FoxO integration of insulin signaling with glucose and lipid metabolism (original) (raw)
. Author manuscript; available in PMC: 2018 May 1.
Published in final edited form as: J Endocrinol. 2017 Feb 17;233(2):R67–R79. doi: 10.1530/JOE-17-0002
Abstract
The forkhead box O family consists of FoxO1, FoxO3, FoxO4 and FoxO6 proteins in mammals. Expressed ubiquitously in the body, the four FoxO isoforms share in common the amino DNA binding domain, known as “forkhead box” domain. They mediate the inhibitory action of insulin or insulin-like growth factor on key functions involved in cell metabolism, growth, differentiation, oxidative stress, senescence, autophagy and aging. Genetic mutations in FoxO genes or abnormal expression of FoxO proteins are associated with metabolic disease, cancer or altered lifespan in humans and animals. Of the FoxO family, FoxO6 is the least characterized member and is shown to play pivotal roles in the liver, skeletal muscle and brain. Altered FoxO6 expression is associated with the pathogenesis of insulin resistance, dietary obesity and type 2 diabetes, and risk of neurodegeneration disease. FoxO6 is evolutionally divergent from other FoxO isoforms. FoxO6 mediates insulin action on target genes in a mechanism that is fundamentally different from other FoxO members. Here we focus our review on the role of FoxO6, in contrast with other FoxO isoforms, in health and disease. We review the distinctive mechanism by which FoxO6 integrates insulin signaling to hepatic glucose and lipid metabolism. We highlight the importance of FoxO6 dysregulation in the dual pathogenesis of fasting hyperglycemia and hyperlipidemia in diabetes. We review the role of FoxO6 in memory consolidation and its contribution to neurodegeneration disease and aging. We discuss the potential therapeutic option of pharmacological FoxO6 inhibition for improving glucose and lipid metabolism in diabetes.
Key terms: FoxO1, FoxO3, FoxO4, FoxO6, Glucose metabolism, Lipid metabolism, Insulin Signaling, Obesity, Diabetes
The forkhead box O family
The forkhead box O (FoxO) family consists of FoxO1 (FKHR), FoxO3 (FKHRL1), FoxO4 (AFX) and FoxO6 proteins, each encoded by a distinct gene in mammalian cells. These four isoforms share a common structural motif, namely the “forkhead box” or “winged helix” domain that is responsible for binding to chromatin DNA in the nucleus of cells (Accili & Arden 2004). FoxO proteins act as nuclear transcription factors that mediate the inhibitory action of insulin or insulin-like growth factor (IGF-1) on key functions in diverse pathways including cell metabolism, proliferation, differentiation, oxidative stress, cell survival and senescence, autophagy and aging in mammals (Accili & Arden 2004). As a result, genetic mutations in FoxO genes or altered expression of FoxO proteins are associated with diseases including diabetes and cancer or with reduced lifespan in mammals. This latter aspect is exemplified by the illustration of role of the FoxO ortholog Daf16 in Caenorhabditis elegans (Hwangbo et al. 2004; Lee et al. 2003). Daf16 mediates the effect of insulin-like receptor Daf-2 on cellular anti-oxidative functions (Essers et al. 2005; Mueller et al. 2014). Increased Daf16 expression, resulting from attenuated insulin-like signaling or increased oxidative stress, promotes anti-oxidative function, contributing to the lifespan extension in Caenorhabditis elegans (Lee et al. 2003). Likewise, the FoxO1 ortholog dFoxO signaling through insulin-like receptor dInR contributes to the regulation of longevity in Drosophila melanogaster (Hwangbo et al. 2004).
Among the four FoxO isoforms in mammalian cells, FoxO6 remains the least characterized member. FoxO6 has garnered less interest, due in part to the misconception that FoxO6 expression is confined in the brain (Jacobs et al. 2003). Subsequent research has rectified this misconception, showing that FoxO6 is expressed in central and peripheral tissues including the liver, intestine, lung, kidney, muscle, and adipose tissues, a broad tissue distribution that is characteristic of other FoxO isoforms in mammals (Kim et al. 2011). Recent studies indicate that FoxO6 plays important roles in integrating insulin signaling to glucose and lipid metabolism. However, unlike other FoxO isoforms, FoxO6 mediates insulin action on target gene expression in cells via a distinct mechanism that is fundamentally different from the FoxO family (Kim et al. 2011). This is consistent with the distinctive feature of FoxO6 protein (Fig. 1). FoxO6 expression becomes deregulated in metabolic disease. Furthermore, altered FoxO6 expression in the brain is associated with impaired cognitive function in mice. In this article, we provide a comprehensive review on the role of FoxO6 in insulin action and carbohydrate metabolism in the liver. We provide mechanistic insight into the distinct mechanism by which insulin differentially modulates the transcriptional activity of FoxO6, as opposed to other members in the FoxO family. We discuss how FoxO6 deregulation in the liver contributes to the development of metabolic abnormalities in animal models with obesity and type 2 diabetes. We also review nascent evidence that FoxO6 plays important roles in neuronal cells with its potential contribution to neurodegeneration and longevity.
Fig. 1. Schematic depiction of the mouse FoxO6 protein.
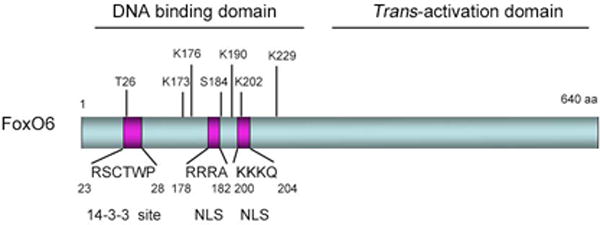
FoxO6 is characterized by its amino DNA binding and carboxyl _trans_-activation domains. Within the DNA binding domain are the bipartite nuclear localization signal (NLS), consensus 14-3-3 protein-binding motif and two conserved Akt/PKB phosphorylation sites T26 and S184. Amino acid residues K173, K176, K190, K202 and K229 denote five distinct acetylation sites that are conserved across the species.
FoxO integration of insulin signaling to target gene expression
FoxO proteins or FoxO orthologs (Daf16 and dFoxO) are the class of transcrption factors that mediates the inhibitory action of insulin or insulin-like growth factor (IGF-1) on target genes in cells. It is well characterized that insulin (or IGF-1) exerts its inhibitory effect on gene expression via a highly conserved sequence (TG/ATTTT/G), known as the “insulin responsive element” (IRE) in target promoters (Accili & Arden 2004). In response to attenuated insulin (or IGF-1) signaling, FoxO proteins or FoxO orthologs become active in the nucleus and bind as trans-activators to the IRE DNA motif within target gene promoters to stimulate target gene expression. In keeping with this action, FoxO proteins or FoxO orthologs possess the amino forkhead box domain for binding to chromatin DNA and the carboxyl trans-activation domain for enhancing promoter activity. Crystallographic studies reveal that FoxO proteins bind as monomers to chromatin DNA (Brent et al. 2008). In response to increased insulin (or IGF-1) signaling, FoxO proteins or FoxO orthologs undergo insulin-mediated phosphorylation and translocation from the nucleus to the cytoplasm, resulting in the inhibition of target gene expression (Accili & Arden 2004). This phosphorylation-dependent nucleocytoplasmic trafficking serves as an effective mechanism by which insulin (or IGF-1) regulate FoxO transcriptional activity in cells. All members of the FoxO family, with the exception of FoxO6, undergo insulin-dependent phosphorylation and nuclear exclusion. Failure in insulin (or IGF-1) -mediated FoxO phosphorylation results in FoxO permanent nuclear localization, accounting for unchecked FoxO activity and constitutive expression of target genes, an anomaly that is associated with defective insulin (or IGF-1) signaling in the pathogenesis of diseases including cancer and diabetes (Accili & Arden 2004; Altomonte et al. 2004; Barthel et al. 2005; Cifarelli et al. 2012; Matsumoto et al. 2006; Qu et al. 2006; Sparks & Dong 2009).
Mechanism of FoxO nucleocytoplasmic trafficking
Although the nucleocytoplasmic shuttling constitutes a compartmental effect for insulin (or IGF-1) to curb FoxO transcriptional activity in cells (Accili & Arden 2004; Kamagate & Dong 2008), the underlying mechanism remains elusive. Using FoxO1 as an example, Kim and co-workers (Kim et al. 2011) show that FoxO1 is capable of interacting with chromosome region maintenance 1 (CRM-1), also known as exportin-1 that is responsible for binding to the nuclear export signal (NES) of a cargo protein and transporting the cargo protein from the nucleus to cytoplasm (Dong et al. 2009; Monecke et al. 2009). Consistent with this observation is the presence of a highly conserved NES motif in the carboxyl domain of FoxO1. Immunocytochemistry reveals that FoxO1 and CRM-1 are co-localized in the nucleus of cultured hepatocytes. In response to insulin, FoxO1 in complex with CRM-1 is translocated from the nucleus to the cytoplasm. This insulin-dependent FoxO1 and CRM-1 translocation is abrogated by leptomycin B, an agent that binds specifically to CRM-1 and disables its cargo-trafficking activity (Kim et al. 2011). These results characterize CRM-1 as a mediator in facilitating insulin-dependent FoxO1 subcellular redistribution. A prevailing view is that all FoxO proteins including its orthologs dFoxO and Daf16, except for FoxO6, undergo insulin (or IGF-1)-mediated nucleocytoplasmic trafficking in cells.
FoxO integration of oxidative stress to cell survival
FoxO proteins and orthologs (dFoxO and Daf16) play important roles in protecting cells from oxidative stress. This cytoprotective effect is attributable to FoxO anti-oxidative function. In response to oxidative stress, FoxO proteins undergo translocation from the cytoplasm to the nucleus, contributing to increased FoxO activity in promoting FoxO target genes encoding anti-oxidative enzymes such as manganese superoxide dismutase (MnSOD) expression, catalase (CAT) and glutathione peroxidase (GPX). This effect contributes to the preservation of cell survival and function in the face of oxidative stress (Kops et al. 2002). However, chronic oxidative stress is also associated with cellular apoptosis. Lehtinen and coworkers show that the oxidative-stress-regulated mammalian sterile 20-like kinas-1 (MST1) is activated in neuronal cells (Lehtinen et al. 2006). Activated MST1 kinases phosphorylate FoxO3 at a conserved site Ser 207 within FoxO3 forkhead box domain. The resulting effect disrupts FoxO3 interaction with 14-3-3 proteins and promotes FoxO3 nuclear translocation and stimulates apoptotic gene expression, thereby inducing death in neurons (Lehtinen et al. 2006). These data characterize MST1 as a significant upstream regulator of FoxO activity and cell surivival in response to oxidative stress.
It is noteworthy that increased MST1 activity, resulting from oxidative stress, also regulates the p38 AMPK and c-Jun N-terminal kinase (JNK) pathway (Graves et al. 1998). Essers and colleagues demonstrate that the small GTPase Ral is activated in cells in the presene of H2O2-induced oxidative stress, and this results in JNK-dependent phosphorylation of FoxO4 on Thr-447 and Thr-451 (Essers et al. 2004). This signalling pathway is also employed by tumor necrosis factor alpha (TNF-α) to activate FoxO4 activity in response to inflammation. As a result, JNK-mediated phosphorylation of FoxO4 promotes its nuclear translocation and enhances its transcriptional activity in driving the expression of anti-oxidative functions including MnSOD, CAT and GPX, protecting mammalian cells from oxidative damage (Essers et al. 2004). Interestingly, such an anti-oxidative function of FoxO1 is conserved from C. elegans to mammals.
Further physiological underpinning for the cytoprotective role of FoxO in defending oxidative stress derives from a number of significant studies in pancreatic β-cells – a cell type that is associated with extremely low expression of anti-oxidative enzymes. This renders β-cells more vulnerable to oxidative stress, accounting in part for β-cell dysfunction in diabetes. In this context, Zhang and co-workers show that FoxO1 stimulates β-cell expression of Sod1, catalase (Cat) and glutathione peroxidase 1 (Gpx1), contributing to improved β-cell survival and function in the presence of oxidative stress (Zhang et al. 2016). Kibbe and colleagues report that FoxO1 protects β-cells from oxidative stress by inhibiting the expression of thioredoxin-interacting protein (TXNIP), a cytosolic factor whose deregulation is associated with β-cell apoptosis (Kibbe et al. 2013). FoxO1 also promotes the expression of DNA repair enzyme GADD45α, protecting β-cells from nitric oxide-elicited DNA damage (Kitamura et al. 2005). Taken together, FoxO confers an evolutionally conserved cytoprotective effect on cell survival and function.
FoxO6 mediates insulin action via a distinct mechanism
FoxO6 is located on chromosome 1 (1p34.1) in humans, which is different from FoxO1 on chromosome 13 (13q14.1), FoxO3 on chromosome 6 (6q21) and chromosome X (Xq13.1). Although catagoried in the FoxO family, FoxO6 has the lowest degree of homology (~ 30%) in amino acid sequence with other FoxO isoforms, consistent with the idea that FoxO6 is functionally divergent from the FoxO subfamily (Wang et al. 2009). FoxO6 is evoluationally conserved across the species ranging from the frogs to humans (Fig. 2), foreboding the physioloigcal importance of FoxO6 in health and disease. Indeed, characterization of FoxO6 in insulin signaling reveals that FoxO6 mediates insulin action on target gene expression via a district mechanism that is fundamentally different from other FoxO isoforms. Unlike other FoxO members that undergo insulin-mediated nucleocytoplasmic shuttling, FoxO6 remains in the nucleus of cultured cells irrespective of the addition of insulin into culture medium (Jacobs et al. 2003; Kim et al. 2011). Likewise, FoxO6 localizes in the nucleus of hepatocytes in mice, independently of fasting or refeeding state (Kim et al. 2011). In accordance with these observations, FoxO6 lacks the consensus NES motif, correlating with the inability to undergo insulin-dependent nucleocytoplasmic trafficking. Nonetheless, FoxO6 is highly sensitive to insulin inhibition in cells. These observations raise a fundamental question as to how FoxO6 mediates the inhibitory effect of insulin on target gene expression.
Fig. 2. FoxO6 is evolutionally conserved across the species.

Amino acid sequences of FoxO6 proteins were aligned in different species. The conserved amino acids corresponding to the nuclear localization signal (NLS) and Akt/PKB phosphorylation sites Thr26 and Ser184 are marked in red box.
To address this issue, Kim and co-workers (Kim et al. 2011) show that FoxO6 interacts with the multifunctional protein 14-3-3 in the nucleus of hepatocytes, coinciding with the conservation of a consensus 14-3-3 binding motif (23RSCTWP28) within the FoxO6 DNA-binding domain (Fig. 1). This effect depends on insulin-stimulated FoxO6 phosphorylation at two consensus Akt/PKB phosphorylation sites Thr26 and Ser184, one of which resides within the 14-3-3 binding motif. Thus, one potential mechanism by which insulin inhibits FoxO6 transcriptional activity in the nucleus is by insulin-stimulated phosphorylation of FoxO6 proteins. This effect distorts the FoxO6 DNA binding domain and prevents FoxO6 from binding to target promoters. As an alternative mechanism, FoxO6, when phosphorylated by Akt/PKB in response to insulin, interacts with 14-3-3, a scaffolding protein that prevents FoxO6 from binding to target promoters. There is evidence that FoxO1 adopts a similar mechanism for mediating the inhibitory action of insulin in cells. When phosphorylated in response to insulin, FoxO1 forms a complex with 14-3-3, which masks FoxO1 DNA binding motif and precludes FoxO1 binding to target promoters. Furthermore, 14-3-3 serves as an anchor to sequester FoxO1 in the cytoplasm, preventing FoxO1 from re-entering into the nucleus (Accili & Arden 2004; Barthel et al. 2005).
An important implication of these findings is that insulin inhibition of FoxO transcriptional activity and FoxO nucleocytoplasmic trafficking are two separate events. In support of this notion, Tsai and co-workers (Tsai et al. 2003) examined insulin-dependent phosphorylation and subcellular localization of a mutant FoxO1 with an altered NES motif. Despite its inability to undergo insulin-mediated nucleocytoplasmic trafficking, such FoxO1 mutant is subject to insulin inhibition to the same extent as its wild-type counterpart. It follows that insulin inhibition of FoxO transcriptional activity can ensue without necessarily altering FoxO subcellular redistribution, but this mechanism depends on insulin-stimulated FoxO phosphorylation.
This raises an interesting question as to why insulin promotes FoxO1, but not FoxO6, translocation from the nucleus to the cytoplasm, an compartmental effect that is sufficient, but not necessary, for insulin inhibition of FoxO1 activity. One plausible interpretation is that FoxO6 serves as a basal mechanism for adjusting the rate of hepatic gluconeogenesis for rapid adaptation to changes in physiological states. This mechanism ensures that constitutively nuclear FoxO6 can immediately prime the liver for augmented gluconeogenesis in the post-absorptive phase, in which FoxO1 is already translocated from the nucleus to the cytoplasm in response to postprandial insulin release.
The alternative explanation is that cytosolic FoxO1 proteins possess secondary functions in cells. Consistent with this interpretation, Zhao and co-workers (Zhao et al. 2010) show that FoxO1 mediates autophagy via its protein-protein interaction with Atg7 (autophagy-related protein 7) in response to cellular stress. This action, which takes place in the cytoplasm, independently of FoxO1 activity in the nucleus, impacts autophagy-mediated cell death in cancer cells (Zhao et al. 2010). This observation also refutes the longstanding notion that FoxO1, after trafficking from the nucleus to the cytoplasm, is destined for proteasome-mediated degradation (Jang et al. 2016; Kato et al. 2008; Matsuzaki et al. 2003).
Insulin-dependent and independent regulation of hepatic gluconeogenesis
Gluconeogenesis is a pivotal metabolic pathway for converting non-carbohydrate metabolites (lactate, glycerol, and amino acids) to glucose in the body. A life-sustaining process, gluconeogenesis provides the sole fuel source for the brain, nervous system, testes and erythrocytes during a prolonged fast or exercise. Gluconeogenesis takes place mainly in liver and to a much lesser extent in kidney (Edgerton et al. 2009; Wahren & Ekberg 2007). In healthy individuals, hepatic gluconeogenesis accounts for up to 80% of total endogenous glucose production during a prolonged fast (Ekberg et al. 1999). Gluconeogenesis is tightly regulated by hormonal and nutritional cues. In response to postprandial insulin secretion, hepatic gluconeogenic activity is suppressed to limit glucose production. This effect serves two purposes; i) to prevent prolonged postprandial glucose excursion and ii) to replenish glycogen content in liver, as increased glucose influx into hepatocytes promotes the synthesis and storage of glycogen in liver after meals. Conversely, in response to reduced insulin action and elevated glucagon secretion during fasting, hepatic gluconeogenesis is stimulated, resulting in increased glucose output from liver. Such a reciprocal mechanism, orchestrated by the two opposing hormones (insulin and glucagon), is crucial for rapid adaptation of liver to metabolic shift between fed and fasting states for maintaining blood sugar levels within the physiological range (Wahren & Ekberg 2007).
Gluconeogenesis is governed by two key enzymes, phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) in liver. PEPCK functions to convert oxaloacetate to phosphoenolpyruvate and G6Pase acts to convert glucose-6-phosphate to glucose. These two hepatic enzymes catalyze the first and last steps in the gluconeogenesis pathway. Consistent with their duties in gluconeogenesis, hepatic production of PEPCK and G6Pase is inhibited by insulin in the fed state and stimulated by glucagon under fasting conditions. However, there is anecdotal evidence that increased hepatic glucose production can occur without the induction of hepatic expression of PEPCK and G6Pase in the liver. Samuel and co-workers (Samuel et al. 2009) report that hepatic expression of both PEPCK and G6Pase mRNA levels remained unchanged, despite a significant induction of endogenous glucose production in the liver of human subjects and rats with diabetes. These results seem at variance with the dogma that hepatic PEPCK and G6Pase enzymes catalyze the rate-limiting steps in the gluconeogenesis pathway. A caveat of this study is that the conclusion dwells on the measurement of hepatic PEPCK and G6Pase expression at the mRNA levels in the liver. Thus, it remains an open question as to whether PEPCK and G6Pase protein levels and their enzymatic activities are correlated with rates of hepatic glucose production in type 2 diabetes.
Aside from its direct inhibitory effect on gluconeogenesis, insulin acts to suppress gluconeogenesis by indirect mechanisms. Consistent with this notion, Olefsky and colleagues show that in obese subjects, suppression of hepatic glucose production can occur in response to insulin infusion without an apparent increase in portal insulin concentrations (Prager et al. 1987). Recently, two independent groups report that hepatic insulin signaling is dispensable for the regulation of glucose production in the liver (I et al. 2015; Titchenell et al. 2015). Insulin is known to exert a potent inhibitory effect on hormone-sensitive lipase, which in turn curbs adipose tissue lipolysis and reduces the availability of free fatty acid (FFA) and glycerol (Holm et al. 2000). In parallel, insulin inhibits muscle proteolysis and restrains the release of amino acids (Liu et al. 2008). FFA is used as an energy source for gluconeogenesis, and glycerol and amino acids are precursors of gluconeogenesis. A significant reduction in FFA influx to liver along with dwindling supplies of ambient gluconeogenic substrates imparts a potent indirect inhibitory effect of insulin on hepatic gluconeogenesis (Ader & Bergman 1990). In addition, insulin acts on pancreatic α-cells to suppress glucagon release, limiting hepatic glucose output (Ishihara et al. 2003; Jiang & Zhang 2003; Unger et al. 1978). Recently, Perry and co-workers report that hepatic acetyl-CoA, derived from white adipose tissue, serves as a significant regulator of hepatic glucose production. Under fasting or insulin resistant condition, hepatic acetyl-CoA levels become higher, fueling hepatic gluconeogenesis. This effect is acutely inhibited by insulin. Insulin suppresses lipolysis in adipose tissues to limit hepatic acetyl-CoA levels, contributing to the inhibition of glucose production in the liver (Perry et al. 2015). Thus, insulin modulates hepatic gluconeogenesis via both direct and indirect mechanisms (Ader & Bergman 1990; Cherrington 2005).
FoxO6 in hepatic glucose metabolism
Although FoxO1 is shown to play a key role in integrating insulin signaling to hepatic gluconeogenesis, FoxO1 depletion in liver does not result in abolition of hormonal regulation of gluconeogenesis in mice (Matsumoto et al. 2007). In keeping with this observation, Kamagate and co-workers (Kamagate et al. 2008) show that mice with FoxO1 loss-of-function in liver are associated with about 50% reduction in gluconeogenic activity in response to fasting. FoxO1 loss-of-function reduces, but does not abrogate the responsiveness of liver to insulin or glucagon (via cAMP) signaling, raising the plausibility that there are additional factors or compensatory mechanisms in mediating hormonal regulation of hepatic gluconeogenesis (Kamagate et al. 2008; Matsumoto et al. 2007).
In this context, Kim and co-workers (Kim et al. 2011) have characterized FoxO6 as an important regulator of hepatic gluconeogenesis in response to insulin. FoxO6 is expressed in both human and rodent livers. Hepatic FoxO6 expression is maintained at low basal levels in fed states. However, in response to prolonged fasting, hepatic FoxO6 expression and transcriptional activity is markedly induced in livers of mice in response to fasting. This effect translates into a significant induction of hepatic gluconeogenesis in fasted mice. Consistent with its role in regulating hepatic gluconeogenesis, FoxO6 targets both PEPCK and G6Pase genes for trans-activation in hepatocytes, and this action is counteracted by insulin. However, unlike FoxO1, FoxO6 is unable to undergo insulin-dependent subcellular redistribution. Instead, insulin inhibits FoxO6 activity in the nucleus by directly stimulates FoxO6 phosphorylation and disables its DNA binding activity. Conversion of the conserved Akt/PKB phosphorylation site at Ser184 to Ala renders FoxO6 refractory to insulin inhibition, contributing to its constitutive transcriptional activity. As a result, transgenic mice expressing the constitutively active FoxO6 variant in the liver are associated with increased gluconeogenesis, contributing to the development of fasting hyperglycemia (Kim et al. 2011). In contrast, hepatic FoxO6 deficiency attenuates hepatic gluconeogenesis, resulting in fasting hypoglycemia in FoxO6 knockout mice (Calabuig-Navarro et al. 2015). Consistent with its independent role in regulating hepatic gluconeogenesis, FoxO6 depletion does not elicit a compensatory induction of hepatic FoxO1 expression or activity (Calabuig-Navarro et al. 2015). Taken together, these data have revealed a distinct mechanism by which insulin signaling through FoxO6 adjusts the rate of hepatic glucose production in response to changes in physiological states. While it is claimed that FoxO3 and FoxO4 also participate in the gluconeogenesis pathway, hepatic FoxO3 and FoxO4 depletion did not result in further significant reduction in fasting blood glucose levels in liver-specific FoxO1-knockout mice (Haeusler et al. 2010). Thus, the independent contributions of FoxO3 or FoxO4 to insulin regulation of hepatic glucose production remain to be determined.
FoxO6 in the pathogenesis of fasting hyperglycemia in diabetes
Excessive endogenous glucose production exerts its deleterious effect on whole-body metabolism in diabetes in three fundamental ways: 1) It causes prolonged postprandial blood glucose excursion, 2) It contributes to the pathogenesis of fasting hyperglycemia, and 3) It accounts in part for diminished glycogen storage in liver. Because of its importance in energy homeostasis in health and its sequelae in disease, the gluconeogenesis pathway has been a major therapeutic target site for pharmacological intervention of hyperglycemia in diabetes. Identification of FoxO6 as a significant transcription factor for conducting insulin-dependent regulation of hepatic glucose production raises the postulation that FoxO6 might be a potential target for improving glycemic control in diabetes. Consistent with this idea are the observations that hepatic FoxO6 activity becomes abnormally higher in insulin resistant livers of dietary obese mice and diabetic db/db mice (Kim et al. 2011). Likewise, hepatic FoxO6 expression is also upregulated in insulin deficient livers of streptozotocin-induced diabetic mice (Kim et al. 2011). These data indicate that insulin deficiency or insulin resistance is invariably associated with FoxO6 dysregulation in the liver. It is anticipated that selective FoxO6 inhibition would curb hepatic glucose production and ameliorate fasting hyperglycemia in diabetes. In support of this notion, Kim and co-workers (Kim et al. 2011) show that siRNA-mediated hepatic FoxO6 knockdown results in a significant reduction in hepatic gluconeogenesis in insulin resistant liver of diabetic db/db mice. This effect contributes to reduced fasting blood glucose levels, enhanced glucose tolerance and improved insulin sensitivity in diabetic db/db mice (Kim et al. 2011). Furthermore, Calabuig-Navarro and co-workers (Calabuig-Navarro et al. 2015) show that FoxO6 deficient mice are protected from developing fasting hyperglycemia and glucose intolerance in response to high fat feeding.
FoxO6 in hepatic lipid metabolism
Apart from its role in mediating insulin action on hepatic gluconeogenesis and glucose metabolism, FoxO6 plays a significant role in regulating hepatic lipid metabolism. Kim and co-workers (Kim et al. 2014) show that FoxO6-transgenic mice with liver-specific FoxO6 production develop hyperlipidemia, characterized by elevated plasma triglyceride (TG) levels. To dissect the underlying mechanism, Kim and co-workers (Kim et al. 2014) demonstrate that FoxO6 promoted lipogenesis and augmented TG-rich very low-density lipoprotein (VLDL-TG) secretion. This action correlates with the ability of FoxO6 to stimulate hepatic production of microsomal triglyceride transfer protein (MTP), a lipid transfer protein (MW, 88 kDa). Produced mainly in liver and to a less extent in the intestine, MTP, when heterodimerized with its small subunit protein disulphide isomerase (PDI, 58 kDa) in the endoplasmic reticulum (ER), catalyzes the transfer of lipid to nascent apolipoprotein B (apoB), a rate-limiting step in the nascent assembly and secretion of VLDL and chylomicrons (Gordon et al. 1995; Hui et al. 2002; Hussain et al. 2003; Manchekar et al. 2004; Swift et al. 2005; Swift et al. 2003). The observation that FoxO6 promotes hepatic MTP expression, which is recapitulated in primary human and mouse hepatocytes, underscores the importance of FoxO6 in regulating hepatic VLDL-TG secretion in response to insulin or nutritional cues. It is noteworthy that FoxO1 operates a similar mechanism for conducting insulin-dependent regulation of VLDL-TG production in the liver (Kamagate et al. 2008). In response to fasting, both FoxO1 and FoxO6 activity is upregulated in the liver. It remains to be determined whether FoxO1 and FoxO6 act in synergy to promote VLDL-TG production in the liver under fasting conditions.
FoxO6 in the etiology of diabetic hypertriglyceridemia
It is well established that insulin resistance is intertwined with hypertriglyceridemia in subjects with obesity and type 2 diabetes. Nonetheless, genetic factors that mechanistically couple insulin resistance with the pathogenesis of hypertriglyceridemia are incompletely characterized (Choi & Ginsberg 2011; Sparks & Dong 2009; Sparks et al. 2012). Characterization of FoxO6 as an integrator of hepatic insulin signaling with VLDL-TG production in the liver provides important insights to the mechanism of hypertriglyceridemia. In healthy individuals, insulin signaling through FoxO6 plays a pivotal role in adjusting the rates of hepatic VLDL-TG production to maintain normal lipid homeostasis. In insulin resistant subjects, this mechanism becomes deregulated, as evidenced by significantly elevated FoxO6 production and augmented FoxO6 activity in the liver (Kim et al. 2011). As a result, hepatic MTP expression is markedly upregulated in insulin resistant liver. This effect along with increased FFA influx into the liver promotes excessive VLDL-TG production, contributing to the development of hypertriglyceridemia in obesity and type 2 diabetes (Kim et al. 2014).
As FoxO6 is the most recently discovered FoxO isoform, there is a lack of clinical studies about the role of FoxO6 in humans. However, there is clinical evidence that FoxO1 contributes to glucose and lipid metabolism in humans. Genetic FOXO1 variants are associated with glucose intolerance, insulin resistance and type 2 diabetes in Finnish men and Pima Indians (Muller et al. 2015; Mussig et al. 2009). In contrast, it is also reported that such a close association between FOXO1 polymorphism and type 2 diabetes is lacking in different ethical groups including Caucasian and African-Americans (Karim et al. 2006). Further clinical studies are needed to characterize the role of FoxO6 and FoxO1 in metabolic diseases.
FoxO6 in skeletal muscle
Although FoxO6 is abundantly expressed in myocytes, its role in cardiac and skeletal muscles remains largely unknown. Chung and co-workers (Chung et al. 2013) report that both FoxO6 expression and transcriptional activity are upregulated in myocytes by peroxisome-proliferator-activated receptor γ co-activator 1α (PGC-1α), a transcriptional co-factor that playing important roles in mitochondria biogenesis and fatty acid oxidation in skeletal muscle (Wu et al. 1999). PGC-1α also plays a key role in muscle fiber subtype conversion from fast-twitch to slow-twitch fibres for adapation to oxidative metabolism in mice (Lin et al. 2002). PGC-1α binds directly to the FoxO6 promoter and stimulate FoxO6 expression in cultured myocytes as well as in skeletal muscle of mice in response to oxidative low-intensity exercise (Chung et al. 2013). Interestingly, increased FoxO6 activity in turns suppresses PGC-1α expression in myocytes, setting a feedback loop in which FoxO6 and PGC-1α interplays functionally for regulating oxidative metabolism in skeletal muscle in response to exercise (Chung et al. 2013).
FoxO6 in the brain
All FoxO proteins are abundantly expressed in the brain. However, unlike other FoxO isoforms (FoxO1, FoxO3 and FoxO4) with a broad distribution in the whole brain (Furuyama et al. 2000; Hoekman et al. 2006), FoxO6 is preferentially enriched in the hippocampus, a region that is critical for learning and memory consolidation (Jacobs et al. 2003). This has inspired the hypothesis that FoxO6 might be involved in learning and memory consolidation. Indeed, Salih and co-workers (Salih et al. 2012) show that FoxO6 knockout mice display normal learning but impaired memory consolidation. These findings suggest that FoxO6 activity in the hippocampus is required for memory consolidation and that reduced FoxO6 expression in the hippocampus may contribute to pathological age-dependent decline in memory. However, this notion seems at odds with the observation that FoxO6 expression is significantly upregulated in the brain of aged mice (Zemva et al. 2012). In contrast, FoxO6 expression is significantly down-regulated in the brain of dietary obese mice. This effect has been implicated in the pathogenesis of obesity and insulin resistance associated with dementia. Nonetheless, the underlying mechanism remains unexplored. Thus, further investigation is warranted to better understand the role of FoxO1 in the brain and its impact on cognitive function in relation with aging and diabetes.
FoxO6 in longevity
The FoxO family of proteins is known to play an important role in aging. Increased FoxO activity, resulting from attenuated insulin or insulin-like growth factor signaling, constitutes a caloric restriction regimen, contributing to lifespan extension in diverse organisms. This is typified by Daf16, the FoxO ortholog that acts downstream of insulin-like signaling to regulate anti-oxidative function and longevity in C. elegans in response to environment cues (Lee et al. 2003; Murphy et al. 2003). Likewise, the FoxO ortholog dFoxO seems to integrate insulin-like growth factor to aging in Drosophila melanogaster. Giannoakou and co-workers (Giannakou et al. 2004) show that dFoxO overexpression in the fat body increases lifespan by 20–50% in female but not in male Drosophila melanogaster. This conclusion was challenged by Tatar (Tatar 2005), who comment that their studies are limited to the experimental condition, under which control female flies are somewhat associated with unusually higher death rates during the time course of studies. Indeed, there is evidence that dFoxO is not required for dietary restriction-induced lifespan extension in Drosophila melanogaster, although dFoxO modulates this process (Giannakou et al. 2008; Hwangbo et al. 2004; Min et al. 2008). In a recent study, Dobson and colleagues took an opposite approach to address whether dFoxO is liable for the overnutrition-elicited reduction of lifespan in Drosophila melanogaster (Dobson et al. 2017). They demonstrate that even transient feeding with a sugar-rich diet in early adulthood could produce a long-lasting detrimental effect on subsequent survival in female flies. This effect could be ascribed to the sugar-rich diet-mediated induction of insulin-like signaling, which in turn inhibits dFoxO activity and suppresses dFoxO target genes encoding epigenetic regulators in the fat body. Their data underscore the importance of dFoxO in linking overnutrition to lifespan attenuation in Drosophila melanogaster (Dobson et al. 2017).
Among the four mammalian FoxO isoforms, only FoxO3 is consistently correlated with associated with longevity. Clinical studies characterize the FOXO3A as a susceptibility gene for human longevity in different ethnic groups including the Japanese from Hawaii (Willcox et al. 2008), Italian (Anselmi et al. 2009), German (Flachsbart et al. 2009), Chinese(Li et al. 2009), American (Pawlikowska et al. 2009) and Jewish (Soerensen et al. 2010). Preclinical studies indicate that FoxO3 is required for the lifespan extension induced by caloric restriction in mice (Shimokawa et al. 2015). In contrast, there is a lack of association between FOXO1, FOXO4 or FOXO6 and life expectancy in humans (Kleindorp et al. 2011). Likewise, transgenic mice with FoxO1 overproduction in skeletal muscle have a similar lifespan as wild-type littermates (Chiba et al. 2009). To date, the mechanism underlying FoxO3-mediated extension of lifespan in humans or animals remains elusive and warrants in-depth investigation.
FoxO functional redundancy and divergence
Despite their structural conservation in the forkhead box domain and functional redundancy to some extent, FoxO proteins possess non-overlapping functions, in line with their variable carboxyl domains and differential tissue distributions (Wang et al. 2009). For example, FoxO1, FoxO3 and FoxO4 are expressed across the brain. In contrast, FoxO6 expression is confined to the hippocampus region. FoxO1-null mice die at embryonic day 11.5 due to defective angiogenesis and vasculogenesis in the embryos (Furuyama et al. 2004). In contrast, homozygous FoxO4 and FoxO6 knockout mice are viable (Calabuig-Navarro et al. 2015; Hosaka et al. 2004). Although FoxO3 null mice are viable, female FoxO3 null mice are associated with age-dependent infertility, due to abnormal ovarian follicular development (Castrillon et al. 2003). FoxO6 knockout mice are protected from developing fat-elicited insulin resistance and metabolic syndrome. In contrast, FoxO1, FoxO3 and FoxO4, all of which are evolutionally diverged from FoxO6, display functional redundancy in regulating hepatic glucose metabolism (Haeusler et al. 2010). These results highlight the functional divergence among the four FoxO isoforms in the body.
Conclusion
FoxO6 has emerged as a key player in glucose and lipid metabolism (Fig. 3). FoxO6 is ubiquitously expressed in mammals, characteristic of the FoxO family. FoxO6 constitutes a distinct route by which the liver orchestrates insulin-dependent regulation of gluconeogenesis and VLDL-TG production. Hepatic FoxO6 activity is increased and this effect primes the liver for promoting glucose production and VLDL-TG secretion in response to fasting. In response to postprandial insulin secretion, hepatic FoxO6 activity is inhibited to suppress glucose production and VLDL-TG assembly in the liver and prevent excessive postprandial glucose and TG excursion. Unlike other FoxO members that undergo insulin-dependent trafficking from the nucleus to the cytoplasm, FoxO6 mediates insulin action in a distinct mechanism without altering its subcellular redistribution. Hepatic insulin signaling through FoxO6 plays pivotal role in regulating glucose and lipid homeostasis. Unchecked FoxO6 activity, resulting from insulin resistance, promotes hepatic overproduction of glucose and VLDL-TG, accounting in part for the dual pathogenesis of fasting hyperglycemia and hypertriglyceridemia in diabetes. These results characterize FoxO6 as a potential therapeutic target for treating metabolic disease. Apart from its role in the liver, FoxO6 in hippocampus seems to play an important role in memory consolidation (Fig. 3). In addition, FoxO6 appears to regulate oxidative metabolism in skeletal muscle (Fig. 3). Further studies are needed to define the distinct roles of FoxO6 in central and peripheral tissues.
Fig. 3. FoxO6 mediates the inhibitory effect of insulin or insulin-like growth factor on target genes in central and peripheral tissues.
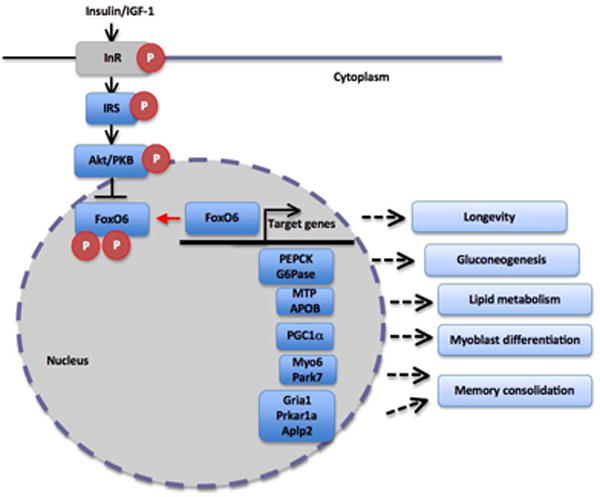
In the liver, FoxO6 contributes to insulin-dependent regulation of gluconeogenesis via PEPCK and G6Pase. Similarly, insulin signaling through FoxO6 regulates hepatic MTP and APOB expression and modulates VLDL-TG production in the liver. FoxO6 also targets PGC1α for _trans_-activation, which in turns suppresses FoxO6 expression in myocytes. Such FoxO6-PGC1α feedback loop plays an important part in regulating myoblast differentiation and oxidative metabolism in skeletal muscle in response to exercise. In the brain, FoxO6 contributes to neuronal regulation of Myosin 6 (Myo6), Parkinson disease protein 7 (Park7), Glutamate receptor 1 (Gria1), cAMP-dependent protein kinase type I-alpha regulatory subunit (Prkar1a) and Amyloid-like protein 2 (Aplp2), key functions in memory consolidation.
Plain Language Summary.
The FoxO transcription factors integrate insulin signaling with glucose and lipid metabolism. Genetic variants of FoxO genes are associated with metabolic disease in humans. We review the role of FoxO factors in central and peripheral tissues to gain insight into the mechanism of metabolic disease associated with FoxO deregulation.
Acknowledgments
This work was supported by NIH grant R01 DK098437.
Footnotes
Disclosure Statement: The authors have nothing to disclose
References
- Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117:421–426. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- Ader M, Bergman RN. Peripheral effects of insulin dominate suppression of fasting hepatic glucose production. Am J Physiol. 1990;258:E1020–1032. doi: 10.1152/ajpendo.1990.258.6.E1020. [DOI] [PubMed] [Google Scholar]
- Altomonte J, Cong L, Harbaran S, Richter A, Xu J, Meseck M, Dong HH. Foxo1 Mediates Insulin Action on ApoC-III and Triglyceride Metabolism. J Clin Invest. 2004;114:1493–1503. doi: 10.1172/JCI19992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselmi CV, Malovini A, Roncarati R, Novelli V, Villa F, Condorelli G, Bellazzi R, Puca AA. Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res. 2009;12:95–104. doi: 10.1089/rej.2008.0827. [DOI] [PubMed] [Google Scholar]
- Barthel A, Schmoll D, Unterman TG. FoxO proteins in insulin action and metabolism. Trends Endocrinol Metab. 2005;16:183–189. doi: 10.1016/j.tem.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Brent MM, Anand R, Marmorstein R. Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure. 2008;16:1407–1416. doi: 10.1016/j.str.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabuig-Navarro V, Yamauchi J, Lee S, Zhang T, Liu YZ, Sadlek K, Coudriet GM, Piganelli JD, Jiang CL, Miller R, et al. Forkhead Box O6 (FoxO6) Depletion Attenuates Hepatic Gluconeogenesis and Protects against Fat-induced Glucose Disorder in Mice. J Biol Chem. 2015;290:15581–15594. doi: 10.1074/jbc.M115.650994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301:215–218. doi: 10.1126/science.1086336. [DOI] [PubMed] [Google Scholar]
- Cherrington AD. The role of hepatic insulin receptors in the regulation of glucose production. J Clin Invest. 2005;115:1136–1139. doi: 10.1172/JCI25152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T, Kamei Y, Shimizu T, Shirasawa T, Katsumata A, Shiraishi L, Sugita S, Ogawa Y, Miura S, Ezaki O. Overexpression of FOXO1 in skeletal muscle does not alter longevity in mice. Mech Ageing Dev. 2009;130:420–428. doi: 10.1016/j.mad.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab. 2011;22:353–363. doi: 10.1016/j.tem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SY, Huang WC, Su CW, Lee KW, Chi HC, Lin CT, Chen ST, Huang KM, Tsai MS, Yu HP, et al. FoxO6 and PGC-1alpha form a regulatory loop in myogenic cells. Biosci Rep. 2013;33 doi: 10.1042/BSR20130031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifarelli V, Lee S, Kim DH, Zhang T, Kamagate A, Slusher S, Bertera S, Luppi P, Trucco M, Dong HH. FOXO1 Mediates the Autocrine Effect of Endothelin-1 on Endothelial Cell Survival. Mol Endocrinol. 2012;26:1213–1224. doi: 10.1210/me.2011-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson AJ, Ezcurra M, Flanagan CE, Summerfield AC, Piper MD, Gems D, Alic N. Nutritional Programming of Lifespan by FOXO Inhibition on Sugar-Rich Diets. Cell Rep. 2017;18:299–306. doi: 10.1016/j.celrep.2016.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, Chook YM. Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature. 2009;458:1136–1141. doi: 10.1038/nature07975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgerton DS, Johnson KM, Cherrington AD. Current strategies for the inhibition of hepatic glucose production in type 2 diabetes. Front Biosci. 2009;14:1169–1181. doi: 10.2741/3301. [DOI] [PubMed] [Google Scholar]
- Ekberg K, Landau BR, Wajngot A, Chandramouli V, Efendic S, Brunerngraber H, Wahren J. Contributions by kidney and liver to glucose production in the postabsorptive state and after 60 h of fasting. Diabetes. 1999;48:292–298. doi: 10.2337/diabetes.48.2.292. [DOI] [PubMed] [Google Scholar]
- Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, Burgering BM. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. Embo J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flachsbart F, Caliebe A, Kleindorp R, Blanche H, von Eller-Eberstein H, Nikolaus S, Schreiber S, Nebel A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci U S A. 2009;106:2700–2705. doi: 10.1073/pnas.0809594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama T, Kitayama K, Shimoda Y, Ogawa M, Sone K, Yoshida-Araki K, Hisatsune H, Nishikawa S, Nakayama K, Ikeda K, et al. Abnormal angiogenesis in Foxo1 (Fkhr)-deficient mice. J Biol Chem. 2004;279:34741–34749. doi: 10.1074/jbc.M314214200. [DOI] [PubMed] [Google Scholar]
- Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem J. 2000;349:629–634. doi: 10.1042/0264-6021:3490629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakou ME, Goss M, Junger MA, Hafen E, Leevers SJ, Partridge L. Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science. 2004;305:361. doi: 10.1126/science.1098219. [DOI] [PubMed] [Google Scholar]
- Giannakou ME, Goss M, Partridge L. Role of dFOXO in lifespan extension by dietary restriction in Drosophila melanogaster: not required, but its activity modulates the response. Aging Cell. 2008;7:187–198. doi: 10.1111/j.1474-9726.2007.00362.x. [DOI] [PubMed] [Google Scholar]
- Gordon DA, Wetterau JR, Gregg RE. Microsomal triglyceride transfer protein: a protein complex required for the assembly of lipoprotein particles. Trends Cell Biol. 1995;5:317–321. doi: 10.1016/s0962-8924(00)89054-6. [DOI] [PubMed] [Google Scholar]
- Graves JD, Gotoh Y, Draves KE, Ambrose D, Han DK, Wright M, Chernoff J, Clark EA, Krebs EG. Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J. 1998;17:2224–2234. doi: 10.1093/emboj/17.8.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler RA, Kaestner KH, Accili D. FoxOs function synergistically to promote glucose production. J Biol Chem. 2010;285:35245–35248. doi: 10.1074/jbc.C110.175851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekman MF, Jacobs FM, Smidt MP, Burbach JP. Spatial and temporal expression of FoxO transcription factors in the developing and adult murine brain. Gene Expr Patterns. 2006;6:134–140. doi: 10.1016/j.modgep.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Holm C, Osterlund T, Laurell H, Contreras JA. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Annu Rev Nutr. 2000;20:365–393. doi: 10.1146/annurev.nutr.20.1.365. [DOI] [PubMed] [Google Scholar]
- Hosaka T, Biggs WH, 3rd, Tieu D, Boyer AD, Varki NM, Cavenee WK, Arden KC. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A. 2004;101:2975–2980. doi: 10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui TY, Olivier LM, Kang S, Davis RA. Microsomal triglyceride transfer protein is essential for hepatic secretion of apoB-100 and apoB-48 but not triglyceride. J Lipid Res. 2002;43:785–793. [PubMed] [Google Scholar]
- Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003;44:22–32. doi: 10.1194/jlr.r200014-jlr200. [DOI] [PubMed] [Google Scholar]
- Hwangbo DS, Gershman B, Tu MP, Palmer M, Tatar M. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429:562–566. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- OS I, Zhang W, Wasserman DH, Liew CW, Liu J, Paik J, DePinho RA, Stolz DB, Kahn CR, Schwartz MW, et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun. 2015;6:7079. doi: 10.1038/ncomms8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H, Maechler P, Gjinovci A, Herrera PL, Wollheim CB. Islet beta-cell secretion determines glucagon release from neighbouring alpha-cells. Nat Cell Biol. 2003;5:330–335. doi: 10.1038/ncb951. [DOI] [PubMed] [Google Scholar]
- Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003;278:35959–35967. doi: 10.1074/jbc.M302804200. [DOI] [PubMed] [Google Scholar]
- Jang H, Lee GY, Selby CP, Lee G, Jeon YG, Lee JH, Cheng KK, Titchenell P, Birnbaum MJ, Xu A, et al. SREBP1c-CRY1 signalling represses hepatic glucose production by promoting FOXO1 degradation during refeeding. Nat Commun. 2016;7:12180. doi: 10.1038/ncomms12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Zhang BB. Glucagon and regulation of glucose metabolism. Am J Physiol Endocrinol Metab. 2003;284:E671–678. doi: 10.1152/ajpendo.00492.2002. [DOI] [PubMed] [Google Scholar]
- Kamagate A, Dong HH. FoxO1 integrates insulin signaling to VLDL production. Cell Cycle. 2008;7:3162–3170. doi: 10.4161/cc.7.20.6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamagate A, Qu S, Perdomo G, Su D, Kim DH, Slusher S, Meseck M, Dong HH. FoxO1 mediates insulin-dependent regulation of hepatic VLDL production in mice. J Clin Invest. 2008;118:2347–2364. doi: 10.1172/JCI32914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim MA, Craig RL, Wang X, Hale TC, Elbein SC. Analysis of FOXO1A as a candidate gene for type 2 diabetes. Mol Genet Metab. 2006;88:171–177. doi: 10.1016/j.ymgme.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Kato S, Ding J, Pisck E, Jhala US, Du K. COP1 functions as a FoxO1 ubiquitin E3 ligase to regulate FoxO1-mediated gene expression. J Biol Chem. 2008;283:35464–35473. doi: 10.1074/jbc.M801011200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbe C, Chen J, Xu G, Jing G, Shalev A. FOXO1 competes with carbohydrate response element-binding protein (ChREBP) and inhibits thioredoxin-interacting protein (TXNIP) transcription in pancreatic beta cells. J Biol Chem. 2013;288:23194–23202. doi: 10.1074/jbc.M113.473082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Perdomo G, Zhang T, Slusher S, Lee S, Phillips BE, Fan Y, Giannoukakis N, Gramignoli R, Strom S, et al. FoxO6 Integrates Insulin Signaling With Gluconeogenesis in the Liver. Diabetes. 2011;60:2763–2774. doi: 10.2337/db11-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Zhang T, Lee S, Calabuig-Navarro V, Yamauchi J, Piccirillo A, Fan Y, Uppala R, Goetzman E, Dong HH. FoxO6 integrates insulin signaling with MTP for regulating VLDL production in the liver. Endocrinology. 2014;155:1255–1267. doi: 10.1210/en.2013-1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, Accili D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Kleindorp R, Flachsbart F, Puca AA, Malovini A, Schreiber S, Nebel A. Candidate gene study of FOXO1, FOXO4, and FOXO6 reveals no association with human longevity in Germans. Aging Cell. 2011;10:622–628. doi: 10.1111/j.1474-9726.2011.00698.x. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- Lee SS, Kennedy S, Tolonen AC, Ruvkun G. DAF-16 target genes that control C. elegans life-span and metabolism. Science. 2003;300:644–647. doi: 10.1126/science.1083614. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang WJ, Cao HQ, Lu JH, Wu C, Hu FY, Guo J, Zhao L, Yang F, Zhang YX, et al. Genetic association of FOXO1A and FOXO3A with longevity trait in Han Chinese populations. Hum Mol Genet. 2009;18:4897–4904. doi: 10.1093/hmg/ddp459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, Milne J, Meyers DJ, Cole P, Yates J, 3rd, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–273. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchekar M, Richardson PE, Forte TM, Datta G, Segrest JP, Dashti N. Apolipoprotein B-containing lipoprotein particle assembly: lipid capacity of the nascent lipoprotein particle. J Biol Chem. 2004;279:39757–39766. doi: 10.1074/jbc.M406302200. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464–2472. doi: 10.1172/JCI27047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor foxo1 in liver. Cell Metab. 2007;6:208–216. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A. 2003;100:11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min KJ, Yamamoto R, Buch S, Pankratz M, Tatar M. Drosophila lifespan control by dietary restriction independent of insulin-like signaling. Aging Cell. 2008;7:199–206. doi: 10.1111/j.1474-9726.2008.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monecke T, Guttler T, Neumann P, Dickmanns A, Gorlich D, Ficner R. Crystal Structure of the Nuclear Export Receptor CRM1 in Complex with Snurportin1 and RanGTP. Science. 2009;324:1087–1091. doi: 10.1126/science.1173388. [DOI] [PubMed] [Google Scholar]
- Mueller MM, Castells-Roca L, Babu V, Ermolaeva MA, Muller RU, Frommolt P, Williams AB, Greiss S, Schneider JI, Benzing T, et al. DAF-16/FOXO and EGL-27/GATA promote developmental growth in response to persistent somatic DNA damage. Nat Cell Biol. 2014;16:1168–1179. doi: 10.1038/ncb3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller YL, Hanson RL, Wiessner G, Nieboer L, Kobes S, Piaggi P, Abdussamad M, Okani C, Knowler WC, Bogardus C, et al. Assessing FOXO1A as a potential susceptibility locus for type 2 diabetes and obesity in American Indians. Obesity (Silver Spring) 2015;23:1960–1965. doi: 10.1002/oby.21236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Mussig K, Staiger H, Machicao F, Stancakova A, Kuusisto J, Laakso M, Thamer C, Machann J, Schick F, Claussen CD, et al. Association of common genetic variation in the FOXO1 gene with beta-cell dysfunction, impaired glucose tolerance, and type 2 diabetes. J Clin Endocrinol Metab. 2009;94:1353–1360. doi: 10.1210/jc.2008-1048. [DOI] [PubMed] [Google Scholar]
- Pawlikowska L, Hu D, Huntsman S, Sung A, Chu C, Chen J, Joyner AH, Schork NJ, Hsueh WC, Reiner AP, et al. Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging Cell. 2009;8:460–472. doi: 10.1111/j.1474-9726.2009.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prager R, Wallace P, Olefsky JM. Direct and indirect effects of insulin to inhibit hepatic glucose output in obese subjects. Diabetes. 1987;36:607–611. doi: 10.2337/diab.36.5.607. [DOI] [PubMed] [Google Scholar]
- Qu S, Altomonte J, Perdomo G, He J, Fan Y, Kamagate A, Meseck M, Dong HH. Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology. 2006;147:5641–5652. doi: 10.1210/en.2006-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih DA, Rashid AJ, Colas D, de la Torre-Ubieta L, Zhu RP, Morgan AA, Santo EE, Ucar D, Devarajan K, Cole CJ, et al. FoxO6 regulates memory consolidation and synaptic function. Genes Dev. 2012;26:2780–2801. doi: 10.1101/gad.208926.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Beddow SA, Iwasaki T, Zhang XM, Chu X, Still CD, Gerhard GS, Shulman GI. Fasting hyperglycemia is not associated with increased expression of PEPCK or G6Pc in patients with Type 2 Diabetes. Proc Natl Acad Sci U S A. 2009;106:12121–12126. doi: 10.1073/pnas.0812547106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokawa I, Komatsu T, Hayashi N, Kim SE, Kawata T, Park S, Hayashi H, Yamaza H, Chiba T, Mori R. The life-extending effect of dietary restriction requires Foxo3 in mice. Aging Cell. 2015;14:707–709. doi: 10.1111/acel.12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soerensen M, Dato S, Christensen K, McGue M, Stevnsner T, Bohr VA, Christiansen L. Replication of an association of variation in the FOXO3A gene with human longevity using both case-control and longitudinal data. Aging Cell. 2010;9:1010–1017. doi: 10.1111/j.1474-9726.2010.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks DJ, Dong HH. FoxO1 and hepatic lipid metabolism. Current Opinion in Lipidology. 2009;20:217–226. doi: 10.1097/MOL.0b013e32832b3f4c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JD, Sparks CE, Adeli K. Selective hepatic insulin resistance, VLDL overproduction, and hypertriglyceridemia. Arterioscler Thromb Vasc Biol. 2012;32:2104–2112. doi: 10.1161/ATVBAHA.111.241463. [DOI] [PubMed] [Google Scholar]
- Swift LL, Jovanovska A, Kakkad B, Ong DE. Microsomal triglyceride transfer protein expression in mouse intestine. Histochem Cell Biol. 2005;123:475–482. doi: 10.1007/s00418-005-0772-7. [DOI] [PubMed] [Google Scholar]
- Swift LL, Zhu MY, Kakkad B, Jovanovska A, Neely MD, Valyi-Nagy K, Roberts RL, Ong DE, Jerome WG. Subcellular localization of microsomal triglyceride transfer protein. J Lipid Res. 2003;44:1841–1849. doi: 10.1194/jlr.M300276-JLR200. [DOI] [PubMed] [Google Scholar]
- Tatar M. Comment on “Long-lived Drosophila with overexpressed dFOXO in adult fat body”. Science. 2005;307:675. doi: 10.1126/science.1104733. author reply 675. [DOI] [PubMed] [Google Scholar]
- Titchenell PM, Chu Q, Monks BR, Birnbaum MJ. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nat Commun. 2015;6:7078. doi: 10.1038/ncomms8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WC, Bhattacharyya N, Han LY, Hanover JA, Rechler MM. Insulin inhibition of transcription stimulated by the forkhead protein Foxo1 is not solely due to nuclear exclusion. Endocrinology. 2003;144:5615–5622. doi: 10.1210/en.2003-0481. [DOI] [PubMed] [Google Scholar]
- Unger RH, Dobbs RE, Orci L. Insulin, glucagon, and somatostatin secretion in the regulation of metabolism. Annu Rev Physiol. 1978;40:307–343. doi: 10.1146/annurev.ph.40.030178.001515. [DOI] [PubMed] [Google Scholar]
- Wahren J, Ekberg K. Splanchnic regulation of glucose production. Annu Rev Nutr. 2007;27:329–345. doi: 10.1146/annurev.nutr.27.061406.093806. [DOI] [PubMed] [Google Scholar]
- Wang M, Zhang X, Zhao H, Wang Q, Pan Y. FoxO gene family evolution in vertebrates. BMC Evol Biol. 2009;9:222. doi: 10.1186/1471-2148-9-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, Masaki KH, Willcox DC, Rodriguez B, Curb JD. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A. 2008;105:13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Zemva J, Schilbach K, Stohr O, Moll L, Franko A, Krone W, Wiesner RJ, Schubert M. Central FoxO3a and FoxO6 Expression is Down-regulated in Obesity Induced Diabetes but not in Aging. Exp Clin Endocrinol Diabetes. 2012;120:340–350. doi: 10.1055/s-0031-1297970. [DOI] [PubMed] [Google Scholar]
- Zhang T, Kim DH, Xiao X, Lee S, Gong Z, Muzumdar R, Calabuig-Navarro V, Yamauchi J, Harashima H, Wang R, et al. FoxO1 Plays an Important Role in Regulating beta-Cell Compensation for Insulin Resistance in Male Mice. Endocrinology. 2016;157:1055–1070. doi: 10.1210/en.2015-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12:665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]