E2‐mediated cathepsin D (CTSD) activation involves looping of distal enhancer elements (original) (raw)
Abstract
Estrogen receptor alpha (ERα) is a ligand dependent transcription factor that regulates the expression of target genes through interacting with cis‐acting estrogen response elements (EREs). However, only a minority of ERα binding sites are located within the proximal promoter regions of responsive genes. Here we report the characterization of an ERE located 9kbp upstream of the TSS of the cathepsin D gene (CTSD) that up‐regulates CTSD expression upon estrogen stimulation in MCF‐7 cells. Using ChIP, we show recruitment of ERα and phosphorylated PolII at the CTSD distal enhancer region. Moreover, we determine the kinetics of transient CpG methylation on the promoter region of CTSD and for the first time, at a distal enhancer element. We show that ERα is crucial for long‐distance regulation of CTSD expression involving a looping mechanism.
Keywords: Estrogen receptor alpha, CTSD, Distal enhancer, Chromatin looping, DNA methylation
Abbreviations
ERα
estrogen receptor alpha
ERE
estrogen response element
E2
estradiol
TSS
transcriptional start site
FCS
fetal calf serum
DMEM
Dulbecco's modified Eagle's medium
ChIP
chromatin immunoprecipitation
PPolII
activated (phosphorylated) RNA polymerase II
3C
chromatin conformation capture
1.
Cathepsin D (CTSD) is a lysosomal aspartyl protease that plays a role in protein degradation and in the processing of protein precursors to generate their biologically active forms (Piwnica et al., 2004). Besides its functional role in the lysosomal compartment, CTSD has been implicated in tumour progression and in metastasis; CTSD is used as a specific biomarker in breast cancer diagnosis (Fusek and Vetvicka, 2005). CTSD over‐expression is a feature of aggressive breast cancer, and is associated with poor clinical outcome in consequence of mitogenic, angiogenicand anti‐apoptotic effects upon tumour growth (Liaudet‐Coopman et al., 2006). Whereas CTSD is constitutively expressed in ERα‐negative cells, expression in ERα‐positive cells is dependent on estrogen (Liaudet‐Coopman et al., 2006). Transcription of CTSD is initiated at five sites (TSS1–5) located between −72 and −20bp upstream of the initiation codon. However, estrogen selectively induces transcription from the TATA box‐dependent start site at −20 (Cavailles et al., 1993). Within the proximal promoter region, binding sites for ERα, SP1 and USF1/2 cooperate in estrogen‐dependent regulation of the CTSD gene (Augereau et al., 1994, 1994, 1995, 1997, 1998, 2001, 1998). Additionally, Shang et al. (2000) have shown that association of ERα with the proximal promoter of CTSD (−295 to −54bp) occurs in a cyclical manner with a periodicity of 90min. The influence of distal enhancer regions in CTSD expression has not been extensively studied. Additional ERα binding sites, located at 9 and 33kbp upstream of the transcription start site, have been reported (Bourdeau et al., 2004; Carroll et al., 2006). Although ERα is recruited to these sites, it is unclear whether these distal EREs contribute to estrogen‐induced activation of CTSD.
Transcription has to overcome a generally repressive environment that acts to prevent inappropriate gene expression. DNA methylation on CpG dinucleotides is one restrictive covalent epigenetic mark associated with transcriptionally silent condensed chromatin. Hypomethylated DNA is frequently found on promoters of transcriptionally competent genes, whereas hypermethylated promoters are inactive. DNA methylation patterns are preserved through cellular replication by DNA methyltransferases (Klose and Bird, 2006). However, dynamic changes in the methylation status have been implicated in tissue‐specific gene expression (Lenz et al., 2006; Lucarelli et al., 2001). Additionally, we reported that the methylation status of the E2‐responsive pS2 gene could be modified on a time‐scale of hours (Reid et al., 2005). Following from this, we recently described minute‐scale changes in the DNA methylation pattern of several promoters, including the pS2 gene (Kangaspeska et al., 2008; Metivier et al., 2008). The impact of DNA methylation on the estrogen regulated expression the CTSD gene has not been characterized. Here, we address the temporal involvement of methylation in the physical interaction of distal and proximal regulatory regions that coordinate CTSD expression.
We show that the 9kbp upstream enhancer physically interacts with the proximal promoter and induces coordinated estrogen‐dependent activation of CTSD in reporter gene assays. Moreover, we assess the recruitment of transcriptional components to the CTSD enhancer and proximal promoter in response to E2‐stimulation and show that coordinated transient changes in CpG methylation occur at both sites. Our data support a model in which ERα and Polymerase II (PolII) are recruited to the distal enhancer and then orchestrate CTSD expression and regulation by interaction with the transcription start site through loop formation in an ERα‐dependent manner.
1. Experimental procedures
1.1. Cell culture
MCF‐7 cells were maintained in DMEM supplemented with 10% FCS, 2mM l‐glutamine (Invitrogen), penicillin/streptomycin (Invitrogen) (described as full medium) at 37°C under 5% CO2. When hormone deprivation was required, cells were cultured in phenol‐red‐free DMEM (Gibco) containing 2.5% charcoal–dextran stripped serum and antibiotics (described as stripped medium) for 72h prior to treatment.
1.2. RT‐PCR and quantitative real‐time PCR
RNA was extracted from cells using the TriZol reagent (Invitrogen). cDNA reverse‐transcription using poly‐dT oligos was performed on 3μg total RNA using Expand Reverse Transcriptase (Roche) according to the manufacturer's instructions. qPCRs were performed on an ABIprism7500 (Applied Biosystems) using SYBRGreen PCR Master Mix (Applied Biosystems) and the CTSD specific primers (fwd: cagaagctggtggaccagaac, rev: tgcgggtgacattcaggtag). β‐actin (fwd: gggtacttcagggtgaggatg, rev: gtcttcccctccatcgtg) was used as internal reference gene.
1.3. Luciferase‐reporter constructs
CTSD promoter regions were amplified using FastStart Polymerase (Roche) and primers introducing restriction sites for cloning. The distal primer set (fwd: at gctagc atttgtgatcctggaaggtcaggt, Nhe1, rev: gt agatct cctcctcttagggctgagtcactg, BglII) amplifies the −9829 to −8856bp segment containing a putative enhancer, the proximal primer set (fwd: gt agatct gagttgacgtgagtggacaaaagg, BglII, rev: ac aagctt gtgcgcttatagccgggatgac, HindIII) amplifies the −886 to −36 segment harboring the transcription start sites 2–5 and the proximal promoter (Cavailles et al., 1993). The PCR products were cloned into pGL3‐basic vector (Promega) both, separate and combined, using the restriction sites introduced by PCR amplification. The ERE in the distal region was disrupted using site directed mutagenesis (fwd: tCCggAATTCggtTgccc cagctctgagagtg, rev: gggcAaccGAATTc cggaagagaaagggggctgcg). All constructs were verified by sequencing. For control experiments, the distal region was also subcloned into the pGL3‐promoter vector (Promega).
1.4. Transient transfections and luciferase assays
Transfections were performed in 24‐well plates at 70% confluency with ExGene™500 (Fermentas). Cells were transfected using 500ng Firefly luciferase‐reporter constructs and 100ng Renilla luciferase reporter (phRL‐TK vector, Promega). Cells were treated immediately after transfection with either EtOH or 10nM E2 (Sigma–Aldrich). Twenty‐four hours later, cells were harvested and cellular extracts were analyzed for luciferase activity using the Dual‐luciferase‐reporter system (Promega). Firefly luciferase‐reporter activities were normalized to Renilla luciferase activities and presented as relative light units (RLU). At least three independent experiments with triplicates were performed for each experiment.
1.5. Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed as described previously (Nelson et al., 2006; Metivier et al., 2003) with minor modifications. Cells were grown in 10cm plates, and, when needed in experimental setups, deprived of hormone for 3 days before stimulation with 10−8M E2 for the times indicated. Alternatively, cells were maintained in full media and treated with the full‐antagonist ICI182,780 (TOCRIS). Cells were crosslinked with 1.5% formaldehyde for 10min at room temperature. The reaction was stopped by addition of glycine to the final concentration of 0.125M. Following two washes with cold PBS cells were scraped into 300μl lysis buffer (1% SDS, 10mM EDTA, 50mM Tris–HCl (pH 8.0), 10mM β‐glycerophosphate, 1mM Na3VO4, proteinase inhibitors) and incubated on ice for at least 10min. Chromatin was sonicated to about 500bp fragment size and cell debris was sedimented by centrifugation at 14,000×g for 10min. The supernatant was diluted to 6–10ml with ChIP dilution buffer (0.01% SDS, 1.1% Triton X‐100, 1.2mM EDTA, 16.7mM Tris–HCl (pH 8.0), 167mM NaCl, 10mM β‐glycerophosphate, 1mM Na3VO4, proteinase inhibitors); 100μl chromatin were saved as input. Per IP, 1ml chromatin was incubated with 1μg antibody over night. To recover the protein–DNA‐complexes 50μl 50% Protein A slurry (pre‐absorbed with sheared salmon sperm DNA) were added, and incubated for 2–3h rotating at 4°C. Subsequent washing steps were performed with 1ml of each TSEI (0.1% SDS, 1% Triton X‐100, 2mM EDTA, 20mM Tris–HCl (pH 8.0), 150mM NaCl), TSEII (0.1% SDS, 1% Triton X‐100, 2mM EDTA, 20mM Tris–HCl (pH 8.0), 500mM NaCl), TSEIII (1mM EDTA, 10mM Tris–HCl (pH 8.0), 1% NP‐40, 1% sodium deoxycholate, 0.25M LiCl), and 3× with TE buffer (10mM Tris (pH 8.0), 1mM EDTA). For DNA recovery 100μl of 10% Chelex (in water) were added to the beads and incubated at 95°C for 10min to reverse the crosslinking. Further release of DNA was achieved by digesting with Proteinase K (Invitrogen) at 55°C for 30min followed by enzyme inactivation at 95°C for 10min. Supernatant was cleared by centrifugation and transferred to a fresh tube. The following primers were used: TSS: fwd: ggagcggagggtccattc, rev: tccagacatcctctctggaa, unspecific −3000: fwd: cctcacaggtgcgtatctca, rev: agcaaggggtgaaagatggt, distal enhancer: fwd, cctcctcaactgctcttgca rev: gcggctgagatgctgagtca, unspecific −18,000: fwd: ttcagcccagcaaaagaaca, rev: cttcaccccatcctcctgtc.
1.6. Chromosome conformation capture (3C) assay
3C assays were performed as described before (Dekker et al., 2002) with minor modifications. MCF‐7 cells were collected and crosslinked for 15min at room temperature in 1.5% formaldehyde. Formaldehyde was quenched with glycine at a final concentration of 0.125M. Cells were collected and lysed in 1ml ice cold lysis buffer (10mM Tris (pH 8.0), 10mM NaCl, 0.2% NP‐40 and 1× CompleteMini protease inhibitors (Roche)) and incubated for 15min on ice. Lysis was completed using a dounce homogenizer (pestle A). Nuclei were collected at 5000rpm for 5min, washed with and resuspended in 1× restriction buffer B (Fermentas) supplemented with 0.1% SDS to obtain 106 cells/25μl. 106 cells were incubated for 10min at 37°C before SDS was sequestered with Triton X‐100 at a final concentration of 1%. Chromatin was digested with 400U Csp6I (Fermentas) for 90min at 37°C in a final volume of 400μl. The restriction enzyme was inactivated by addition of 1.6% SDS and incubation at 65°C for 20min.
The reaction was diluted with ligase buffer (50mM Tris (pH 7.5), 10mM MgCl2, 10mM DTT, 0.1mg/ml bovine serum albumin, 1mM ATP) supplemented with Triton X‐100 (final concentration 1%) and ligation was performed with 4000U of T4 DNA Ligase (NEB) for 2h at 16°C. Ligation was stopped with EDTA (10mM final concentration). Proteinase K and incubation at 65°C over night were used to reverse the crosslinking. DNA was purified using phenol–chloroform extraction and ethanol precipitation. As controls, the procedure was also performed omitting either crosslinking, digest or ligation.
Control template for PCR was generated through cloning of the promoter regions (region A–E, primers are available on request) analyzed, followed by digest with Csp6I and ligation. Primers used are: 1 fwd: ttcagcccagcaaaagaaca, 2 fwd: tgcagctccctcctctgtg, 3 fwd: agcatttgcacgggtagtcc, 4 fwd: tgatcactgatgcaaggatgc, 5 fwd: ggcgttgtcagtgttttgga, 6 fwd: ttacaggcatctgccaccac, 7 fwd: agatgggcaagtctgggcta, 8 fwd: agaggcgccagatgcctctccc, 9 fwd: gaggacctgattgccaaagg, 10 fwd: ccctcttactctctgcctcctg. PCR products were run on 2% agarose gel.
1.7. GST‐MBD pull‐down and HpaII digest
The methylation status of CTSD promoter was analyzed by GST‐MBD pull‐down and methylation sensitive restriction digestion as described recently (Kangaspeska et al., 2008; Metivier et al., 2008). Briefly, the methyl binding domain (MBD) of human MeCP2 (Nan et al., 1993) was cloned into the pGEX‐6p1 expression vector (Invitrogen) using the following primers fwd: ggatcctctgcctcccccaaacagcgccgct, rev: gaattctcatctcccagttaccgtgaagtca, and purified from bacterial cultures as GST‐MBD fusion protein. For kinetic methylation analysis cells were synchronized by hormone deprivation and subsequent addition of 10−8M E2 for the times indicated. Genomic DNA from ∼2×106 cells was then prepared according to the method described by Nelson and Krawetz (Nelson and Krawetz, 1992) and sonicated to an average size of 300bp. Sonicated DNA (5μg) was incubated with 5μg of GST‐MBD fusion protein or GST alone in pull‐down buffer (20mM Tris (pH 8.0), 100mM NaCl, 1mM EDTA, 2mM DTT) for 2h at 4°C with constant rotation. DNA–protein complexes were then captured by the addition of 100μl 50% slurry of glutathione agarose beads, with a further incubation at 4°C for 1h. Beads were washed 4 times with 500μl of pull‐down buffer. Bound GST‐ or GST‐MBD–DNA complexes were eluted with 10mM glutathione and eluted DNA was purified using the Qiaquick PCR Purification Kit (Qiagen). DNA was then subject to quantitative PCR (qPCR; ABI Prism®7000 System, Applied Biosciences) to amplify regions of interest (primers as used for ChIP). To analyze the methylation status of specific CpG dinucleotides, 5μg of genomic DNA was digested for 2h at 37°C with 10U of the methylation sensitive restriction enzyme HpaII or the methylation insensitive isoschizomere MspI (Roche). Undigested samples were used as controls. All samples were then precipitated and subject to PCR with primer sets that flank 5′ CCGG 3′ HpaII/MspI recognition sites. Primers were as follows: TSS HpaII fwd: ccgaaacgggaatcctccag, TSS HpaII rev: ctgggcggggcaacct, unspecific −3000 HpaII fwd: actggccatccgcactac, unspecific −3000 HpaII rev tgagatacgcacctgtgagg, enhancer HpaII fwd: catttgtgatcctggaaggt, enhancer HpaII rev cccgagtcctgagcttactg.
2. Results and discussion
Previous studies showed that the precursor of cathepsin D, procathepsin D, is one of the most abundant proteins found in the ER expressing breast carcinoma cell line MCF‐7. It is secreted solely in response to estrogen treatment (Rochefort et al., 1987), while it is secreted constitutively in ER negative cell lines. Moreover, it has been shown that secreted procathepsin D can provoke proliferation of cancer cells and also promote metastases (Vetvicka et al., 1994). Although a prognostic value for cathepsin D exists for breast and ovarian cancer, it remains elusive whether it can be used as a prognostic factor for other types of malignancies, despite the fact that procathepsin D is detectable in both, pre‐malignant conditions and developed malignancies (Rochefort, 1992; Leto et al., 2004). Given the invasive and prometastatic properties of procathepsin D it is considered as a potential target for cancer therapy. In fact, downregulation of CTSD significantly reduced proliferation and metastatic potential of breast cancer cells in an athymic nude mice model (Ohri et al., 2007). Therefore, it is of high importance to understand the endogenous regulation of this breast cancer marker.
2.1. Stimulation of CTSD expression upon E2 treatment
We first examined the extent of estrogen‐induction of CTSD in MCF‐7 cells. E2 treatment following a 3‐day starvation of serum and E2 resulted in an increase of CTSD mRNA, reaching a 2.4‐fold increase after 24h (Figure 1A). These results are in concordance with gene expression profiling, which shows 2.3‐fold increase in CTSD transcript levels after estrogen treatment for 24h (data not shown).
Figure 1.
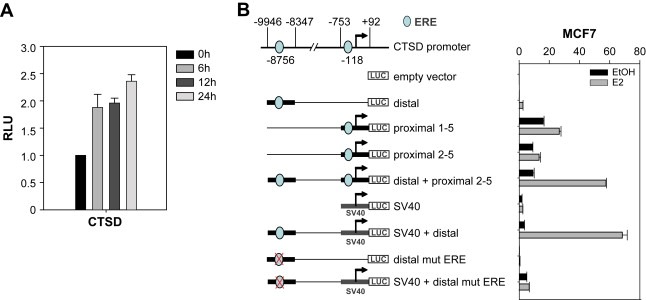
CTSD induction by estrogen. (A) RT‐PCR. Time course of induction of CTSD expression by E2 in MCF‐7 cells. MCF‐7 cells stripped for 3 days were treated with hormone for the times indicated. cDNA was prepared and transcript levels were determined by qPCR and normalized to β‐actin expression. (B) Luciferase assay. Promoter analysis of CTSD using luciferase‐reporter constructs. Schematic overview of CTSD promoter and generated luciferase‐reporter constructs. MCF‐7 cells hormone deprived for 2 days were transfected with the indicated constructs and treated with either EtOH or E2. Luciferase activity was determined 24h later and normalized to Renilla luciferase.
2.2. The estrogen receptor binding site 9kbp upstream of CTSD regulates expression
The ERE located 9kbp upstream of the transcription start site differs from the consensus ERE in only 1 base position (agGGTCAtggTG**g**CCcc) (Bourdeau et al., 2004). To evaluate if this site modulates CTSD gene expression in response to estrogen, luciferase‐reporter constructs were generated (Figure 1B). These constructs contain either the distal ERE (−9829 to −8856bp, “distal”), the proximal promoter either covering TSS 1–5 (−365 to −10bp, “proximal 1–5), or covering TSS 2–5 (−886 to −36bp, “proximal 2–5”), or both (“distal+proximal 2–5”). Given the distance between enhancer and TSS, intermediate sequences were omitted.
Transient transfection experiments in MCF‐7 cells with the proximal promoter (TSS 1–5) showed basal expression with estrogen‐mediated induction after 24h. This is in agreement with TSS1 being the major estrogen‐responsive site of the proximal promoter (Cavailles et al., 1993).
The distal element is strongly responsive to estrogen (12‐fold upregulation with E2) but basal expression is low, as this region in isolation does not contain minimal promoter elements. The basal expression of the distal element was enhanced by positioning it either upstream of an SV40 minimal promoter (“distal+SV40) or upstream of the CTSD proximal promoter, which lacked the estrogen‐responsive TSS1 (“distal+proximal 2–5”). The latter construct determines the influence of the enhancer on the endogenous CTSD promoter, without incorporating the estrogen regulation of proximal promoter elements. Both constructs had significant E2‐dependent induction (19‐ and 6‐fold, respectively). This induction was abolished by disruption of the distal ERE motif (“distal mut ERE” and “distal mut ERE+SV40”). Nevertheless, it cannot be excluded that further regulatory elements contribute to CTSD gene regulation within the chromosomal context. In conclusion, the distal estrogen receptor binding site, 9kbp upstream, confers estrogen responsiveness and contributes to estrogen‐induced upregulation of the CTSD gene in MCF‐7 cells.
2.3. ERα and PolII bind to the distal ERE
Association of ERα and RNA Polymerase II with the proximal CTSD promoter has been reported (Shang et al., 2000; Bourdeau et al., 2004). However, the dynamics of association of the distal enhancer with ERα and PPolII have not been analyzed (Figure 2A). We performed ChIP analysis using E2 starved MCF‐7 cells treated for 4h, or not, with E2 (Figure 2B) and found an increased association of ERα with the upstream enhancer in the presence of E2. In agreement with previous reports (Shang et al., 2000), recruitment of phosphorylated PolII (PPolII) to the TSS was apparent, however, occupancy of ERα predominantly occured on the enhancer. Recruitment of ERα to the enhancer was E2‐dependent, as treatment with the antiestrogen ICI182,780 abolished this interaction. In kinetic ChIP experiments, stimulation with E2 resulted in rapid recruitment of ERα and PPolII to the enhancer (Figure 2C). No recruitment of PPolII was observed to the intermediate region at −3kbp and to a distal region at −18kbp. In contrast to ERα, treatment with E2 nor ICI182,780 did not affect PolII recruitment to the proximal promoter which is in concordance with the reported usage of the other E2‐independent transcription start sites (Cavailles et al., 1993).
Figure 2.
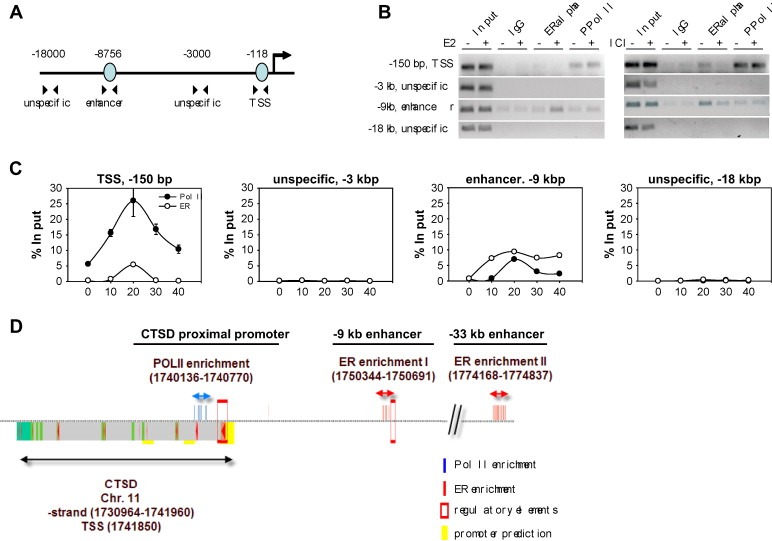
ERα can associate with the distal ERE in vivo. (A) Schematic representation of the CTSD promoter indicating the regions tested in ChIP. (B) Conventional ChIP analysis. MCF‐7 cells were either hormone deprived for 3 days and treated with EtOH or E2 for 4h or maintained in full media (DMEM, NM) and treated with EtOH and ICI182,780. Chromatin was submitted to ChIP procedure using mouse and rabbit IgG, and antibodies against ERα (HC20, Santa Cruz) and PPolII (CTD4H8, Upstate). Primers enclosing the distal ERE (enhancer) and the TSS were used for PCR. (C) Kinetic ChIP analysis to study the dynamics at the CTSD promoter in terms of ERα and PPolII recruitment. Cells were hormone deprived for 3 days, then treated with E2 and collected every 10min up to 90min before being subjected to ChIP procedure. Primers enclosing two unspecific regions (−3 and −18kb) were also included. (D) Bioinformatic analysis of the CTSD promoter superimposed with ChIP‐on‐chip data for ERα and PolII reported by Carroll et al. (2006).
In summary, kinetic ChIP data suggest that the −9kbp ERE has a role as a functional enhancer in CTSD gene expression.
Bioinformatic analysis of the CTSD promoter sequence using PromoterInspector (Genomatix Software GmbH) predicts regulatory elements at the transcriptional start site and in the enhancer region (Figure 2D, red boxes). Superimposed on the figure are significant enriched probes from ChIP‐on‐chip experiments performed by the group of Myles Brown (Carroll et al., 2006) for PolII (blue) and ERα (red). Probe enrichment for PolII and ERα at the CTSD TSS, −9kbp enhancer and −33kbp enhancer correlates well with the data presented here (Figure 2B) and by Bourdeau et al. (2004). Absence of PPolII at the enhancer in the ChIP‐on‐chip data might be due to kinetic changes after 4h treatment with E2 in comparison to 90min or due to cellular differences.
2.4. The −9kbp upstream enhancer and transcription start site associate with each other
Enhancer sequences have been identified more than 100kbp upstream of the genes they regulate (Carroll et al., 2006; Kleinjan and van, 2005). Three models have been proposed to explain how long‐range interactions impinge upon regulation (Bondarenko et al., 2003). A looping model proposes the assembly of transcription factors and co‐factors at the distal enhancer followed by looping out of the intervening chromatin which subsequently results in a physical interaction between the enhancer and TSS to promote transcriptional activation or repression. In line with this model, long‐range interactions between ERα binding sites have been reported for the estrogen‐responsive gene GREB1 (Deschenes et al., 2007; Sun et al., 2007). In the tracking model, key factors and PolII assemble on the enhancer and then PPolII tracks along the DNA towards the TSS. The final model proposes both spreading and looping. It is based on the assumption that proteins distributed between the enhancer and the TSS polymerize thereby forming small chromatin loops and moving the enhancer closer to the TSS.
As PPolII is recruited to the enhancer, this suggests either that PPolII is independently recruited to both sites or that the distal enhancer and the transcription start site are, at least for a proportion of time during transcription, in close proximity to each other through chromatin loop formation.
This question was addressed by chromosome conformation capture (3C) assays (Dekker et al., 2002). In this procedure, chromatin conformation is preserved by crosslinking with formaldehyde. Physical interaction between specific sites are then analyzed by restriction digest followed by intramolecular ligation of crosslinked fragments. Consequently, interactions between regions result in ligation products that can be detected by PCR.
In summary, primers (Figure 3; black arrows) spread over 22kbp (−18,728 to +3456bp) and primer 4 (−8544bp, blue arrow) located in the fragment containing the enhancer were designed to probe interactions between different regions of the CTSD gene. A strong enrichment for the ligation product between the −9kbp enhancer and the TSS (primer 4 and 8) was observed, with no ligation products derived from intervening regions detected (Figure 3). Furthermore, no PCR products were observed in control experiments using samples from uncrosslinked chromatin, undigested crosslinked chromatin or digested chromatin without subsequent ligation, indicating that the interaction between the −9kbp enhancer and the TSS is specific (data not shown).
Figure 3.
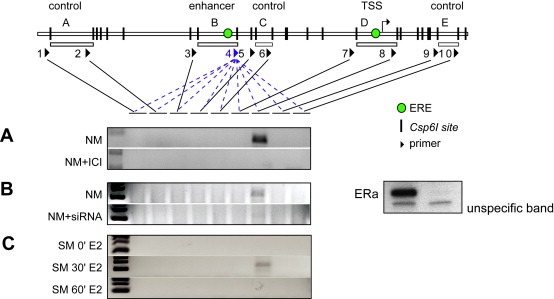
Loop formation between the enhancer and the TSS. Chromosome conformation capture (3C) assay was applied to analyze chromatin loops between the enhancer and other regions of the CTSD promoter spread over 22kbp. Cells were either maintained in full media (NM) and treated with ICI182,780 (A) or siRNA against ERα (B) for 24h, or cells were hormone deprived for 3 days and treated with E2 for indicated times (C).
The central role of ERα in loop formation is reflected by the effect of treatment with the ERα antagonist ICI182,780 or by knockdown experiments using siRNA directed against ERα. In each case, abrogating ERα prevented looping. Furthermore, when cells were released from synchronization by estrogen deprivation for 3 days, physical interaction between the −9kbp enhancer and the TSS was only observed at 30min following E2 treatment. This is interesting in view of the synchronous presence of ERα and PPolII at these sites. Both approaches indicate a specific interaction between enhancer and TSS and support the temporarily limited formation of a chromatin loop between both regions. The other two models would predict interactions with the intervening regions and therefore are less likely.
2.5. DNA methylation pattern changes in response to E2
Methylation of cytosine bases in CpG dinucleotides has conventionally been considered as a stable epigenetic modification associated with transcriptional repression (Costello and Plass, 2001). Although CpGs are under‐represented in the mammalian genome, they are enriched in proximal promoters and are usually hypomethylated (Robertson, 2005), a state that is associated with transcriptionally active genes. A regulatory role of CpG methylation might not be restricted to proximal promoter elements, and indeed the methylation status of an upstream enhancer has been shown to regulate expression of the retinoic acid‐responsive Ucp1 gene (Kiskinis et al., 2007).
We analyzed the methylation status of the CTSD proximal promoter and −9kbp enhancer region by affinity purification of methylated DNA fragments using a GST‐MBD fusion protein (Kangaspeska et al., 2008). CpG density maps of the TSS and −9kbp regions are shown in Figure 4A. In unsynchronized MCF‐7 cells, the proximal promoter and the distal enhancer have low levels of methylation whereas the intermediate region around −3kbp was found to have high levels of methylation (Figure 4B). We also addressed the methylation state of specific CpG dinucleotides in the proximity of these regions through the use of the methylation sensitive restriction enzyme HpaII. This analysis confirmed that individual CpGs were either completely (proximal) or partially (−9kbp enhancer) unmethylated (Figure 4C).
Figure 4.
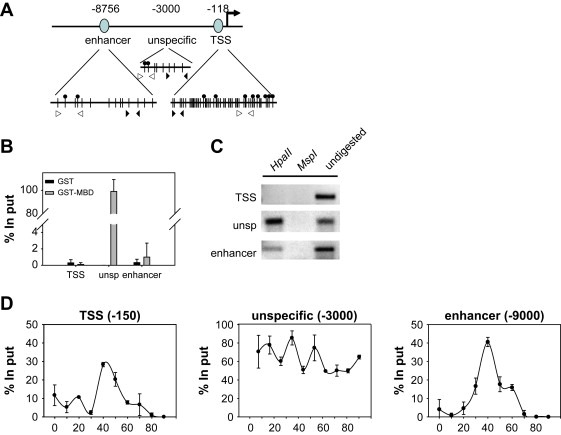
Dynamic changes in the methylation of the CTSD promoter. (A) Schematic representation of the CTSD promoter indicating the CpGs, HpaII sites and the primers used for PCR (vertical lines, filled circles and arrow heads (⊲: HpaII digest, ▶: GST‐MBD pull‐down), respectively). (B) GST‐MBD fusion protein utilizing the MBD of human MeCP2 (Nan et al., 1993) was used to selectively pull‐down the methylated DNA in unsynchronized MCF‐7 cells. The methylation status of CTSD promoter in indicated regions was monitored by qPCR. (C) The methylation status of selected CpGs (indicated in A) was monitored using the methylation sensitive restriction enzyme HpaII or its methylation insensitive isoschizomere MspI followed by semi‐quantitative PCR. (D) MCF‐7 cells were hormone deprived for 3 days and treated with 10−8M E2 for indicated times. GST‐MBD pull‐down was used to monitor the methylation status of CTSD promoter in indicated regions.
Recent work has shown minute‐scale changes in the methylation status of the proximal promoter of the pS2 gene upon E2‐stimulation (Kangaspeska et al., 2008; Metivier et al., 2008). Given these observations and the dynamic association and dissociation of transcription factors with the CTSD (Figure 2 and Shang et al., 2000) and pS2 promoters (Shang et al., 2000; Metivier et al., 2003), we determined the CpG methylation status of the CTSD promoter around the TSS and the distal (−9kbp) regulatory elements after E2‐stimulation. Kinetic affinity isolation of methylated DNA demonstrates that the proximal promoter and −9kbp enhancer change their methylation status on a scale of tens of minutes (Figure 4D). The initial unmethylated state of these loci became methylated, peaking at 40min after E2‐stimulation, after which the sites became unmethylated again. In contrast, the unspecific region −3000 remained methylated throughout the stimulation. These results raise the possibility that the proximal and distal regions are either subject to common regulatory mechanisms that do not influence the constantly methylated −3000bp region, or that these sites physically interact and concordant modifications occur. The peak of methylation on the TSS and −9kbp enhancer occurs after peak occupancy by PPolII and coincides with times when the CTSD gene is transcriptionally unproductive. Rapid methylation of the enhancer and the proximal promoter occurs after recruitment of ERα and PPolII to these regions, suggesting that similar to recruitment of histone deacetylases and repressive complexes (Metivier et al., 2003), methylation might act to restrict transcription. Recently, experiments using different promoter synchronization methods (Kangaspeska et al., 2008; Metivier et al., 2008) revealed generally similar kinetics of dynamic methylation on several promoters, suggesting that cell cycle‐independent methylation/demethylation might be a common feature of at least a subset of promoters. These reports concentrated on the description of short‐term methylation events on proximal promoter regions; the finding that enhancer regions can also undergo dynamic fluctuations in methylation status extends the functional influence of enhancer elements in the regulation of transcription. These results demonstrate that dynamic changes in transcription factor occupancy at the TSS and −9kbp enhancer regions correlate with dynamic changes to their methylation status and are localized to these functional elements as the DNA region between them remains methylated. Moreover, the coordinated physical association of the TSS and −9kbp enhancer element at the time when the CTSD gene is actively transcribed indicates that the enhancer element contributes to active transcription for restricted periods of time.
3. Conclusions
Many genes are regulated through distal motifs but to date, little knowledge exists on their detailed involvement in gene transcription. Here we investigated the role of a 9kbp upstream enhancer of the CTSD gene. We provide evidence for (a) simultaneous recruitment of PPolII to the enhancer and the TSS, but not to the intermediate region (−3kbp) or upstream regions (−18kbp); (b) accumulation of ERα on the distal enhancer in an E2‐dependent manner; (c) spatial proximity between enhancer and TSS which occurs immediately after recruitment of ERα to the enhancer as determined by 3C assays; and (d) coordinate dynamic methylation and demethylation of the −9kbp enhancer and the TSS elements that contrast with constant high levels of CpG methylation in the −3kbp intermediate region. These findings suggest that recruitment of ERα to the −9kbp enhancer provokes loop formation with the proximal promoter. Mechanistically, these processes dynamically promote and then restrict the influence of the −3kbp enhancer upon the TSS of the CTSD promoter. We propose that the upstream enhancer region actively participates in estrogen receptor‐mediated regulation of the CTSD gene most likely through temporary chromatin loop formation (Figure 5) and show for the first time that dynamic methylation occurs on an upstream enhancer element. These new insights improve our understanding of the complex processes involved in the CTSD regulation.
Figure 5.
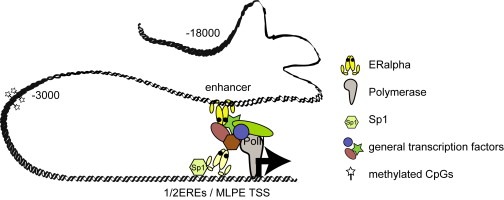
Model for chromatin loop formation between the distal enhancer and the TSS after estrogen stimulation.
Acknowledgement
This work was supported by EC 6th framework program grant CRESCENDO (FP6‐018652), the European Molecular Biology Laboratory (EMBL) and the European Molecular Biology Organization (EMBO). The authors are grateful to Dr. Chiara Lanzuolo and Luke Dillon for their technical contribution.
Bretschneider Nancy, Kangaspeska Sara, Seifert Martin, Reid George, Gannon Frank, Denger Stefanie, (2008), E2‐mediated cathepsin D (CTSD) activation involves looping of distal enhancer elements, Molecular Oncology, 2, doi: 10.1016/j.molonc.2008.05.004.
References
- Augereau, P. , Miralles, F. , Cavailles, V. , Gaudelet, C. , Parker, M. , Rochefort, H. , 1994. Mol. Endocrinol. 8, 693–703. [DOI] [PubMed] [Google Scholar]
- Bourdeau, V. , Deschenes, J. , Metivier, R. , Nagai, Y. , Nguyen, D. , Bretschneider, N. , Gannon, F. , White, J.H. , Mader, S. , 2004. Mol. Endocrinol. 18, 1411–1427. [DOI] [PubMed] [Google Scholar]
- Bondarenko, V.A. , Liu, Y.V. , Jiang, Y.I. , Studitsky, V.M. , 2003. Biochem. Cell Biol. 81, 241–251. [DOI] [PubMed] [Google Scholar]
- Cavailles, V. , Augereau, P. , Rochefort, H. , 1993. Proc. Natl. Acad. Sci. U.S.A 90, 203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, J.S. , Meyer, C.A. , Song, J. , Li, W. , Geistlinger, T.R. , Eeckhoute, J. , Brodsky, A.S. , Keeton, E.K. , Fertuck, K.C. , Hall, G.F. , Wang, Q. , Bekiranov, S. , Sementchenko, V. , Fox, E.A. , Silver, P.A. , Gingeras, T.R. , Liu, X.S. , Brown, M. , 2006. Nat. Genet. 38, 1289–1297. [DOI] [PubMed] [Google Scholar]
- Costello, J.F. , Plass, C. , 2001. J. Med. Genet. 38, 285–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker, J. , Rippe, K. , Dekker, M. , Kleckner, N. , 2002. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Deschenes, J. , Bourdeau, V. , White, J.H. , Mader, S. , 2007. J. Biol. Chem [DOI] [PubMed] [Google Scholar]
- Fusek, M. , Vetvicka, V. , 2005. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech. Repub. 149, 43–50. [DOI] [PubMed] [Google Scholar]
- Krishnan, V. , Wang, X. , Safe, S. , 1994. J. Biol. Chem. 269, 15912–15917. [PubMed] [Google Scholar]
- Krishnan, V. , Porter, W. , Santostefano, M. , Wang, X. , Safe, S. , 1995. Mol. Cell Biol. 15, 6710–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose, R.J. , Bird, A.P. , 2006. Trends Biochem. Sci. 31, 89–97. [DOI] [PubMed] [Google Scholar]
- Kangaspeska, S., Stride, B., Metivier, R., Polycarpou-Schwarz, M., Ibberson, D., Carmouche, R.P., Benes, V., Gannon, F., Reid, G., 2008. Nature 452, 112–115. [DOI] [PubMed]
- Kleinjan, D.A. , van, H.V. , 2005. Am. J. Hum. Genet. 76, 8–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis, E. , Hallberg, M. , Christian, M. , Olofsson, M. , Dilworth, S.M. , White, R. , Parker, M.G. , 2007. EMBO J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaudet-Coopman, E. , Beaujouin, M. , Derocq, D. , Garcia, M. , Glondu-Lassis, M. , Laurent-Matha, V. , Prebois, C. , Rochefort, H. , Vignon, F. , 2006. Cancer Lett. 237, 167–179. [DOI] [PubMed] [Google Scholar]
- Lenz, B. , Bleich, S. , Beutler, S. , Schlierf, B. , Schwager, K. , Reulbach, U. , Kornhuber, J. , Bonsch, D. , 2006. Exp. Cell Res. 312, 4049–4055. [DOI] [PubMed] [Google Scholar]
- Lucarelli, M. , Fuso, A. , Strom, R. , Scarpa, S. , 2001. J. Biol. Chem. 276, 7500–7506. [DOI] [PubMed] [Google Scholar]
- Leto, G. , Tumminello, F.M. , Crescimanno, M. , Flandinna, C. , Gebbia, N. , 2004. Clin. Exp. Metastasis 21, 91–106. [DOI] [PubMed] [Google Scholar]
- Metivier, R. , Gallais, R. , Tiffoche, C. , Le Péron, C. , Jurkowska, R.Z. , Carmouche, R.P. , Ibberson, D. , Barath1, P. , Demay, F. , Reid, G. , Benes, V. , Jeltsch, A. , Gannon, F. , Salbert, G. , 2008. Nature 452, 45–50. [DOI] [PubMed] [Google Scholar]
- Metivier, R. , Penot, G. , Hubner, M.R. , Reid, G. , Brand, H. , Kos, M. , Gannon, F. , 2003. Cell 115, 751–763. [DOI] [PubMed] [Google Scholar]
- Nelson, J.D. , Denisenko, O. , Bomsztyk, K. , 2006. Nat. Protoc. 1, 179–185. [DOI] [PubMed] [Google Scholar]
- Nan, X. , Meehan, R.R. , Bird, A. , 1993. Nucleic Acids Res. 21, 4886–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, J.E. , Krawetz, S.A. , 1992. Anal. Biochem. 207, 197–201. [DOI] [PubMed] [Google Scholar]
- Ohri, S.S. , Vashishta, A. , Proctor, M. , Fusek, M. , Vetvicka, V. , 2007. Cancer Biol. Ther. 20, (7) 6 [DOI] [PubMed] [Google Scholar]
- Piwnica, D. , Touraine, P. , Struman, I. , Tabruyn, S. , Bolbach, G. , Clapp, C. , Martial, J.A. , Kelly, P.A. , Goffin, V. , 2004. Mol. Endocrinol. 18, 2522–2542. [DOI] [PubMed] [Google Scholar]
- Reid, G. , Metivier, R. , Lin, C.Y. , Denger, S. , Ibberson, D. , Ivacevic, T. , Brand, H. , Benes, V. , Liu, E.T. , Gannon, F. , 2005. Oncogene 24, 4894–4907. [DOI] [PubMed] [Google Scholar]
- Rochefort, H. , Capony, F. , Garcia, M. , Cavailles, V. , Freiss, G. , Chambon, M. , Morriset, M. , Vignon, F. , 1987. J. Cell. Biochem. 35, 17–29. [DOI] [PubMed] [Google Scholar]
- Rochefort, H. , 1992. Eur. J. Cancer 28A, 1780–1783. [DOI] [PubMed] [Google Scholar]
- Robertson, K.D. , 2005. Nat. Rev. Genet. 6, 597–610. [DOI] [PubMed] [Google Scholar]
- Shang, Y. , Hu, X. , DiRenzo, J. , Lazar, M.A. , Brown, M. , 2000. Cell 103, 843–852. [DOI] [PubMed] [Google Scholar]
- Sun, J. , Nawaz, Z. , Slingerland, J.M. , 2007. Mol. Endocrinol. 21, 2651–2662. [DOI] [PubMed] [Google Scholar]
- Vetvicka, V. , Vetvickova, J. , Fusek, M. , 1994. Cancer Lett. 79, 131–135. [DOI] [PubMed] [Google Scholar]
- Wang, F. , Porter, W. , Xing, W. , Archer, T.K. , Safe, S. , 1997. Biochemistry 36, 7793–7801. [DOI] [PubMed] [Google Scholar]
- Wang, F. , Hoivik, D. , Pollenz, R. , Safe, S. , 1998. Nucleic Acids Res. 26, 3044–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, F. , Samudio, I. , Safe, S. , 2001. Mol. Cell Endocrinol. 172, 91–103. [DOI] [PubMed] [Google Scholar]
- Xing, W. , Archer, T.K. , 1998. Mol. Endocrinol. 12, 1310–1321. [DOI] [PubMed] [Google Scholar]