AKT Inhibition in Solid Tumors With AKT1 Mutations (original) (raw)
Abstract
Purpose
AKT1 E17K mutations are oncogenic and occur in many cancers at a low prevalence. We performed a multihistology basket study of AZD5363, an ATP-competitive pan-AKT kinase inhibitor, to determine the preliminary activity of AKT inhibition in _AKT_-mutant cancers.
Patients and Methods
Fifty-eight patients with advanced solid tumors were treated. The primary end point was safety; secondary end points were progression-free survival (PFS) and response according to Response Evaluation Criteria in Solid Tumors (RECIST). Tumor biopsies and plasma cell-free DNA (cfDNA) were collected in the majority of patients to identify predictive biomarkers of response.
Results
In patients with AKT1 E17K–mutant tumors (n = 52) and a median of five lines of prior therapy, the median PFS was 5.5 months (95% CI, 2.9 to 6.9 months), 6.6 months (95% CI, 1.5 to 8.3 months), and 4.2 months (95% CI, 2.1 to 12.8 months) in patients with estrogen receptor–positive breast, gynecologic, and other solid tumors, respectively. In an exploratory biomarker analysis, imbalance of the AKT1 E17K–mutant allele, most frequently caused by copy-neutral loss-of-heterozygosity targeting the wild-type allele, was associated with longer PFS (hazard ratio [HR], 0.41; P = .04), as was the presence of coincident PI3K pathway hotspot mutations (HR, 0.21; P = .045). Persistent declines in AKT1 E17K in cfDNA were associated with improved PFS (HR, 0.18; P = .004) and response (P = .025). Responses were not restricted to patients with detectable AKT1 E17K in pretreatment cfDNA. The most common grade ≥ 3 adverse events were hyperglycemia (24%), diarrhea (17%), and rash (15.5%).
Conclusion
This study provides the first clinical data that AKT1 E17K is a therapeutic target in human cancer. The genomic context of the AKT1 E17K mutation further conditioned response to AZD5363.
INTRODUCTION
Phosphoinositide 3-kinase (PI3K)/AKT is one of the most frequently activated pathways in cancer.1,2 Activation can occur through mutation of multiple signaling nodes including PTEN, PIK3R1, PIK3CA, AKT, and mTOR.3-5 Clinical development of drugs targeting this pathway has focused primarily on inhibitors of PI3K isoforms and mammalian target of rapamycin (mTOR).6-8 The AKT kinase family includes three structurally related serine-threonine kinases that serve as critical downstream effectors of PI3K signaling. Large-scale genomic profiling of human cancers has identified gain-of-function mutations in AKT1 in a broad range of tumor types, with AKT1 E17K being, by far, the most frequent hotspot.9-14 This mutation promotes pathologic localization of AKT1 to the plasma membrane, thereby stimulating constitutive downstream signaling.15
AKT inhibitors have been in clinical testing for several years but have not been specifically evaluated in _AKT1_-mutant tumors.16 Testing these inhibitors in _AKT1_-mutant patients using traditional clinical trial designs is challenging because, unlike many other oncogenes, AKT1 E17K is infrequent in all individual tumor lineages. To determine whether _AKT1_-mutant cancers are sensitive to direct AKT inhibition and whether tumor lineage influences drug sensitivity, we performed a multicohort basket study of the orally administered pan-AKT inhibitor AZD536317 in patients with _AKT1_-mutant solid tumors. Tumor biopsies and analyses of tumor-derived DNA in plasma were performed to identify genomic determinants of response and to guide future combination studies.
PATIENTS AND METHODS
Study Oversight
The study (ClinicalTrials.gov identifier: NCT01226316) was designed by AstraZeneca (Cambridge, United Kingdom) with the principal investigators and conducted in accordance with the provision of the Declaration of Helsinki and Good Clinical Practice guidelines. Institutional review boards at each center approved the protocol. Funding was provided by AstraZeneca.
Patients
Eligible patients had histologically confirmed advanced solid tumors refractory to standard therapies, no prior exposure to catalytic AKT inhibitors, and tumors harboring AKT1 mutations but no known concurrent RAS/RAF mutations as determined by local tumor testing. Complete eligibility criteria are available in the Data Supplement. Written informed consent was obtained for all participants.
Study Design, Treatment, and End Points
This was a multicohort basket study of patients with solid tumors harboring AKT1 mutations. The phase I component of this study has been presented previously and defined the safety, optimal dose, and schedule of AZD5363 and its limited efficacy in patients with _PIK3CA_-mutant estrogen receptor (ER) –positive or human epidermal growth factor receptor 2–positive breast cancer (one of 26 partial responses) and _PIK3CA_-mutant gynecologic cancers (two of 25 partial responses).18,19 Here, patients were enrolled onto part D of the study into one of the following three cohorts: ER-positive breast cancer, gynecologic cancers, and all other solid tumors. Patients were treated on a 21-day cycle of AZD5363 480 mg twice daily for 4 days followed by 3 days off, repeated weekly. The primary end point was safety; secondary end points included investigator-assessed response according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 and progression-free survival (PFS). Patient-level clinical data are available in the Data Supplement.
Assessments
Disease assessments with computed tomography or magnetic resonance imaging were performed at baseline, every 6 weeks for 6 months, and then every 12 weeks until disease progression, death, or withdrawal. Adverse events were graded by the investigator according to the Common Terminology Criteria for Adverse Events (version 4.0) until day 28 after discontinuation of study treatment.
Biomarker Studies
Tumor tissue samples and tumor-derived cell-free DNA (cfDNA) in plasma were collected for retrospective exploratory biomarker analyses. Next-generation sequencing was performed using both targeted and whole-exome sequencing on pretreatment DNA from formalin-fixed paraffin embedded tumor and matched blood specimens (Data Supplement).20,21 Droplet digital polymerase chain reaction analysis using an allele-specific assay was performed on cfDNA from pretreatment and longitudinally collected plasma samples. Complete sequencing and data analysis methods are described in the Data Supplement. Given small sample sizes, biomarker analyses were not preplanned. Individual associations among genomic changes and response were assessed using either the Fisher’s exact test or χ2 test (where appropriate).
Statistical Analysis
Analysis was initially planned after enrollment of 20 patients to each cohort; however, as a result of slow enrollment onto the gynecologic cohort because of a low incidence of AKT1 mutations in this population, accrual was halted after 58 patients (ER-positive breast, n = 20; gynecologic, n = 18; other, n = 20). All patients receiving at least one dose of AZD5363 (n = 58) were analyzed. One patient with an AKT1 wild-type tumor was mistakenly enrolled (not eligible) and is annotated as “Not Detected” in Table 1. As exploratory expansion cohorts within a phase I study, no formal hypotheses for efficacy were tested and no early stopping rules for futility were prespecified. PFS was estimated using the Kaplan-Meier method. Patients who missed two or more response assessments after discontinuing study drug for reasons other than documented progression were censored at the time of the latest evaluable RECIST assessment.
Table 1.
Patient Demographic and Baseline Disease Characteristics

RESULTS
Patients, Efficacy, and Safety
Fifty-eight patients were treated (E17K, n = 52; non-E17K, n = 5; AKT1 mutation not detected, n = 1; Table 1). Patients were heavily pretreated (median of five prior regimens). In AKT1 E17K–mutant patients, confirmed partial responses were observed in ER-positive breast and endometrial cancers (n = 4 and n = 2, respectively), as well as cervical cancer, triple-negative breast cancer, and lung adenocarcinoma (n = 1 each; Fig 1). Additional unconfirmed partial responses occurred in ER-positive breast cancer (n = 2), triple-negative breast cancer (n = 1), and anal adenocarcinoma (n = 1). In AKT1 non-E17K patients, a tumor regression qualifying as RECIST stable disease was observed in one patient with AKT1 Q79K mutations (ovarian cancer), lasting 14 months. Median PFS in the AKT1 E17K–mutant ER-positive breast, gynecologic, and other solid cancer cohorts was 5.5 months (95% CI, 2.9 to 6.9 months), 6.6 months (95% CI, 1.5 to 8.3 months), and 4.2 months (95% CI, 2.1 to 12.8 months), respectively. There was no apparent relationship between tumor type and likelihood of response.
Fig 1.

Integrated treatment outcome and genomics of AKT1 E17K–mutant solid tumors. Data are shown for 58 patients and grouped by membership in the AKT1 E17K–mutant breast, gynecologic, and other solid tumor cohorts followed by patients with non-E17K AKT1 mutations. From top to bottom: Best change from baseline in the target lesion diameter according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (gradient arrows reflect not evaluable); duration of therapy (days); and genomic annotation from pretreatment tumor tissue or cell-free DNA (cfDNA) sequencing. All genes in the mutational heat map, including AKT1, reflect results from pretreatment centrally determined genomic data rather than local testing. Twelve patients enrolled lacked genomic data (track: Genomic data). Individual mutations are shown as annotated in the accompanying legend, and subclonality was determined as described in the Data Supplement. Individual annotation tracks annotate the cancer type, best response, the detection of AKT1 E17K by droplet digital polymerase chain reaction (ddPCR) analysis in baseline plasma samples, and the existence of pretreatment genomic data from either tumor tissue or cfDNA sequencing. ER, estrogen receptor; NSCLC, non–small-cell lung cancer; PR, partial response; TNBC, triple-negative breast cancer.
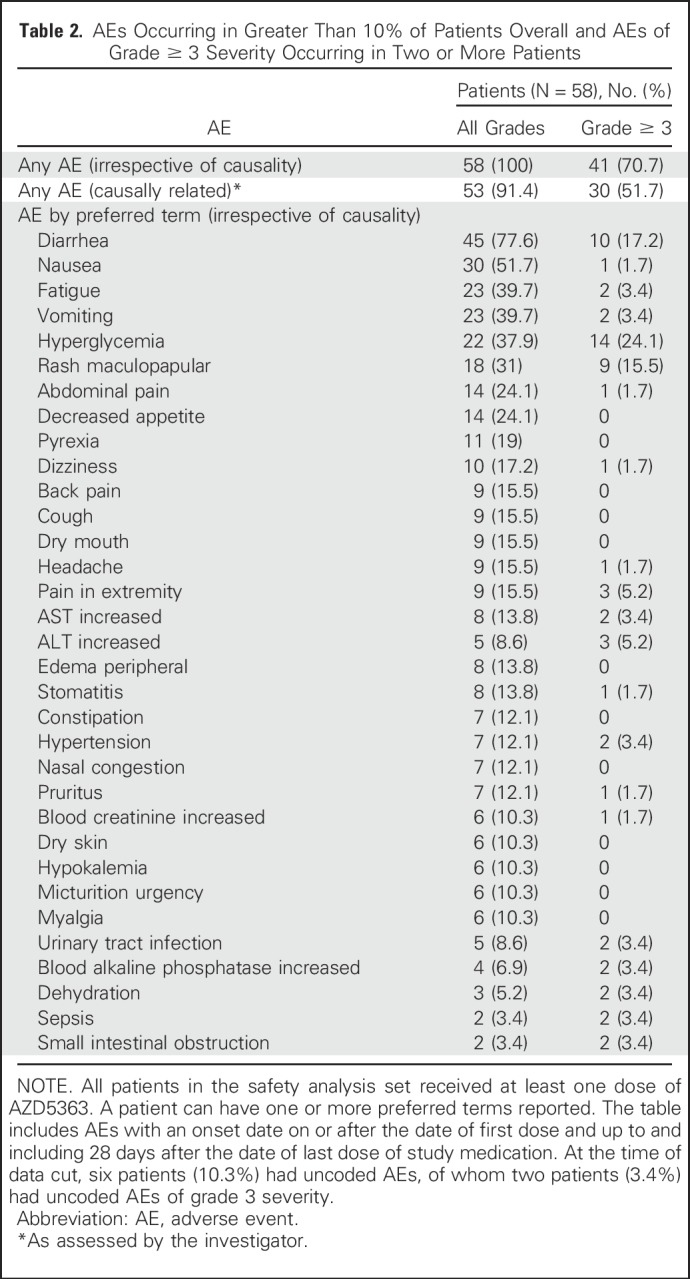
The most common grade ≥ 3 adverse events were hyperglycemia (24%), diarrhea (17%), and maculopapular rash (15.5%; Table 2). Overall, 34% of patients required a dose reduction, with diarrhea, maculopapular rash, and hyperglycemia being the most common indications. AZD5363 was permanently discontinued in 12% of patients as a result of adverse events. The median and mean administered total daily doses were 943.7 mg and 871.4 mg, respectively. Drug-related serious adverse events occurred in 15.5% of patients and were consistent with the overall adverse effect profile of AZD5363 (Data Supplement).
Table 2.
AEs Occurring in Greater Than 10% of Patients Overall and AEs of Grade ≥ 3 Severity Occurring in Two or More Patients
Noninvasive Monitoring of Circulating Biomarker in cfDNA
Because patients were enrolled based on local archival tumor sequencing, we sought to determine the presence of AKT1 E17K in cfDNA from plasma collected at the time of enrollment. Notably, AKT1 E17K was detected in pretreatment plasma by droplet digital polymerase chain reaction analysis in only 81.4% of patients (35 of 43 patients) with evaluable samples (Fig 1). Among patients with undetectable AKT1 E17K in cfDNA (n = 8), archival tumor was available for central sequencing in six patients and confirmed the presence of the E17K mutation in five patients. Two of these patients had partial responses, and a third patient had a durable tumor regression lasting more than 8 months. Broader analysis of plasma from these three _AKT1_-mutant patients using a capture-based cfDNA assay also identified no tumor-derived mutations. The only patient in whom AKT1 E17K could not be confirmed in either cfDNA or in archival tumor experienced rapid disease progression.
To determine whether tumor-derived cfDNA could be used as an early surrogate of drug response and to explore the dynamics of the circulating biomarker under the selective pressure of AKT inhibition, longitudinal plasma samples were tested in 23 patients (Fig 2A). A decrease in AKT1 E17K–mutant allele fraction of ≥ 50% from baseline during cycle 1 was observed in 95.5% of patients (22 of 23 patients) but did not correlate with outcome (Fig 2B). Conversely, persistent decreases maintained into cycle 2 were associated with longer PFS when compared with patients in whom cfDNA decreases were not achieved or did not persist (median PFS, 5.6 v 2.6 months, respectively; hazard ratio [HR], 0.18; P = .004; Fig 2C). Persistent clearance of circulating AKT1 E17K (>21 days) correlated with objective response, with all five patients meeting this criteria achieving partial responses lasting ≥18 weeks (P = .025). Progression by cfDNA, defined as an increase in the circulating AKT1 E17K–mutant allele fraction of ≥ 50% greater than nadir, preceded radiographic progression in all but one patient by a median of 42 days (95% CI, 31 to 68 days; Fig 2D). Broader next-generation sequencing of pretreatment cfDNA also captured the complete mutational profile of genetically heterogeneous individual tumor sites (Fig 2E).
Fig 2.

Noninvasive monitoring of treatment response in cell-free DNA (cfDNA). (A) Imaging at baseline and 6 weeks after treatment initiation indicates a response (in red) to AZD5363 in an E17K-mutant, estrogen receptor (ER) –positive, human epidermal growth factor receptor 2 (HER2) –negative breast cancer that was confirmed molecularly with an initial decrease in, and persistently low levels of, the AKT1 E17K burden in cfDNA. (B) Tumor burden, indicated by a greater than 50% decrease in AKT1 E17K–mutant allele fraction in circulating cfDNA, was evident in all but one patient (95.5%) by day 11 of treatment cycle 1 but did not correlate with outcome as measured by a duration of therapy of greater than 12 weeks (left). Data are shown for 23 patients with longitudinal cfDNA samples collected throughout treatment and who were positive for AKT1 E17K by droplet digital polymerase chain reaction analysis at baseline. (C) A decline of circulating AKT1 E17K of greater than 50% at day 21 compared with before treatment was correlated with response to AKT inhibition (hazard ratio [HR], 0.18; P = .004). (D) In evaluable patients (Data Supplement), cfDNA progression (increase in AKT1 E17K allele fraction of > 50% above nadir) preceded radiographic progression by a median of 42 days (range, 0 to 113 days) Each line is a patient, all cfDNA collection time points are shown normalized to the date of Response Evaluation Criteria in Solid Tumors (RECIST) progression, and the arrowheads indicate the start of therapy. Filled circles correspond to the time point of cfDNA progression, as defined earlier, and the vertical line indicates median lead time of cfDNA progression relative to radiologic progression (shaded area is the 95% CI of lead times). The bottom-most patient had a radiologic progression without AKT1 E17K increase in cfDNA. (E) Broader next-generation sequencing of pretreatment cfDNA in one patient captured the complete mutational profile of genetically heterogeneous individual tumor sites. NS, not significant; VUS, variant of unknown significance.
Genomic Correlates of Response to AKT Inhibition
To determine whether the genomic configuration of AKT1 (number of mutant and wild-type copies) or coincident tumor mutations influenced AZD5363 response, we performed whole-exome or targeted sequencing of archival and fresh pretreatment tumors in a subset of patients. In the 37 patients with adequate material for this analysis, 57% (21 of 37 patients) exhibited allelic imbalance of the AKT1 E17K mutation. Here, the frequency of the E17K allele was higher than expected for a heterozygous oncogenic mutation and higher than the allele frequency of other clonal somatic mutations in the corresponding tumors (Fig 3A). This finding could not be explained by focal amplification of the E17K allele, which was present in only two tumors. To determine the etiology of this allelic imbalance, we performed allele-specific copy number analysis of the sequencing data, which revealed that 48% of patients (10 of 21 patients) had copy-neutral loss of heterozygosity (CN-LOH). This duplication of the mutant AKT1 allele with concomitant loss of the remaining wild-type copy ultimately resulted in two mutant AKT1 E17K copies and no wild-type copies (Fig 3B; Data Supplement). CN-LOH occurred in molecular time shortly after acquisition of the E17K mutation and, in some patients, was followed by genomic gains of the locus. Notably, patients whose tumors exhibited allelic imbalance of AKT1 E17K had a longer PFS than those without it (median PFS, 8.2 v 4.1 months, respectively; HR, 0.41; P = .04; Fig 3C). In the study cohort, AKT1 E17K allelic imbalance was associated with tumor lineage, arising more commonly in breast and gynecologic cancers compared with all other cancers enrolled (90% v 10% respectively; Fig 1).
Fig 3.

Clonality of the sensitizing biomarker. (A) For 37 patients with sufficient baseline sequencing data, AKT1 E17K–mutant allele frequency is shown (filled circle), as is the median allele frequency of all somatic mutations detected in each patient (horizontal line; vertical line is the median absolute deviation) from pretreatment tumor tissue or cell-free DNA (cfDNA) sequencing. Patients with focal amplification of AKT1 E17K are indicated by upward-pointing triangles, whereas patients possessing subclonal AKT1 E17K are indicated by downward-pointing triangles. Patients are grouped as having a heterozygous AKT1 E17K (left) or possessing high mutant allele fraction (right). (B) Schematic of the acquisition of AKT1 E17K–mutant (thick line) allele imbalance in this study cohort, beginning from a heterozygous mutation in a diploid genome and chromosome 14 (leftmost; maternal and paternal chromosomes are indicated). Allelic imbalance is in the form of copy-neutral loss of heterozygosity that duplicates the mutant allele (top) and can be followed by other serial genetic changes including genomic gains and whole-genome duplication (WGD) or either heterozygous loss of the wild-type copy (bottom left) or whole chromosome or more focal gains of the mutant allele (Data Supplement). (C) AKT1 E17K–mutant allele imbalance by any of the mechanisms described in panel B is associated with improved progression-free survival (PFS) in response to AKT inhibition (median PFS, 8.2 v 4.1 months in patients whose tumors exhibited allelic imbalance of AKT1 E17K v those without it, respectively; hazard ratio [HR], 0.41; P = .04). (D) A patient with an ovarian granulosa cell tumor received AZD5363 for 8 months and achieved a best response of 24% tumor regression (right pelvic tumor regression shown), a notable response that was far greater than what would have been predicted based on the frequency of the sensitizing AKT1 mutation. (E) Sequencing of eight metastatic sites sampled before therapy revealed that whereas the earliest occurring lesions were clonal (FOXL2 and TERT), the AKT1 mutation was variably subclonal across the lesions and was present at highest cellular fraction (67%, subclonal) in the right pelvic tumor that achieved the best response to AZD5363 therapy (labeled E in panel D). (F) The presence of coincident activating mutations in either up- or downstream effectors of PI3K/mTOR signaling in AKT1 E17K–mutant tumors was associated with improved PFS (median PFS, not reached v 4.3 months without such lesions; HR, 0.21; P = .045).
We also explored how clonality of the AKT1 E17K mutation within the tumor site sequenced influenced AZD5363 response. In total, 92% of patients (34 of 37 patients) had clonal (present in all tumor cells) AKT1 mutations (Fig 3A). Two of the three patients with subclonal AKT1 E17K mutations had rapid disease progression. The third patient, with ovarian granulosa cell cancer, had a mixed response, with an overall tumor regression of 24% lasting 253 days (Fig 3D). To understand the basis of this durable tumor regression despite the presence of a subclonal AKT1 E17K mutation, we sequenced nine metastatic sites sampled before the initiation of AZD5363 treatment (Fig 3E) and found that although the AKT1 mutation was subclonal across the lesions, the resected right pelvic tumor that subsequently recurred and achieved the best response (−42.5%) had the highest cellular fraction (67% of cancer cells) of the AKT1 E17K mutation (Fig 3E). These results suggest that later acquisition of AKT1 E17K driver mutations may not entirely preclude response to AZD5363.
Leveraging the broader-based sequencing we performed here, we investigated whether particular comutations were associated with intrinsic sensitivity or resistance to AKT inhibition. Notably, five patients had coincident activating mutations in either up- or downstream effectors of PI3K/mTOR signaling. The presence of coincident PI3K pathway alterations was associated with improved PFS compared with patients without these alterations (median PFS, not reached v 4.3 months, respectively; HR, 0.21; P = .045). Importantly, concurrently mutated genes that would be expected to activate parallel signaling pathways did not necessarily preclude response to AZD5363. Two of five patients with loss-of-function NF1 mutations (cervical and breast cancer) achieved durable partial responses, one of whom also had a subclonal FGFR3 S249C hotspot mutation. In a patient with nonresponding colorectal cancer, a subclonal KRAS A146T hotspot mutation not detected by local tumor profiling was identified in pretreatment cfDNA, a mutation that preclinically is associated with resistance to AZD5363.17 Mutational hotspots in the ligand-binding domain of ESR1, which are associated with acquired resistance to endocrine therapy and poor prognosis,22 were identified in metastatic tumor tissue or cfDNA in seven (35%) of 20 patients with ER-positive breast cancer and were associated with a shorter median PFS compared with patients without these mutational hotspots (P = .004; Fig 1; Data Supplement).
DISCUSSION
To our knowledge, this study provides the first robust clinical evidence that AKT1 E17K is a targetable oncogene in human cancer. Treatment with AZD5363 yielded durable responses and tumor regressions across a variety of tumor types harboring the mutation including breast (ER positive and triple negative), endometrial, cervical, and lung cancers. The degree of activity observed here is greater than that seen with AZD5363 in PIK3CA mutants, even among similar patient populations with breast and gynecologic cancer, further emphasizing that AKT1 E17K mutants are a distinct genomic subpopulation and more broadly suggesting that different genomic mechanisms of activating the PI3K pathway may be associated with unique pharmacologic dependencies.
The breadth and depth of pretreatment sequencing data available allowed us to perform exploratory analyses to determine how different facets of these patients’ tumors further conditioned response to AKT inhibition. We unexpectedly found that tumors harboring AKT1 E17K mutations frequently exhibit selection against the remaining wild-type allele, most often as a result of duplication of the mutant allele via CN-LOH, resulting in allelic imbalance. This genomic configuration, surprising for an oncogene, seems to be lineage specific because it was enriched in AKT1 E17K–mutant breast and endometrial cancers but not observed in other tumor lineages (Data Supplement). This AKT1 E17K allelic imbalance was associated with a statistically and clinically significant improvement in PFS. Although this finding requires prospective confirmation in a larger patient cohort, it suggests that classifying genomic biomarkers as simply present or absent may overlook additional informative factors, such as genomic configuration, that are relevant to patient selection and lineage dependence. Similarly, we found that although two patients with tumors bearing subclonal AKT1 mutations did not respond to AZD5363, one patient with granulosa cell cancer with extensive intratumoral heterogeneity had durable tumor regression at disease sites harboring the highest cellular fraction of AKT1 E17K. This finding suggests that limiting targeted therapy to patients only with clonal AKT1 mutations may not be entirely appropriate.
Surprisingly, we identified five patients whose tumors harbored activating mutations in other effectors of PI3K/mTOR signaling in addition to AKT1 E17K, a finding we confirmed in 12.5% of _AKT1_-mutant patients from an independent genomic data set (Data Supplement). Again, the statistically and clinically significant longer PFS observed in these dual-mutant patients argues that rather than implying functional redundancy, coincident mutations in effectors of the same pathway may result in distinct signaling phenotypes with important therapeutic implications. Further biologic investigation of whether such coincident drivers further sensitize tumors to PI3K pathway inhibition is warranted.
The analysis of cfDNA within the context of this early-phase study also yielded several findings with broad implications. Importantly, we observed responses in patients with undetectable AKT1 E17K in pretreatment cfDNA. Our findings emphasize how low tumor burden and insufficient shedding of cfDNA into plasma can impact detection of actionable biomarkers in plasma and can have downstream implications for genomic screening strategies that rely on this technology for patient selection. We also demonstrate how cfDNA can be used to detect intratumoral heterogeneity unappreciated by single-site tissue biopsies and how serial monitoring cfDNA for AKT1 mutations can serve as a surrogate for response and progression.
Although E17K is the most common AKT1 mutation and was the focus of this study, other activating mutations in AKT1, AKT2, and AKT3 have been identified.23 Among these, AKT1 Q79K is the second most recurrent hotspot mutation after E17K (Data Supplement). Of the patients with non-E17K mutations in this study, only those with AKT1 Q79K demonstrated tumor regressions. Looking beyond AKT1 E17K mutations to other mutant alleles in all three AKT isoforms might therefore broaden the population of _AKT_-mutant patients who could benefit from AKT inhibitors.
Despite the promising PFS achieved with AZD5363 in patients with heavily pretreated AKT1 E17K–mutant breast and gynecologic cancers, the observed response rate was lower than with therapies targeting EGFR, ALK, ROS1, and BRAF.24-26 Realizing the full potential of AZD5363 in _AKT1_-mutant cancers may require drug combinations. Overall, the strongest signal of activity was observed in ER-positive breast cancer as well as endometrial cancers of the subtype associated with sensitivity to antiestrogens. Studies combining antiestrogen therapy with AKT inhibition in ER-positive, _AKT1-_mutant cancers are ongoing.
In summary, we demonstrate that mutant AKT1 is a rational therapeutic target for AZD5363 in diverse cancers. Unlike prior basket studies that sought to expand the indication of a US Food and Drug Administration–approved drug previously studied extensively using traditional trial designs,27 we show that a drug can be successfully studied in a mutation-specific context even when the mutation is consistently rare across all populations. By incorporating comprehensive tissue- and plasma-based correlative studies, we elucidate the multifaceted genomic basis of response in a manner that facilitates simultaneous translational genomic discoveries and clinical hypothesis validation to inform future studies.
ACKNOWLEDGMENT
We thank patients and their families for participating in this study and the members of the Taylor Laboratory and Marie-Josée and Henry R. Kravis Center for Molecular Oncology for discussions and support. AZD5363 was discovered by AstraZeneca subsequent to a collaboration with Astex Therapeutics (and its collaboration with the Institute of Cancer Research and Cancer Research Technology Limited).
Footnotes
Supported by grants from the National Institutes of Health (Grants No. P30 CA008748, P50 CA092629, R01 CA204749, and R01 CA207244), Cycle for Survival, Conquer Cancer Foundation of ASCO, and AstraZeneca.
Presented in part at the 2015 National Cancer Institute–European Organisation for Research and Treatment of Cancer–American Association for Cancer Research International Conference on Molecular Targets and Cancer Therapeutics, Boston, MA, November 5-9, 2015, and the 52nd Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, June 3-7, 2016.
AUTHOR CONTRIBUTIONS
Conception and design: David M. Hyman, Helen Ambrose, Andrew Foxley, Justin P.O. Lindemann, Martin Pass, David B. Solit, Udai Banerji, José Baselga, Barry S. Taylor
Financial support: David M. Hyman, Lillian M. Smyth, Sarat Chandarlapaty, David B. Solit, José Baselga, Barry S. Taylor
Administrative support: David M. Hyman, Helen Ambrose, Martin Pass, David B. Solit, José Baselga, Barry S. Taylor
Provision of study materials or patients: David M. Hyman, Lillian M. Smyth, Shannon N. Westin, Philippe L. Bedard, Emma J. Dean, Hideaki Bando, Anthony B. El-Khoueiry, José A. Perez-Fidalgo, Alain Mita, Jan H.M. Schellens, Tara E. Soumerai, José Baselga
Collection and assembly of data: David M. Hyman, Lillian M. Smyth, Mark T.A. Donoghue, Shannon N. Westin, Philippe L. Bedard, Emma J. Dean, Hideaki Bando, Anthony B. El-Khoueiry, José A. Perez-Fidalgo, Alain Mita, Jan H.M. Schellens, Matthew T. Chang, Jonathan B. Reichel, Nancy Bouvier, S. Duygu Selcuklu, Jean Torrisi, Joseph P. Erinjeri, Helen Ambrose, J. Carl Barrett, Justin P.O. Lindemann, Robert McEwen, Michael F. Berger, Sarat Chandarlapaty, David B. Solit, Udai Banerji, Barry S. Taylor
Data analysis and interpretation: David M. Hyman, Lillian M. Smyth, Mark T.A. Donoghue, Philippe L. Bedard, Alain Mita, Matthew T. Chang, Jonathan B. Reichel, Tara E. Soumerai, Helen Ambrose, J. Carl Barrett, Brian Dougherty, Justin P.O. Lindemann, Robert McEwen, Gaia Schiavon, Michael F. Berger, Sarat Chandarlapaty, David B. Solit, Udai Banerji, Barry S. Taylor
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
AKT Inhibition in Solid Tumors With AKT1 Mutations
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
David M. Hyman
Consulting or Advisory Role: Atara Biotherapeutics, Chugai Pharma, CytomX Therapeutics, Boehringer Ingelheim
Research Funding: AstraZeneca, Puma Biotechnology, Loxo
Lillian M. Smyth
Consulting or Advisory Role: AstraZeneca, Genentech
Research Funding: AstraZeneca (Inst), Genentech (Inst)
Mark T.A. Donoghue
No relationship to disclose
Shannon N. Westin
Consulting or Advisory Role: Amgen (I), Spectrum Pharmaceuticals (I), Novartis (I), Roche, AstraZeneca, Ovation Sciences, Medivation, Genentech, Vermillion, Casdin Capital, Medscape, Clovis Oncology
Research Funding: AstraZeneca, Novartis, Merck, Biomarin, GlaxoSmithKline, ProNAi (I), Karyopharm Therapeutics (I), Spectrum Pharmaceuticals (I), Celgene (I), Millennium (I), Janssen (I), Critical Outcome Technologies
Philippe L. Bedard
Research Funding: Astra Zeneca (Inst)
Emma J. Dean
Consulting or Advisory Role: Theradex, Aptus Clinical (Inst)
Travel, Accommodations, Expenses: AstraZeneca
Hideaki Bando
Research Funding: AstraZeneca, Sysmex
Anthony B. El-Khoueiry
Honoraria: Merrimack, Bayer, Novartis, Bristol-Myers Squibb, AstraZeneca, Genentech
Consulting or Advisory Role: CytomX Therapeutics, Bristol-Myers Squibb, AstraZeneca, Bayer
Speakers' Bureau: Merrimack
José A. Perez-Fidalgo
No relationship to disclose
Alain Mita
No relationship to disclose
Jan H.M. Schellens
No relationship to disclose
Matthew T. Chang
No relationship to disclose
Jonathan B. Reichel
Stock or Other Ownership: Illumina
Nancy Bouvier
No relationship to disclose
S. Duygu Selcuklu
No relationship to disclose
Tara E. Soumerai
No relationship to disclose
Jean Torrisi
No relationship to disclose
Joseph P. Erinjeri
Consulting or Advisory Role: Galil Medical
Helen Ambrose
Employment: AstraZeneca
Stock or Other Ownership: AstraZeneca
J. Carl Barrett
Employment: AstraZeneca
Stock or Other Ownership: AstraZeneca
Brian Dougherty
Employment: AstraZeneca
Stock or Other Ownership: AstraZeneca
Andrew Foxley
Employment: AstraZeneca
Stock or Other Ownership: AstraZeneca
Justin P.O. Lindemann
Employment: AstraZeneca
Stock or Other Ownership: AstraZeneca
Robert McEwen
Employment: AstraZeneca
Stock or Other Ownership: AstraZeneca
Martin Pass
Employment: AstraZeneca, AstraZeneca (I)
Stock or Other Ownership: AstraZeneca, AstraZeneca (I)
Patents, Royalties, Other Intellectual Property: During course of current employment with AstraZeneca, I have been named inventor on a number of patents (Inst)
Travel, Accommodations, Expenses: AstraZeneca, AstraZeneca (I)
Gaia Schiavon
Employment: AstraZeneca
Michael F. Berger
Consulting or Advisory Role: Cancer Genetics, Sequenom
Sarat Chandarlapaty
Honoraria: Oncothyreon, Chugai Pharma, Foresite Capital, AstraZeneca, Macrogenics, Sermonix Pharmaceuticals, Agendia
Research Funding: Eli Lilly (Inst), Novartis (Inst)
Travel, Accommodations, Expenses: Macrogenics, AstraZeneca, Chugai Pharma
David B. Solit
Honoraria: Loxo, Pfizer
Consulting or Advisory Role: Pfizer, Loxo
Udai Banerji
Employment: Institute of Cancer Research
Consulting or Advisory Role: Novartis, Astex Pharmaceuticals
Research Funding: AstraZeneca (Inst), Onyx (Inst), Chugai Pharma (Inst)
José Baselga
Leadership: Infinity Pharmaceuticals, Varian, GRAIL
Stock or Other Ownership: PMV Pharma, Juno Therapeutics, Infinity Pharmaceuticals, GRAIL, Varian Medical Systems, Tango, Aura Biomedical, Apogen, Northern Biologics
Honoraria: PMV Pharma, Juno Therapeutics, Infinity Pharmaceuticals, GRAIL, Northern Biologics
Consulting or Advisory Role: Eli Lilly, Novartis
Research Funding: Roche/Genentech
Patents, Royalties, Other Intellectual Property: Combination therapy using PDK1 and PI3K inhibitors. Pending. Memorial Sloan Kettering (MSK) owned, listed as investigator. Use of phosphoinositide 3-kinase inhibitors for treatment of vascular malformations. Licensed. MSK owned, listed as investigator.
Travel, Accommodations, Expenses: Roche/Genentech, Daiichi
Barry S. Taylor
No relationship to disclose
REFERENCES
- 1.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 2.Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 3.Engelman JA. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 4.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 5.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–2963. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 6.Rodon J, Dienstmann R, Serra V, et al. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 7.Yap TA, Bjerke L, Clarke PA, et al. Drugging PI3K in cancer: Refining targets and therapeutic strategies. Curr Opin Pharmacol. 2015;23:98–107. doi: 10.1016/j.coph.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bleeker FE, Felicioni L, Buttitta F, et al. AKT1(E17K) in human solid tumours. Oncogene. 2008;27:5648–5650. doi: 10.1038/onc.2008.170. [DOI] [PubMed] [Google Scholar]
- 10.Cohen Y, Shalmon B, Korach J, et al. AKT1 pleckstrin homology domain E17K activating mutation in endometrial carcinoma. Gynecol Oncol. 2010;116:88–91. doi: 10.1016/j.ygyno.2009.09.038. [DOI] [PubMed] [Google Scholar]
- 11.Do H, Solomon B, Mitchell PL, et al. Detection of the transforming AKT1 mutation E17K in non-small cell lung cancer by high resolution melting. BMC Res Notes. 2008;1:14. doi: 10.1186/1756-0500-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim MS, Jeong EG, Yoo NJ, et al. Mutational analysis of oncogenic AKT E17K mutation in common solid cancers and acute leukaemias. Br J Cancer. 2008;98:1533–1535. doi: 10.1038/sj.bjc.6604212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chang MT, Asthana S, Gao SP, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34:155–163. doi: 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rudolph M, Anzeneder T, Schulz A, et al. AKT1 (E17K) mutation profiling in breast cancer: Prevalence, concurrent oncogenic alterations, and blood-based detection. BMC Cancer. 2016;16:622. doi: 10.1186/s12885-016-2626-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carpten JD, Faber AL, Horn C, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 16.Tamura K, Hashimoto J, Tanabe Y, et al. Safety and tolerability of AZD5363 in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;77:787–795. doi: 10.1007/s00280-016-2987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies BR, Greenwood H, Dudley P, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: Pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11:873–887. doi: 10.1158/1535-7163.MCT-11-0824-T. [DOI] [PubMed] [Google Scholar]
- Elvin P, Palmer A, Womack C, et al: Pharmacodynamic activity of the AKT inhibitor AZD5363 in patients with advanced solid tumors. J Clin Oncol 32, 2014 (suppl; abstr 2541)
- Banerji U, Dean EJ, Perez-Fidalgo JA, et al: A pharmacokinetically (PK) and pharmacodynamically (PD) driven phase I trial of the pan-AKT inhibitor AZD5363 with expansion cohorts in PIK3CA mutant breast and gynecological cancers. J Clin Oncol 33, 2015 (suppl; abstr 2500)
- 20.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandarlapaty S, Chen D, He W, et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: A secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol. 2016;2:1310–1315. doi: 10.1001/jamaoncol.2016.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parikh C, Janakiraman V, Wu WI, et al. Disruption of PH-kinase domain interactions leads to oncogenic activation of AKT in human cancers. Proc Natl Acad Sci USA. 2012;109:19368–19373. doi: 10.1073/pnas.1204384109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 25.Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–1971. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373:726–736. doi: 10.1056/NEJMoa1502309. [DOI] [PMC free article] [PubMed] [Google Scholar]