Mesothelin Immunotherapy for Cancer: Ready for Prime Time? (original) (raw)
Abstract
Mesothelin is a tumor antigen that is highly expressed in many human cancers, including malignant mesothelioma and pancreatic, ovarian, and lung adenocarcinomas. It is an attractive target for cancer immunotherapy because its normal expression is limited to mesothelial cells, which are dispensable. Several antibody-based therapeutic agents as well as vaccine and T-cell therapies directed at mesothelin are undergoing clinical evaluation. These include antimesothelin immunotoxins (SS1P, RG7787/LMB-100), chimeric antimesothelin antibody (amatuximab), mesothelin-directed antibody drug conjugates (anetumab ravtansine, DMOT4039A, BMS-986148), live attenuated _Listeria monocytogenes_–expressing mesothelin (CRS-207, JNJ-64041757), and chimeric antigen receptor T-cell therapies. Two antimesothelin agents are currently in multicenter clinical registration trials for malignant mesothelioma: amatuximab in the first-line setting and anetumab ravtansine as second-line therapy. Phase II randomized clinical trials of CRS-207 as a boosting agent and in combination with immune checkpoint inhibition for pancreatic cancer are nearing completion. These ongoing studies will define the utility of mesothelin immunotherapy for treating cancer.
INTRODUCTION
Mesothelin (MSLN) is a tumor-differentiation antigen discovered by Ira Pastan and Mark Willingham at the National Cancer Institute more than 20 years ago.1,2 It is a cell-surface glycoprotein with normal expression limited to mesothelial cells lining the pleura, peritoneum, and pericardium but is also highly expressed in many cancers, including malignant mesothelioma, pancreatic cancer, ovarian cancer, lung adenocarcinoma, endometrial cancer, biliary cancer, gastric cancer, and pediatric acute myeloid leukemia.3-11 There are few tumor-specific antigens that are used to therapeutically target solid tumors, because most of them are also expressed on critical tissues. Because MSLN is expressed only on dispensable tissues, the risk of nonspecific toxicity is decreased. Since our initial report in 1999 that indium-111–labeled anti-MSLN monoclonal antibody K1 localizes to MSLN-expressing tumor xenografts in mice, it has been shown that anti-MSLN monoclonal antibodies do localize to MSLN-positive solid tumors in patients.12-14 In addition, preclinical studies as well as results from initial clinical trials have validated MSLN as an attractive target for cancer therapy with antibody-based approaches as well as tumor vaccines.15
The first patient to receive an MSLN-targeted therapy was a patient with ovarian cancer treated by Raffit Hassan in 2000 with the anti-MSLN agent SS1P.16 Since then, there has been a remarkable proliferation of drugs targeting MSLN. Many are in advanced stages of clinical testing, including randomized trials for mesothelioma and pancreatic cancer. The discovery of MSLN and its validation as a target for cancer therapy have been previously described.15 This review will focus on important therapeutic approaches as well as ongoing clinical trials and discuss the broad implications of targeting MSLN for cancer therapy.
MSLN
MSLN is a glycophosphatidylinositol (GPI) –linked cell-surface glycoprotein. It is synthesized as a 71-kD precursor protein and is then cleaved by the endoprotease furin to release the secreted N-terminal region, called megakaryocyte potentiating factor (MPF), whereas the 41-kD mature MSLN remains attached to the membrane.2,17 The remaining GPI-linked mature MSLN can also be shed from the cell through the action of the tumor necrosis factor α–converting enzyme protease.18 The normal physiologic distribution of MSLN identifies it as a differentiation factor for mesothelial cells, but the biologic role that MSLN plays in these cells remains unclear. Database searches reveal that MSLN is remotely homologous to two inner ear proteins of unknown structure and contains no conserved consensus domains.19 Three-dimensional structure prediction programs have determined that MSLN consists of a superhelical structure with armadillo-type repeats.20 No crystal structure has yet been determined for the whole protein, but the structure of an N-terminal fragment bound to a Fab of the SS1 antibody has been obtained.21 Furthermore, MSLN knockout mice grow and reproduce normally and have no detectable phenotype.22
MSLN is expressed by many solid tumors, with particularly robust expression in mesothelioma, epithelial ovarian cancer, and pancreatic adenocarcinoma (Table 1).23-46 Higher expression of MSLN has been correlated with poorer prognosis for patients with ovarian cancer,35 cholangiocarcinoma,41,42 lung adenocarcinoma,8,9 triple-negative breast cancer,10,45 and resectable pancreatic adenocarcinoma.32,33 In the neoplastic setting, MSLN is known to bind to the ovarian cancer antigen MUC16 (cancer antigen 125).47 The two proteins are frequently coexpressed, and binding of MSLN and MUC16 has been shown to induce cell-to-cell adhesion in these cell types.48 MUC16 expressed on cancer cells can also facilitate cancer cell attachment to the MSLN-expressing serosal surfaces in the pleura and peritoneum, possibly contributing to peritoneal seeding and metastatic spread. In addition, signaling mediated by MSLN and MUC16 binding has been reported to increase cellular resistance to anoikis,49 upregulate matrix metalloproteinases important in cellular invasion and metastasis,50-52 and induce secretion of autocrine growth factors by constitutively activating nuclear factor kappa B (NF-κB).53,54 However, it seems that MSLN expression may also trigger signaling events independent of MUC16 binding.51 The exact mechanics of these pathways and how MSLN interacts with components of the tumor microenvironment, including stromal cells, the extracellular matrix, and immune-cell populations, are not known.
Table 1.
MSLN Expression in Solid Tumors
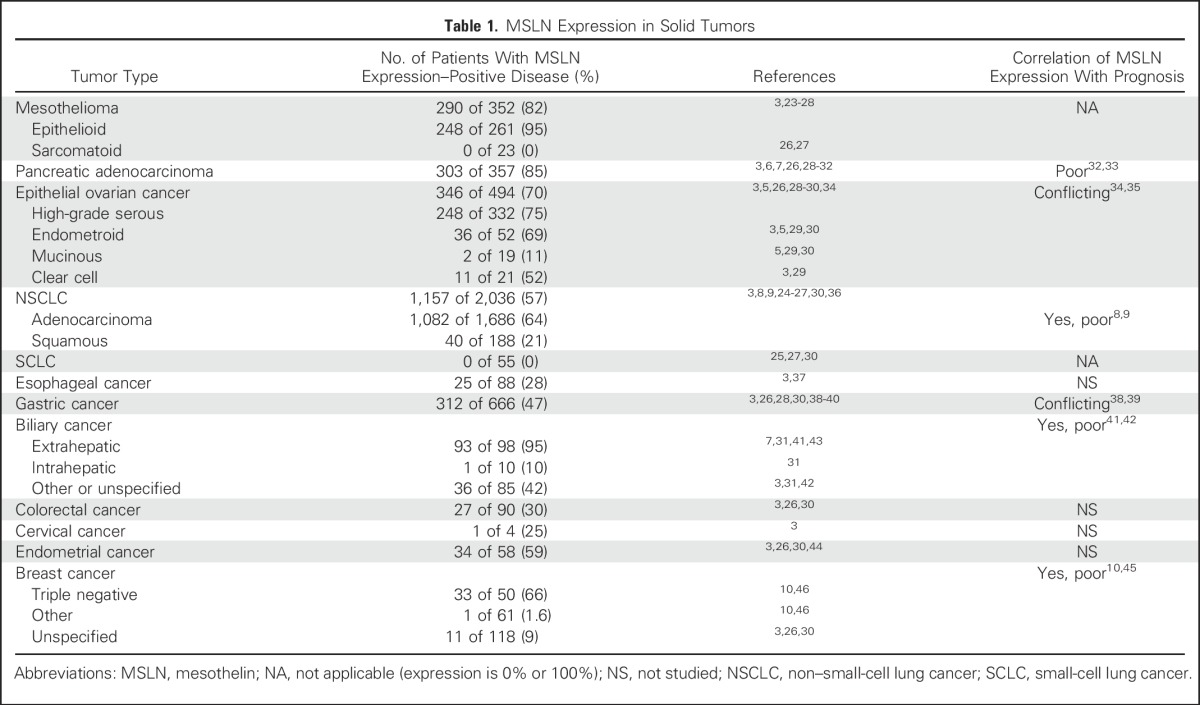
Why MSLN, a glycoprotein normally restricted to serosal cells of the pleura, peritoneum, and pericardium, is expressed in a wide variety of adenocarcinomas is not clear at this time. However, regulation of MSLN expression in tumors has been assessed in several studies and seems to be cell-type specific. At the epigenetic level, it was found that hypomethylation did not correlate with MSLN expression in ovarian or endometrial cancer specimens44 or in mesothelioma.27 However, hypomethylation of the promoter was noted in MSLN-expressing pancreatic cancer specimens, and treatment of a nonexpressing pancreatic cancer cell line with demethylating agents could induce expression of MSLN, suggesting that epigenetic mechanisms may regulate MSLN expression in this cell type.55 Transcription of MSLN is driven by a TATA-less promoter located upstream of the transcriptional start site. Enhancers responsible for initiating strong expression in normal serosal cells and cancers derived from them (eg, mesothelioma and ovarian cancer) are unknown. Cancer-specific ectopic upregulation in pancreatic and cervical cancers has been attributed to the transcriptional enhancer factor 1 (TEF-1) transcription factor binding to a conventional MCAT sequence within an upstream enhancer region called CanScript. However, TEF-1 expression itself is necessary but not sufficient to induce MSLN expression, suggesting an unknown cofactor is also required.56 Similarly, the yes-associated protein 1 (YAP1) transcription factor binds to an SP-1 motif within the CanScript and is also required but insufficient for MSLN expression.57 Further study will be required to delineate this mechanism. More recently, it was discovered that MSLN is reciprocally regulated at the post-transcriptional level by mIR-198 as part of a feedback loop that involves NF-kB and the homeobox transcription factors octamer transcription factor 2 (OCT-2), pre–B cell leukemia homeobox 1 (PBX-1), and valosin-containing protein (VCP).58
STRATEGIES TO TARGET MSLN
The high cell-surface expression of MSLN in many cancers lends itself to tumor-specific targeting using monoclonal antibodies, by themselves or carrying protein toxins or low–molecular weight cytotoxic agents, as well as by chimeric T cells containing Fv fragments that recognize MSLN (Fig 1). Other approaches in advanced clinical trials include vaccines that can induce T-cell immune response to MSLN.
Fig 1.

Approaches used to target MSLN in clinical trials. APC, antigen-presenting cell; CAR, chimeric antigen receptor; DM4, ravtansine; mAb, monoclonal antibody; PE, pseudomonas exotoxin.
Immunotoxins
SS1P is a recombinant protein therapeutic that consists of a high-affinity disulfide-bonded Fv that targets MSLN fused to a Pseudomonas exotoxin A (PE) payload.59,60 SS1P binds to MSLN and is internalized by the cell through endocytosis. The toxin is delivered to the cytosol, where it irreversibly modifies elongation factor-2 to halt protein synthesis and induce apoptosis.61 SS1P has been tested as a single agent as well as in combination with chemotherapy.16,62,63 The dose-limiting toxicities (DLTs) are capillary leak syndrome, which is a class effect of immunotoxin therapies, and pleuritis, from on-target, but off-tumor, SS1P binding to mesothelial cells causing inflammation of the normal pleura. The single-agent maximum-tolerated dose (MTD) is 45 mcg/kg when administered as a bolus infusion on days 1, 3, and 5.16 Additive toxicity was not observed in combination with cisplatin plus pemetrexed chemotherapy.63 Efficacy of SS1P is limited by antidrug antibody formation, which typically occurs by the end of the first cycle (three doses) of treatment. For this reason, SS1P is now being administered in combination with a lymphocyte-depleting conditioning regimen of pentostatin and cyclophosphamide. This delays antidrug antibody formation and allows patients to receive multiple effective cycles of SS1P.64
LMB-100 (previously RG7787 and Ro6927005) is a recently developed recombinant immunotoxin that also targets MSLN. It consists of a humanized anti-MSLN Fab fragment with a newly designed PE (PE24) engineered to be less immunogenic than SS1P.65 It also has decreased toxicity in animal models and shows broad activity against different MSLN-expressing cancers.66-68 Phase I clinical trials of LMB-100 have recently opened for treatment of patients with mesothelioma and pancreatic cancer.
Vaccine
Elizabeth Jaffee’s laboratory at Johns Hopkins was the first to identify MSLN as a vaccine-induced CD8+ T-cell target using immunized lymphocytes from patients responding to granulocyte-macrophage colony-stimulating factor–secreting whole pancreatic tumor cell vaccine (GVAX), which expresses multiple antigens. Using an unbiased genomics-based screening approach, they identified MSLN as a target of vaccine-induced T cells in patients who were treated with GVAX who also demonstrated long-term disease-free survival.69,70 Subsequently, in collaboration with scientists at Aduro Biotech, they developed a Listeria monocytogenes vaccine expressing MSLN (LM-mesothelin) and demonstrated in preclinical models that GVAX was the best priming vaccine and LM-mesothelin was the best boosting vaccine for the treatment of pancreatic cancer. These studies led to the clinical development of an LM-mesothelin for other cancers expressing MSLN.
CRS-207 is a recombinant live-attenuated, double-deleted L monocytogenes engineered to secrete MSLN into the cytosol of infected antigen presentation cells.71 The bacteria retain the potency of the fully virulent pathogen but have vastly reduced toxicity as compared with wild-type Listeria because of the selective deletion of two virulence factors: ActA (ΔactA) and internalin B (ΔinlB). This blocks the direct internalin B–mediated infection of nonphagocytic cells, such as hepatocytes, and the indirect ActA-mediated infection by cell-to-cell spread from adjacent phagocytic cells. LM provides potent stimulation of innate immunity and also stimulates an adaptive immune response through recruitment and activation of CD4+ and CD8+ T cells specific for encoded heterologous antigens. Listeria ΔactA/ΔinlB-based vaccines were rapidly cleared from mice after immunization and induced potent and durable effector and memory T-cell responses with no measurable liver toxicity. In a phase I study, CRS-207 was well tolerated at doses up to a maximum planned dose of 1 × 1010 colony-forming units.72 Immune activation and induction of MSLN-specific T-cell responses were observed.
Chimeric Monoclonal Antibody
Amatuximab (previously MORAb-009) is a chimeric high-affinity monoclonal immunoglobulin G1/k antibody targeting MSLN.73 In vitro, amatuximab mediates inhibition of MSLN-dependent cell adhesion and antibody-dependent cellular cytotoxicity. Although it has moderate antitumor activity as a monotherapy against MSLN-expressing tumor xenografts in vivo, this effect is markedly increased in combination with chemotherapy. In a phase I trial, the DLTs were transaminitis and serum sickness; the MTD was determined to be 200 mg/m2.74
Antibody–Drug Conjugates
Anetumab ravtansine (previously BAY 94-9343) is an antibody–drug conjugate consisting of a human anti-MSLN antibody conjugated to the maytansinoid tubulin inhibitor DM4 via a disulfide-containing linker. In preclinical studies, anetumab ravtansine was shown to be both potent and highly selective in killing MSLN-expressing tumor cells compared with MSLN-negative cells.75 The drug is bound and internalized by MSLN-expressing tumor cells75; degradation of the BAY 94-9343 disulfide-based linker releases a cell-permeable DM4 metabolite with bystander killing potential. In vivo, anetumab ravtansine localizes specifically to MSLN-expressing tumors and inhibits tumor growth in pancreatic, ovarian, and mesothelioma cancer models. The MTD of anetumab ravtansine in a phase I trial was 6.5 mg/kg administered once every 3 weeks intravenously.76 DLTs were keratitis and peripheral neuropathy. These adverse events were reversible and not life threatening, but they required dose reduction in approximately half of all patients. DMOT4039A, a humanized immunoglobulin G1 anti-MSLN monoclonal antibody conjugated to the antimitotic agent MMAE has completed phase I testing in patients with pancreatic and ovarian cancers (ClinicalTrials.gov identifier NCT01469793). The MTD was found to be 2.4 mg/kg when administered once every 3 weeks and 1 mg/kg when administered on a once-per-week schedule. DLTs were hyperglycemia, hypophosphatemia, and ileus.77
Chimeric Antigen Receptor T Cells
To make anti-MSLN chimeric antigen receptor (CAR) T cells, autologous patient T cells are modified to express an MSLN-binding T-cell receptor linked to appropriate cosignaling molecules, such that binding of these modified T cells to MSLN on cancer cells activates the cells to attack the tumor.78,79 Given concerns that targeting MSLN could cause serositis, as is seen with the immunotoxin SS1P, an mRNA electroporation technique was used to engineer the anti-MSLN CAR T cells used in the initial clinical study, because this method produces only transient expression of the CAR (ClinicalTrials.gov identifier NCT1355965). Data from two patients in this study have been reported; both patients experienced transient responses, and neither developed serositis.80 Given these promising results, two trials of anti-MSLN CAR T cells engineered by typical viral transduction methods have been initiated (ClinicalTrials.gov identifiers NCT01583686 and NCT02159716). More recently, it was also shown in preclinical studies that intrapleural infusion of anti-MSLN CAR T cells could enhance cell persistence and induce more durable responses81; a clinical trial of this delivery method is now open (ClinicalTrials.gov identifier NCT02414269). Because there have been recent reviews describing results from ongoing clinical trials as well as future directions of these therapeutics,78,79 CAR T-cell clinical trials are not discussed in the remainder of this review.
CLINICAL DEVELOPMENT OF MSLN-TARGETED THERAPIES
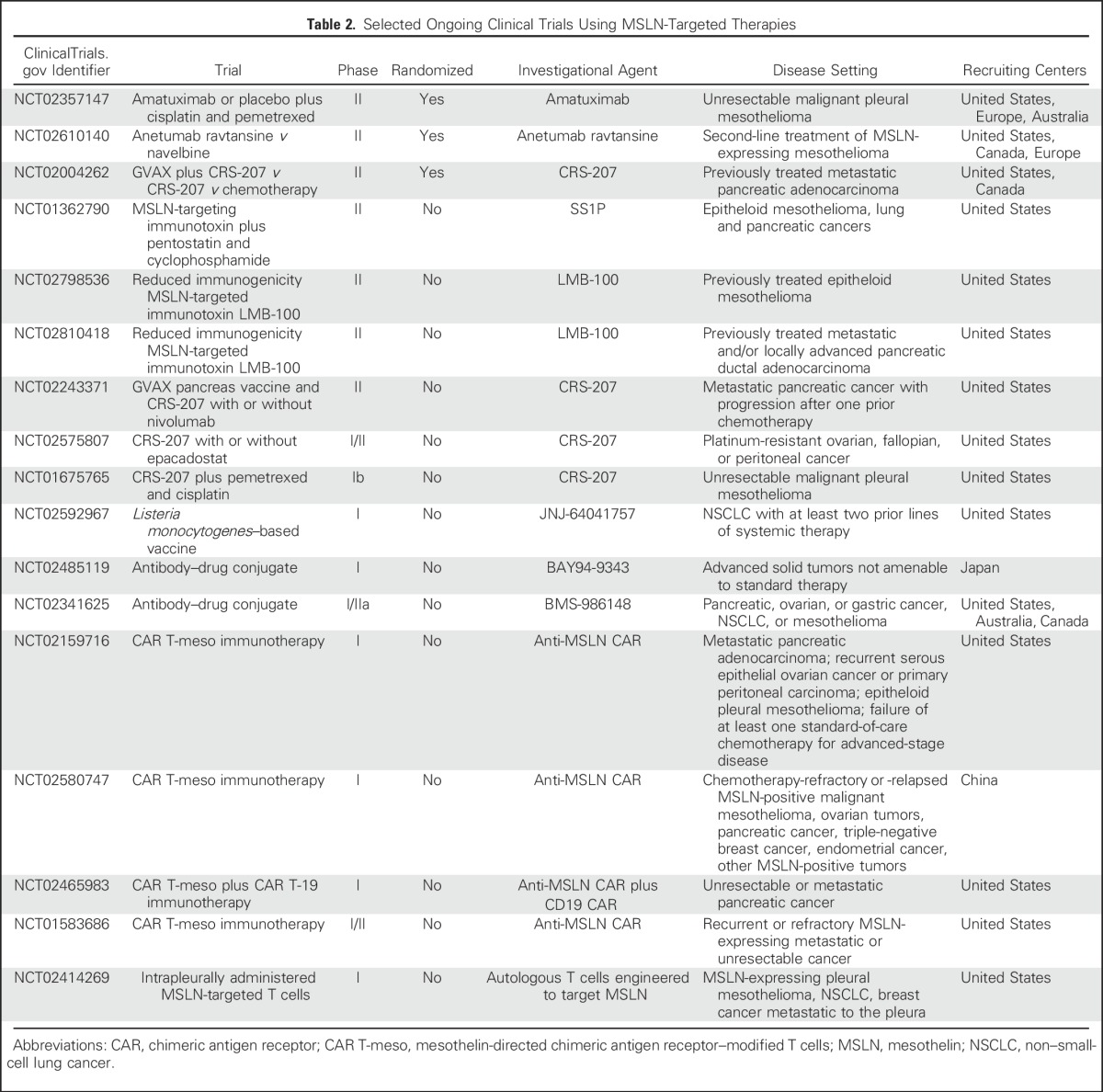
Multiple clinical trials are currently evaluating different anti-MSLN agents for cancer therapy. Most of the efforts thus far have focused on malignant mesothelioma, pancreatic cancer, and ovarian cancer (Table 2).
Table 2.
Selected Ongoing Clinical Trials Using MSLN-Targeted Therapies
Mesothelioma
Because a majority of malignant mesotheliomas have high and uniform cell-surface expression of MSLN, this disease is an especially good target for MSLN-directed therapies. However, sarcomatoid mesotheliomas, which constitute approximately 10% to 15% of all mesotheliomas, are not eligible for these therapies, because they lack MSLN expression. There are currently more than 10 clinical trials of anti-MSLN agents for treating mesothelioma, including one first-line registration trial and one registration trial in the second-line setting.
SS1P clinical trials.
The anti-MSLN immunotoxin SS1P has been extensively evaluated in this tumor type. In the phase I clinical trials of SS1P, patients with mesothelioma were treated on an every-other-day bolus schedule16 and on a continuous-infusion schedule.62 These studies established the safety of MSLN as a target for cancer therapy. The DLT was pleuritis because of expression of MSLN on mesothelial cells lining the pleura. Surprisingly, patients did not develop pericarditis. However, antitumor activity was limited because most patients developed neutralizing antibodies against the toxin portion of SS1P and could not receive additional effective treatment cycles. On the basis of laboratory studies, two different approaches were pursued to increase the efficacy of SS1P. Given the remarkable antitumor synergy between SS1P and chemotherapy in vivo, SS1P was tested in combination with pemetrexed and cisplatin as first-line therapy for pleural mesothelioma.63 Patients who were not candidates for curative surgical resection received pemetrexed 500 mg/m2 and cisplatin 75 mg/m2 on day 1 of a 21-day cycle for a maximum of six cycles; SS1P 45 μg/kg was administered on days 1, 3, and 5 during cycles one and two only. This study demonstrated the safety of the combination. The most common SS1P-related toxicities included hypoalbuminemia, fatigue, hypotension, and edema. Of the 20 evaluable patients treated in this study, 12 (60%) experienced objective tumor responses, and three had stable disease. This study also showed that serum MSLN and serum MPF levels correlated with radiologic tumor response. However, the addition of chemotherapy did not delay the formation of anti-SS1P neutralizing antibodies.
Because neutralizing antibodies to immunotoxins limited their clinical efficacy, strategies were developed to circumvent this. Single-agent immunosuppressive agents, such as steroids, cyclosporine, and rituximab, were not successful.82 A preclinical study showed that coadministration of pentostatin and cyclophosphamide ended anti-SS1P antibody formation in immunocompetent mice.83 On the basis of these results, a pilot study was designed in which patients with mesothelioma for whom standard therapies had failed received pentostatin and cyclophosphamide before SS1P. Using this approach, only two of 10 patients developed anti-SS1P antibodies after the first cycle compared with 88% of patients in prior studies of SS1P alone.64 This regimen induced lymphopenia without neutropenia, and none of the patients developed opportunistic infections. More importantly, three of 10 evaluable patients experienced major cancer regressions that were long lasting. To avoid use of an immunosuppressive regimen with immunotoxin therapy, a less immunogenic anti-MSLN immunotoxin, LMB-100 (RG7787), was developed and is now being evaluated in a clinical trial for treatment of patients with mesothelioma for whom prior chemotherapy has failed (ClinicalTrials.gov identifier NCT02798536).
Amatuximab for pleural mesothelioma.
On the basis of the phase I study of amatuximab in patients with advanced solid tumors that established its safety74 and laboratory data showing synergy with chemotherapy in preclinical in vivo models, a phase II study of amatuximab was initiated in patients with mesothelioma.84 In this nonrandomized trial, 89 patients with malignant pleural mesothelioma who had received no prior chemotherapy and were not candidates for surgical resection were treated with pemetrexed 500 mg/m2 and cisplatin 75 mg/m2 on day 1 of a once-every-3-weeks cycle for six cycles. Amatuximab 5 mg/kg was administered on days 1 and 8 of a 21-day cycle. The primary end point of this study was improvement of progression-free survival (PFS). The objective response rate by independent radiologic review was 40%, and 51% of patients had stable disease, for an overall disease control rate of 91%. Although the study did not meet its primary end point of a 6-month PFS response rate of 62%, the overall survival of patients was 14.8 months, which is better than historical controls with pemetrexed and cisplatin alone. Analysis of the pharmacokinetic data showed that serum amatuximab trough concentrations greater than the population median of 38.2 μg/mL were associated with significant improvement of both PFS as well as overall survival. Overall survival was 583 days for patients with amatuximab trough concentrations greater than 38.2 μg/mL versus 375 days for patients with amatuximab trough concentration less than 38.2 μg/mL.85 Pharmacodynamic modeling shows that administering amatuximab 5 mg/kg once per week will allow 80% of patients to achieve amatuximab trough concentrations greater than 38.2 μg/mL. On the basis of these data, a randomized phase II registration clinical trial of amatuximab plus pemetrexed and cisplatin versus pemetrexed and cisplatin for patients with newly diagnosed unresectable mesothelioma has been initiated (ClinicalTrials.gov identifier NCT02357147).
Anetumab ravtansine trials.
Anetumab ravtansine was evaluated in a phase I clinical trial in patients with MSLN-expressing cancers that included an expansion cohort of patients with mesothelioma.76 In this trial, anetumab ravtansine was administered to patients in increasing doses once every 3 weeks; the MTD of anetumab ravtansine with this schedule was 6.5 mg/kg. The DLTs at 7.5 mg/kg were keratitis and neuropathy. The activity of anetumab ravtansine in patients with MSLN-positive malignant mesothelioma was evaluated in an expansion cohort. Of 16 patients with mesothelioma treated at the MTD, five patients (31%) experienced objective tumor responses, and seven (44%) had stable disease. However, in patients with pleural mesothelioma who received anetumab ravtansine as second-line therapy, five (50%) of 10 experienced objective partial responses, and four (40%) had stable disease. These results have led to a randomized phase II trial of anetumab ravtansine as second-line therapy for patients with mesothelioma (ClinicalTrials.gov identifier NCT02610140). In this registration clinical trial, patients are randomly assigned to anetumab ravtansine 6.5 mg/kg once every 3 weeks versus vinorelbine 30 mg/m2 once per week, with progression-free survival as the primary end point.
CRS-207 study in pleural mesothelioma.
On the basis of a phase I study showing the safety of CRS-207 and induction of MSLN-specific T-cell immune response in patients with mesothelioma,72 a phase Ib clinical trial of CRS-207 plus pemetrexed and cisplatin was initiated in patients with pleural mesothelioma who were not candidates for surgical resection. Patients received two doses of intravenous CRS-207 2 weeks apart, followed 2 weeks later by standard doses of pemetrexed and cisplatin for four to six cycles. Patients achieving a response or stable disease received two more doses of CRS-207 once every 3 weeks as maintenance. As of December 2015, 38 patients had been enrolled in the study. The most common CRS-207 toxicities were similar to those seen in the phase I trial, consisting mostly of grade 1 fever (79%) and chills or rigor (82%). Of the 34 evaluable patients treated in the study, 20 (59%) experienced objective partial responses, and 12 (35%) had stable disease, for a disease control rate of 94%.86
Pancreatic Cancer
Because several studies have shown high expression of MSLN in pancreatic cancer, different approaches to targeting it are being evaluated in the clinic, including MSLN vaccine, chimeric antibody, and antibody–drug conjugate.
Clinical trials of CRS-207 in pancreatic cancer.
A heterologous prime–boost strategy using cyclophosphamide with GVAX pancreas followed by CRS-207 was tested in previously treated patients with metastatic pancreatic cancer. Cyclophosphamide was administered the day before GVAX to inhibit regulatory T cells. Patients in the comparator arm received cyclophosphamide plus GVAX alone. Despite a heavily pretreated patient population, there was an overall survival benefit favoring the prime–boost arm (6.1 v 3.9 months; hazard ratio, 0.59; P = .02).87 A follow-up study comparing the efficacy of CRS-207 alone versus prime–boost vaccine versus chemotherapy in patients who had received two or more prior regimens for metastatic disease was recently completed (ClinicalTrials.gov identifier NCT02004262). The primary end point examining overall survival was not reached, with a median overall survival of 3.8 months for patients treated with CRS-207 plus GVAX pancreas, 5.4 months for patients treated with CRS-207 alone, and 4.6 months for patients treated with chemotherapy (D. Le, personal communication, September 2016). Ongoing and future strategies are aimed at combinatorial strategies with other immune modulators. A second study testing the prime–boost strategy with or without programmed death 1 (PD-1) inhibition is almost complete (ClinicalTrials.gov identifier NCT02243371). Results of these studies will inform further development of these agents.
Antibody-based therapies for pancreatic cancer.
The antibody–drug conjugate DMOT4039A was recently tested in a phase I study that included patients with previously treated advanced pancreatic cancer. In this study, MSLN expression was not prospectively assessed in the majority of enrolled patients; however, the last 10 patients enrolled were required to have 2+ or 3+ expression to join. Responses were seen in two of 26 patients treated at the recommended phase II dose.77
On the basis of antitumor efficacy of the antimesothelin immunotoxin SS1P administered in combination with pentostatin and cyclophosphamide in patients with malignant mesothelioma, one study is currently enrolling patients with advanced previously treated pancreatic cancer (ClinicalTrials.gov identifier NCT01362790). In addition, based on preclinical studies showing remarkable synergy between nab-paclitaxel and LMB-100, a phase I clinical trial of this combination has just started accrual for patients with previously treated pancreatic cancer with progressive disease (ClinicalTrials.gov identifier NCT02810418).
Non–Small-Cell Lung Cancer
MSLN mRNA and protein are present in a substantial number of lung adenocarcinomas.8,9,25,30,36 Recent studies have identified MSLN expression in more than 50% of both early-stage and advanced lung adenocarcinomas. Kachala et al9 found MSLN expression in 69% of early-stage (stages I to III) lung adenocarcinomas, with approximately 20% of patients strongly expressing MSLN. Thomas et al8 observed MSLN expression in 53% of advanced (stages IIIB to IV) lung adenocarcinomas, with high expression found in approximately 25% of patients. The studies also indicated that MSLN expression might be an independent driver of an aggressive tumor phenotype; in both series, high MSLN expression was independently associated with inferior overall survival. Tumors with high MSLN expression were also more likely to have KRAS mutations, compared with tumors with low MSLN expression. Given its frequent expression and possibly important role in lung adenocarcinoma growth and dissemination, MSLN is being actively investigated as a potential therapeutic target in patients with therapy-resistant lung adenocarcinoma. A phase I study is evaluating the safety and immunogenicity of JNJ-64041757, a live attenuated L monocytogenes vaccine in patients with non–small-cell lung cancer (ClinicalTrials.gov identifier NCT02592967).
Ovarian Cancer
The antibody–drug conjugate DMOT4039A was recently tested in a phase I study that included patients with platinum-resistant ovarian cancer.77 In this study, MSLN expression was prospectively assessed, and 3+ expression by immunohistochemistry was required for eligibility. Responses were seen in three of 10 patients treated once every 3 weeks and one of 12 patients treated on the once-per-week administration schedule.
FUTURE DIRECTIONS
There has been substantial progress in the development of different approaches to target MSLN for cancer therapy. Although there was initial concern that pericardial toxicity could be limiting in the development of MSLN-targeted agents because of mesothelin expression on mesothelial cells lining the pericardium, this has not been seen in patients. Because MSLN is highly expressed in many cancers, these therapies could have broad implications for treatment of patients with solid tumors. In the case of malignant mesothelioma, two drugs are currently in multicenter registration clinical trials. These include amatuximab plus pemetrexed and cisplatin versus chemotherapy alone in untreated patients with unresectable pleural mesothelioma. In the second-line setting, a randomized clinical trial of anetumab ravtansine versus vinorelbine is currently ongoing in patients with pleural mesothelioma who experienced progression with prior pemetrexed and cisplatin therapy. Clinical trials of anti-MSLN immunotoxins are also ongoing for previously treated patients with mesothelioma. In the case of pancreatic cancer, a randomized clinical trial of GVAX pancreas vaccine and CRS-207 with or without nivolumab is ongoing in patients with metastatic disease. In addition, the combination of LMB-100 with nab-paclitaxel is being evaluated in patients with locally advanced and metastatic pancreatic cancers for whom prior therapies have failed. A new clinical trial of an attenuated _L monocytogenes_–expressing MSLN is currently ongoing in lung cancer.
Because some of the MSLN-directed therapies may have nonoverlapping toxicity with chemotherapy and immunotherapy agents and could potentially result in synergistic activity, combination clinical trials have just been initiated. These include anetumab ravtansine with chemotherapy for treatment of patients with mesothelioma and lung cancer and clinical trials of CRS-207 plus immune checkpoint inhibitors for pancreatic cancer. However, with regard to combination therapy trials, it will be important to have good scientific rationale before conducting large-scale clinical trials. For example, in the case of lung adenocarcinoma, to combine MSLN-directed agents with immune checkpoint therapy, it would be important to have a good understanding of coexpression of MSLN as well as programmed death ligand 1 (PD-L1). Furthermore, additional clinical trials for some of the agents we have described are being planned for assessment in additional cancers with MSLN expression, such as gastric cancer, endometrial cancer, cholangiocarcinoma, and triple-negative breast cancer. Finally, it is imperative to identify and validate companion assays to detect tumor MSLN expression.
Footnotes
Supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute (NCI), Center for Cancer Research, and in part by Grant No. 1K23CA163672 from NCI (D.T.L.).
Authors’ disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
AUTHOR CONTRIBUTIONS
Conception and design: All authors
Collection and assembly of data: All authors
Data analysis and interpretation: All authors
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Mesothelin Immunotherapy for Cancer: Ready for Prime Time?
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Raffit Hassan
Research Funding: Aduro Biotech (Inst), Roche (Inst), Morphotek (Inst), Bayer HealthCare Pharmaceuticals (Inst)
Patents, Royalties, Other Intellectual Property: Royalties for megakaryocyte potentiating factor assay from Morphotek (approximately $700 per year)
Anish Thomas
No relationship to disclose
Christine Alewine
No relationship to disclose
Dung T. Le
Honoraria: Merck
Research Funding: Merck, Bristol-Myers Squibb, Aduro Biotech
Elizabeth M. Jaffee
No relationship to disclose
Ira Pastan
No relationship to disclose
REFERENCES
- 1.Chang K, Pastan I, Willingham MC. Isolation and characterization of a monoclonal antibody, K1, reactive with ovarian cancers and normal mesothelium. Int J Cancer. 1992;50:373–381. doi: 10.1002/ijc.2910500308. [DOI] [PubMed] [Google Scholar]
- 2.Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci USA. 1996;93:136–140. doi: 10.1073/pnas.93.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ordóñez NG. Application of mesothelin immunostaining in tumor diagnosis. Am J Surg Pathol. 2003;27:1418–1428. doi: 10.1097/00000478-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Chang K, Pai LH, Pass H, et al. Monoclonal antibody K1 reacts with epithelial mesothelioma but not with lung adenocarcinoma. Am J Surg Pathol. 1992;16:259–268. doi: 10.1097/00000478-199203000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Hassan R, Kreitman RJ, Pastan I, et al. Localization of mesothelin in epithelial ovarian cancer. Appl Immunohistochem Mol Morphol. 2005;13:243–247. doi: 10.1097/01.pai.00000141545.36485.d6. [DOI] [PubMed] [Google Scholar]
- 6.Argani P, Iacobuzio-Donahue C, Ryu B, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: Identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE) Clin Cancer Res. 2001;7:3862–3868. [PubMed] [Google Scholar]
- 7.Hassan R, Laszik ZG, Lerner M, et al. Mesothelin is overexpressed in pancreaticobiliary adenocarcinomas but not in normal pancreas and chronic pancreatitis. Am J Clin Pathol. 2005;124:838–845. [PubMed] [Google Scholar]
- 8.Thomas A, Chen Y, Steinberg SM, et al. High mesothelin expression in advanced lung adenocarcinoma is associated with KRAS mutations and a poor prognosis. Oncotarget. 2015;6:11694–11703. doi: 10.18632/oncotarget.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- doi: 10.1158/1078-0432.CCR-13-1862. Kachala SS, Bograd AJ, Villena-Vargas J, et al: Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin Cancer Res 20:1020-1028, 2014 [Erratum 20:3896, 2014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tozbikian G, Brogi E, Kadota K, et al. Mesothelin expression in triple negative breast carcinomas correlates significantly with basal-like phenotype, distant metastases and decreased survival. PLoS One. 2014;9:e114900. doi: 10.1371/journal.pone.0114900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinbach D, Onda M, Voigt A, et al. Mesothelin, a possible target for immunotherapy, is expressed in primary AML cells. Eur J Haematol. 2007;79:281–286. doi: 10.1111/j.1600-0609.2007.00928.x. [DOI] [PubMed] [Google Scholar]
- 12.Hassan R, Wu C, Brechbiel MW, et al. 111Indium-labeled monoclonal antibody K1: Biodistribution study in nude mice bearing a human carcinoma xenograft expressing mesothelin. Int J Cancer. 1999;80:559–563. doi: 10.1002/(sici)1097-0215(19990209)80:4<559::aid-ijc13>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 13.Lindenberg L, Thomas A, Adler S, et al. Safety and biodistribution of 111In-amatuximab in patients with mesothelin expressing cancers using single photon emission computed tomography-computed tomography (SPECT-CT) imaging. Oncotarget. 2015;6:4496–4504. doi: 10.18632/oncotarget.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamberts LE, Menke-van der Houven van Oordt CW, ter Weele EJ, et al. ImmunoPET with anti-mesothelin antibody in patients with pancreatic and ovarian cancer before anti-mesothelin antibody-drug conjugate treatment. Clin Cancer Res. 2016;22:1642–1652. doi: 10.1158/1078-0432.CCR-15-1272. [DOI] [PubMed] [Google Scholar]
- 15.Pastan I, Hassan R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. 2014;74:2907–2912. doi: 10.1158/0008-5472.CAN-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassan R, Bullock S, Premkumar A, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13:5144–5149. doi: 10.1158/1078-0432.CCR-07-0869. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi N, Hattori K, Oh-eda M, et al. A novel cytokine exhibiting megakaryocyte potentiating activity from a human pancreatic tumor cell line HPC-Y5. J Biol Chem. 1994;269:805–808. [PubMed] [Google Scholar]
- 18.Zhang Y, Chertov O, Zhang J, et al. Cytotoxic activity of immunotoxin SS1P is modulated by TACE-dependent mesothelin shedding. Cancer Res. 2011;71:5915–5922. doi: 10.1158/0008-5472.CAN-11-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zwaenepoel I, Mustapha M, Leibovici M, et al. Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. Proc Natl Acad Sci USA. 2002;99:6240–6245. doi: 10.1073/pnas.082515999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sathyanarayana BK, Hahn Y, Patankar MS, et al. Mesothelin, stereocilin, and otoancorin are predicted to have superhelical structures with ARM-type repeats. BMC Struct Biol. 2009;9:1. doi: 10.1186/1472-6807-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma J, Tang WK, Esser L, et al. Characterization of crystals of an antibody-recognition fragment of the cancer differentiation antigen mesothelin in complex with the therapeutic antibody MORAb-009. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2012;68:950–953. doi: 10.1107/S1744309112028229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bera TK, Pastan I. Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol. 2000;20:2902–2906. doi: 10.1128/mcb.20.8.2902-2906.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galloway ML, Murray D, Moffat DF. The use of the monoclonal antibody mesothelin in the diagnosis of malignant mesothelioma in pleural biopsies. Histopathology. 2006;48:767–769. doi: 10.1111/j.1365-2559.2005.02279.x. [DOI] [PubMed] [Google Scholar]
- 24.Kushitani K, Takeshima Y, Amatya VJ, et al. Immunohistochemical marker panels for distinguishing between epithelioid mesothelioma and lung adenocarcinoma. Pathol Int. 2007;57:190–199. doi: 10.1111/j.1440-1827.2007.02080.x. [DOI] [PubMed] [Google Scholar]
- 25.Miettinen M, Sarlomo-Rikala M. Expression of calretinin, thrombomodulin, keratin 5, and mesothelin in lung carcinomas of different types: An immunohistochemical analysis of 596 tumors in comparison with epithelioid mesotheliomas of the pleura. Am J Surg Pathol. 2003;27:150–158. doi: 10.1097/00000478-200302000-00002. [DOI] [PubMed] [Google Scholar]
- 26.Ordóñez NG. Value of mesothelin immunostaining in the diagnosis of mesothelioma. Mod Pathol. 2003;16:192–197. doi: 10.1097/01.MP.0000056981.16578.C3. [DOI] [PubMed] [Google Scholar]
- 27.Tan K, Kajino K, Momose S, et al. Mesothelin (MSLN) promoter is hypomethylated in malignant mesothelioma, but its expression is not associated with methylation status of the promoter. Hum Pathol. 2010;41:1330–1338. doi: 10.1016/j.humpath.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Illei PB, Alewine C, Zahurak M, et al. Mesothelin expression in advanced gastroesophageal cancer represents a novel target for immunotherapy. Appl Immunohistochem Mol Morphol. 2016;24:246–252. doi: 10.1097/PAI.0000000000000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scales SJ, Gupta N, Pacheco G, et al. An antimesothelin-monomethyl auristatin e conjugate with potent antitumor activity in ovarian, pancreatic, and mesothelioma models. Mol Cancer Ther. 2014;13:2630–2640. doi: 10.1158/1535-7163.MCT-14-0487-T. [DOI] [PubMed] [Google Scholar]
- 30.Frierson HF, Jr, Moskaluk CA, Powell SM, et al. Large-scale molecular and tissue microarray analysis of mesothelin expression in common human carcinomas. Hum Pathol. 2003;34:605–609. doi: 10.1016/s0046-8177(03)00177-1. [DOI] [PubMed] [Google Scholar]
- 31.Swierczynski SL, Maitra A, Abraham SC, et al. Analysis of novel tumor markers in pancreatic and biliary carcinomas using tissue microarrays. Hum Pathol. 2004;35:357–366. doi: 10.1016/j.humpath.2003.10.012. [DOI] [PubMed] [Google Scholar]
- doi: 10.1371/journal.pone.0040157. Winter JM, Tang LH, Klimstra DS, et al: A novel survival-based tissue microarray of pancreatic cancer validates MUC1 and mesothelin as biomarkers. PLoS One 7:e40157, 2012 [Erratum: PLoS One 7, 2012] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimizu A, Hirono S, Tani M, et al. Coexpression of MUC16 and mesothelin is related to the invasion process in pancreatic ductal adenocarcinoma. Cancer Sci. 2012;103:739–746. doi: 10.1111/j.1349-7006.2012.02214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yen MJ, Hsu CY, Mao TL, et al. Diffuse mesothelin expression correlates with prolonged patient survival in ovarian serous carcinoma. Clin Cancer Res. 2006;12:827–831. doi: 10.1158/1078-0432.CCR-05-1397. [DOI] [PubMed] [Google Scholar]
- 35.Cheng WF, Huang CY, Chang MC, et al. High mesothelin correlates with chemoresistance and poor survival in epithelial ovarian carcinoma. Br J Cancer. 2009;100:1144–1153. doi: 10.1038/sj.bjc.6604964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ho M, Bera TK, Willingham MC, et al. Mesothelin expression in human lung cancer. Clin Cancer Res. 2007;13:1571–1575. doi: 10.1158/1078-0432.CCR-06-2161. [DOI] [PubMed] [Google Scholar]
- 37.Alvarez H, Rojas PL, Yong KT, et al. Mesothelin is a specific biomarker of invasive cancer in the Barrett-associated adenocarcinoma progression model: Translational implications for diagnosis and therapy. Nanomedicine (Lond) 2008;4:295–301. doi: 10.1016/j.nano.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baba K, Ishigami S, Arigami T, et al. Mesothelin expression correlates with prolonged patient survival in gastric cancer. J Surg Oncol. 2012;105:195–199. doi: 10.1002/jso.22024. [DOI] [PubMed] [Google Scholar]
- 39.Einama T, Homma S, Kamachi H, et al. Luminal membrane expression of mesothelin is a prominent poor prognostic factor for gastric cancer. Br J Cancer. 2012;107:137–142. doi: 10.1038/bjc.2012.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ito T, Kajino K, Abe M, et al. ERC/mesothelin is expressed in human gastric cancer tissues and cell lines. Oncol Rep. 2014;31:27–33. doi: 10.3892/or.2013.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawamata F, Kamachi H, Einama T, et al. Intracellular localization of mesothelin predicts patient prognosis of extrahepatic bile duct cancer. Int J Oncol. 2012;41:2109–2118. doi: 10.3892/ijo.2012.1662. [DOI] [PubMed] [Google Scholar]
- 42.Nomura R, Fujii H, Abe M, et al. Mesothelin expression is a prognostic factor in cholangiocellular carcinoma. Int Surg. 2013;98:164–169. doi: 10.9738/INTSURG-D-13-00001.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao H, Davydova L, Mandich D, et al. S100A4 protein and mesothelin expression in dysplasia and carcinoma of the extrahepatic bile duct. Am J Clin Pathol. 2007;127:374–379. doi: 10.1309/37RTWYAEPABYY410. [DOI] [PubMed] [Google Scholar]
- 44.Obulhasim G, Fujii H, Matsumoto T, et al. Mesothelin gene expression and promoter methylation/hypomethylation in gynecological tumors. Eur J Gynaecol Oncol. 2010;31:63–71. [PubMed] [Google Scholar]
- 45.Li YR, Xian RR, Ziober A, et al. Mesothelin expression is associated with poor outcomes in breast cancer. Breast Cancer Res Treat. 2014;147:675–684. doi: 10.1007/s10549-014-3077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tchou J, Wang LC, Selven B, et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat. 2012;133:799–804. doi: 10.1007/s10549-012-2018-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rump A, Morikawa Y, Tanaka M, et al. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol Chem. 2004;279:9190–9198. doi: 10.1074/jbc.M312372200. [DOI] [PubMed] [Google Scholar]
- 48.Gubbels JA, Belisle J, Onda M, et al. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol Cancer. 2006;5:50. doi: 10.1186/1476-4598-5-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uehara N, Matsuoka Y, Tsubura A. Mesothelin promotes anchorage-independent growth and prevents anoikis via extracellular signal-regulated kinase signaling pathway in human breast cancer cells. Mol Cancer Res. 2008;6:186–193. doi: 10.1158/1541-7786.MCR-07-0254. [DOI] [PubMed] [Google Scholar]
- 50.Servais EL, Colovos C, Rodriguez L, et al. Mesothelin overexpression promotes mesothelioma cell invasion and MMP-9 secretion in an orthotopic mouse model and in epithelioid pleural mesothelioma patients. Clin Cancer Res. 2012;18:2478–2489. doi: 10.1158/1078-0432.CCR-11-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen SH, Hung WC, Wang P, et al. Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Sci Rep. 2013;3:1870. doi: 10.1038/srep01870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang MC, Chen CA, Chen PJ, et al. Mesothelin enhances invasion of ovarian cancer by inducing MMP-7 through MAPK/ERK and JNK pathways. Biochem J. 2012;442:293–302. doi: 10.1042/BJ20110282. [DOI] [PubMed] [Google Scholar]
- 53.Bharadwaj U, Marin-Muller C, Li M, et al. Mesothelin overexpression promotes autocrine IL-6/sIL-6R trans-signaling to stimulate pancreatic cancer cell proliferation. Carcinogenesis. 2011;32:1013–1024. doi: 10.1093/carcin/bgr075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bharadwaj U, Marin-Muller C, Li M, et al. Mesothelin confers pancreatic cancer cell resistance to TNF-α-induced apoptosis through Akt/PI3K/NF-κB activation and IL-6/Mcl-1 overexpression. Mol Cancer. 2011;10:106. doi: 10.1186/1476-4598-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sato N, Maitra A, Fukushima N, et al. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res. 2003;63:4158–4166. [PubMed] [Google Scholar]
- 56.Hucl T, Brody JR, Gallmeier E, et al. High cancer-specific expression of mesothelin (MSLN) is attributable to an upstream enhancer containing a transcription enhancer factor dependent MCAT motif. Cancer Res. 2007;67:9055–9065. doi: 10.1158/0008-5472.CAN-07-0474. [DOI] [PubMed] [Google Scholar]
- 57.Ren YR, Patel K, Paun BC, et al. Structural analysis of the cancer-specific promoter in mesothelin and in other genes overexpressed in cancers. J Biol Chem. 2011;286:11960–11969. doi: 10.1074/jbc.M110.193458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marin-Muller C, Li D, Bharadwaj U, et al. A tumorigenic factor interactome connected through tumor suppressor microRNA-198 in human pancreatic cancer. Clin Cancer Res. 2013;19:5901–5913. doi: 10.1158/1078-0432.CCR-12-3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chowdhury PS, Viner JL, Beers R, et al. Isolation of a high-affinity stable single-chain Fv specific for mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc Natl Acad Sci USA. 1998;95:669–674. doi: 10.1073/pnas.95.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chowdhury PS, Pastan I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat Biotechnol. 1999;17:568–572. doi: 10.1038/9872. [DOI] [PubMed] [Google Scholar]
- 61.Pastan I, Hassan R, Fitzgerald DJ, et al. Immunotoxin therapy of cancer. Nat Rev Cancer. 2006;6:559–565. doi: 10.1038/nrc1891. [DOI] [PubMed] [Google Scholar]
- 62.Kreitman RJ, Hassan R, Fitzgerald DJ, et al. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin Cancer Res. 2009;15:5274–5279. doi: 10.1158/1078-0432.CCR-09-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hassan R, Sharon E, Thomas A, et al. Phase 1 study of the antimesothelin immunotoxin SS1P in combination with pemetrexed and cisplatin for front-line therapy of pleural mesothelioma and correlation of tumor response with serum mesothelin, megakaryocyte potentiating factor, and cancer antigen 125. Cancer. 2014;120:3311–3319. doi: 10.1002/cncr.28875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hassan R, Miller AC, Sharon E, et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci Transl Med. 2013;5:208ra147. doi: 10.1126/scitranslmed.3006941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hassan R, Alewine C, Pastan I. New life for immunotoxin cancer therapy. Clin Cancer Res. 2016;22:1055–1058. doi: 10.1158/1078-0432.CCR-15-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hollevoet K, Mason-Osann E, Liu XF, et al. In vitro and in vivo activity of the low-immunogenic antimesothelin immunotoxin RG7787 in pancreatic cancer. Mol Cancer Ther. 2014;13:2040–2049. doi: 10.1158/1535-7163.MCT-14-0089-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alewine C, Xiang L, Yamori T, et al. Efficacy of RG7787, a next-generation mesothelin-targeted immunotoxin, against triple-negative breast and gastric cancers. Mol Cancer Ther. 2014;13:2653–2661. doi: 10.1158/1535-7163.MCT-14-0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu W, Onda M, Lee B, et al. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc Natl Acad Sci USA. 2012;109:11782–11787. doi: 10.1073/pnas.1209292109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jaffee EM, Hruban RH, Biedrzycki B, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: A phase I trial of safety and immune activation. J Clin Oncol. 2001;19:145–156. doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]
- 70.Thomas AM, Santarsiero LM, Lutz ER, et al. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med. 2004;200:297–306. doi: 10.1084/jem.20031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brockstedt DG, Giedlin MA, Leong ML, et al. Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc Natl Acad Sci USA. 2004;101:13832–13837. doi: 10.1073/pnas.0406035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Le DT, Brockstedt DG, Nir-Paz R, et al. A live-attenuated Listeria vaccine (ANZ-100) and a live-attenuated Listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: Phase I studies of safety and immune induction. Clin Cancer Res. 2012;18:858–868. doi: 10.1158/1078-0432.CCR-11-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hassan R, Ebel W, Routhier EL, et al. Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 2007;7:20. [PMC free article] [PubMed] [Google Scholar]
- 74.Hassan R, Cohen SJ, Phillips M, et al. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin-expressing cancers. Clin Cancer Res. 2010;16:6132–6138. doi: 10.1158/1078-0432.CCR-10-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Golfier S, Kopitz C, Kahnert A, et al. Anetumab ravtansine: A novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther. 2014;13:1537–1548. doi: 10.1158/1535-7163.MCT-13-0926. [DOI] [PubMed] [Google Scholar]
- 76.Hassan R, Blumenschein G, Jr, Kindler HL, et al. Phase I study of anti-mesothelin antibody drug conjugate anetumab ravtansine. Presented at the 16th World Conference on Lung Cancer, Denver, CO, September 6-9, 2015. [Google Scholar]
- 77.Weekes CD, Lamberts LE, Borad MJ, et al. Phase I study of DMOT4039A, an antibody-drug conjugate targeting mesothelin, in patients with unresectable pancreatic or platinum-resistant ovarian cancer. Mol Cancer Ther. 2016;15:439–447. doi: 10.1158/1535-7163.MCT-15-0693. [DOI] [PubMed] [Google Scholar]
- 78.Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: Driving T cells to solid tumors. Cancer Discov. 2016;6:133–146. doi: 10.1158/2159-8290.CD-15-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.O’Hara M, Stashwick C, Haas AR, et al. Mesothelin as a target for chimeric antigen receptor-modified T cells as anticancer therapy. Immunotherapy. 2016;8:449–460. doi: 10.2217/imt.16.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adusumilli PS, Cherkassky L, Villena-Vargas J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hassan R, Williams-Gould J, Watson T, et al. Pretreatment with rituximab does not inhibit the human immune response against the immunogenic protein LMB-1. Clin Cancer Res. 2004;10:16–18. doi: 10.1158/1078-0432.ccr-1160-3. [DOI] [PubMed] [Google Scholar]
- 83.Mossoba ME, Onda M, Taylor J, et al. Pentostatin plus cyclophosphamide safely and effectively prevents immunotoxin immunogenicity in murine hosts. Clin Cancer Res. 2011;17:3697–3705. doi: 10.1158/1078-0432.CCR-11-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hassan R, Kindler HL, Jahan T, et al. Phase II clinical trial of amatuximab, a chimeric antimesothelin antibody with pemetrexed and cisplatin in advanced unresectable pleural mesothelioma. Clin Cancer Res. 2014;20:5927–5936. doi: 10.1158/1078-0432.CCR-14-0804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gupta A, Hussein Z, Hassan R, et al. Population pharmacokinetics and exposure-response relationship of amatuximab, an anti-mesothelin monoclonal antibody, in patients with malignant pleural mesothelioma and its application in dose selection. Cancer Chemother Pharmacol. 2016;77:733–743. doi: 10.1007/s00280-016-2984-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hassan R, Antonia S, Alley E, et al. Anti-mesothelin vaccine CRS-207 plus chemotherapy as front-line treatment for malignant pleural mesothelioma (MPM). Presented at the European Cancer Congress, Vienna, Austria, September 25-29, 2015. [Google Scholar]
- 87.Le DT, Wang-Gillam A, Picozzi V, et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes–expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015;33:1325–1333. doi: 10.1200/JCO.2014.57.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]