Improved de novo structure prediction in CASP11 by incorporating coevolution information into Rosetta (original) (raw)
. Author manuscript; available in PMC: 2017 Sep 1.
Published in final edited form as: Proteins. 2016 Feb 24;84(Suppl 1):67–75. doi: 10.1002/prot.24974
Abstract
We describe CASP11 de novo blind structure predictions made using the Rosetta structure prediction methodology with both automatic and human assisted protocols. Model accuracy was generally improved using coevolution derived residue–residue contact information as restraints during Rosetta conformational sampling and refinement, particularly when the number of sequences in the family was more than three times the length of the protein. The highlight was the human assisted prediction of T0806, a large and topologically complex target with no homologs of known structure, which had unprecedented accuracy—<3.0 Å root-mean-square deviation (RMSD) from the crystal structure over 223 residues. For this target, we increased the amount of conformational sampling over our fully automated method by employing an iterative hybridization protocol. Our results clearly demonstrate, in a blind prediction scenario, that coevolution derived contacts can considerably increase the accuracy of template-free structure modeling.
Keywords: protein structure prediction, rosetta, ab initio prediction, coevolution, contact prediction
INTRODUCTION
The Eleventh Critical Assessment of Techniques for Protein Structure Prediction (CASP11) had considerably more free modeling targets (45 domains) than recent CASP experiments (three times more than in CASP10) and hence provided a significantly larger benchmark for template-free modeling methods. In this article, we describe the methods used to generate structure models for these targets by our fully automated structure prediction server, Robetta1 (BAKER-ROSETTASERVER), and the human assisted approaches used to generate models in cases we thought could significantly improve with human intervention (BAKER). As in previous CASP experiments, our general approach was to sample a diverse set of conformations using the Rosetta fragment assembly method2 followed by all-atom refinement,3 and select final models based on clustering and Rosetta all-atom energy. Due to the sheer size of the conformational search space, the success of this approach for high accuracy models has been limited to small proteins (<100 residues). In CASP11, we used residue–residue coevolution derived restraints4–6 during sampling and refinement to help guide the search toward the native conformation. The results clearly indicate that given sufficient numbers of aligned sequences, coevolution derived contacts can considerably increase structure prediction accuracy. For one example (T0806), the predicted structure had significantly higher accuracy than any previous prediction in the 20 years of CASP experiments for proteins >100 amino acids lacking homologs of known structure.
MATERIALS AND METHODS
Robetta de novo pipeline
Figure 1 outlines the automated Robetta protocol used for difficult targets without clearly detectable homologs of known structure. The individual steps are described in the following sections. At the very beginning of the protocol (not shown in Fig. 1), Robetta scans the query sequence for signal sequences using SignalP7 and removes them if found as they are unlikely to be present in the final structure.
Figure 1.
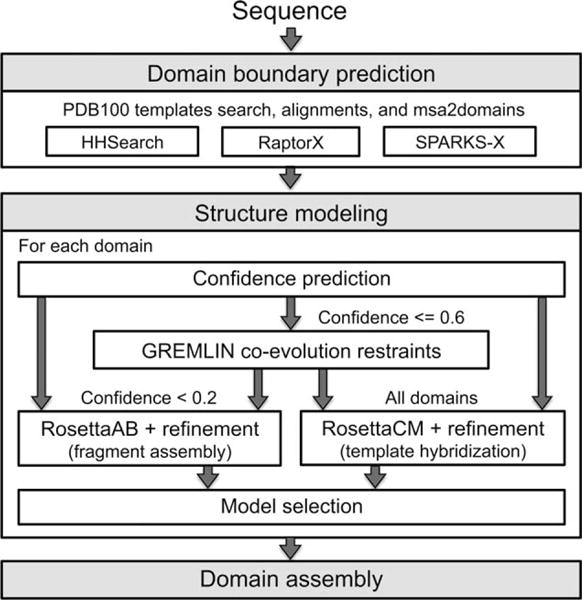
Fully automated Robetta structure prediction protocol.
Domain boundary prediction
Domain boundaries were predicted from PDB structures with sequence similarity to the query using an iterative process. For each iteration, HHSearch,8 Sparks-X,9 and RaptorX10 were used to identify templates from the PDB100 database and generate alignments. The sequence was threaded onto the template structures to generate partial models, which were then clustered to identify distinct topologies that were ranked based on the likelihood of the alignments (RosettaCM supplementary methods11). Structural similarity in the top ranked cluster was determined within a 30 residue sliding window using MaxSub12 with a 4 Å threshold and the closest coil positions to sites where the MaxSub value was <10, 20, or 30% (in order of precedence) were selected as potential domain boundaries. Potential domains of at least 35 residues, including regions that were not covered by the partial-threads or not similar in structure (<30% average windowed MaxSub), were passed on to a next template search iteration. A confidence score11 was estimated for each domain using the degree of structural consensus between the top ranked partial threads from each alignment method. Since templates may be incorrect for regions with low confidence, domain boundaries were also identified in regions with confidence scores less than 0.2 using the multiple sequence alignment (MSA)-based method “msa2domains.”13 Through this iterative process, the most confident non-overlapping clusters were identified that, together, covered the full length of the target sequence, and domain boundaries were assigned at the transitions between the clusters. Final domains with confidence scores less than 0.2 were classified in the “twilight” regime,14 and modeled using the Rosetta comparative modeling (RosettaCM11) and ab initio (RosettaAB2,15) protocols.
GREMLIN coevolutionary restraints
For each domain with a confidence score equal to or less than 0.6, Robetta determined if there were enough homologous sequences to accurately predict residue–residue coevolution contacts. High confidence targets were excluded since additional information was likely not gained over that contained in the template coordinates. Due to the three day time constraint for servers, a fast alignment tool, HHblits,16 was used with an e-value cutoff of 1E-20 and a 90% identity redundancy cutoff. If there were less than 2_L_ sequences with 75% alignment coverage, HHblits was rerun with progressively less stringent settings (down to an e-value and cutoff of 1E-4 and 50% alignment coverage). During CASP11, the latest clustered uniprot database available for HHblits was from 2013, therefore, if <2_L_ sequences were found, Jackhmmer17 was used to search against the latest uniref90 sequence database18 with the same search criteria. If at least 1_L_ sequences were identified, GREMLIN5 was used to learn a global statistical model of the MSA using a pseudo likelihood approach,4 and contacts were predicted using the residue–residue coupling values obtained from the model fitting procedure. The top 3 L/2 contacts with sequence separation of at least three were converted to restraints, and the weights of these restraints were normalized by the average of the coupling scores. Distance restraints were implemented as sigmoidal functions of the form: restraint(d) = 3 * weight/(1 + exp(−slope(d − cutoff)) − 1 where d is the distance between the constrained Cβ atoms (C_α_ for glycine) and the cutoffs and slopes are amino acid pair specific (Supporting Information Table III in Ref.5). These distance restraints supplement the Rosetta energy function; the combination ensures the sampling of physically realistic structures consistent with the contact predictions. Given that both the number of contacts and the Rosetta energy function are roughly proportional to the length of the protein, we found that simple scaling of the restraint score by threefold gave a dynamic range similar to that of the Rosetta energy roughly independent of the protein length.
Residue pairs with strong coevolution signals are not always in contact, some interactions may be intermediate, ligand mediated, or between copies of a homo-oligomeric protein. When the number of homologous sequences is low, more predicted contacts may be incorrect due to noise. To overcome these inaccuracies, we use a sigmoidal restraint with no penalty after a certain distance, as opposed to a linear (bounded) or quadratic (harmonic) penalty [Fig. 2(A)]. The disadvantage of using sigmoidal restraints is that there is no gradient past the distance cutoff, and, therefore, a large amount of sampling is required (see sections below). The advantage is the ability to sample within protein-like space while maximizing self-consistent contacts [Fig. 2(B)]; inconsistent contacts, which are likely to be incorrect, do not contribute to the restraint score.
Figure 2.
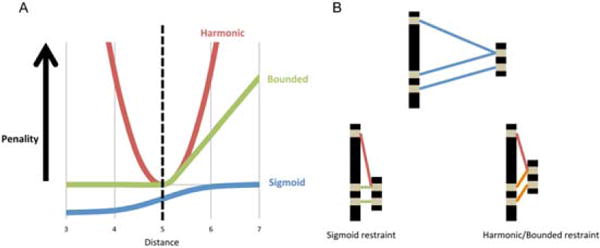
Choice of restraint functional form. A) Alternative functional form for restraint penalty functions. B) Use of sigmoidal restraints with no gradient beyond 8 Å requires a large amount of sampling, but reduces the impact of incorrect contact predictions on the resulting models, which maximize satisfaction of internally consistent sets of contacts.
Rosetta ab initio modeling
RosettaAB has previously been described in detail.2,15 Conformational sampling is carried out using a Monte Carlo fragment replacement strategy guided by a low-resolution score function that favors protein-like features. Bond angles and bond lengths are kept fixed, and side-chains are represented by a single “centroid” interaction center; the only degrees of freedom are the backbone phi, psi, and omega torsion angles. Conformational sampling proceeds, starting from an extended chain, by random replacement of backbone torsion angles with torsion angles from fragments selected from PDB templates using the Rosetta fragment picker.19 Previously described sequence profile, secondary structure (SS), and Ramachandran-based score terms were used for fragment selection, in addition to depth dependent structure profile,20 phi and psi torsion,21 and solvent accessibility21 score terms. Score term weights were optimized from a previous unpublished benchmark and used as default in the “make_fragments.pl” wrapper script available in the Rosetta software package. The previously described SS quota mechanism19 was not used in favor of a single predictor, PSIPRED.22 Variable fragment lengths of 3–19 residues were used as previously described.23 If GREMLIN contacts were predicted, they were used as sigmoidal restraints for sampling and refinement, and only 3 and 9 residue fragments were used.
Rosetta comparative modeling
RosettaCM has previously been described in detail.11 In summary, the protocol recombines structural elements from clustered partial-threads and models missing segments using a combination of fragment insertion and mixed torsion-Cartesian space minimization. Conformational sampling is performed using the Rosetta low-resolution score function2 with spatial restraints that are generated separately from each cluster.24 If GREMLIN contacts were predicted, the clusters were reranked using this information, and the template-based spatial restraints were supplemented with the coevolution restraints for sampling and refinement. The weighted fraction of top contacts made in each partial-thread was used to modify the p-correct scores11 for re-ranking clusters. The contribution of the GREMLIN term was scaled so that the score of the best scoring template was zero.
Rosetta refinement
All models were relaxed in the Rosetta full atom energy function first in torsion space and then in Cartesian space.3 If GREMLIN contacts were predicted, the GREMLIN score was added to the all atom energy during full-atom refinement; in both the RosettaCM and RosettaAB protocols, the Cβ-Cβ amino acid specific GREMLIN restraints were replaced with ambiguous distance restraints between side-chain heavy atoms (cutoff 5.5, slope 4).
Model sampling and selection
Robetta uses the BOINC25 distributed computing project, Rosetta@home (R@h; http://boinc.bakerlab.org/rosetta), to generate models. Each R@h work unit runs as many independent sampling trajectories as possible within a 4 hour average run time limit on a host computer. RosettaAB models were generated from 12,000 work units for the query sequence, and 4000 each for up to two sequence homologs and two query sequence variants with at least 90% coverage whose termini were trimmed based on predicted disorder.26 Around 71,000 models were generated for each query sequence and 28,000 for each homolog and trimmed variant, on average. If GREMLIN contacts were predicted, models were only generated for the query sequence. The top 10,000 all-atom scoring (and restraint fit if GREMLIN contacts existed) query models were clustered with the top 2000 scoring models for each homolog and trimmed variant. The top scoring query model from each of the top four clusters ranked by cluster size and the overall top scoring query model not represented in the clusters were selected, and the top 3 scoring models from this subset were submitted as models 2, 3, and 5, ordered by score. The remaining models, 1 and 4, were selected from among the best scoring 100 RosettaCM models by identifying low-energy clusters and averaging. For each model, we compute a neighbor-weighted probability, where the Bolzmann weight of each structure is calculated, and then—for each conformation—the weight of nearby neighbors is added, scaled by the similarity of the two structures using maxsub with a 3 Å radius. Using this metric, the highest-probability structure and its ten nearest neighbors are selected, and they are averaged in Cartesian space by superimposing and computing the mean coordinates for each atom. Positions with too much variability (positional B factor > 100) are not averaged, but instead are assigned the coordinates of the selected model. These averaged models are then refined using Rosetta relax with coordinate constraints. The order of these final models was determined by the rank of the alignment cluster from which they belonged to. RosettaCM models were generated from up to 3000 work units; the number of work units was a function of L* log(L) where L is the sequence length. On average, 5100 models were generated for up to five partial-thread clusters each.
Domain assembly
The method used for domain assembly for multiple domain targets depended on whether an alignment was generated to a template that contained all of the domains. If no such template was found, the final selected models for each domain were combined into a full-length model using the Rosetta domain assembly method,27 which assembles domains by performing fragment insertion within the linkers. If domain models superimposed with a full-length partial thread, RosettaCM was used to guide assembly based on the orientation in the full length model by recombining the domain models with the full-length partial thread.
Human intervention
We decided to manually intervene only for targets we thought would significantly improve in prediction accuracy using either methods not yet implemented in Robetta or with more sampling (7 FM domains). For the remaining targets, we simply submitted the Robetta models. The general protocol we used is shown in Figure 3. The majority of human targets were chosen if GREMLIN contacts were predicted. If a large fraction of the top coevolving contacts were not made in the Robetta models, more RosettaAB models were produced (T0824, T0826, and T0836). If most contacts were made, further optimization of the Rosetta all-atom energy and coevolution restraints was carried out through multiple RosettaCM iterations starting with the Robetta models as initial templates (T0806). Fragment insertions were also made in template regions (10% of the total moves) to introduce more diversity. Some targets were also chosen if there were templates suggesting possible homo-oligomeric states that Robetta did not detect. For these targets (T0763, T0785, and T0799), RosettaCM was used to generate models in the oligomeric states inferred from templates. The final submissions for these targets were selected from the oligomeric state runs and the original Robetta server models ranked by GOAP score.28
Figure 3.
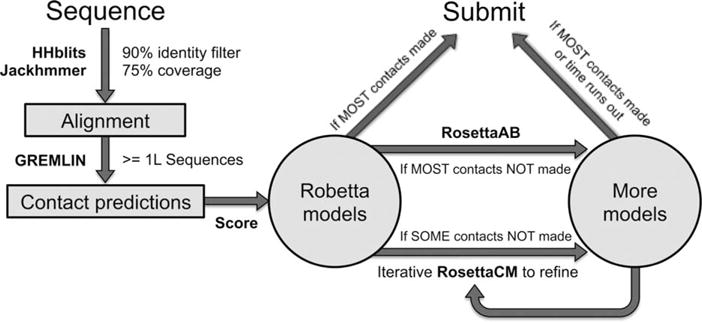
Human assisted structure prediction protocol.
RESULTS
As described in the methods, for CASP11 we sought to automate as much of the prediction process as possible, and for many targets the “human” group submitted Robetta server models. We first describe automated Robetta results and second, results for targets where additional human-guided calculations were carried out.
Robetta
A significant development in Robetta since last CASP was the use of predicted coevolution contacts to generate restraints for conformational sampling. To evaluate the contribution of the predicted contacts in our automated protocol, we reran the CASP FM targets for which they were used through the standard automated protocol without predicted contact information. As shown in Figure 4, nine out of 13 domains have improved GDT-TS29 quality scores in the original run with contact information compared to this control, and 5 improve by >6 GDT-TS units. It is clear that coevolutionary information can significantly improve modeling accuracy. Figure 5 shows a number of notable Robetta submitted models. T0790-D2 (domain 2; 130 residues alpha/beta; 2.9 L sequences) model 3 has 3.9 Å C_α_-RMSD over 92 residues to the native [Fig. 5(A)]. The modeled region (101–293) covering the assessor’s official domain (136–265) includes 63 extra residues due to an incorrect domain parse and missing coordinates at the C-terminus in the native structure. Despite modeling the incorrectly parsed domain, a significant portion of the native topology consisting of helices packed against an anti-parallel beta-sheet was correctly modeled. The most confident predicted contacts are within the anti-parallel beta-sheet [right structure in Fig. 5(A); contacts are represented as lines in the native structure]. The prediction accuracy is better by 13.1 GDT-TS units than the no contact control. Another highlight was the correctly predicted helical region in T0789-D1 model 2 (residues 6–81; 2.7 L sequences), which has 2.8 Å C_α_-RMSD over 71 residues to the native structure [Fig. 5(B)]. The predicted contacts are consistent with the overall topology [right structure in Fig. 5(B)], and the prediction accuracy is better than the control by 6.5 GDT-TS units.
Figure 4.
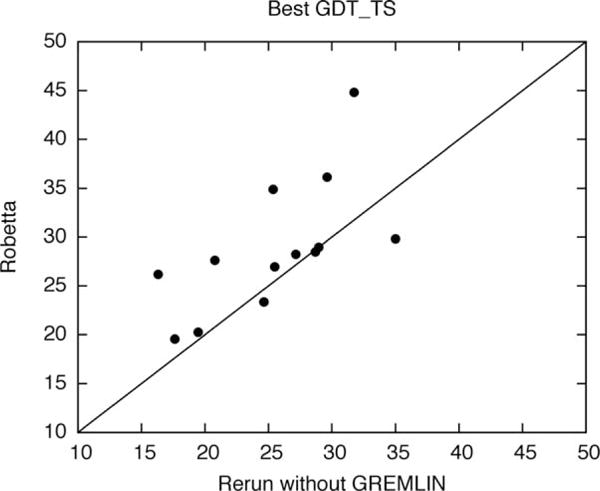
Comparison of Robetta results with (during CASP11) and without (post CASP control) GREMLIN predicted contacts.
Figure 5.
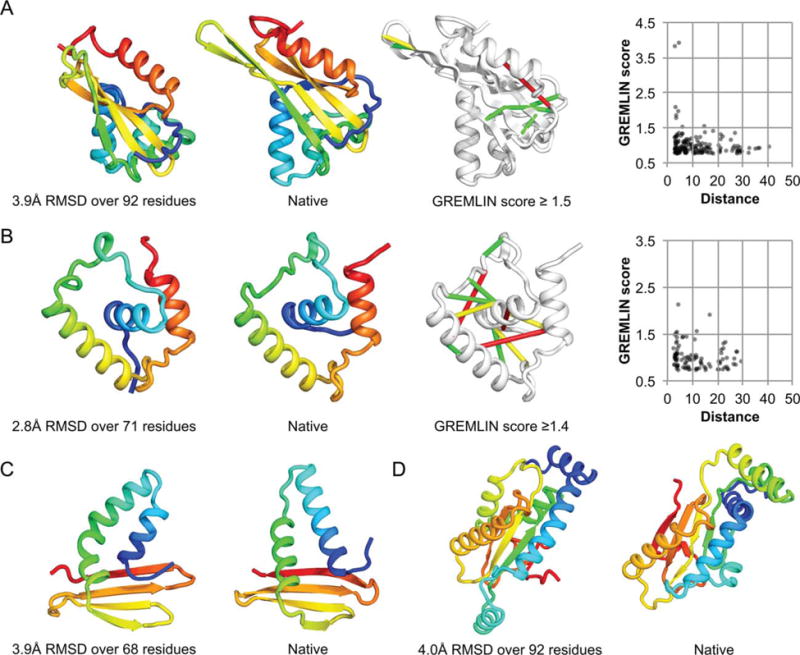
Robetta highlights. A) T0790-D2 (residues 136–265); B) T0789-D1 (residues 6–81); C) T0761-D2 (residues 202–285); D) T0767-D2 (residues 133– 312). All the GREMLIN contact predictions used in modeling are shown in the scatter plot; most of the high scoring residue pairs were indeed in contact in the native structure (_x_-axis). Lines connect residue pairs with GREMLIN score ≥1.5 (for cases with more sequences, more contacts are shown, with cutoff as indicated in figure); green, closest heavy atom distance <5 Å; yellow, distance closest <10 Å and red, >10 Å.
Robetta also made notable predictions for T0761-D2 [model 4; Fig. 5(C)] and T0767-D2 [model 3; Fig. 5(D)], which do not use coevolution information. Although Robetta incorrectly predicted the domain boundary for T0761-D2 (residues 150–285), it modeled a shorter region spanning residues 202–285 with 3.9 Å C_α_-RMSD over 68 residues to the native. RosettaCM was used for this prediction and improves upon the best input template (4fnq) in TM-score30 from 0.43 to 0.57. For T0767-D2 (residues 133–312), Robetta correctly assigns the domain boundary and models a 188 residue region (residues 131–318) with 4.0 Å C_α_-RMSD over 92 residues to the native. Although the quality of models is generally low for most FM domains due to their difficulty, length, and/or lack of homologous information, Robetta’s best models are top ranked among servers for a significant number of domains (13). Robetta ranked 3rd overall considering the best submitted models among humans and servers according to the official assessment for FM domains; two human groups (BAKER and Kiharalab) rank better.
Human intervention
For the four targets which had sufficient sequences for GREMLIN contact predictions, the human protocol yielded better models than the automated Robetta protocol (Table I). The primary differences between the human and the Robetta protocol were increased sampling in the first topology level search, and iterative recombination of the resulting models (this makes it possible for accurately modeled features in different models to be combined; the chance that all regions are correctly modeled in one fold level Monte Carlo trajectory is very small). For two of the four targets, the submitted models were the best for these targets in CASP11 by a considerable margin. These include T0806 (34.4 vs. 60.7 GDT-TS) and T0824 (41.2 vs. 55.3 GDT-TS) (Fig. 6A–B). Both targets had no detectable structural homolog in the PDB, but had a large number of homologous sequences for accurate coevolution contact prediction (T0806 had 5 L and T0824 had 3 L sequences). The second best model for both targets came from David Jones’ group, which also used coevolutionary information in their modeling protocol. The two other targets were trans-membrane domains and had less sequences (T0826-D1 with 2 L and T0836 with 1 L). For these targets, David Jones’ group submissions were better (T0826-D1: 37.7 vs. 29.1 and T0836: 44.1 vs. 39.0 GDT-TS). For T0836, we incorrectly excluded the first 30 residues as a signal peptide, which hurt performance (Fig. 6C). Our successful predictions and those of David Jones’ clearly show that coevolutionary contact information can significantly improve structure prediction for difficult targets provided there are sufficient numbers of sequences in their families.
Table I.
BAKER Human Targets
| Target | Parameters for MSAa | Best GDT-TS | ||
|---|---|---|---|---|
| Human | Robetta server | Others | ||
| T0806 | 4.60_L_, 1E-40, iter8, jackhmmer | 60.7 | 26.2 | 34.7 |
| T0824 | 2.46_L_, 1E-06, iter8, jackhmmer | 55.3 | 28.5 | 41.2 |
| T0826-D1 | 1.80_L_, 1E-40, iter8, jackhmmer | 29.1 | 23.4 | 37.7 |
| T0836 | 1.10_L_, 1E-06, iter4, hhblits | 39.0 | 27.0 | 44.1 |
| T0763 | N/A | 20.8 | 20.8 | 39.0 |
| T0785 | N/A | 25.7 | 25.7 | 30.1 |
| T0799-D1 | N/A | 16.0 | 19.9 | 21.6 |
| T0799-D2 | N/A | 23.5 | 27.9 | 31.9 |
Figure 6.
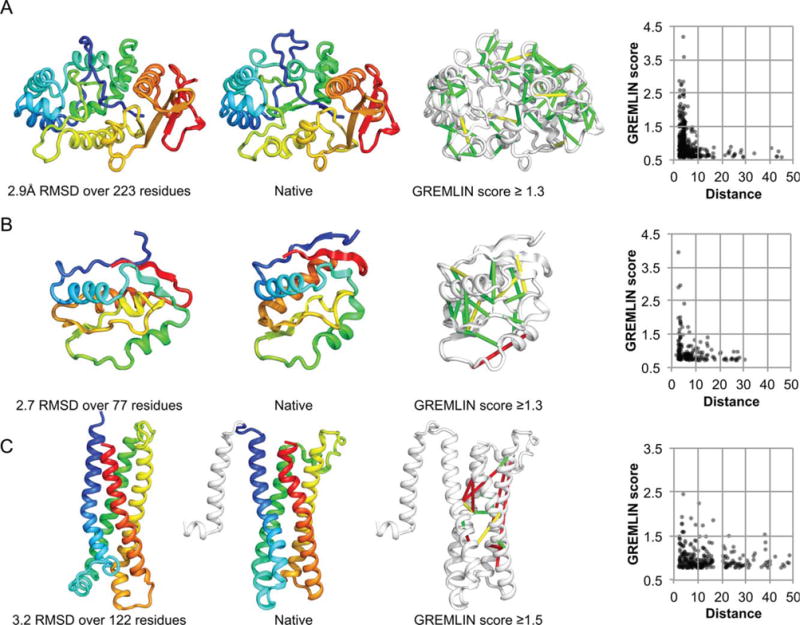
Human assisted prediction highlights. A) T0806; B) T0824; C) T0836.
The predictions made using oligomeric states inferred from detected templates (T0763, T0785, and T0799) were no better than the Robetta models, which is not surprising given that the templates were incorrect.
What went wrong
As in previous CASP experiments, correct domain parsing and classification remains challenging yet important for accurate modeling. The assessors parsed 19 out of 30 full-length FM targets into multiple domains accounting for 34 domains from multi-domain targets, and Robetta reasonably parsed only 5 domains. 2 out of 11 single domain targets were incorrectly parsed into multiple domains, and 4 multi-domain targets were modeled as single domains. It is not surprising that domain prediction was difficult for CASP11 FM targets due to the lack of detectable structure homologs and, for some targets, the number of homologous sequences was also limited. In addition to incorrect domain parsing, some targets were modeled using the Robetta CM protocol only (T0793-D5, T0824, and T0827), and would likely have improved using the de novo RosettaAB protocol.
The model ranking by Robetta was quite poor; only six of 45 FM domains had model 1 submissions that were the best of the five submitted models. To make the bookkeeping simple, Robetta during CASP11 always selected template-based RosettaCM structures for submitted model 1 and 4, and de novo structures for 2, 3, and 5 submitted models. This was clearly a very poor strategy for producing good model 1 predictions, since, in the difficult target regime, the de novo models were generally better and the templates for comparative modeling were quite distant from the actual structure.
Because the CASP11 Robetta ranking scheme was so poor, it can be improved (retrospectively) very easily. Model Quality Assessment (MQA) methods use features such as evolutionary information, residue environment, statistical and knowledge-based potentials, and various predicted structural features to score models and have been shown to improve model ranking in previous CASP experiments.31,32 Simply re-ranking the five submitted Robetta models using the MQA method, ProQ2,33 significantly improves model 1 accuracy for the FM domains (Fig. 7). Still better results could likely be obtained by re-ranking a larger set of models from both RosettaCM and RosettaAB. We will implement an improved model selection scheme in the public Robetta server and test it in the ongoing CAMEO34 structure prediction evaluation project.
Figure 7.
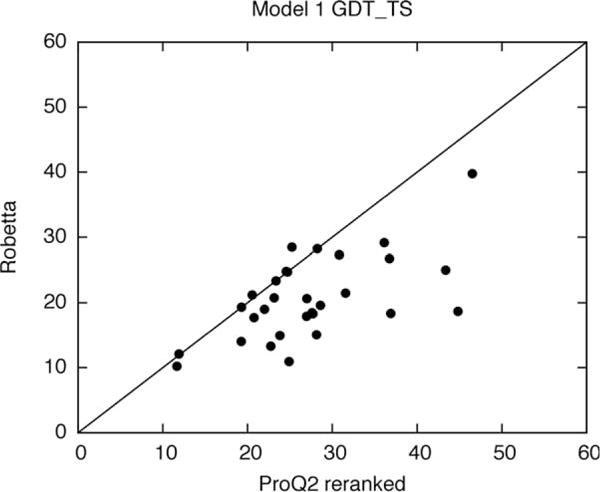
Re-ranking the five submitted models using a single-model MQA method, ProQ2, improves model 1 accuracy for FM domains.
Among our human submissions, three (T0806, T0824, and T0826) of the four targets that used GREMLIN restraints had regions with poor fragment quality due to incorrect SS predictions. It is likely that using GREMLIN restraints directly in fragment selection would yield improved models. We did not improve upon our server submissions for the three targets that were modeled using homo-oligomeric states due to the use of incorrect templates.
DISCUSSION
Compared to CASP10 and CASP9, CASP11 had substantially more FM domains and overall many challenging targets. Although most targets were difficult to model accurately, there were a few outstanding highlights owing to the use of accurately predicted coevolutionary contacts. The results suggest that provided there are enough homologous sequences available, large topologically complex proteins with no homologous structures in the PDB can be modeled with close to atomic-level accuracy using Rosetta with coevolutionary restraints; T0806 is the first blind prediction of a relatively large (258 residues) topologically complex protein structure predicted with such accuracy (<3.0 Å RMSD over 223 residues).
Our results and those of others clearly show that predicted contacts are useful at the conformational sampling stage. Predicted contacts may also be useful at other steps in the structure prediction process. Many FM domains were from multi-domain targets that were difficult to accurately parse and MSA-based domain prediction methods, with GREMLIN contacts could help to improve domain prediction and accurately model domain orientation. Predicted contacts likely can also improve fragment selection for Rosetta de novo folding calculations, help validate oligomeric states inferred from templates and model symmetric oligomers de novo (as illustrated by T0680 from CASP10, modeling with the correct oligomeric state in addition to sparse contact information can improve prediction accuracy).
The major limitation to methods such as GREMLIN is the dependence on the availability of homologous sequences, and indeed, half of the CASP11 FM targets had <1 _L_ homologous sequences. Although 5 _L_ sequences provide consistent high accuracy contacts,5 we reasoned that less accurate contacts from at least 1 _L_ would still be better than no predicted contacts for free-modeling targets. However, it was the targets with close to 5 _L_ sequences, T0806 and T0824, for which the most accurate models were produced. Further analysis on a benchmark of 13 free-modeling targets with >5 L sequences, showed similar results. 11 of the 13 targets converged and the C_α_-RMSD of the predicted structures to the experimentally determined structures over the converged residues were below 4.0 Å.35 This study also showed that the number of non-redundant (at 80% identity cutoff) sequences over the square-root of length is a better predictor of accuracy than sequences per length when there are <5 L sequences. The proteins that had many sequences typically are from families that are found in prokaryotic organisms. The amount of available sequence data continues to expand through sequencing efforts covering many different organisms, and there is also potential for using in vitro evolution involving “directed evolution” selection schemes36 and deep sequencing37 to identify coupled residue pairs. Large scale genome and metagenomic sequencing is having an unanticipated impact on protein structure modeling, enabling accurate protein structure modeling using coevolution-based predicted contacts. As more simple yet diverse eukaryotic organisms are sequenced, through projects such as the recent Tara Ocean expedition,38,39 it should become possible to accurately model not just prokaryotic protein families but also a large number of eukaryote specific protein families.
AVAILABILITY
Robetta and GREMLIN are available for non-commercial use at http://robetta.bakerlab.org and http://gremlin.bakerlab.org, respectively. The Rosetta software suite can be downloaded from http://www.rosettacommons.org.
Acknowledgments
The authors thank Keith Laidig and Darwin Alonso for developing the computational and network infrastructure and Rosetta@home participants for providing the computing resources necessary for this work. They also thank the CASP11 organizers, the structural biologists who generously provided targets, and Jinbo Xu (RaptorX), Johannes Söding (HHSearch), and Yaoqi Zhou (Sparks-X) for making their excellent software available and providing technical support. The work was supported by the NIH.
References
- 1.Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–531. doi: 10.1093/nar/gkh468. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simons KT, Ruczinski I, Kooperberg C, Fox BA, Bystroff C, Baker D. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins. 1999;34:82–95. doi: 10.1002/(sici)1097-0134(19990101)34:1<82::aid-prot7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 3.Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014;23:47–55. doi: 10.1002/pro.2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balakrishnan S, Kamisetty H, Carbonell JG, Lee SI, Langmead CJ. Learning generative models for protein fold families. Proteins. 2011;79:1061–1078. doi: 10.1002/prot.22934. [DOI] [PubMed] [Google Scholar]
- 5.Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence-and structure-rich era. Proc Natl Acad Sci USA. 2013;110:15674–15679. doi: 10.1073/pnas.1314045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ovchinnikov S, Kamisetty H, Baker D. Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. eLife. 2014;3:e02030. doi: 10.7554/eLife.02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 8.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, Faraggi E, Zhao H, Zhou Y. Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics. 2011;27:2076–2082. doi: 10.1093/bioinformatics/btr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. Template-based protein structure modeling using the RaptorX web server. Nat Protoc. 2012;7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, Baker D. High-resolution comparative modeling with RosettaCM. Structure. 2013;21:1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siew N, Elofsson A, Rychlewski L, Fischer D. MaxSub: an automated measure for the assessment of protein structure prediction quality. Bioinformatics. 2000;16:776–785. doi: 10.1093/bioinformatics/16.9.776. [DOI] [PubMed] [Google Scholar]
- 13.Kim DE, Chivian D, Malmstrom L, Baker D. Automated prediction of domain boundaries in CASP6 targets using Ginzu and Rosetta-DOM. Proteins. 2005;61(Suppl 7):193–200. doi: 10.1002/prot.20737. [DOI] [PubMed] [Google Scholar]
- 14.Doolittle RF. Of URFs and ORFs: a primer on how to analyze derived amino acid sequences. Mill Valley, California: University Science Books; 1986. [Google Scholar]
- 15.Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 16.Remmert M, Biegert A, Hauser A, Soding J. HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat Methods. 2012;9:173–175. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 17.Eddy SR. Accelerated profile HMM searches. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31:926–932. doi: 10.1093/bioinformatics/btu739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gront D, Kulp DW, Vernon RM, Strauss CE, Baker D. Generalized fragment picking in Rosetta: design, protocols and applications. PLoS One. 2011;6:e23294. doi: 10.1371/journal.pone.0023294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou H, Zhou Y. Fold recognition by combining sequence profiles derived from evolution and from depth-dependent structural alignment of fragments. Proteins. 2005;58:321–328. doi: 10.1002/prot.20308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faraggi E, Xue B, Zhou Y. Improving the prediction accuracy of residue solvent accessibility and real-value backbone torsion angles of proteins by guided-learning through a two-layer neural network. Proteins. 2009;74:847–856. doi: 10.1002/prot.22193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 23.Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, Kim D, Kellogg E, DiMaio F, Lange O, Kinch L, Sheffler W, Kim BH, Das R, Grishin NV, Baker D. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins. 2009;77(Suppl 9):89–99. doi: 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson J, Baker D. Incorporation of evolutionary information into Rosetta comparative modeling. Proteins. 2011;79:2380–2388. doi: 10.1002/prot.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson DP. IEEE/ACM International Workshop on Grid Computing. Pittsburgh, PA: 2004. BOINC: a system for public-resource computing and storage; pp. 4–10. [Google Scholar]
- 26.Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–2139. doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- 27.Wollacott AM, Zanghellini A, Murphy P, Baker D. Prediction of structures of multidomain proteins from structures of the individual domains. Protein Sci. 2007;16:165–175. doi: 10.1110/ps.062270707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou H, Skolnick J. GOAP: a generalized orientation-dependent, all-atom statistical potential for protein structure prediction. Biophys J. 2011;101:2043–2052. doi: 10.1016/j.bpj.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zemla A. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J, Zhang Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 2010;26:889–895. doi: 10.1093/bioinformatics/btq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kryshtafovych A, Fidelis K, Tramontano A. Evaluation of model quality predictions in CASP9. Proteins. 2011;79(Suppl10):91–106. doi: 10.1002/prot.23180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kryshtafovych A, Barbato A, Fidelis K, Monastyrskyy B, Schwede T, Tramontano A. Assessment of the assessment: evaluation of the model quality estimates in CASP10. Proteins. 2014;82(Suppl2):112–126. doi: 10.1002/prot.24347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ray A, Lindahl E, Wallner B. Improved model quality assessment using ProQ2. BMC Bioinformatics. 2012;13:224. doi: 10.1186/1471-2105-13-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haas J, Roth S, Arnold K, Kiefer F, Schmidt T, Bordoli L, Schwede T. The protein model portal–a comprehensive resource for protein structure and model information. Database. 2013;2013:bat031. doi: 10.1093/database/bat031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ovchinnikov S, Kinch L, Park H, Liao Y, Pei J, Kim DE, Kamisetty H, Grishin N, Baker D. Large-scale determination of previously unsolved protein structures using evolutionary information. eLife. 2015;4:e09248. doi: 10.7554/eLife.09248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Romero PA, Arnold FH. Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol. 2009;10:866–876. doi: 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- 38.Bork P, Bowler C, de Vargas C, Gorsky G, Karsenti E, Wincker P. Tara Oceans. Tara Oceans studies plankton at planetary scale. Introduction. Science. 2015;348:873. doi: 10.1126/science.aac5605. [DOI] [PubMed] [Google Scholar]
- 39.Sunagawa S, Karsenti E, Bowler C, Bork P. Computational ecosystems biology in Tara Oceans: translating data into knowledge. Mol Syst Biol. 2015;11:809. doi: 10.15252/msb.20156272. [DOI] [PMC free article] [PubMed] [Google Scholar]