Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma (original) (raw)
Abstract
Acquired resistance to BRAF inhibitors often involves MAPK re‐activation, yet the MEK inhibitor trametinib showed minimal clinical activity in melanoma patients that had progressed on BRAF‐inhibitor therapy. Selective ERK inhibitors have been proposed as alternative salvage therapies. We show that ERK inhibition is more potent than MEK inhibition at suppressing MAPK activity and inhibiting the proliferation of multiple BRAF inhibitor resistant melanoma cell models. Nevertheless, melanoma cells often failed to undergo apoptosis in response to ERK inhibition, because the relief of ERK‐dependent negative feedback activated RAS and PI3K signalling. Consequently, the combination of ERK and PI3K/mTOR inhibition was effective at promoting cell death in all resistant melanoma cell models, and was substantially more potent than the MEK/PI3K/mTOR inhibitor combination. Our data indicate that a broader targeting strategy concurrently inhibiting ERK, rather than MEK, and PI3K/mTOR may circumvent BRAF inhibitor resistance, and should be considered during the clinical development of ERK inhibitors.
Keywords: ERK inhibitors, BRAF inhibitors, MEK inhibitors, Melanoma, Acquired resistance
Highlights
- We examine the activity of the MEK and ERK inhibition in models of melanoma BRAF inhibitor resistance.
- ERK inhibitors more efficiently suppress MAPK signalling in these models.
- Despite cell cycle arrest ERK inhibitors can only induce apoptosis in a subset of resistance models.
- Survival is due to ERK‐dependent negative feedback activated RAS and PI3K signalling.
- Combined ERK and PI3K/mTOR inhibition was effective at promoting cell death in all models.
1. Introduction
Constitutive signalling through the mitogen activated protein kinase (MAPK) pathway is common in melanoma and often driven by activating mutations in the BRAF kinase (Davies et al., 2002). Potent inhibitors of the BRAFV600 mutant protein, dabrafenib and vemurafenib, have produced response rates of 50–60% and improved progression‐free and overall survival, compared to dacarbazine, in patients with BRAFV600E mutant metastatic melanoma (Chapman et al., 2011; Falchook et al., 2012b; Flaherty et al., 2010; Hauschild et al., 2012). The use of these targeted therapies is limited, however by the development of drug resistance which occurs in the majority of patients (Falchook et al., 2012b; Flaherty et al., 2010).
Multiple mechanisms of resistance to BRAF inhibition have been identified, and the majority involve MAPK pathway re‐activation via alternate BRAF transcript splicing, BRAF amplification (Poulikakos et al., 2011; Shi et al., 2012), activating mutations in N‐RAS or MEK1 (Nazarian et al., 2010; Wagle et al., 2011), over expression of the kinases COT1 and CRAF (Johannessen et al., 2010; Montagut et al., 2008) or activation of receptor tyrosine kinases (RTKs) (Girotti et al., 2013; Shi et al., 2011; Villanueva et al., 2010). Despite the prevalence of persistent MAPK signalling in resistant metastases, inhibition downstream of BRAF using the MEK inhibitor trametinib, as a single agent, had minimal clinical activity in melanoma patients that had progressed on BRAF‐inhibitor therapy (Kim et al., 2013). Selective ERK inhibitors have been proposed as alternative salvage therapies in BRAF inhibitor resistant cell models (Hatzivassiliou et al., 2012; Morris et al., 2013).
To examine the potency of ERK inhibition, we compared MEK and ERK inhibitors in nine melanoma cell models with acquired resistance to BRAF inhibition. Resistance in these models was driven by the expression of BRAF splice variants, BRAF amplification, mutant N‐RAS, mutant MEK1 or RTK signalling. Our studies confirm that inhibiting ERK suppressed MAPK signalling and the proliferation of all nine BRAF‐inhibitor resistant melanoma models. Nevertheless, ERK inhibition did not induce substantial cell death in five of nine resistant melanoma cell models, and this was often due to the activity and induction of PI3K survival signals. Consequently, the combination of ERK inhibition with PI3K/mTOR inhibition promoted cell death in these BRAF inhibitor resistant melanoma cells, and this combination was more potent than simultaneously co‐inhibiting MEK and the PI3K/mTOR cascade. Our data indicate that inhibition of ERK is more effective than inhibiting MEK but both inhibitors fail to consistently induce melanoma cell death. Thus, ERK inhibition alone is unlikely to show sufficient clinical activity in patients with BRAF inhibitor resistant disease. Instead, inhibiting the MAPK pathway at the ERK, rather than MEK node, in combination with PI3K/mTOR inhibition, efficiently and consistently overcomes acquired resistance to BRAF inhibitors.
2. Materials and methods
2.1. Patients, cell culture and compounds
Informed consent was obtained for each patient under approved Human Research Ethics Committee protocols. Melanoma cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Carlsbad, CA) with 10% fetal bovine serum, HEPES and l‐glutamine and cultured in a 37 °C incubator with 5% CO2. Patient derived short‐term cultures with acquired resistance to BRAF ± MEK inhibition were generated in the absence of inhibitor (Carlino et al., 2013) and BRAF inhibitor resistant cells derived after chronic drug exposure were maintained in drug. Drug resistance was regularly monitored and confirmed using viability assays (Carlino et al., 2013; Lai et al., 2012). BRAFV600 genotype of melanoma cultures was confirmed using PCR‐based capillary sequencing (data not shown).
Stocks of dabrafenib (Active Biochem, Maplewood, NJ), trametinib (Selleck Chemicals, Houston, TX), VX‐11e (Active Biochem), MEK162 (Selleck Chemicals), SCH772984 (Active Biochem), BEZ235 (Selleck Chemicals), LY294002 (Selleck Chemicals) and RAD001 (Selleck Chemicals), were made in DMSO. Cell authentication was confirmed using the StemElite ID system from Promega (Madison, WI).
2.2. Resistance screen
An RT‐PCR resistance screen was used to examine the expression of BRAF splice variants, the complete coding sequence of MEK1, MEK2 and N‐RAS cDNAs and the 5′ half of the AKT1 cDNA (encompassing amino acids 1–200) in all BRAF‐inhibitor resistant cell lines (Table 1). Reverse transcription reactions were performed using the Superscript III First‐Strand Synthesis kit (Life Technologies, Carlsbad, CA) with the oligo dT primer. The MEK1, MEK2, N‐RAS and AKT1 gene products were each amplified from cDNA using Taq polymerase (Fisher Bioteh, Wembley, WA, Australia) and BRAF cDNA was amplified with Pfx polymerase (Life Technologies). PCR products were purified using QIAquick PCR purification kit (Qiagen, Limburg, Netherlands) followed by Sanger sequencing on the 3730xl DNA Analyser (AGRF, Westmead, NSW, Australia). Amplification and sequencing primers are listed in Table S1. The identity of mutations was confirmed using an independent RT‐PCR product. B‐RAF relative copy number was determined by quantitative PCR using the Corbett Rotor‐Gene 6000 as previously described (Corcoran et al., 2010) (Table 1).
Table 1.
Summary of resistance screen.
| Gene | Resistance mechanism | Screening method |
|---|---|---|
| BRAF | Amplification | Quantitative PCR |
| BRAF | Splice variant | RT‐PCR and partial sequence of splice variants |
| MEK1 | Mutation | RT‐PCR and complete ORF sequence |
| MEK2 | Mutation | RT‐PCR and complete ORF sequence |
| N‐RAS | Mutation | RT‐PCR and complete ORF sequence |
| AKT1 | Mutation | RT‐PCR and partial ORF sequence |
| RTK | Activation | RTK array |
Phosphorylation of RTKs was determined with human phospho‐RTK array kits (ART001; R&D Systems, Minneapolis, MN) (Table 1). Two hundred and fifty μg of protein extract was analysed as per the manufacturer's protocol.
2.3. Pharmacological growth inhibition assays
Cultured cells were seeded into 96‐well plates (1‐2E3 cells per well) and serial dilutions of each inhibitor, prepared in media, were added to cells 24 h after seeding. Cells were incubated for 72 h following addition of drug and cell viability was determined as previously described (Carlino et al., 2013).
2.4. Cell cycle and apoptosis analysis
Both adherent and floating cells were harvested and cell cycle analyses were performed as previously described (Gallagher et al., 2005).
2.5. Immunoblotting
Total cellular proteins were extracted and resolved as previously described (Carlino et al., 2013). Western blots were probed with antibodies against DUSP6 (Abcam, Cambridge UK), p‐p90RSKS363 (Santa Cruz Biotechnologies, Santa Cruz, CA); p‐pRbS807/811, p‐AKTS473, p‐RPS6S235/236, p‐MEKS217/221, p‐CRAFS338, BRAF (Cell Signalling, Danvers, MA); β‐actin (AC‐74; Sigma–Aldrich); p27Kip1, cyclin D1, PARP (Becton Dickinson, St. Louis, MO); and BRAFV600E (VE1 clone (Long et al., 2013)).
Activated GTP‐bound RAS was measured using an activated RAS detection kit (Cell Signalling), which employs a CRAF RAS‐binding domain fusion protein pull‐down assay as described in the manufacturer's instructions.
2.6. Statistical analysis
All data are presented as the mean values and standard deviation of at least two independent experiments. Statistical significance (p value < 0.05) was assessed using the Student's _t_‐test for paired values.
3. Results
3.1. Models of BRAF inhibitor resistance
To determine the potency of ERK inhibition, we compared the activity of MEK and ERK inhibitors in a series of BRAF inhibitor resistant melanoma cell models. We included melanoma cells expressing BRAF splice variants, oncogenic N‐RAS, mutant MEK1, amplified BRAF or activated RTK signalling (Table 2). Four resistant models were generated in vitro and five short‐term patient‐derived melanoma cell lines with acquired resistance to either BRAF inhibition or combined BRAF and MEK inhibition were analysed (Table 2, Figure S1) (Carlino et al., 2013; Gowrishankar et al., 2012; Lai et al., 2012; Nazarian et al., 2010).
Table 2.
Details of BRAF inhibitor resistant melanoma cell models.
| Cell line | Mechanism of resistance | Inhibitor |
|---|---|---|
| In‐vitro | ||
| BR2 | NRASQ61H | Dabrafenib |
| BR4 | BRAF splice (exon 4–10Δ) | Dabrafenib |
| M238R1 | PDGFRβ activation | Vemurafenib |
| CR201 | RTK activation | Dabrafenib + trametinib |
| Patient‐derived | ||
| WMD009 | BRAF splice (exon 2–10Δ) | Dabrafenib |
| WMD013 | Unknownb | Dabrafenib |
| SMU030Ra | BRAF amplification | Dabrafenib + trametinib |
| Patient 1 | MEK1E203K | Vemurafenib |
| Patient 3 | Unknownb | Vemurafenib |
3.2. Selection of MEK inhibitor and ERK inhibitor concentrations
Suitable concentrations of the allosteric MEK inhibitor trametinib and the ATP‐competitive ERK inhibitor VX‐11e (Aronov et al., 2009) were initially selected by comparing drug activity in a panel of sensitive BRAFV600E‐mutant melanoma cell lines (Figure S2). The median GI50 for trametinib was 0.93 nM (range 0.17–2.35 nM) and GI50 for VX‐11e was 0.77 μM (range 0.47–0.93 μM). We selected drug concentrations approximately 10‐fold the median GI50 values, and used 10 nM trametinib (Carlino et al., 2013) and 10 μM VX‐11e. These MEK and ERK inhibitor concentrations produced similar cell cycle‐inhibitory and apoptotic effects in sensitive melanoma cell lines (Figures 1 and 2).
Figure 1.
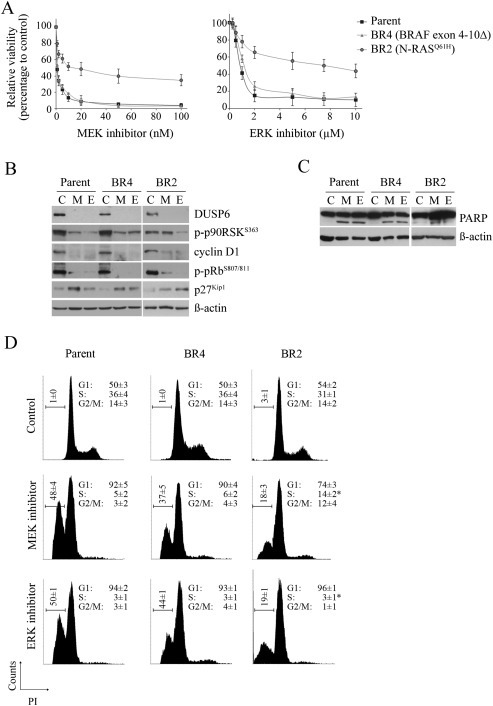
ERK and MEK inhibitors display variable activity towards BRAF inhibitor resistant sublines expressing a BRAF splice variant or mutant N‐RAS A. Viability curves of the parental SKMel28 and isogenic sublines, BR2 and BR4, treated with indicated concentrations of the MEK inhibitor trametinib and ERK inhibitor VX‐11e for 72 h (relative to DMSO‐treated controls; mean ± SD; n = 2). B. SKMel28 parental and dabrafenib‐resistant sublines were treated with DMSO (Control, C), 10 nM MEK inhibitor trametinib (M) or 10 μM ERK inhibitor VX‐11e (E) for 24 h. Western blots of lysates showing protein markers of MAPK activity and cell cycle progression. C. PARP cleavage was determined 72 h after treating melanoma cells with inhibitors as described above. Both full length and major cleaved PARP proteins shown. D. Cell cycle distribution of indicated cell lines treated with DMSO (control), 10 nM trametinib (MEK inhibitor) or 10 μM VX‐11e (ERK inhibitor) for 72 h (mean ± SD; n = 4). *Significant differences for subG1 and S phase when MEK compared to ERK inhibitor treatment (p < 0.05).
Figure 2.
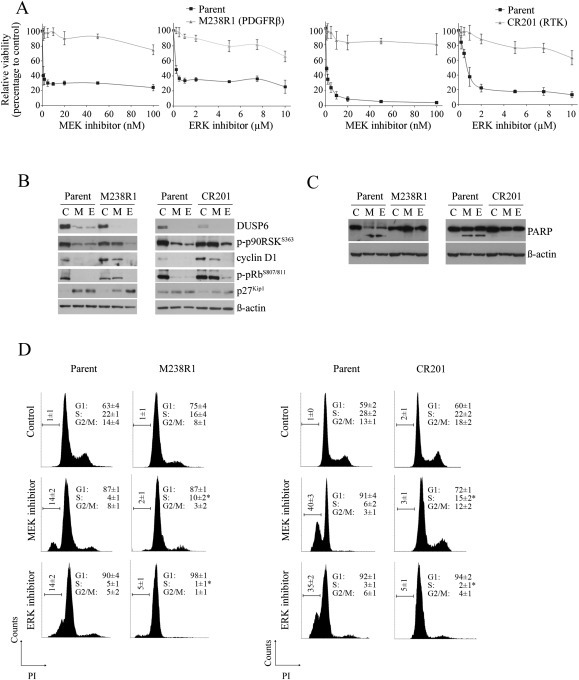
Resistant melanomas with RTK activation retain anti‐proliferative sensitivity to ERK but not MEK inhibition with no apoptotic sensitivity to either inhibitor A. Viability curves of the parental and isogenic melanoma sublines treated with indicated concentrations of the MEK inhibitor trametinib and ERK inhibitor VX‐11e for 72 h (relative to DMSO‐treated controls; mean ± SD; n = 2). B. Parental and BRAF inhibitor‐resistant sublines were treated with DMSO (C), 10 nM MEK inhibitor trametinib (M) or 10 μM ERK inhibitor VX‐11e (E) for 24 h. Western blots of lysates showing protein markers of MAPK activity and cell cycle progression. C. PARP cleavage was determined 72 h after treating sublines with inhibitors as described above. Both full length and major cleaved PARP proteins shown. D. Cell cycle distribution of the indicated cell lines treated with either DMSO (control), 10 nM trametinib (MEK inhibitor) or 10 μM VX‐11e (ERK inhibitor) for 72 h (mean ± SD; n = 4). *Significant differences for subG1 and S phase when MEK compared to ERK inhibitor treatment (p < 0.05).
3.3. Activity of MEK and ERK inhibition in BRAF inhibitor resistance cell lines
Both MEK and ERK inhibitors overcame BRAF inhibitor‐resistance conferred by BRAF splice variants. The response of the BR4 subline (expresses BRAF variant lacking exons 4–10) (Figure S1A), to MEK and ERK inhibition, was analogous to the response of the sensitive, parental SKMel28 cells (Figure 1A). Both inhibitors produced similar anti‐proliferative effects as demonstrated by reduced accumulation of cyclin D1, activation of the retinoblastoma protein (pRb; i.e, loss of hyperphosphorylated pRb) and accumulation of the cyclin‐dependent kinase inhibitor p27Kip1 (Figure 1B). Moreover, both inhibitors induced comparable levels of apoptosis as shown by an increase in the subG1 population and cleaved PARP (poly (ADP‐ribose) polymerase‐1), a biomarker of apoptosis (Figure 1C and D). Similarly, the patient‐derived cell line WMD009 (expresses BRAF variant lacking exons 2–10, Figure S1A) was sensitive to both MEK and ERK inhibition (Figure S3).
The N‐RASQ61H‐driven BR2 subline (Carlino et al., 2013) displayed intermediate sensitivity to MEK and ERK inhibition (Figure 1A), and this was associated with weak induction of apoptosis (Figure 1C and D). The ERK inhibitor consistently induced greater proliferative arrest in this N‐RAS mutant cell line, and this was associated with stronger MAPK inhibition (Figure 1B and D). In particular, although accumulation of the ERK transcriptional target, dual specificity phosphatase 6 (DUSP6) was inhibited by both MEK and ERK inhibition, only the ERK inhibitor suppressed the ERK‐dependent phosphorylation of p90 ribosomal S6 kinase (p90RSK) and induced the activation of pRb and accumulation of p27Kip1 (Figure 1B).
Two models of RTK activation driven resistance, the PDGFRβ overexpressing M238R1 (Nazarian et al., 2010) and CR201 with activation of several RTKs, including EGFR (Figure S1B) (Carlino et al., 2013) were examined. MEK and ERK inhibitors did not circumvent BRAF inhibitor resistance mediated by RTK activation (Figure 2A). The level of inhibitor activity was not identical however; ERK inhibition was more effective in suppressing MAPK activity in both the RTK‐activated melanoma cell models. This was most evident when comparing p90RSK phosphorylation and p27Kip1 accumulation, which were substantially affected by ERK, but not MEK inhibition. Consequently, only the ERK inhibitor promoted loss of cyclin D1, diminished pRb phosphorylation and induced proliferative arrest (Figure 2B and D). Nevertheless, cell cycle arrest did not lead to apoptosis in these RTK‐activated models and there was no evidence of PARP cleavage or accumulation of a subG1 cell population in response to ERK inhibition (Figure 2C, D). The MEK inhibitor promoted only modest changes in MAPK targets, and induced minimal cell cycle inhibition and no apoptosis (Figure 2B–D).
Melanoma cells with mutant MEK1 (MEK1E203K mutation in the patient 1‐derived cell line, Figure S1C) and amplified BRAF (SMU030 patient‐derived cell line, Figure S1D) were also more responsive to ERK inhibition, compared to MEK inhibition. Single agent MEK inhibitor partially suppressed MAPK signalling but was unable to induce substantial proliferative arrest or apoptosis. In contrast, ERK inhibition effectively suppressed MAPK activity, and promoted proliferative arrest and apoptosis in both of these models (Figure 3A–C).
Figure 3.
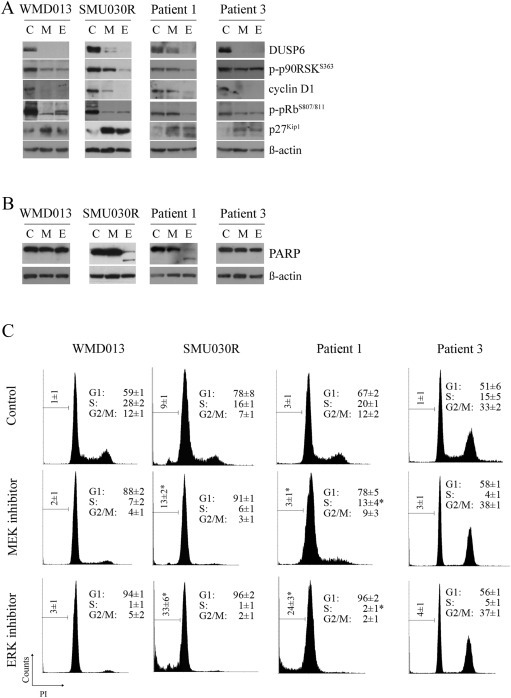
Patient‐derived short‐term melanoma cell lines with acquired resistance to BRAF inhibitors arrest in response to ERK inhibition A. Patient derived MAPK resistant cell lines were treated with DMSO (C), 10 nM MEK inhibitor trametinib (M) or 10 μM ERK inhibitor VX‐11e (E) for 24 h. Western blots of lysates showing protein markers of MAPK activity and cell cycle progression. B. PARP cleavage was determined 72 h after treating cells with inhibitors as described above. Both full length and major cleaved PARP proteins shown. C. Cell cycle distribution of the indicated cell lines treated with either DMSO (control), 10 nM trametinib (MEK inhibitor) or 10 μM VX‐11e (ERK inhibitor) for 72 h (mean ± SD; n = 4). *Significant differences for subG1 and S phase when MEK compared to ERK inhibitor treatment (p < 0.05).
The WMD013 and the patient 3‐derived cell lines have undefined mechanisms of resistance. These cell lines displayed pronounced proliferative arrest in response to ERK inhibition, moderate cell cycle inhibition in response to MEK inhibition, but no cell death when exposed to either ERK or MEK inhibitors (Figure 3A–C).
To ensure that the differential activities of the MEK and ERK inhibitors did not reflect differences in effective concentrations, we exposed the CR201, RTK‐activated cells to increasing MEK inhibitor concentrations, up to 100 nM (100‐fold the GI50 of the parental cell line). The MEK inhibitor failed to suppress p90RSK or pRb phosphorylation at these increased doses, and was less effective than lower doses (approximately three times the parental GI50) of the ERK inhibitor (Figure S4a). Furthermore, elevated concentrations of MEK inhibitor failed to potently arrest the proliferation of CR201 cells (Figure S4b).
To determine whether the activity of trametinib and VX‐11e was comparable to other MEK and ERK inhibitors we examined the clinically active MEK inhibitor MEK162 (Ascierto et al., 2013), and the ERK inhibitor SCH772984, which has derivatives in early phase clinical trials (Morris et al., 2013). As expected, MEK162 and SCH772984 promoted proliferative arrest and cell death in the sensitive cell line SKMel28 (Figure S5). In the resistant SMU030R cells with amplified BRAF, both types of MEK and ERK inhibitors promoted potent proliferative arrest, but only the ERK inhibitors induced significant cell death (Figure S5). In contrast, no inhibitor promoted death in RTK‐activated CR201 cells or in the patient 3‐derived cell line, which has an unknown mechanism of resistance (Figure S5). These data confirm that MEK162 and SCH772984 show comparable activity to trametinib and VX‐11e, respectively in BRAF inhibitor resistant melanoma cell models.
3.4. ERK inhibition in combination with BRAF and/or MEK inhibition is unable to induce apoptosis in models resistant to single agent ERK inhibitor
The combined inhibition of BRAF and MEK has superior clinical activity to single agent therapy (Flaherty et al., 2012) and preclinical evidence confirms that pathway inhibition at multiple nodes is more effective and can partially overcome single agent resistance in a subset of melanoma cell models (Gowrishankar et al., 2012; Shi et al., 2012). Accordingly, the concurrent inhibition of ERK with BRAF and/or MEK promoted significantly more cell death than single agent therapy in the sensitive cells (Figure S6). Nevertheless, the combination of MAPK inhibitors did not significantly increase cell death in the RTK‐activated CR201 cells or the patient 3‐derived melanoma cells (Figure S6).
3.5. Resistant melanoma cell models survive via activated PI3K signalling
ERK inhibition effectively inhibited proliferation of all nine BRAF‐inhibitor resistant melanoma cell lines, without promoting substantial cell death in five of these models, including those with activated RTK or N‐RAS. The PI3K/AKT signalling pathway is known to modulate cell survival in response to BRAF inhibitors (Shi et al., 2011; Villanueva et al., 2010), and this pathway was examined in these five cell models. Four of these five sublines accumulated high basal levels of activated, phosphorylated AKT (p‐AKT) and/or showed induced AKT phosphorylation in response to ERK inhibition (Figure 4A). Induction of AKT activation was associated with increased phosphorylation of CRAF and/or MEK (Figure 4A). Relief of ERK‐dependent feedback can induce phosphorylation of CRAF and MEK, along with RAS activation (Lito et al., 2012) and we confirmed that ERK inhibition led to RAS‐GTP induction in these resistant cell models (Figure 4B). RAS induction was weakest in the WMD013 cell line, the only subline with relatively low phosphorylated AKT levels (Figure 4A and B).
Figure 4.
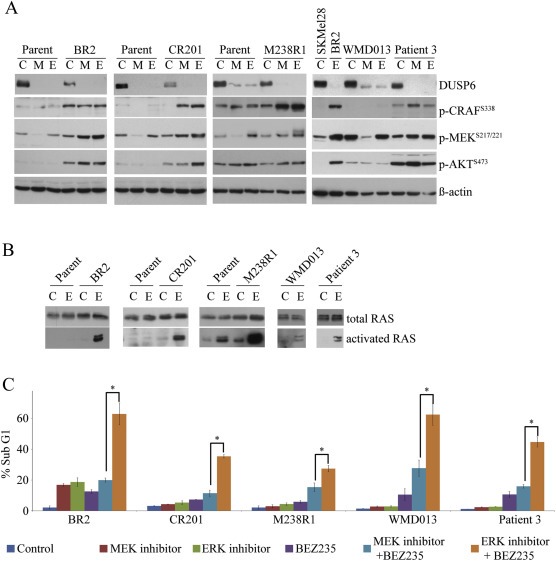
Combination ERK inhibitor with BEZ235 is more active than the MEK inhibitor/BEZ235 combination at promoting cell death A. The indicated cell lines were treated DMSO (C), 10 nM trametinib (M) or 10 μM VX‐11e (E) for 24 h. Western blots of lysates showing protein markers of MAPK and AKT activity. WMD013 and Patient 3 cell lines were directly compared to SKMel28 and BR2 cell lines. B. Whole cell lysates from the indicated cell lines were subjected to pull‐down (PD) assays with GST‐bound CRAF RAS‐binding domain after 24 h treatment with DMSO (C) or 10 μM VX‐11e (E). Whole cell lysates (total RAS) and pull down products (activated RAS) were immunoblotted with a pan‐RAS antibody. C. Histogram showing the percentage sub‐G1 population of cell lines treated with either DMSO (control), 10 nM trametinib (MEK inhibitor), 10 μM VX‐11e (ERK inhibitor), 2 uM BEZ235 or combinations as indicated for 72 h (mean ± SD; n = 4). *Significant differences between the ERK/BEZ235 inhibitor combination compared with the MEK/BEZ235 inhibitor combination (p < 0.05).
The significance of RAS‐PI3K signalling in BRAF‐inhibitor resistance was confirmed using the dual PI3K and mTOR inhibitor BEZ235. As expected, BEZ235 inhibited AKT and downstream ribosomal protein S6 (RPS6) activation in all resistant models (Figure S7). The combination of ERK inhibitor and BEZ235 led to the simultaneous inhibition of MAPK and PI3K signalling and marked induction of cell death (Figure 4C). Importantly, when combined with trametinib, BEZ235 was less active and the trametinib combination consistently promoted lower levels of cell death compared to the ERK inhibitor/BEZ235 combination (Figure 4C).
It is worth noting that combination of BEZ235 and ERK inhibition did not significantly enhance ERK inhibitor activity in four of the five melanoma cells that were initially sensitive to ERK inhibitor alone (i.e, SKMel28, BR4, SMU030R, Patient 1 derived cell line) (Figure S8). In the M238 cells, which show low levels of ERK inhibitor induced cell death (Figure 2), the addition of BEZ235 substantially increased cell death (Figure S8). In keeping with this response to the addition of BEZ235 M238 cells show high basal p‐AKT and RAS‐GTP levels, and an adaptive response to MAPK inhibition involving PDGFRβ up regulation (Shi et al., 2013a) and RAS activation (Figure 4A/B and Figure S8).
Considering the potent activity of the dual PI3K/mTOR inhibitor BEZ235, we examined the contribution of PI3K and mTOR in resistance by utilising selective inhibitors. As shown in Figure S9, the PI3K inhibitor LY294002 was inactive as a single agent, however the combination of LY294002 and VX‐11e increased levels of cell death in the RTK activated CR201 model and the patient 3‐derived cells. In contrast, the mTORC1 inhibitor RAD001, alone or in combination, did not lead to increased levels of cell death, and as expected promoted the phosphorylation of AKT (Deng et al., 2012; O'Reilly et al., 2006) (Figure S9).
4. Discussion
BRAF and MEK inhibitor therapy has substantially advanced the management of BRAFV600‐mutant metastatic melanoma (Chapman et al., 2011; Falchook et al., 2012a; Flaherty et al., 2012; Hauschild et al., 2012). However despite high response rates, resistance to these drugs develops in the majority of patients and represents a major challenge in melanoma clinical practice. Given the frequency of ERK reactivation in resistant tumours (Nazarian et al., 2010; Poulikakos et al., 2011; Shi et al., 2013b) it has been suggested that targeting ERK directly may be effective in acquired BRAF and MEK inhibitor resistance. Preclinical studies have shown activity of ERK inhibitors in BRAF/MEK inhibitor resistance (Hatzivassiliou et al., 2012; Morris et al., 2013). However the apoptotic activity of ERK inhibitors and their relative potency compared to MEK inhibitors has not been examined (Hatzivassiliou et al., 2012; Morris et al., 2013). This comparison is clinically relevant given that the MEK inhibitor trametinib had minimal clinical activity in melanoma patients that progressed on BRAF inhibitor therapy, despite preclinical evidence suggesting MEK inhibitors would be active in a subset of resistance models (Gowrishankar et al., 2012; Kim et al., 2013).
We show that inhibition of ERK, rather than MEK, is more effective at suppressing MAPK activity and inhibiting the proliferation of multiple BRAF inhibitor resistant melanoma cell models. These differences in activity are not due to differences in effective concentration of MEK and ERK inhibitors as the selected doses have comparable anti‐proliferative and apoptotic activity in sensitive melanoma cell lines, and dose escalation of the MEK inhibitor, trametinib did not improve pathway inhibition or alter cell line responses.
The strategy of treating BRAF inhibitor‐resistant melanomas with downstream kinase inhibitors has been disappointing, with no confirmed responses using single‐agent trametinib (Kim et al., 2013). Although, our preclinical data suggests that a subset of resistance mechanisms, particularly BRAF splice variants, should respond to MEK inhibitors, it is clear that BRAF‐inhibitor resistance cannot be overcome by MEK inhibition in the clinical setting (Kim et al., 2013). It is uncertain whether the discrepancy between pre‐clinical and clinical activity of MEK inhibitors is due to limitations in delivering effective MEK inhibitor concentrations and/or intra‐patient heterogeneity of resistance (Romano et al., 2013; Shi et al., 2013b; Van Allen et al., 2013; Wilmott et al., 2012) and if the same discrepancy will be seen with ERK inhibitors.
Clinical studies are underway to determine whether ERK inhibitors can be delivered at concentrations that are clinically effective (NCT01781429, NCT01358331). Given that ERK inhibitors target wild‐type kinases they are likely to have a narrow therapeutic index, and toxicities attributable to MEK inhibition may also be dose limiting with ERK inhibitors. Further, although the combination of BRAF/MEK and ERK inhibitors did not salvage ERK‐inhibitor resistance in vitro, these combinations may still be clinically useful. As seen with BRAF and MEK inhibitor combinations, the addition of an ERK inhibitor to a BRAF inhibitor may be less toxic than either drug alone (Flaherty et al., 2012). Conversely MEK and ERK inhibitors are likely to have overlapping and additive toxicities and as such appear less rational combination partners.
Although ERK inhibition suppressed MAPK activity and produced strong cytostatic effects in all nine BRAF inhibitor‐resistant models tested, it did not induce substantial apoptosis in five of these resistant cell models. Instead the inhibition of ERK caused the relief of ERK‐dependent negative feedback, reactivation of RAS and PI3K/AKT signalling, and the survival of growth‐arrested melanoma cells. The combination of the ERK and PI3K inhibition was therefore effective at promoting cell death in all resistant melanoma cell models, and was substantially more potent than the MEK/PI3K inhibitor combination. This contrasts with the lack of activity seen with combined ERK and mTORC1 inhibition, presumably because inhibition of an mTORC1 mediated negative feedback loop hyperactivates AKT (reviewed in (Kwong and Davies, 2013)).
Our study suggests that ERK inhibitors are likely to only have limited efficacy as sequential therapy in patients previously treated with a BRAF inhibitor. Although MAPK signalling remains critical for the proliferation of BRAF inhibitor resistant melanoma cells, it is not consistently required for the survival of these tumour cells. We found that, in many MAPK resistant models, compensatory PI3K survival networks are active and often induced in response to MAPK inhibition. The combined inhibition of ERK/PI3K and mTOR effectively overcame all models of ERK‐inhibitor resistance in this study, and if clinically deliverable, may be effective against the multiple, heterogeneous resistance mechanisms identified within BRAF‐mutant melanoma patients (Van Allen et al., 2013). Trials are underway combining PI3K inhibitors (NCT01512251, NCT01616199, NCT01363232) or AKT inhibitors (NCT01902173, NCT01519427) with BRAF and/or MEK inhibitors, however, our data suggest that combinations using ERK inhibitors rather than MEK inhibitors may be more active and should be considered during the clinical development of ERK inhibitors.
Disclosures
M.S Carlino: Honoraria to Novartis
P. Hersey: Consultancies and honoraria to GlaxoSmithKline, Roche and Merck
R.F. Kefford: Consultancies to GlaxoSmithKline, Roche, and Merck. Honoraria from Merck
G.V. Long: Consultancies to GlaxoSmithKline, Roche, Novartis. Honoraria from Roche.
All other authors state no conflicts of interest.
Supporting information
The following are the supplementary data related to this article:
Figure S1: Analysis of resistance mechanisms in melanoma cell models. A. Western blots of melanoma cell lysates showing BRAFV600E mutant protein detected by the mutant‐specific antibody VE1. The following melanoma cells were analysed. MelMS (wild type BRAF), SKMel28 (full‐length BRAFV600E), WMD009 (expresses BRAF transcripts lacking exons 2–10) and BR4 (expresses BRAF transcripts lacking exons 4–10) (upper panel). Representative Sanger sequencing traces of truncated BRAF RT‐PCR products derived from the WMD009 cell line, showing the exon 1 splicing junction and BR4 subline, showing the exon 3 splicing junction (lower panel). B. Phosphorylation of receptor tyrosine kinases (RTKs) was analysed in the parent and resistant CR201 subline using phospho‐RTK antibody arrays. The position of phosphorylated EGFR, ErbB3 and AXL are indicated (upper panel). Viability curves of the parent and the resistant CR201 subline treated with the indicated drug doses for 72 h (relative to DMSO‐treated controls; mean ± SD; n = 2) (lower panel). C. Sanger sequencing trace of MEK1 RT‐PCR products isolated from the vemurafenib‐resistant patient 1‐derived melanoma cell line. D. Two short‐term cell cultures were generated from a single patient (SMU030) treated with dabrafenib and trametinib; a pre‐treatment SMU030P and resistant, progressing SMU030R cell line (with 21 ± 1 BRAF gene copies; data not shown). Western blot analyses showing increased BRAF protein levels in the resistant SMU030R cell lines compared to the SMU030P and SKMel28 melanoma cells (left panel). Cell cycle distribution of the SMU030R cell line treated with DMSO (control), 100 nM dabrafenib, or 100 nM dabrafenib combined with 10 nM trametinib (mean ± SD; n = 4) (right panel).
Figure S2: MEK and ERK inhibitor activity in a panel of sensitive BRAF‐mutant melanoma cell lines Viability curves of BRAF‐mutant melanoma cell lines treated with the indicated concentrations of trametinib or VX‐11e for 72 h (relative to DMSO‐treated controls; mean ± SD; n = 2).
Figure S3: Response of patient derived WMD009 (expresses BRAF transcripts lacking exons 2–10) cell line to MEK and ERK inhibitor A. WMD009 cells were treated with DMSO (C), 10 nM trametinib (M) or 10 μM VX‐11e (E) for 24 h. Western blots of lysates showing protein markers of MAPK activity and cell cycle progression. B. PARP cleavage was determined 72 h after treating with inhibitors as described above. Both full length and major cleaved PARP proteins shown. C. Cell cycle distribution of WMD009 cells treated with DMSO (control), 10 nM trametinib (MEK inhibitor) or 10 μM VX‐11e (ERK inhibitor) for 72 h (mean ± SD; n = 4). Differences in subG1 and S phase between MEK and ERK treatment are not statistically different.
Figure S4: Impact of MEK inhibitor dose escalation A. CR201 cells were treated with DMSO (C), trametinib (M) or VX‐11e (E) at the indicated concentrations for 24 h. Western blots of lysates showing protein markers of MAPK activity and cell cycle progression. B. Cell cycle distribution of CR201 cells treated with DMSO (control), trametinib (MEK inhibitor) or VX‐11e (ERK inhibitor) at the indicated concentrations for 72 h (mean ± SD; n = 4).
Figure S5: MEK162 and SCH772984 have comparable activity to that seen with trametinib and VX‐11e A. Indicated cell lines were treated with DMSO (control), MEK162 (500 nM) or SCH772984 (SCH984, 500 nM) for 24 h. Western blots of lysates showing protein markers of MAPK activity and cell cycle progression B. Cell cycle distribution of indicated cells treated with DMSO (control), MEK162 (500 nM) or SCH772984 (500 nM) for 72 h (mean ± SD; n = 4). *Significant differences for subG1 and S phase when MEK162 compared to SCH772984 inhibitor treatment (p < 0.05).
Figure S6: ERK inhibition in combination with BRAF and/or MEK inhibitor(s) is unable to induce cell death in models resistant to single agent ERK inhibition Histogram showing the percentage subG1 population of cell lines treated with either DMSO (control), 100 nM dabrafenib (BRAF inhibitor), 10 nM trametinib (MEK inhibitor), 10 μM VX‐11e (ERK inhibitor) or the indicated combination for 72 h (mean ± SD; n = 4). *Significant differences between single agent ERK inhibitor and combinations (p < 0.05).
Figure S7: BEZ235 suppresses PI3K/AKT pathway activity in ERK inhibitor resistance models MAPK‐inhibitor resistant cell lines were treated with DMSO (−), 10 nM trametinib (MEK inhibitor), 10 μM VX‐11e (ERK inhibitor), 2 μM BEZ235 or the combinations indicated for 24 h. Western blots of lysates showing protein markers of MAPK and AKT activity.
Figure S8: Addition of BEZ235 does not result in significant increases in cell death in models sensitive to single agent ERK inhibition. Histogram showing the percentage subG1 population of cell lines treated with either DMSO (control), 10 nM trametinib (MEK inhibitor), 10 μM VX‐11e (ERK inhibitor), 2uM BEZ235 or combinations as indicated for 72 h (mean ± SD; n = 4). *Significant differences between the MEK or ERK inhibitor compared to combinations with BEZ235 (p < 0.05).
Figure S9: PI3K inhibition cooperates with ERK inhibition to increase cell death in ERK inhibitor resistant melanoma cell A. CR201 and the patient 3 derived cell lines treated with DMSO (control), 10 μM VX‐11e, 10 μM LY294002, or 20 nM RAD001 or the indicated combinations indicated for 24 h. Western blots of lysates showing protein markers of MAPK and AKT activity. B. Histogram showing the percentage subG1 population of CR201 and the patient 3 derived cell line treated with either DMSO (control), 10 μM VX‐11e, 10 μM LY294002, or 20 nM RAD001 or combinations as indicated for 72h (mean ± SD; n = 4). *Significant differences between single agent ERK inhibitor and combinations (p < 0.05).
Acknowledgements
We thank Therese Becker, Mal Irvine and Suzanah Boyd for technical support and are grateful to Roger Lo for providing the M238 and M238R1 cell lines and Professor Andreas von Deimling (University of Heidelberg) who kindly provided the VE1 antibody. The support of the Melanoma Foundation of the University of Sydney and colleagues from Melanoma Institute Australia and the Department of Anatomical Pathology at the Royal Prince Alfred Hospital are also gratefully acknowledged.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.01.003.
Carlino Matteo S., Todd Jason R., Gowrishankar Kavitha, Mijatov Branka, Pupo Gulietta M., Fung Carina, Snoyman Stephanie, Hersey Peter, Long Georgina V., Kefford Richard F. and Rizos Helen, (2014), Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.01.003.
†
This work was supported by Program Grant 633004 and project grants of the National Health and Medical Research Council of Australia (NHMRC) and an infrastructure grant to Westmead Millennium Institute by the Health Department of NSW through Sydney West Area Health Service. This work was supported by the Sydney West Translational Cancer Research Centre, which is funded by the Cancer Institute NSW. Westmead Institute for Cancer Research is the recipient of capital grant funding from the Australian Cancer Research Foundation. H. Rizos and G.V. Long are recipients of Cancer Institute New South Wales, Research Fellowship and H. Rizos is supported by an NHMRC Senior Research Fellowship. M.S. Carlino is supported by a Rotary Health Australia scholarship.
References
- Aronov, A.M. , Tang, Q. , Martinez-Botella, G. , Bemis, G.W. , Cao, J. , Chen, G. , Ewing, N.P. , Ford, P.J. , Germann, U.A. , Green, J. , Hale, M.R. , Jacobs, M. , Janetka, J.W. , Maltais, F. , Markland, W. , Namchuk, M.N. , Nanthakumar, S. , Poondru, S. , Straub, J. , ter Haar, E. , Xie, X. , 2009. Structure-guided design of potent and selective pyrimidylpyrrole inhibitors of extracellular signal-regulated kinase (ERK) using conformational control. J. Med. Chem.. 52, 6362–6368. [DOI] [PubMed] [Google Scholar]
- Ascierto, P.A. , Schadendorf, D. , Berking, C. , Agarwala, S.S. , van Herpen, C.M. , Queirolo, P. , Blank, C.U. , Hauschild, A. , Beck, J.T. , St-Pierre, A. , Niazi, F. , Wandel, S. , Peters, M. , Zubel, A. , Dummer, R. , 2013. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol.. 14, 249–256. [DOI] [PubMed] [Google Scholar]
- Carlino, M.S. , Gowrishankar, K. , Saunders, C.A.B. , Pupo, G.M. , Snoyman, S. , Zhang, X.D. , Saw, R. , Becker, T.M. , Kefford, R.F. , Long, G.V. , Rizos, H. , 2013. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol. Cancer Ther.. 12, 1332–1342. [DOI] [PubMed] [Google Scholar]
- Chapman, P.B. , Hauschild, A. , Robert, C. , Haanen, J.B. , Ascierto, P. , Larkin, J. , Dummer, R. , Garbe, C. , Testori, A. , Maio, M. , Hogg, D. , Lorigan, P. , Lebbe, C. , Jouary, T. , Schadendorf, D. , Ribas, A. , O'Day, S.J. , Sosman, J.A. , Kirkwood, J.M. , Eggermont, A.M.M. , Dreno, B. , Nolop, K. , Li, J. , Nelson, B. , Hou, J. , Lee, R.J. , Flaherty, K.T. , McArthur, A.,G. , 2011. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med.. 364, 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran, R.B. , Dias-Santagata, D. , Bergethon, K. , Iafrate, A.J. , Settleman, J. , Engelman, J.A. , 2010. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAFV600E mutation. Sci. Signal. 3, ra84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, H. , Bignell, G.R. , Cox, C. , Stephens, P. , Edkins, S. , Clegg, S. , Teague, J. , Woffendin, H. , Garnett, M.J. , Bottomley, W. , Davis, N. , Dicks, E. , Ewing, R. , Floyd, Y. , Gray, K. , Hall, S. , Hawes, R. , Hughes, J. , Kosmidou, V. , Menzies, A. , Mould, C. , Parker, A. , Stevens, C. , Watt, S. , Hooper, S. , Wilson, R. , Jayatilake, H. , Gusterson, B.A. , Cooper, C. , Shipley, J. , Hargrave, D. , Pritchard-Jones, K. , Maitland, N. , Chenevix-Trench, G. , Riggins, G.J. , Bigner, D.D. , Palmieri, G. , Cossu, A. , Flanagan, A. , Nicholson, A. , Ho, J.W. , Leung, S.Y. , Yuen, S.T. , Weber, B.L. , Seigler, H.F. , Darrow, T.L. , Paterson, H. , Marais, R. , Marshall, C.J. , Wooster, R. , Stratton, M.R. , Futreal, P.A. , 2002. Mutations of the BRAF gene in human cancer. Nature. 417, 949–954. [DOI] [PubMed] [Google Scholar]
- Deng, W. , Gopal, Y.N. , Scott, A. , Chen, G. , Woodman, S.E. , Davies, M.A. , 2012. Role and therapeutic potential of PI3K-mTOR signaling in de novo resistance to BRAF inhibition. Pigment Cell Melanoma Res.. 25, 248–258. [DOI] [PubMed] [Google Scholar]
- Falchook, G.S. , Lewis, K.D. , Infante, J.R. , Gordon, M.S. , Vogelzang, N.J. , DeMarini, D.J. , Sun, P. , Moy, C. , Szabo, S.A. , Roadcap, L.T. , Peddareddigari, V.G. , Lebowitz, P.F. , Le, N.T. , Burris, H.A. , Messersmith, W.A. , O'Dwyer, P.J. , Kim, K.B. , Flaherty, K. , Bendell, J.C. , Gonzalez, R. , Kurzrock, R. , Fecher, L.A. , 2012. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol.. 13, 782–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchook, G.S. , Long, G.V. , Kurzrock, R. , Kim, K.B. , Arkenau, T.H. , Brown, M.P. , Hamid, O. , Infante, J.R. , Millward, M. , Pavlick, A.C. , O'Day, S.J. , Blackman, S.C. , Curtis, C.M. , Lebowitz, P. , Ma, B. , Ouellet, D. , Kefford, R.F. , 2012. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 379, 1893–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty, K.T. , Infante, J.R. , Daud, A. , Gonzalez, R. , Kefford, R.F. , Sosman, J. , Hamid, O. , Schuchter, L. , Cebon, J. , Ibrahim, N. , Kudchadkar, R. , Burris, H.A. , Falchook, G. , Algazi, A. , Lewis, K. , Long, G.V. , Puzanov, I. , Lebowitz, P. , Singh, A. , Little, S. , Sun, P. , Allred, A. , Ouellet, D. , Kim, K.B. , Patel, K. , Weber, J. , 2012. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med.. 367, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty, K.T. , Puzanov, I. , Kim, K.B. , Ribas, A. , McArthur, G.A. , Sosman, J.A. , O'Dwyer, P.J. , Lee, R.J. , Grippo, J.F. , Nolop, K. , Chapman, P.B. , 2010. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med.. 363, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher, S. , Kefford, R.F. , Rizos, H. , 2005. Enforced expression of p14ARF induces p53-dependent cell cycle arrest but not apoptosis. Cell Cycle. 4, 465–472. [DOI] [PubMed] [Google Scholar]
- Girotti, M.R. , Pedersen, M. , Sanchez-Laorden, B. , Viros, A. , Turajlic, S. , Niculescu-Duvaz, D. , Zambon, A. , Sinclair, J. , Hayes, A. , Gore, M. , Lorigan, P. , Springer, C. , Larkin, J. , Jorgensen, C. , Marais, R. , 2013. Inhibiting EGF receptor or SRC Family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov.. 3, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar, K. , Snoyman, S. , Pupo, G.M. , Becker, T.M. , Kefford, R.F. , Rizos, H. , 2012. Acquired resistance to BRAF inhibition can confer cross-resistance to combined BRAF/MEK inhibition. J. Invest. Dermatol.. 132, 1850–1859. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou, G. , Liu, B. , O'Brien, C. , Spoerke, J.M. , Hoeflich, K.P. , Haverty, P.M. , Soriano, R. , Forrest, W.F. , Heldens, S. , Chen, H. , Toy, K. , Ha, C. , Zhou, W. , Song, K. , Friedman, L.S. , Amler, L.C. , Hampton, G.M. , Moffat, J. , Belvin, M. , Lackner, M.R. , 2012. ERK inhibition overcomes acquired resistance to MEK inhibitors. Mol. Cancer Ther.. 11, 1143–1154. [DOI] [PubMed] [Google Scholar]
- Hauschild, A. , Grob, J.-J. , Demidov, L.V. , Jouary, T. , Gutzmer, R. , Millward, M. , Rutkowski, P. , Blank, C.U. , Miller, W.H. , Kaempgen, E. , Martín-Algarra, S. , Karaszewska, B. , Mauch, C. , Chiarion-Sileni, V. , Martin, A.-M. , Swann, S. , Haney, P. , Mirakhur, B. , Guckert, M.E. , Goodman, V. , Chapman, P.B. , 2012. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 380, 358–365. [DOI] [PubMed] [Google Scholar]
- Johannessen, C.M. , Boehm, J.S. , Kim, S.Y. , Thomas, S.R. , Wardwell, L. , Johnson, L.A. , Emery, C.M. , Stransky, N. , Cogdill, A.P. , Barretina, J. , Caponigro, G. , Hieronymus, H. , Murray, R.R. , Salehi-Ashtiani, K. , Hill, D.E. , Vidal, M. , Zhao, J.J. , Yang, X. , Alkan, O. , Kim, S. , Harris, J.L. , Wilson, C.J. , Myer, V.E. , Finan, P.M. , Root, D.E. , Roberts, T.M. , Golub, T. , Flaherty, K.T. , Dummer, R. , Weber, B.L. , Sellers, W.R. , Schlegel, R. , Wargo, J.A. , Hahn, W.C. , Garraway, L.A. , 2010. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 468, 968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.B. , Kefford, R. , Pavlick, A.C. , Infante, J.R. , Ribas, A. , Sosman, J.A. , Fecher, L.A. , Millward, M. , McArthur, G.A. , Hwu, P. , Gonzalez, R. , Ott, P.A. , Long, G.V. , Gardner, O.S. , Ouellet, D. , Xu, Y. , DeMarini, D.J. , Le, N.T. , Patel, K. , Lewis, K.D. , 2013. Phase II study of the MEK1/MEK2 inhibitor trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J. Clin. Oncol.. 31, 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong, L.N. , Davies, M.A. , 2013. Navigating the therapeutic complexity of PI3K pathway inhibition in melanoma. Clin. Cancer Res.. 19, 5310–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, F. , Jiang, C.C. , Farrelly, M.L. , Zhang, X.D. , Hersey, P. , 2012. Evidence for upregulation of Bim and the splicing factor SRp55 in melanoma cells from patients treated with selective BRAF inhibitors. Melanoma Res.. 22, 244–251. [DOI] [PubMed] [Google Scholar]
- Lito, P. , Pratilas, Christine A. , Joseph, Eric W. , Tadi, M. , Halilovic, E. , Zubrowski, M. , Huang, A. , Wong, Wai L. , Callahan, Margaret K. , Merghoub, T. , Wolchok, Jedd D. , de Stanchina, E. , Chandarlapaty, S. , Poulikakos, Poulikos I. , Fagin, James A. , Rosen, N. , 2012. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 22, 668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, G.V. , Wilmott, J.S. , Capper, D. , Preusser, M. , Zhang, Y.E. , Thompson, J.F. , Kefford, R.F. , von Deimling, A. , Scolyer, R.A. , 2013. Immunohistochemistry is highly sensitive and specific for the detection of V600E BRAF mutation in melanoma. Am. J. Surg. Pathol.. 37, 61–65. [DOI] [PubMed] [Google Scholar]
- Montagut, C. , Sharma, S.V. , Shioda, T. , McDermott, U. , Ulman, M. , Ulkus, L.E. , Dias-Santagata, D. , Stubbs, H. , Lee, D.Y. , Singh, A. , Drew, L. , Haber, D.A. , Settleman, J. , 2008. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res.. 68, 4853–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, E.J. , Jha, S. , Restaino, C.R. , Dayananth, P. , Zhu, H. , Cooper, A. , Carr, D. , Deng, Y. , Jin, W. , Black, S. , Long, B. , Liu, J. , Dinunzio, E. , Windsor, W. , Zhang, R. , Zhao, S. , Angagaw, M.H. , Pinheiro, E.M. , Desai, J. , Xiao, L. , Shipps, G. , Hruza, A. , Wang, J. , Kelly, J. , Paliwal, S. , Xiaolei, G. , Boga, S.B. , Zhu, L. , Daublain, P. , Zhang, L. , Lutterbach, B.A. , Pelletire, M.R. , Philippar, U. , Siliphaivanh, P. , Witter, D. , Kirschmeier, P. , Bishop, W.R. , Hicklin, D. , Gilliland, G. , Jayaraman, L. , Zawel, L. , Fawell, S.E. , Samatar, A.A. , 2013. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov.. 29, 29 [DOI] [PubMed] [Google Scholar]
- Nazarian, R. , Shi, H. , Wang, Q. , Kong, X. , Koya, R.C. , Lee, H. , Chen, Z. , Lee, M.K. , Attar, N. , Sazegar, H. , Chodon, T. , Nelson, S.F. , McArthur, G. , Sosman, J.A. , Ribas, A. , Lo, R.S. , 2010. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 468, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly, K.E. , Rojo, F. , She, Q.B. , Solit, D. , Mills, G.B. , Smith, D. , Lane, H. , Hofmann, F. , Hicklin, D.J. , Ludwig, D.L. , Baselga, J. , Rosen, N. , 2006. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res.. 66, 1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos, P.I. , Persaud, Y. , Janakiraman, M. , Kong, X. , Ng, C. , Moriceau, G. , Shi, H. , Atefi, M. , Titz, B. , Gabay, M.T. , Salton, M. , Dahlman, K.B. , Tadi, M. , Wargo, J.A. , Flaherty, K.T. , Kelley, M.C. , Misteli, T. , Chapman, P.B. , Sosman, J.A. , Graeber, T.G. , Ribas, A. , Lo, R.S. , Rosen, N. , Solit, D.B. , 2011. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 480, 387–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano, E. , Pradervand, S. , Paillusson, A. , Weber, J. , Harshman, K. , Muehlethaler, K. , Speiser, D.E. , Peters, S. , Rimoldi, D. , Michielin, O. , 2013. Identification of multiple mechanisms of resistance to vemurafenib in a patient with BRAFV600E-mutated cutaneous melanoma successfully rechallenged after progression. Clin. Cancer Res. 19, 5749–5757. [DOI] [PubMed] [Google Scholar]
- Shi, H. , Hong, A. , Kong, X. , Koya, R.C. , Song, C. , Moriceau, G. , Hugo, W. , Yu, C.C. , Ng, C. , Chodon, T. , Scolyer, R.A. , Kefford, R.F. , Ribas, A. , Long, G.V. , Lo, R.S. , 2013. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 4, 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, H. , Hugo, W. , Kong, X. , Hong, A. , Koya, R.C. , Moriceau, G. , Chodon, T. , Guo, R. , Johnson, D.B. , Dahlman, K.B. , Kelley, M.C. , Kefford, R.F. , Chmielowski, B. , Glaspy, J.A. , Sosman, J.A. , van Baren, N. , Long, G.V. , Ribas, A. , Lo, R.S. , 2013. Acquired resistance and Clonal Evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 4, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, H. , Kong, X. , Ribas, A. , Lo, R.S. , 2011. Combinatorial treatments that overcome PDGFR -Driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res.. 71, 5067–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, H. , Moriceau, G. , Kong, X. , Lee, M.-K. , Lee, H. , Koya, R.C. , Ng, C. , Chodon, T. , Scolyer, R.A. , Dahlman, K.B. , Sosman, J.A. , Kefford, R.F. , Long, G.V. , Nelson, S.F. , Ribas, A. , Lo, R.S. , 2012. Melanoma whole-exome sequencing identifies V600EB-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun.. 3, 724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen, E.M. , Wagle, N. , Sucker, A. , Treacy, D.J. , Johannessen, C.M. , Goetz, E.M. , Place, C.S. , Taylor-Weiner, A. , Whittaker, S. , Kryukov, G.V. , Hodis, E. , Rosenberg, M. , McKenna, A. , Cibulskis, K. , Farlow, D. , Zimmer, L. , Hillen, U. , Gutzmer, R. , Goldinger, S.M. , Ugurel, S. , Gogas, H.J. , Egberts, F. , Berking, C. , Trefzer, U. , Loquai, C. , Weide, B. , Hassel, J.C. , Gabriel, S.B. , Carter, S.L. , Getz, G. , Garraway, L.A. , Schadendorf, D. , on behalf of the Dermatologic Cooperative Oncology Group of, G2013. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 4, 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva, J. , Vultur, A. , Lee, J.T. , Somasundaram, R. , Fukunaga-Kalabis, M. , Cipolla, A.K. , Wubbenhorst, B. , Xu, X. , Gimotty, P.A. , Kee, D. , Santiago-Walker, A.E. , Letrero, R. , D'Andrea, K. , Pushparajan, A. , Hayden, J.E. , Brown, K.D. , Laquerre, S. , McArthur, G.A. , Sosman, J.A. , Nathanson, K.L. , Herlyn, M. , 2010. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 18, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle, N. , Emery, C. , Berger, M.F. , Davis, M.J. , Sawyer, A. , Pochanard, P. , Kehoe, S.M. , Johannessen, C.M. , Macconaill, L.E. , Hahn, W.C. , Meyerson, M. , Garraway, L.A. , 2011. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol.. 29, 3085–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmott, J.S. , Tembe, V. , Howle, J.R. , Sharma, R. , Thompson, J.F. , Rizos, H. , Lo, R.S. , Kefford, R.F. , Scolyer, R.A. , Long, G.V. , 2012. Intratumoral molecular heterogeneity in a BRAF-mutant, BRAF inhibitor-resistant melanoma: a case illustrating the challenges for personalized medicine. Mol. Cancer Ther.. 11, 2704–2708. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Figure S1: Analysis of resistance mechanisms in melanoma cell models. A. Western blots of melanoma cell lysates showing BRAFV600E mutant protein detected by the mutant‐specific antibody VE1. The following melanoma cells were analysed. MelMS (wild type BRAF), SKMel28 (full‐length BRAFV600E), WMD009 (expresses BRAF transcripts lacking exons 2–10) and BR4 (expresses BRAF transcripts lacking exons 4–10) (upper panel). Representative Sanger sequencing traces of truncated BRAF RT‐PCR products derived from the WMD009 cell line, showing the exon 1 splicing junction and BR4 subline, showing the exon 3 splicing junction (lower panel). B. Phosphorylation of receptor tyrosine kinases (RTKs) was analysed in the parent and resistant CR201 subline using phospho‐RTK antibody arrays. The position of phosphorylated EGFR, ErbB3 and AXL are indicated (upper panel). Viability curves of the parent and the resistant CR201 subline treated with the indicated drug doses for 72 h (relative to DMSO‐treated controls; mean ± SD; n = 2) (lower panel). C. Sanger sequencing trace of MEK1 RT‐PCR products isolated from the vemurafenib‐resistant patient 1‐derived melanoma cell line. D. Two short‐term cell cultures were generated from a single patient (SMU030) treated with dabrafenib and trametinib; a pre‐treatment SMU030P and resistant, progressing SMU030R cell line (with 21 ± 1 BRAF gene copies; data not shown). Western blot analyses showing increased BRAF protein levels in the resistant SMU030R cell lines compared to the SMU030P and SKMel28 melanoma cells (left panel). Cell cycle distribution of the SMU030R cell line treated with DMSO (control), 100 nM dabrafenib, or 100 nM dabrafenib combined with 10 nM trametinib (mean ± SD; n = 4) (right panel).
Figure S2: MEK and ERK inhibitor activity in a panel of sensitive BRAF‐mutant melanoma cell lines Viability curves of BRAF‐mutant melanoma cell lines treated with the indicated concentrations of trametinib or VX‐11e for 72 h (relative to DMSO‐treated controls; mean ± SD; n = 2).
Figure S3: Response of patient derived WMD009 (expresses BRAF transcripts lacking exons 2–10) cell line to MEK and ERK inhibitor A. WMD009 cells were treated with DMSO (C), 10 nM trametinib (M) or 10 μM VX‐11e (E) for 24 h. Western blots of lysates showing protein markers of MAPK activity and cell cycle progression. B. PARP cleavage was determined 72 h after treating with inhibitors as described above. Both full length and major cleaved PARP proteins shown. C. Cell cycle distribution of WMD009 cells treated with DMSO (control), 10 nM trametinib (MEK inhibitor) or 10 μM VX‐11e (ERK inhibitor) for 72 h (mean ± SD; n = 4). Differences in subG1 and S phase between MEK and ERK treatment are not statistically different.
Figure S4: Impact of MEK inhibitor dose escalation A. CR201 cells were treated with DMSO (C), trametinib (M) or VX‐11e (E) at the indicated concentrations for 24 h. Western blots of lysates showing protein markers of MAPK activity and cell cycle progression. B. Cell cycle distribution of CR201 cells treated with DMSO (control), trametinib (MEK inhibitor) or VX‐11e (ERK inhibitor) at the indicated concentrations for 72 h (mean ± SD; n = 4).
Figure S5: MEK162 and SCH772984 have comparable activity to that seen with trametinib and VX‐11e A. Indicated cell lines were treated with DMSO (control), MEK162 (500 nM) or SCH772984 (SCH984, 500 nM) for 24 h. Western blots of lysates showing protein markers of MAPK activity and cell cycle progression B. Cell cycle distribution of indicated cells treated with DMSO (control), MEK162 (500 nM) or SCH772984 (500 nM) for 72 h (mean ± SD; n = 4). *Significant differences for subG1 and S phase when MEK162 compared to SCH772984 inhibitor treatment (p < 0.05).
Figure S6: ERK inhibition in combination with BRAF and/or MEK inhibitor(s) is unable to induce cell death in models resistant to single agent ERK inhibition Histogram showing the percentage subG1 population of cell lines treated with either DMSO (control), 100 nM dabrafenib (BRAF inhibitor), 10 nM trametinib (MEK inhibitor), 10 μM VX‐11e (ERK inhibitor) or the indicated combination for 72 h (mean ± SD; n = 4). *Significant differences between single agent ERK inhibitor and combinations (p < 0.05).
Figure S7: BEZ235 suppresses PI3K/AKT pathway activity in ERK inhibitor resistance models MAPK‐inhibitor resistant cell lines were treated with DMSO (−), 10 nM trametinib (MEK inhibitor), 10 μM VX‐11e (ERK inhibitor), 2 μM BEZ235 or the combinations indicated for 24 h. Western blots of lysates showing protein markers of MAPK and AKT activity.
Figure S8: Addition of BEZ235 does not result in significant increases in cell death in models sensitive to single agent ERK inhibition. Histogram showing the percentage subG1 population of cell lines treated with either DMSO (control), 10 nM trametinib (MEK inhibitor), 10 μM VX‐11e (ERK inhibitor), 2uM BEZ235 or combinations as indicated for 72 h (mean ± SD; n = 4). *Significant differences between the MEK or ERK inhibitor compared to combinations with BEZ235 (p < 0.05).
Figure S9: PI3K inhibition cooperates with ERK inhibition to increase cell death in ERK inhibitor resistant melanoma cell A. CR201 and the patient 3 derived cell lines treated with DMSO (control), 10 μM VX‐11e, 10 μM LY294002, or 20 nM RAD001 or the indicated combinations indicated for 24 h. Western blots of lysates showing protein markers of MAPK and AKT activity. B. Histogram showing the percentage subG1 population of CR201 and the patient 3 derived cell line treated with either DMSO (control), 10 μM VX‐11e, 10 μM LY294002, or 20 nM RAD001 or combinations as indicated for 72h (mean ± SD; n = 4). *Significant differences between single agent ERK inhibitor and combinations (p < 0.05).