pairwise comparison:
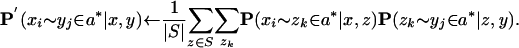
In matrix form, the transformation may be written as
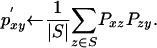
Since most values in the Pxz and Pzy matrices will be near zero, the transformation is computed efficiently using sparse matrix multiplication by ignoring all entries smaller than a threshold ω. This step may be repeated as many times as desired.
Step 4: Computation of guide tree
Construct a guide tree for S through hierarchical clustering. As a measure of similarity between two sequences x and y use E(x, y) as computed in Step 2. Define the similarity of two clusters by a weighted average of the pairwise similarities between sequences of the clusters.
Step 5: Progressive alignment
Align sequence groups hierarchically according to the order specified in the guide tree. Alignments are scored using a sum-of-pairs scoring function in which aligned residues are assigned the transformed match quality scores P′(xi ∼ yj ∈ a*| x, y) and gap penalties are set to zero.
Post-processing step: Iterative refinement
Randomly partition alignment into two groups of sequences and realign. This step may be repeated as many times as desired.
In addition to the steps shown, we also experimented with the generation of automatic column reliability annotations for the alignment based on the posterior matrix formulation above (see Methods).
Testing methodology
To test the empirical performance of ProbCons, we used three different multiple alignment benchmarking suites, including BAliBASE 2.01 (Thompson et al. 1999a), PREFAB 3.0 (Edgar 2004), and SABmark 1.63 (Van Walle et al. 2004). Tests were performed on a 3.3-GHz Pentium IV with 2 GB RAM.
The BAliBASE 2.01 benchmark alignment database is a collection of 141 reference protein alignments, consisting of structural alignments from the FSSP (Holm and Sander 1994) and HOMSTRAD (Mizuguchi et al. 1998) databases and hand-constructed alignments from the literature. The database is organized into five reference sets: Reference 1 consists of a few equidistant sequences of similar length; Reference 2, families of closely related sequences with up to three distant “orphan” sequences; Reference 3, equidistant divergent families; Reference 4, sequences with large N/C-terminal extensions; and Reference 5, sequences with large internal insertions. Test alignments are scored with respect to BAliBASE core blocks, regions for which reliable alignments are known to exist.
The PREFAB 3.0 database is an automatically generated database consisting of 1932 alignments averaging 49 sequences of length 240. Each test consists of a pair of protein sequences supplemented with homologs found through PSI-BLAST (Altschul et al. 1997) queries over the NCBI nonredundant protein sequence database (Pruitt et al. 2003). The accuracy of a multiple sequence alignment is then evaluated with respect to the pairwise structural alignments of the original two protein sequences using the consensus of FSSP and CE alignments. Note that the pairwise structural alignments in PREFAB only cover some regions of the sequences; we treated these like BAliBASE core blocks.
The SABmark 1.63 database consists of two sets of consensus regions based on SOFI (Boutonnet et al. 1995) and CE (Shindyalov and Bourne 1998) structural alignments of sequences from the ASTRAL (Brenner et al. 2000) database. The “Twilight Zone” set contains 1994 domains sorted into 236 subsets representing SCOP folds (Murzin et al. 1995), where each subset contains sequences within no more than 25% identity. The “Superfamily” set contains 3645 domains sorted into 462 subsets representing SCOP superfamilies, where each subset contains sequences within no more than 50% identity. Unlike BAliBASE, SABmark uses all-pairs pairwise reference structural alignments for evaluating multiple alignment quality.
While no universally accepted accuracy measure exists for protein alignments, we chose to score each alignment according to the original benchmarking measures proposed for its respective database. In the BAliBASE data set, we scored alignments according to the sum-of-pairs score (SP), defined as the number of correctly aligned residue pairs found in the test alignment divided by the total number of aligned residue pairs in core blocks of the reference alignment (Thompson et al. 1999b). Additionally, we measured the column score (CS), defined as the number of correctly aligned columns found in the test alignment divided by the total number of aligned columns in core blocks of the reference alignment. On the PREFAB alignments, we measured the quality (Q) score (Edgar 2004), which is equivalent to the SP score. Finally, for SABmark, we used the developer (fD) score, which is also equivalent to the SP score (where all residues in the reference alignment are treated as being in core blocks), and the modeler (fM) score, defined as the number of correctly aligned residue pairs found in the test alignment divided by the total number of aligned residue pairs in the test alignment (Sauder et al. 2000). For each type of scoring metric used, we averaged the scores per multiple alignment (or average score per subset in the case of SABmark) over all multiple alignment tests in the database.
Comparison to other aligners
We compared the results of ProbCons on the above databases to those of six leading multiple alignment systems: (1) CLUSTALW 1.83 (Thompson et al. 1994), currently the most popular progressive alignment method; (2) DIALIGN 2.2.1 (Morgenstern et al. 1998), a local aligner using segment-based homology; (3) T-Coffee 1.37 (Notredame et al. 2000), a heuristic consistency-based aligner that combines global and local alignments; (4) MAFFT 3.88 (Katoh et al. 2002), a set of six scripts for performing multiple alignment with a variety of iterative refinement techniques; (5) MUSCLE 3.3 (Edgar 2004), a new aligner reporting the best published results on BAliBASE to date; and (6) Align-m 1.0 (Van Walle et al. 2004), a consistency-based method for computing all-pairs pairwise alignments of multiple sequences. Of the six scripts comprising the MAFFT alignment utilities, we chose to test nw-ns-i, the most accurate script. For Align-m 1.0, we used the parameter settings picked for testing the program in Van Walle et al. (2004). All other programs were run with default parameters.
Emission probabilities for the ProbCons HMM were adapted from the BLOSUM62 scoring matrix (Henikoff and Henikoff 1992). The default transition parameters of ProbCons were trained via unsupervised Expectation-Maximization (EM) on unaligned sequences from the BAliBASE benchmark database; thus, the tests on the PREFAB and SABmark databases provide external validation of the results shown on BAliBASE. The default options for the ProbCons program included applying two iterations of the consistency transformation and 100 rounds of iterative refinement for every alignment. We also experimented with a modified version of ProbCons (ProbCons-ext) in which the HMM model was extended to include an extra pair of insertion states (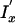 and
and 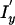 ) to model long or terminal insertions.
) to model long or terminal insertions.
The results of testing on the BAliBASE benchmark alignments database are shown in Table 1. To assess the significance of the differences in overall SP and CS scores, we performed a Friedman rank test for all pairs of programs; these results are summarized in Table 2. A typical BAliBASE alignment and its corresponding plot of column reliability are shown in Figure 2. The correlation between predicted and actual column reliability scores as shown in the diagram demonstrates the ability of pairwise posterior matrices to predict the expected proportion of correctly aligned residue pairs per column.
Table 1.
Performance of aligners on the BAliBASE benchmark alignments database
| Ref 1 (82) | Ref 2 (23) | Ref 3 (12) | Ref 4 (12) | Ref 5 (12) | Overall (141) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aligner | SP | CS | SP | CS | SP | CS | SP | CS | SP | CS | SP | CS | Time (mm:ss) |
| Align-m | 76.6 | n/a | 88.4 | n/a | 68.4 | n/a | 91.1 | n/a | 91.7 | n/a | 80.4 | n/a | 19:25 |
| DIALIGN | 81.1 | 70.9 | 89.3 | 35.9 | 68.4 | 34.4 | 89.7 | 76.2 | 94.0 | 84.3 | 83.2 | 63.7 | 2:53 |
| CLUSTALW | 86.1 | 77.3 | 93.2 | 56.8 | 75.3 | 46.0 | 83.4 | 52.2 | 85.9 | 63.8 | 86.1 | 68.0 | 1:07 |
| MAFFT | 86.7 | 78.1 | 92.4 | 50.2 | 78.8 | 50.4 | 91.6 | 72.7 | 96.3 | 85.9 | 88.2 | 71.4 | 1:18 |
| T-Coffee | 86.6 | 77.4 | 93.4 | 56.1 | 78.5 | 48.7 | 91.8 | 73.0 | 95.8 | 90.3 | 88.3 | 72.2 | 21:31 |
| MUSCLE | 88.7 | 80.8 | 93.5 | 56.3 | 82.5 | 56.4 | 87.6 | 60.9 | 96.8 | 90.2 | 89.6 | 73.9 | 1:05 |
| ProbCons | 90.1 | 82.6 | 94.4 | 61.3 | 84.1 | 61.3 | 90.1 | 72.3 | 97.9 | 91.9 | 91.0 | 77.2 | 5:32 |
| ProbCons-ext | 90.0 | 82.5 | 94.2 | 59.1 | 84.3 | 61.1 | 93.8 | 81.0 | 98.1 | 92.2 | 91.2 | 77.6 | 8:02 |
Table 2.
Significance test for differences in BAliBASE performance
| Align-M | DIALIGN | CLUSTALW | MAFFT | T-Coffee | MUSCLE | ProbCons | ProbCons-ext |
|---|---|---|---|---|---|---|---|
| Align-M | −(0.61) | −8.2 × 10−6 | −<10−10 | −<10−10 | −<10−10 | −<10−10 | −<10−10 |
| DIALIGN | −1.9 × 10−5 | −<10−10 | −<10−10 | −<10−10 | −<10−10 | −<10−10 | |
| CLUSTALW | +2.4 × 10−3 | −1.0 × 10−3 | −3.0 × 10−5 | −4.9 × 10−8 | −6.1 × 10−10 | −<10−10 | |
| MAFFT | +1.2 × 10−9 | +1.0 × 10−3 | −(0.65) | −1.7 × 10−5 | −2.6 × 10−9 | −4.9 × 10−8 | |
| T-Coffee | +<10−10 | +8.4 × 10−6 | − (0.92) | −7.0 × 10−3 | −1.5 × 10−6 | −8.4 × 10−6 | |
| MUSCLE | +<10−10 | +1.9 × 10−8 | +9.6 × 10−6 | +1.7 × 10−3 | −3.0 × 10−3 | −6.6 × 10−3 | |
| ProbCons | +<10−10 | +<10−10 | +1.6 × 10−7 | +1.9 × 10−6 | +0.012 | +0.043 | |
| ProbCons-ext | +<10−10 | +<10−10 | +8.3 × 10−6 | +3.2 × 10−5 | +(0.092) | −(0.088) |
Figure 2.

Column reliability plot for 1csy_ref1 from BAliBASE, Reference 1. The red line and solid regions indicate the predicted and actual proportion of correct pairwise matches at each alignment position, respectively. All column reliability values have been multiplied by 100. Below, the actual ProbCons alignment is shown with core block residues highlighted in green. Note that only pairwise matches in core block regions of the BAliBASE alignment are considered correct when computing the “actual” proportion of correct pairwise matches; however, some residues outside of the core block regions may also be alignable. Thus, regions in which predicted homology exceeds actual homology do not necessarily indicate overprediction of homology by the aligner.
With the exception of Reference 4, ProbCons achieves the strongest performance in both SP and CS scores in all references. Reference 4 sequences are marked by long N/C-terminal extensions in which local alignment methods tend to be more successful, suggesting that incorporation of a local alignment probabilistic model into ProbCons might improve its performance on such sequences. Alternatively, we found that extending the HMM model with an extra pair of insertion states (ProbCons-ext) did improve BAliBASE performance in Reference 4; however, this addition roughly doubled the running time, with variable performance benefit in the other databases.3
The results of testing six of the methods on the PREFAB database are shown in Table 3. Results for the Align-m program are omitted, since the program failed to complete all alignments in the PREFAB database. Again, ProbCons and ProbCons-ext demonstrate a strong lead in SP score although their running times are longer than those of the other aligners except for T-Coffee. This is due to the computation of all-pairs pairwise posterior probability matrices in the first step of the algorithm; other schemes for formulating probabilistic consistency that avoid this need for a quadratic number of initial alignments may be possible. The significance results for these values are given in Table 4.4
Table 3.
Performance of aligners on the PREFAB protein reference alignment benchmark
| Aligner | Overall (1927) | Time |
|---|---|---|
| DIALIGN | 57.2 | 12 h, 25 min |
| CLUSTALW | 58.9 | 2 h, 57 min |
| T-Coffee | 63.6 | 144 h, 51 min |
| MUSCLE | 64.8 | 3 h, 11 min |
| MAFFT | 64.8 | 2 h, 36 min |
| ProbCons | 66.9 | 19 h, 41 min |
| ProbCons-ext | 68.0 | 37 h, 46 min |
Table 4.
Significance test for differences in PREFAB performance
| DIALIGN | CLUSTALW | T-Coffee | MUSCLE | MAFFT | ProbCons | ProbCons-ext |
|---|---|---|---|---|---|---|
| DIALIGN | −1.06 × 10−9 | − <10−10 | − <10−10 | − <10−10 | − <10−10 | − <10−10 |
| CLUSTALW | − <10−10 | − <10−10 | − <10−10 | − <10−10 | − <10−10 | |
| T-Coffee | − <10−10 | − <10−10 | − <10−10 | − <10−10 | ||
| MUSCLE | +2.3 × 10−9 | − <10−10 | − <10−10 | |||
| MAFFT | − <10−10 | − <10−10 | ||||
| ProbCons | −0.031 |
The results of testing of the SABmark benchmark alignment database are shown in Table 5. Many of the same trends as found in the BAliBASE alignments are seen in SABmark, with the difference between ProbCons and the next best aligner in terms of fD (SP) scores even more exaggerated. It should be noted, however, that while the Align-m aligner lags far behind in SP score5 (which may be thought of as a measure of sensitivity), its fM scores, which are the proportion of correctly predicted amino acid matches among all predicted matches (and which may be regarded as a measure of specificity) are the highest. Due to this disparity, it is difficult to make a precise quantitative statement regarding the relative performance of Align-m compared to the other methods without characterizing the sensitivity/specificity trade-off of each method, such as performed in a ROC analysis (Metz 1978).6 Nevertheless, compared to all other aligners, ProbCons demonstrates significantly higher fD and fM scores overall, as seen in Table 6.
Table 5.
Performance of aligners on the SABmark sequence and structure alignment benchmark
| Superfamily (462) | Twilight zone (236) | Overall (698) | |||||
|---|---|---|---|---|---|---|---|
| Aligner | fD | fM | fD | fM | fD | fM | Time (mm:ss) |
| Align-m | 44.4 | 58.9 | 17.1 | 43.0 | 35.2 | 53.5 | 56:44 |
| DIALIGN | 50.3 | 42.5 | 22.5 | 19.2 | 41.0 | 34.6 | 8:28 |
| CLUSTALW | 53.7 | 38.7 | 24.8 | 15.2 | 43.9 | 30.8 | 2:16 |
| MAFFT | 54.1 | 40.0 | 24.8 | 16.0 | 44.2 | 31.9 | 7:33 |
| T-Coffee | 55.4 | 41.8 | 26.4 | 18.0 | 45.6 | 33.7 | 59:10 |
| MUSCLE | 55.9 | 40.1 | 27.6 | 17.5 | 46.4 | 33.0 | 20:42 |
| ProbCons | 59.9 | 45.0 | 32.1 | 21.7 | 50.5 | 37.1 | 17:20 |
| ProbCons-ext | 59.9 | 45.3 | 32.0 | 22.1 | 50.5 | 37.5 | 23:10 |
Table 6.
Significance test for differences in SABmark performance
| Align-M | DIALIGN | CLUSTALW | MAFFT | T-Coffee | MUSCLE | ProbCons | ProbCons-ext |
|---|---|---|---|---|---|---|---|
| Align-M | −<10−10 | −<10−10 | −<10−10 | −<10−10 | −<10−10 | −<10−10 | −<10−10 |
| DIALIGN | −<10−10 | −<10−10 | −<10−10 | −<10−10 | −<10−10 | −<10−10 | −<10−10 |
| CLUSTALW | −<10−10 | −<10−10 | −0.02 | −0.01 | −7.5 × 10−6 | −<10−10 | −<10−10 |
| MAFFT | −<10−10 | −<10−10 | +(0.083) | −1.5 × 10−5 | −<10−10 | −<10−10 | −<10−10 |
| T-Coffee | −<10−10 | −2.5 × 10−3 | +<10−10 | +<10−10 | −0.052 | −<10−10 | −<10−10 |
| MUSCLE | −<10−10 | −1.2 × 10−7 | +<10−10 | +1.2 × 10−4 | −1.5 × 10−5 | −<10−10 | −<10−10 |
| ProbCons | −<10−10 | +<10−10 | +<10−10 | +<10−10 | +<10−10 | +<10−10 | +6.4 × 10−4 |
| ProbCons-ext | −<10−10 | +<10−10 | +<10−10 | +<10−10 | +<10−10 | +<10−10 | +(0.31) |
Comparison of ProbCons variants
To understand the features of ProbCons that give it a strong increase in performance, we compared several ProbCons variants on the “Twilight Zone” set from the SABmark alignment database. In particular, we examined the effects of four main algorithmic changes: (1) using the Viterbi algorithm to compute the highest probability alignment, instead of the highest expected accuracy alignment that is computed by ProbCons; (2) using the posterior probability matrices generated by ProbCons to produce all-pairs pairwise alignments instead of full multiple alignments; (3) varying the number of applications of consistency transformation applied before alignment; and (4) omitting the application of iterative refinement to optimize the alignment with respect to the sum-of-pairs probabilistic consistency metric. In this article, we have omitted a full comparison of expected accuracy guide tree construction to more popular methods such as neighbor-joining or UPGMA, though preliminary results indicate ProbCons to be relatively insensitive to tree topology (R.C. Edgar, pers. comm.).
The results for each of these tests are shown in Table 7. Note that we tested the Viterbi algorithm only on pairwise alignments, as the HMM used in the ProbCons algorithm is strictly for pairwise comparisons; properly extending it to handle progressive profile alignment is beyond the scope of this study. As seen by a comparison of the first two rows of the table, alignments that optimize expected accuracy were significantly more accurate than Viterbi alignments.
Table 7.
Performance of ProbCons Variants on SABmark “Twilight Zone” set
| Algorithm | c | Ir | Output | fD | fM | Time (mm:ss) |
|---|---|---|---|---|---|---|
| 1. Viterbi | 0 | 0 | Pairwise | 27.5 | 17.2 | 0:42 |
| 2. Posterior | 0 | 0 | Pairwise | 29.6 | 18.5 | 2:54 |
| 3. Posterior | 1 | 0 | Pairwise | 32.5 | 20.4 | 3:15 |
| 4. Posterior | 2 | 0 | Pairwise | 33.2 | 21.0 | 3:47 |
| 5. Posterior | 0 | 0 | Multiple | 29.1 | 19.8 | 2:57 |
| 6. Posterior | 1 | 0 | Multiple | 30.9 | 20.8 | 3:17 |
| 7. Posterior | 2 | 0 | Multiple | 31.5 | 21.3 | 3:50 |
| 8. Posterior | 0 | 100 | Multiple | 30.6 | 20.8 | 4:14 |
| 9. Posterior | 2 | 100 | Multiple | 32.1 | 21.7 | 5:50 |
The numbers also show that pairwise methods (rows 2-4) tend to generate alignments with slightly higher fD (SP) scores and slightly lower fM scores than their multiple alignment counterparts (rows 5-7). However, a stronger trend is that in both the pairwise and multiple alignment cases, iterated applications of consistency lead to simultaneous improvements in fD and fM, thus showing that the consistency does help incorporate multiple sequence information into pairwise alignments. Using 100 rounds of iterative refinement helps optimize the alignment, as reflected in the difference between rows 5 and 8 of the table. Employing both iterated consistency and iterative refinement thus gives the default parameter settings for the ProbCons program (row 9).
Interestingly, computing multiple alignments using the expected accuracy criterion alone generates significantly more accurate alignments in terms of both fD and fM scores than those produced by current leading alignment methods. To check the validity of this claim, we applied the expected accuracy criterion for multiple alignment to the entire SABmark database, achieving an fD score of 0.479 and an fM score of 0.355, again significantly better than all other methods except for the full ProbCons method itself. Therefore expected accuracy alignments give better sensitivity in terms of predicting true matches and better specificity in terms of predicting a higher proportion of true matches. This observation suggests that posterior-based approaches are a powerful general approach for improving alignment accuracy. Additionally, among the added features, using the probabilistic consistency transformation provided the largest accuracy improvement.
Discussion
Though the problem of protein multiple sequence alignment is hardly new, the computation of high accuracy multiple sequence alignments is still an open problem. In this article, we presented ProbCons, a practical tool for protein multiple sequence alignment, which has demonstrated dramatic improvements in alignment accuracy over several leading methods on the BAliBASE, PREFAB, and SABmark benchmark alignment databases while maintaining competitive running times.
Despite its strong performance on empirical tests, the ProbCons algorithm uses an extremely simple model of sequence similarity (a three-state pair-HMM) and makes no attempt to incorporate biological knowledge such as position-specific gap scoring, rigorous evolutionary tree construction, and other features used by aligners such as CLUSTALW. ProbCons does not use protein-specific alignment information other than the amino acid alphabet and the BLOSUM emission probability matrices. Replacing these with equivalent values for nucleotides may give a DNA alignment procedure with improved accuracy over standard Needleman-Wunsch-based aligners. In addition, the parameters used in the model are transparent, and include the probability δ of transition from the match/mismatch state to the insertion states (corresponding to a gap-open penalty) the probability ε of self-transition in an insertion state (corresponding to a gap-extend penalty), and the initial probability πinsert of starting with an insertion. Since all training for the program was done automatically on unaligned sequences using Expectation-Maximization without human guidance, it is thus possible to retrain ProbCons on specific sequence types to obtain parameters that would be more appropriate for particular alignment tasks.
Our results in comparing different variations of ProbCons indicate that the two main features that contribute to its accuracy are the use of maximum expected accuracy as an objective function and the application of the probabilistic consistency transformation. The methodology employed in developing the ProbCons algorithm is straightforward and widely applicable: (1) specify an appropriate quality measure and (2) maximize its expected value according to the probability distribution given by the model. For example, the accuracy measure used in this article maximizes the expected number of correct matches in an alignment; if one is concerned about overprediction of matches, one may use an alternative objective function that penalizes overprediction of matches and, provided it is easily decomposable, derive the corresponding optimization algorithm. Exploring this framework provides a novel and exciting direction for future work in pursuing even higher accuracy alignment approaches.
The principles employed, however, are not unique to sequence alignment alone. As an example, consider the related problem of motif finding among a set of divergent sequences. Consistency-based approaches have previously been applied to motif-finding tasks with strong empirical results (Heger et al. 2003). A more principled algorithm based on probabilistic consistency may further increase the sensitivity of motif detection methods. Comparative gene finding and RNA or protein structural prediction methods may also benefit from a probabilistic consistency-based approach.
Methods
The ProbCons algorithm works by (1) computing posterior-probability matrices, (2) computing expected accuracies for each pairwise comparison, (3) applying the probabilistic consistency transformation, (4) computing an expected accuracy guide tree, and (5) performing progressive alignment. As a default, we also perform iterative refinement as a post-processing step. In the subsections that follow, we consider each of these steps in greater detail, describe the EM training procedure used to obtain parameters for the ProbCons HMM, and present a novel technique for estimating column reliability scores based on the alignment scoring matrices.
1. Posterior probability matrices
Let x and y be two proteins represented as character strings in which xi is the _i_th amino acid of x. Consider the pair-HMM given in Figure 1, where A is the space of all possible _x_-y alignments. An alignment a corresponds uniquely to a sequence of state-emission pairs, 〈_s_1, _o_1〉,..., 〈_sn, on_〉. The probability of a is given by
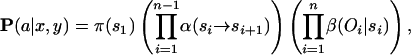
where π(s) is the initial probability of starting in state s, α(si → si+1) is the transition probability from si to si+1, and β(oi| si) is the emission probability for either a single letter or aligner residue pair oi in the state si.
In the derivation which follows, let a* be the (unknown) alignment from A that most nearly represents the “true” biological alignment of x and y. Ideally, we wish to determine a* based on the sequence information in x and y alone. To do this we use the distribution P(A| x, y) to represent our beliefs regarding a*, i.e., we assume that P(a| x, y) is the probability that an alignment a is equal to a*.
Let the notation xi ∼ yj ∈ a denote the event that two positions xi and yj are matched in an alignment a. Formally, the posterior probability of xi ∼ yj ∈ a* is
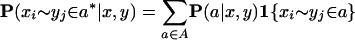
where the common indicator notation 1{condition} is used to define a function that evaluates to 1 whenever condition is true and 0 otherwise. Then, the posterior probability matrix Pxy for the alignment of x and y is a table of P(xi ∼ yj ∈ a*| x, y) values for 1 ≤ i ≤|x|, 1 ≤ j ≤|y|. The ProbCons algorithm begins by calculating these posterior probability matrices using a modification of the Forward and Backward algorithms for computing posterior probabilities in pair-HMMs as described in Durbin et al. (1998). This computation step takes time O(_m_2_L_2), where m is the number of sequences and L is the length of each sequence.
2. Maximal expected accuracy alignment
Most alignment schemes build an “optimal” pairwise alignment by finding the highest probability alignment using the Viterbi algorithm. In this approach, one computes arg max_a_ P(a| x, y), which may be alternatively written as arg max_a_ Ea*[1{a = a*}| _x, y_]; that is, the Viterbi algorithm finds the alignment whose probability of being exactly equal to a* is optimal. When the odds of recovering the exact correct alignment is low but partially correct alignments are still useful, this is not necessarily the best choice.
In this work, we explore an alternative strategy that finds the alignment a that does not maximize the probability of a = a* but rather tries to guarantee high accuracy for a, which we define with respect to the alignment a* as

During the alignment process, however, a* is not known, so we instead maximize the expected accuracy of the reported alignment. Computing this quantity is straightforward since
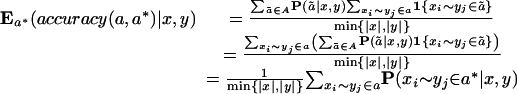
Using this decomposition, we compute the maximal expected accuracy alignment by a simple variant of the Needleman-Wunsch algorithm, where all match/mismatch scores are given by the posterior probability terms for corresponding letters and gap penalties are set to zero. This form of alignment bears strong resemblance to the problem of finding the maximum weight trace of a matrix (Kececioglu 1993), and a similar scheme is used to compute final progressive alignments in the T-Coffee program.
3. Probabilistic consistency transformation
In the previous section, we described a method for performing pairwise sequence alignment of two sequences x and y based on computing P(xi ∼ yj ∈ a*| x,y) values for all positions in x and y, and subsequently using these posterior probabilities as match/mismatch scores in a Needleman-Wunsch-like alignment procedure. In this section we introduce probabilistic consistency, a method for obtaining more accurate substitution scores when a third homologous sequence z is available.
One way to use sequence z is to generalize the pair-HMM given in Figure 1 to a triple-HMM that parameterizes a conditional distribution over three-sequence alignments of x, y, and z, and similarly generalize the previous formulas for expected accuracy to handle three-way alignments. Such an approach, however, leads to impractical O(_L_3) algorithms for computing posterior matrices of sequences of length L. Here, we follow a heuristic approach that allows us to derive an algorithm with an approximately O(L2) running time.
For a sequence z, let z(k,k+1) denote the interletter regions (or gaps) between amino acids k and k + 1 of z for 0 ≤ k ≤|z| (where z(0,1) and z(|z|,| z|+1) denote the gaps at the beginning and ends of z). Generalizing our notation for posterior probabilities of matches, an alternative estimate for the quality of an xi ∼ yj match is given by marginalized probability,
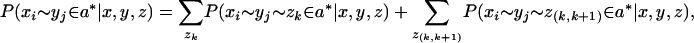
where a* now refers to a three-sequence alignment of x, y, and z. We refer to the concept of re-estimating pairwise alignment match quality scores based on three-sequence information as probabilistic consistency.
As stated, computing P(xi ∼ yj ∈ a*| x, y, z) values for each _xi_-yj pair requires O(_L_3) time for the Forward and Backward algorithms (given an appropriate three-sequence HMM); to avoid this, we simplify the computation as follows. First, we heuristically ignore the second summation over gaps in z to get
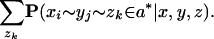
Second, we change the inner condition to an equivalent expression,
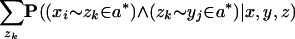
Then, we use the chain rule to factorize each inner term of the summation to obtain
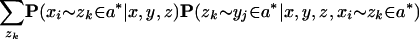
Finally, we make heuristic independence assumptions to get
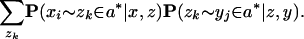
This latter expression still requires O(_L_3) time to be computed. Now, however, we transform the Pxz and Pzy matrices into sparse matrices by discarding all values smaller than a threshold ω (by default, ω = 0.01). For alignable sequences, posterior probability alignment matrices tend to be sparse, with most entries near zero, so this step is justified. This effectively reduces the probabilistic consistency re-estimation step to sparse matrix multiplication; therefore, Pxy is re-estimated in time O(c_2_L), where c is the average number of nonzero elements per row (typically 1 ≤ c ≤ 5 in practice).
With the procedure described above, we can align two sequences given information from a third sequence. To align two sequences x and y given a set of sequences, S, we would ideally like to estimate P(xi ∼ yj ∈ a*| S). In practice, we use the following heuristic decomposition:

where we set P(xi ∼ xj| x) to 1 if i = j and 0 otherwise.
In this sense, the approximate probabilistic consistency calculation may be viewed as a transformation that, given a set of all-pairs pairwise match quality scores, produces a new set of all-pairs pairwise match quality scores that have been adjusted to account for a single intermediate sequence. By iterated applications of the transformation, then, we can informally approximate the effect of accounting for more than one intermediate sequence at a time. As a default, ProbCons uses two iterated applications, which works well in practice (see Results).
In the derivations above, it is clear that several unjustified assumptions were needed in order to obtain an efficiently computable form for probabilistic consistency. In the first step, the simplification of not considering gapped positions in a sequence z is problematic. In the fourth step, the independence assumptions required for the transformation clearly do not hold for sets of related sequences. Furthermore, the decomposition of P(xi ∼ yj ∈ a*| S) into an average over the different intermediate sequences in S is also not well grounded. Nevertheless, these methods work well in practice.
As a sanity check, ignoring gapped positions in the first simplification hurts only when xi is aligned to yj through a gap in z; for reliably alignable regions in which all sequences are present, this has little effect. Averaging P(xi ∼ yj ∈ a*| z) values in the final step can be interpreted as a linear regression-like method for predicting P(xi ∼ yj ∈ a*| S) where all inputs are given identical weight. Finally, to assess the reasonableness of the independence assumptions used in deriving the factorized form of probabilistic consistency, we implemented a version of ProbCons using the full O(_L_3) consistency algorithm. Because this algorithm is slow, we tested it only on a set of 74 alignments with at most five sequences and length at most 100 residues from the Twilight Zone subset of SABmark. The full O(_L_3) consistency algorithm achieved an average fD score of 0.431 compared to 0.403 when no iterations of approximate probabilistic consistency were used, 0.422 when one iteration was used, and 0.427 when two iterations were used. In contrast to the other methods that completed all tests in under 2 sec, however, the O(_L_3) method took nearly 10 min to finish. We decided not to support the O(_L_3) version because it is inherently much slower even in the smallest examples, while it provides only modest improvements on the Twilight Zone alignments where we tested it.
4. Guide tree computation
Most progressive multiple sequence alignment programs use evolutionary distances estimated from pairwise alignments or _k_-mer statistics to build an approximate evolutionary tree via neighbor joining (Saitou and Nei 1987) or UPGMA (Sneath and Sokal 1973). In contrast, ProbCons does not attempt to build an evolutionarily correct tree but rather uses a greedy heuristic method reminiscent of UPGMA to construct a tree with high expected alignment reliability.
Given a set S of sequences to be aligned, denote the expected accuracy for aligning any two sequences x and y as E(x, y). Initially, each sequence is placed in its own cluster. Then, the two clusters x and y with the highest expected accuracy are merged to form a new cluster xy; we then define the expected accuracy of aligning xy with any other cluster z as E(x, y)(E(x, z) + E(y, z))/2. This process is repeated until only a single cluster remains.
Like UPGMA, the guide-tree computation procedure used here relies on modified arithmetic averaging to estimate the “distance” of newly created clusters to other clusters. However, the important distinction is that the computation here has the goal of finding clusters that can be reliably aligned, i.e., have high expected accuracy, rather than ones that may appear evolutionarily closer.
5. Progressive alignment
The final progressive alignment step in ProbCons is a routine extension of maximal expected accuracy alignment to an unweighted sum-of-pairs model. Since the alignments within each group are fixed, we may ignore matches between sequences in each group. Thus, for each progressive alignment step, we run a profile-profile Needleman-Wunsch alignment procedure in which the score for matching a column containing _n_1 non-gap letters to one with _n_2 non-gap letters is computed by summing _n_1_n_2 values from the corresponding pairwise posterior matrices. Note that no gap penalties are used in this final step, thus greatly simplifying the task of profile-profile alignment.
Post-processing: Iterative refinement
While incorporating consistency helps to reduce the chances of errors during the hierarchical merging of groups of sequences, the progressive alignment procedure still does not produce optimal alignments with respect to the sum-of-pairs probabilistic consistency objective function. To improve the alignment, we employ a randomized iterative improvement strategy (Berger and Munson 1991).
In this approach, the sequences of the existing multiple alignment are randomly partitioned into two groups of possibly unequal size by randomly assigning each sequence to one of the two groups to be realigned. Subsequently, the same dynamic programming procedure used for progressive alignment is employed to realign the two projected alignments. This refinement procedure can be iterated either for a fixed number of iterations or until convergence; for simplicity, only the former of these options is implemented in ProbCons, where 100 rounds of iterative refinement are applied in the default setting. Because gap penalties are not used during each realignment step, the sum-of-pairs alignment score is guaranteed to increase monotonically.
Unsupervised EM training
The ProbCons approach to alignment is simple in that the only parameters in the program are the ones specific to the HMM used to model the distribution over alignments. If one keeps the emission probabilities fixed, the HMM in Figure 1 is completely specified by three parameters, which fully determine the initial state and transition probabilities: the initial insertion probability πinsert, the insertion start probability δ, and the insertion extension probability ε. To train ProbCons via Expectation-Maximization (EM), then, we applied 20 iterations of the Baum-Welch algorithm on unaligned BAliBASE sequences, starting from random initial parameters. The resulting parameters (δ = 0.019931, ε = 0.79433, πinsert = 0.19598) were used as the default for the program. The low number of parameters for the probabilistic model here distinguishes ProbCons from profile-HMM approaches (Durbin et al. 1998), which have a much richer alignment model but consequently face a tougher training task.
Estimating column reliability
Many applications that make use of protein sequence alignments need the ability to assess which parts of an alignment are likely to be correct. Previous approaches to quantifying alignment quality have included using suboptimal alignments to locate reliable regions of alignments (Vingron and Argos 1990; Chao et al. 1993) or using a fuzzy “winner-takes-most” version of Needleman-Wunsch dynamic programming in order to “predict” the probability that a pair of residues are correctly aligned (Schlosshauer and Ohlsson 2002). It is clear that both of these approaches deal with many of the questions answered by match posterior probabilities (Miyazawa 1995, Kschischo and Lässing 2000), which represent the likelihood that specific pairs of residues are aligned.
In the multiple alignment case, one possible generalization is to estimate the expected proportion of correct pairwise matches in each column of the alignment. Given a set C of the aligned residues in a particular column, this expected proportion of correct pairwise matches ψ(C) is given by
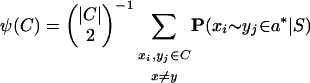
which we approximate using the pairwise posterior matrices calculated in Step 1. Though this is certainly not the only possible measure of column reliability based on posterior probabilities, we leave extensions of this method as future work.
Acknowledgments
The authors thank Arend Sidow and Robert Edgar for useful discussions and Sandhya Kunnatur for help in program development. C.B.D. was partly supported by a Siebel Fellowship. M.B. was partly supported by an NSF Graduate Fellowship. Work in the Batzoglou laboratory is supported in part by NSF grant EF-0312459, NIH grant U01-HG003162, the NSF CAREER Award, and the Alfred P. Sloan Fellowship.
Footnotes
[Supplemental material is available online at www.genome.org. Source code and executables are available as public domain software at http://probcons.stanford.edu.\]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2821705.
3
Previous results on the BAliBASE 2.01 benchmark alignments database reported in an abstract (Do et al. 2004), which correspond to the ProbCons-ext program, differ slightly from those shown in the text. These small differences are attributable to (1) a change in the methods used for extracting BAliBASE core blocks as suggested by Robert C. Edgar (pers. comm.), and (2) minor changes in the HMM model and training procedure for the current version of ProbCons.
4
The results for the nw-ns-i script from MAFFT on the PREFAB database given in Edgar (2004) contain an editing error (R.C. Edgar, pers. comm.); the values shown here are correct. Interestingly, although MAFFT achieves a slightly higher overall average SP score than MUSCLE, a Friedman rank test indicates that MUSCLE consistently produces better alignments than MAFFT (see Table 4).
5
The numbers reported for the Align-m aligner are similar to those given in Edgar (2004), but differ from the results reported in Van Walle et al. (2004). The primary reason for this difference is that the averages in the latter study were computed across all SABmark pairwise alignments; this fails to account for dependencies within each subset, so the weight of each subset scales quadratically with the number of sequences present. We avoid this by averaging pairwise alignment scores within each subset before averaging all subset scores.
6
While a ROC analysis would better characterize aligner performance, properly defining sensitivity and specificity measures for alignment accuracy involves subtle issues regarding the alignability of particular positions in sequences. Furthermore, the appropriate manner for adjusting program parameters so as to observe the sensitivity/specificity trade-off for the expected accuracy alignment algorithm is also an open problem. We leave these questions for future work.
References
- Altschul, S.F. 1991. Amino acid substitution matrices from an information theoretic perspective. J. Mol. Biol. 219**:** 555-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Carroll, R.J., and Lipman, D.J. 1989. Weights for data related by a tree. J. Mol. Biol. 207**:** 647-653. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25**:** 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood, T.K. 2002. The PRINTS database: A resource for identification of protein families. Brief. Bioinform. 3**:** 252-263. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R.D., Hollich, V., Griffiths-Jones, S., Khanna, A., Moxon, M.M., Sonnhammer, E.L., Studholme, D.J., et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32**:** D138-D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, M.P. and Munson, P.J. 1991. A novel randomized iterative strategy for aligning multiple protein sequences. Comput. Appl. Biosci. 7**:** 479-484. [DOI] [PubMed] [Google Scholar]
- Boutonnet, N.S., Rooman, M.J., Ochagavia, M.E., Richelle, J., and Wodak, S.J. 1995. Optimal protein structure alignments by multiple linkage clustering: Application to distantly related proteins. Protein Eng. 8**:** 647-662. [DOI] [PubMed] [Google Scholar]
- Brenner, S.E., Koehl, P., and Levitt, M. 2000. The ASTRAL compendium for protein structure and sequence analysis. Nucleic Acids Res. 28**:** 254-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo, H. and Lipman, D. 1988. The multiple sequence alignment problem in biology. SIAM J. Appl. Math. 48**:** 1073-1082. [Google Scholar]
- Castillo-Davis, C.I., Kondrashov, F.A., Hartl, D.L., and Kulathinal, R.J. 2004. The functional genomic distribution of protein divergence in two animal phyla: Coevolution, genomic conflict, and constraint. Genome Res. 14**:** 802-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, K.-M., Hardison, R.C., and Miller, W. 1993. Locating well-conserved regions within a pairwise alignment. Comput. Appl. Biosci. 9**:** 387-396. [DOI] [PubMed] [Google Scholar]
- Dayhoff, M.O., Schwartz, R.M., and Orcutt, B.C. 1978. A model of evolutionary change in proteins. In Atlas of proteins sequences and structure, vol. 5, Suppl. 2, pp. 345-352. National Biomedical Research Foundation, Washington, D.C. [Google Scholar]
- Do, C.B., Brudno, M., and Batzoglou, S. 2004. ProbCons: Probabilistic consistency-based multiple alignment of amino acid sequences. In Proceedings of the Thirteenth National Conference on Artificial Intelligence, pp. 703-708. AAAI Press, San Jose, CA.
- Durbin, R., Eddy, S., Krogh, A., and Mitchison, G. 1998. Biological sequence analysis. Cambridge University Press, Cambridge, UK.
- Eddy, S.R. 1995. Multiple alignment using hidden Markov models. In Proceedings of the Third International Conference on Intelligent Systems in Molecular Biology, pp. 114-120. AAAI Press, Cambridge, UK. [PubMed]
- Edgar, R.C. 2004. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32**:** 1792-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, D.F. and Doolittle, R.F. 1987. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol. 25**:** 351-360. [DOI] [PubMed] [Google Scholar]
- Gonnet, G.H., Cohen, M.A., and Benner, S.A. 1992. Exhaustive matching of the entire protein sequence database. Science 256**:** 1443-1445. [DOI] [PubMed] [Google Scholar]
- Gotoh, O. 1982. An improved algorithm for matching biological sequences. J. Mol. Biol. 162**:** 705-708. [DOI] [PubMed] [Google Scholar]
- ____. 1990. Consistency of optimal sequence alignments. Bull. Math. Biol. 52**:** 509-525. [DOI] [PubMed] [Google Scholar]
- ____. 1996. Significant improvement in accuracy of multiple protein sequence alignments by iterative refinement as assessed by reference to structural alignments. J. Mol. Biol. 264**:** 823-838. [DOI] [PubMed] [Google Scholar]
- Heger, A., Lappe, M., and Holm, L. 2003. Accurate detection of very sparse sequence motifs. In Proceedings of the Seventh Annual International Conference on Computational Molecular Biology, pp. 139-147. ACM Press, Berlin, Germany. [DOI] [PubMed]
- Henikoff, S. and Henikoff, J.G. 1992. Amino acid substitution matrices from protein blocks. Proc. Nat. Acad. Sci. 89**:** 10915-10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm, L. and Sander, C. 1994. The FSSP database of structurally aligned protein fold families. Nucleic Acids Res. 22**:** 3600-3609. [PMC free article] [PubMed] [Google Scholar]
- Holmes, I. and Durbin, R. 1998. Dynamic programming alignment accuracy. J. Comput. Biol. 5**:** 493-504. [DOI] [PubMed] [Google Scholar]
- Huang, X. and Miller, W. 1991. A time-efficient, linear space local similarity algorithm. Adv. Appl. Math. 12**:** 337-357. [Google Scholar]
- Jaroszewski, L., Li, W., and Godzik, A. 2002. In search for more accurate alignments in the twilight zone. Protein Sci. 11**:** 1702-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, J.M. and Church, G.M. 1999. Alignment and structure prediction of divergent protein families: Periplasmic and outer membrane proteins of bacterial efflux pumps. J. Mol. Biol. 287**:** 695-715. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292**:** 195-202. [DOI] [PubMed] [Google Scholar]
- Katoh, K., Misasa, K., Kuma, K., and Miyata, T. 2002. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30**:** 3059-3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kececioglu, J. 1993. The maximum weight trace problem in multiple sequence alignment. In Proceedings of the Fourth Symposium on Combinatorial Pattern Matching, Springer-Verlag Lecture Notes in Computer Science, vol. 684, pp. 106-119. London. [Google Scholar]
- Kim, J., Pramanik, S., and Chung, M.J. 1994. Multiple sequence alignment using simulated annealing. Comput. Appl. Biosci. 10**:** 419-426. [DOI] [PubMed] [Google Scholar]
- Krogh, A., Brown, M., Mian, I.S., Sjolander, K., and Haussler, D. 1994. Hidden Markov models in computational biology: Applications to protein modeling. J. Mol. Biol. 235**:** 1501-1531. [DOI] [PubMed] [Google Scholar]
- Kschischo, M., and Lässing, M. 2000. Finite-temperature sequence alignment. Pac. Symp. Biocomput. 5**:** 621-632. [DOI] [PubMed] [Google Scholar]
- Metz, C.E. 1978. Basic principles of ROC analysis. Semin. Nucl. Med. 8**:** 283-298. [DOI] [PubMed] [Google Scholar]
- Mizuguchi, K., Deane, C.M., Blundell, T.L., and Overington, J.P. 1998. HOMSTRAD: A database of protein structure alignments for homologous families. Prot. Sci. 7**:** 2469-2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa, S. 1995. A reliable sequence alignment method based on probabilities of residue correspondences. Protein Eng. 8**:** 999-1009. [DOI] [PubMed] [Google Scholar]
- Morgenstern, B., Dress, A., and Werner, T. 1996. Multiple DNA and protein sequence alignment based on segment-to-segment comparison. Proc. Nat. Acad. Sci. 93**:** 12098-12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern, B., Frech, K., Dress, A., and Werner, T. 1998. DIALIGN: Finding local similarities by multiple sequence alignment. Bioinformatics 14**:** 290-294. [DOI] [PubMed] [Google Scholar]
- Murzin, A.G., Brenner, S.E., Hubbard, T., and Chothia, C. 1995. SCOP: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247**:** 536-540. [DOI] [PubMed] [Google Scholar]
- Myers, E.W. and Miller, W. 1988. Optimal alignments in linear space. Comput. Appl. Biosci. 4**:** 11-17. [DOI] [PubMed] [Google Scholar]
- Needleman, S.B. and Wunsch, C.D. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. BIol. 48**:** 443-453. [DOI] [PubMed] [Google Scholar]
- Notredame, C. and Higgins, D.G. 1996. SAGA: Sequence alignment by genetic algorithm. Nucleic Acids Res. 24**:** 1515-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame, C., Holm, L., and Higgins, D.G. 1998. COFFEE: An objective function for multiple sequence alignments. Bioinformatics 14**:** 407-422. [DOI] [PubMed] [Google Scholar]
- Notredame, C., Higgins, D.G., and Heringa, J. 2000. T-Coffee: A novel method for multiple sequence alignments. J. Mol. Biol. 302**:** 205-217. [DOI] [PubMed] [Google Scholar]
- Phillips, A., Janies, D., and Wheeler, W. 2000. Multiple sequence alignments in phylogenetic analysis. Mol. Phylogenet. Evol. 16**:** 317-330. [DOI] [PubMed] [Google Scholar]
- Pruitt, K.D., Tatusova, T., and Maglott, D.R. 2003. NCBI Reference Sequence project: Update and current status. Nucleic Acids Res. 31**:** 34-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost, B. 1999. Twilight zone of protein sequence alignments. Protein Eng. 12**:** 85-94. [DOI] [PubMed] [Google Scholar]
- Rost, B. and Sander, C. 1994. Combining evolutionary information and neural networks to predict protein secondary structure. Proteins 19**:** 55-77. [DOI] [PubMed] [Google Scholar]
- Saitou, N. and Nei, M. 1987. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4**:** 406-425. [DOI] [PubMed] [Google Scholar]
- Sauder, J.M., Arthur, J.W., and Dunbrack Jr., R.L. 2000. Large-scale comparison of protein sequence alignment algorithms with structure alignments. Proteins 40**:** 6-22. [DOI] [PubMed] [Google Scholar]
- Schlosshauer, M. and Ohlsson, M. 2002. A novel approach to local reliability of sequence alignments. Bioinformatics 18**:** 847-854. [DOI] [PubMed] [Google Scholar]
- Shindyalov, I.N. and Bourne, P.E. 1998. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 11**:** 739-747. [DOI] [PubMed] [Google Scholar]
- Smith, T.F. and Waterman, M.S. 1981. Identification of common molecular subsequences. J. Mol. Biol. 147**:** 195-197. [DOI] [PubMed] [Google Scholar]
- Sneath, P.H.A. and Sokal, R.R. 1973. Numerical Taxonomy. Freeman, San Francisco, CA.
- Sonnhammer, E.L.L., Eddy, S.R., Birney, E., Bateman, A., and Durbin, R. 1998. Pfam: Multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 26**:** 320-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22**:** 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Plewniak, F., and Poch, O. 1999a. BAliBASE: A benchmark alignment database for the evaluation of multiple alignment programs. Bioinformatics 15**:** 87-88. [DOI] [PubMed] [Google Scholar]
- ____. 1999b. A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res. 27**:** 2682-2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Walle, I., Lasters, I., and Wyns, L. 2004. Align-m—A new algorithm for multiple alignment of highly divergent sequences. Bioinformatics 20**:** 1428-1435. [DOI] [PubMed] [Google Scholar]
- Vingron, M. and Argos, P. 1989. A fast and sensitive multiple sequence alignment algorithm. Comput. Appl. Biosci. 5**:** 115-121. [DOI] [PubMed] [Google Scholar]
- ____. 1990. Determination of reliable regions in protein sequence alignments. Protein Eng. 3**:** 565-569. [DOI] [PubMed] [Google Scholar]
- ____. 1991. Motif recognition and alignment for many sequences by comparison of dot matrices. J. Math. Biol. 218**:** 34-43. [DOI] [PubMed] [Google Scholar]
- Vingron, M. and Waterman, M.S. 1994. Sequence alignment and penalty choice: Review of concepts, case studies and implications. J. Mol. Biol. 235**:** 1-12. [DOI] [PubMed] [Google Scholar]
- Viterbi, A.J. 1967. Error bounds for convolutional codes and an asymptotically optimal decoding algorithm. IEEE Trans. Inf. Theory IT-13**:** 260-269. [Google Scholar]
WEB SITE REFERENCES
- http://probcons.stanford.edu; ProbCons alignment tool.