Non-Coding Transcription Instructs Cohesin-Dependent Chromatin Folding and Compartmentalization to Dictate Enhancer-Promoter Communication and T Cell Fate (original) (raw)
. Author manuscript; available in PMC: 2018 Sep 21.
SUMMARY
It is now established that Bcl11b specifies T cell fate. Here we show that in developing T-cells the Bcl11b enhancer repositioned from the lamina to the nuclear interior. Our search for factors that relocalized the Bcl11b enhancer identified a non-coding RNA named ThymoD (Thymocyte Differentiation Factor). ThymoD-deficient mice displayed a block at the onset of T cell development and developed lymphoid malignancies. We found that ThymoD transcription promoted demethylation at CTCF bound sites and activated cohesin-dependent looping to reposition the Bcl11b enhancer from the lamina to the nuclear interior and to juxtapose the Bcl11b enhancer and promoter into a single loop domain. These large-scale changes in nuclear architecture were associated with the deposition of activating epigenetic marks across the loop domain, plausibly facilitating phase separation. These data indicate how during developmental progression and tumor suppression non-coding transcription orchestrates chromatin folding and compartmentalization to direct with high precision enhancer-promoter communication.
Graphical Abstract
An enhancer RNA called ThymoD facilitates transcription of T cell specific genes by bringing to close proximity the locus control region and promoter of a key lineage-specifying transcription factor.
INTRODUCTION
The differentiation of T cells is orchestrated in the thymus. Upon exposure to Delta-Notch signaling, early T cell progenitors (ETPs) differentiate into multipotent DN2a cells, which in turn, develop into committed DN2b cells. DN2b cells subsequently progress into DN3a cells in which TCRβ VDJ rearrangement is initiated. Once a productive TCRβ chain has been assembled, DN3b cells expand and differentiate into CD4+CD8+ double positive (DP) thymocytes. In the DP compartment, thymocytes die by either neglect or negative selection or persist through positive selection to differentiate into CD4 single positive (CD4SP) or CD8SP cells (Klein et al., 2014; Naito et al., 2011).
The developmental progression of T cells is regulated by the combined activities of an ensemble of transcriptional regulators. T-lineage development is initiated by the E-proteins that activate the expression of genes encoding components involved in Notch signaling (Bain et al., 1998; Ikawa et al., 2006; Miyazaki et al., 2017). Once instructed to respond to Notch signaling T cell progenitors activate the expression of Bcl11b, GATA-3 and TCF1 (Yui and Rothenberg, 2014). Specifically, Bcl11b expression is initiated at the DN2a cell stage to promote developmental progression to the DN2b cell stage. At the DN2b cell stage Bcl11b expression is further elevated and in concert with E2A activates a T-lineage specific program of gene expression and suppresses the expression of genes associated with alternative cell fates (Liu et al., 2010; Ikawa et al., 2010; Li et al., 2011; Longabaugh et al., 2017). The activation of Bcl11b expression in DN2 cells involves Notch signaling, GATA-3, TCF1 and RUNX1 that bind to an enhancer, named Major Peak, located in the Bcl11b intergenic locus control region (Guo et al., 2008; Weber et al., 2013; Garcia-Ojedo et al., 2013; Li et al., 2013). Recent elegant studies indicated that full activation of Bcl11b expression in developing T cell progenitors requires a rate-limiting transition from an inactive to an active chromatin state (Kueh et al., 2016).
Here we have examined how Bcl11b expression is activated to establish T cell fate and suppress the development of lymphoid malignancies. We found that in developing T cell progenitors the Bcl11b locus control region, containing a well-characterized enhancer, repositioned from the lamina to the nuclear interior. The repositioning of the Bcl11b enhancer was orchestrated by a non-coding RNA, named ThymoD (Thymocyte Differentiation Factor). ThymoD transcription promoted demethylation at sites associated with CTCF occupancy across the transcribed region and activated cohesin-dependent looping, plausibly involving loop extrusion, to bring the Bcl11b promoter and enhancer into a single loop domain. These results are consistent with a model in which non-coding transcription dictates enhancer-promoter communication at multiple levels: (i) demethylation of CpG residues across the ThymoD transcribed region to permit CTCF occupancy, (ii) recruitment of the cohesin complex to the transcribed region to activate cohesin-dependent looping, (iii) loop extrusion to juxtapose with great precision the enhancer and promoter into a single loop domain, (iv) repositioning the enhancer from a heterochromatic to an euchromatic environment and (v) permitting the deposition of activating epigenetic marks across the loop domain to facilitate phase separation.
RESULTS
The Bcl11b Locus Control Region Repositions in Developing T Cell Progenitors
In previous studies we demonstrated that in multipotent progenitors the Bcl11b intergenic region was associated with the transcriptionally repressive compartment (Lin et al., 2012). To explore the possibility that the Bcl11b locus repositioned in developing thymocytes, adult hematopoietic progenitors were isolated from the bone marrow and cultured on an OP9-DL1 stromal layer in the presence of IL7 and FLT3L (Schmitt et al., 2002). Cells were harvested upon reaching the DN2 cell stage and examined for nuclear architecture using HiC (Table S1) (Lieberman-Aiden et al., 2009; Rao et al., 2014). To identify genomic regions that repositioned in developing T cell progenitors, we segregated the DN2 genome into transcriptionally permissive (A) and transcriptionally repressive (B) compartments using principal component analysis. We found that during the transition from the multipotent progenitor to the committed DN2 cell stage a wide spectrum of genomic regions switched compartments (data not shown). Conspicuous among the genomic regions that repositioned was an intergenic region containing the Bcl11b enhancer (Figure 1A) (Li et al., 2013). Specifically, we found that the Bcl11b intergenic region repositioned from the transcriptionally repressive compartment B to the transcriptionally permissive compartment A (Figure 1A). Notably, the repositioning of the Bcl11b intergenic region was associated with de novo genomic interactions involving the Bcl11b enhancer and promoter regions (Figure 1B). To validate these findings, formaldehyde fixed cells were hybridized with fluorescently labeled fosmid probes that span the Bcl11b enhancer. As predicted by the HiC analysis we found that in a large majority of multipotent progenitor cells the Bcl11b intergenic region was sequestered at the nuclear envelope whereas in DN2 cells it was predominantly localized in the nuclear interior (Figure 1C). These data indicate that in developing thymocytes the Bcl11b intergenic region harboring the enhancer repositions from the lamina to the nuclear interior to direct the Bcl11b enhancer to the Bcl11b promoter.
Figure 1. Large-Scale Changes in Nuclear Architecture in Developing T Cell Progenitors.

(A) Normalized genome-wide contact matrices for multipotent progenitors, pro-B cells and DN2 cells of a genomic region located on chromosome 12 are displayed. Top panel shows coding elements (blue) associated with chromosome 12. Positive PC1 values are indicated in black whereas negative PC1 values are displayed in gray. Heatmap indicates the ratio of observed versus expected frequencies for genomic interactions as revealed by HiC. Numerical and statistical details are described in the Methods Section.
(B) Circos diagrams representing genomic interactions across the Bcl11b intergenic region in multipotent progenitors, pro-B cells as well as DN2 cells. Indicated are the Vrk1 and Bcl11b coding regions as well as the Bcl11b intergenic lcous control region. The enhancer is shown in green. Thickness of connecting lines reflects p-values associated with the indicated interactions. Numerical and statistical details are described in the Methods section.
(C) The Bcl11b intergenic region repositions during the developmental progression from multipotent progenitors to DN2 cells from the nuclear lamina to the nuclear interior. Images represent 3D-FISH analysis using a fosmid probe corresponding to the Bcl11b locus control region. The nuclear envelope was visualized using antibodies directed against the lamina and is indicated in red. The two left images show localization for the Bcl11b enhancer at the nuclear lamina in multipotent progenitors. Right images indicate location of the Bcl11b enhancer in the nuclear interior of DN2 cells. DAPI staining is shown in blue. Bar graph shows fraction of spatial distances (<500nm) separating the Bcl11b super-enhancer from the nuclear lamina in multipotent progenitor cells (n=175) and WT DN2 cells (n=135). Images were digitally magnified. Original magnification was ×100. White bar represents 1 μm.
Non-Coding RNA Transcription Initiated within the Bcl11b Intergenic Region Activates Bcl11b Transcription
Previous studies have suggested that intergenic transcription is associated with the repositioning of genomic regions from heterochromatic to euchromatic genomic regions (Schmitt et al., 2005). As a first approach to identify such transcripts in developing T cell progenitors we employed ATAC-Seq and analyzed RNA-Seq reads to detect nucleosome depletion and non-coding transcripts across the Bcl11b intergenic region (Table S1) (Buenrosto et al., 2013; Bossen et al., 2015). This analysis revealed a cluster of ATAC-sensitive sites centered on a previously identified enhancer known to regulate Bcl11b expression (Li et al., 2013) (Figure 2A). Four ATAC-sensitive regions associated with this region were identified as CR1-CR4. CR1 corresponds to the Bcl11b enhancer previously named Major Peak (Li et al., 2013). The roles of CR2-4 were unknown. CR1-4 was depleted of nucleosomes in DN2 cells but not in pro-B cells indicating lineage specificity (Figure S1A). We next examined this region for developmentally regulated non-coding transcripts whose expression preceded and/or overlapped with Bcl11b expression (Zhang et al. 2012). Two distinct non-coding RNAs, previously designated as GM16084, initiated from either CR1 (Major Peak) or CR2 (Figure S1B). We will hereafter refer to GM16084 as ThymoD (Thymocyte Differentiation Factor). At the DN1 cell stage ThymoD transcription was initiated from CR2 immediately prior to the induction of Bcl11b expression (Figure S1B; data not shown). In the DN2 and DN3 cell stages ThymoD transcription was initiated from both CR1 and CR2 (Figure S1B).
Figure 2. Non-Coding RNA ThymoD Activates Bcl11b Expression Across Vast Genomic Distances.

(A) Schematic diagram depicting the ThymoD region. Upper panel shows ATAC seq reads for wild-type DN2 cells across the Bcl11b locus control region. CR1 (Major Peak) indicates the Bcl11b enhancer. CR2-4 refers to genomic regions depleted for nucleosomes in DN2 cells. Positions of pAS insertions are indicated in red.
(B) ThymoD transcription activates with high precision and selectivity Bcl11b expression. ThymoD+/+, ThymoD+/p(A) and ThymoDp(A)/p(A) DN2 cells were examined for chromatin accessibility and nascent transcription using ATAC-Seq and GRO-Seq. ThymoD region is indicated. pAS insertion is shown by red arrow.
(C) pAS insertion abolishes ThymoD transcription. Both GRO-Seq and RNA-Seq tracks are shown for wild-type, ThymoD+/p(A) and ThymoDp(A)/p(A) DN2 cells.
As a first approach to examine potential roles for ThymoD in modulating Bcl11b expression we inserted a poly(A) site upstream of either CR1 or CR2. As a recipient cell line we used SCIDadh representing the DN3 cell stage (Carleton et al., 1999). Upon electroporation of the repair template and gRNAs targeting non-conserved genomic regions either upstream of CR1 or CR2, clones were isolated, expanded and examined for proper poly(A) site insertion. Clones that harbored the poly(A) insertion on both alleles, flanking either CR1 or CR2, were expanded and examined for ThymoD and Bcl11b expression using real-time PCR. We found that insertion of a poly(A) site upstream of CR1 did not interfere with either ThymoD or Bcl11b transcription (Figure S1C and S1D). In contrast, when inserted upstream of CR2 ThymoD as well as Bcl11b transcript abundance significantly declined (Figure S1D). Thus the insertion of a poly(A) site upstream of CR2 but not CR1 interfered with ThymoD and Bcl11b expression.
Generation of ThymoD p(A)/p(A) Mice
To examine the role of ThymoD in thymocyte development we generated mice that carried a poly(A) cassette inserted immediately upstream of CR2 (Figure 2A). gRNAs targeting this region in conjunction with the repair template and Cas9 mRNA were injected into C57Bl/6 zygotes (Figure S2A) (Yang et al., 2013). Mice that carried at least three poly(A) cassettes in tandem in the ThymoD locus were viable and generated offspring at the expected Mendelian ratios (data not shown). To validate for proper insertion, PCR primers were generated that were located adjacent and within the targeting construct (Figure S2A). Fragment size of amplified products was as expected (Figure S2B). Sequenced PCR products verified proper insertion of the p(A) sites (Figure S2B; data not shown). DNA sequencing revealed that the putative regulatory elements CR1-CR4 were not mutated by insertion of the poly(A) cassette (data not shown). To validate that the insertion of poly(A) sites interfered with ThymoD transcription, RNA was isolated from ThymoD +/+, ThymoD +/p(A) and ThymoD p(A)/p(A) thymi. We found that ThymoD transcript levels were severely reduced in thymi isolated from ThymoD p(A)/p(A) mice beyond the p(A) insertion site (Figures S2C and data not shown). To examine how ThymoD transcripts were localized in nuclei derived from DN2 cells, we performed RNA-FISH. ThymoD transcripts were localized in nuclei derived from the majority of wild-type DN2 cells but as expected expressed at low abundance and not detectable in ThymoD poly(A)/poly(A) DN2 cells (data shown).
To determine how p(A) insertion affected the transcription of ThymoD and Bcl11b, we performed GRO-Seq. We found that Bcl11b transcripts as well as transcripts associated with nearby genes, including Vrk1 and Setd3, were readily detectable in wild-type DN2 cells (Figure 2B). To define the transcriptional start sites associated with ThymoD, RNA polymerase II occupancy was analyzed in DN2 cells. Both CR1 and CR2 were enriched for RNA polymerase II occupancy (Figure 2C; lowest panel). Using 3′-RACE we confirmed that CR1 and CR2 act as promoter elements (data not shown). ThymoD sense- and anti-sense transcripts were also detected in wild-type DN2 cells, albeit at relatively low abundance (Figure 2B). However, ThymoD sense- and anti-sense transcript levels were severely decreased in ThymoD poly(A)/poly(A) DN2 cells, indicating that ThymoD transcription is essential to generate a transcriptionally permissive environment (Figure 2C–D). Notably, Bcl11b, but not Setd3 and Vrk1, transcripts were sharply reduced in ThymoD p(A)/p(A) DN2 cells as measured by RNA-Seq and GRO-Seq (Figure 2B; lowest panel). We conclude that ThymoD transcription activates Bcl11b expression across vast genomic distances with great precision and specificity.
ThymoD Specifies T Cell Fate
To determine whether ThymoD expression specifies T cell fate, thymi were isolated from ThymoD +/+, ThymoD +/p(A) and ThymoD p(A)/p(A) mice and analyzed for developmental abnormalities. We found that thymocyte cellularity in ThymoD p(A)/p(A) mice was reduced two-fold when compared to thymi derived from wild-type mice (Figure 3A). The proportion and number of the DN1 and DN2 compartments were increased in thymi isolated from ThymoD p(A)/p(A) mice as compared to wild-type mice, whereas the DN3 and D4 compartments were reduced (Figures 3B and 3C). ThymoD p(A)/p(A) thymi also displayed abnormal ratios of DP and SP cells (Figure S3A). The γδ T-cell compartment was reduced in ThymoD p(A)p(A) thymi as compared to wild-type thymi (Figure S3B). We also observed increases in the percentages of NK1.1+, as well as, NK1.1+B220+ cells in thymi isolated from ThymoD p(A)/p(A) mice (Figures S3C and S3D). Aged ThymoD p(A)/p(A) mice showed more pronounced defects that varied and frequently were accompanied by skewed DN ratios (Figure S3E). Ultimately the majority of aged ThymoD p(A)/p(A) mice succumbed from leukemia and lymphoma (Figures S3F and S3G). The lymphomas and leukemias that developed in ThymoD p(A)/p(A) mice were largely composed of CD4+CD8+ double positive TCRβ+ T-lineage cells (data not shown). Since the developmental defects and the development of lymphoid malignancies observed in ThymoD p(A)/p(A) mice closely resembled those observed in radiation-induced lymphoma in Bcl11b heterozygous mice, DN1-3 cells were examined for Bcl11b expression (Liu et al., 2010; Ikawa et al., 2010; Li et al., 2011; Matsumoto et al., 1998; Kamimura et al., 2017; Gutierrez et al., 2011). Indeed, Bcl11b expression in DN2-DN3 cells sorted from ThymoD p(A)/p(A) mice was significantly reduced, as was observed upon insertion of a poly(A) cassette into the ThymoD locus of the SCIDadh cell line (Figure 3D). To further validate the notion that ThymoD and Bcl11b expression were linked, DN2 cells were generated in vitro using OP9-DL1 cells. We found that hematopoietic progenitors derived from ThymoD p(A)/p(A) mice failed to develop beyond the DN2 stage upon culture on OP9-DL1 monolayers, consistent with the developmental arrest observed in ThymoD p(A)/p(A) thymi in vivo (Figure 3E). Likewise, when cultured on OP9-DL1 cells, ThymoD p(A)/p(A) progenitors also displayed a severe block in the developmental progression from the DN to the DP cell stage and an accumulation of NK1.1+ and NK1.1+;B220+ cells (Figure S4A). To examine Bcl11b expression levels in cultured DN2 cells derived from ThymoD p(A)/p(A) mice, RNA was extracted and analyzed by real-time PCR. As was observed in progenitors derived from ThymoD p(A)/p(A) mice, Bcl11b levels were significantly decreased in ThymoD p(A)/p(A) DN2 cells cultured in vitro (Figure S4B). To determine the extent of overlap between the transcription signatures between ThymoD p(A)/p(A) and Bcl11b-deficient DN2 cells RNA-Seq profiles were compared (Longabaugh et al., 2017). We found that many of the differentially expressed genes in ThymoD p(A)/p(A) were modulated during the DN2 to DN3 transition and the vast majority overlapped with those differentially expressed in Bcl11b-deficient DN2 cells (Figure 3F). This set of differentially expressed genes was closely linked with specification and commitment of adaptive and innate immune cells (Figure 3G and 3H and Figurer S4C). Prominent among the regulators that characterize adaptive and innate immune cell development were Id2, CD3ε, Runx2, Sox13, Il2rb, Zbtb7b, Fcgr3, Ikzf3, c-kit, Notch3 and Dntt (Figure 3H) (Liu et al., 2010; Ikawa et al., 2010; Li et al., 2011; Rothenberg et al., 2016). These data directly link ThymoD and Bcl11b into a common pathway to specify T cell fate.
Figure 3. ThymoD Specifies T Cell Fate.

(A) Reduced cellularity in thymi derived from 5-week-old ThymoD+/+, ThymoD+/p(A) and ThymoDp(A)/p(A) mice. Bar graphs show thymocyte cell numbers in 5-week-old ThymoD+/+, ThymoD+/− and ThymoD−/− mice.
(B) Thymocyte development is partially blocked at the DN2 cell stage in ThymoD p(A)/p(A) mice. Thymocytes derived from 5-week-old ThymoD+/+, ThymoD+/p(A) and ThymoDp(A)/p(A) mice were stained for the expression of CD25 versus CD117 and CD44 versus CD117. Cells were gated on the DN compartment. Right panels show bar graphs that display percentages of DN1, DN2, DN3 and DN4 cells in thymi derived from 5-week-old ThymoD+/+, ThymoD+/p(A) and ThymoD p(A)/p(A) mice.
(C) Left panels show increased numbers of DN1 and DN2 cells in thymi derived from ThymoD p(A)/p(A) mice when compared to thymi isolated from ThymoD+/+ and ThymoD+/p(A) mice. Right panels show decreased numbers of DN3 and DN4 cells in thymi derived from ThymoD p(A)/p(A) mice when compared to thymi isolated from ThymoD+/+ and ThymoD+/p(A) mice.
(D) Bar graph indicating relative levels of Bcl11b expression in the DN1-DN3b cells derived from 5-week-old ThymoD+/+ and ThymoD−/− thymi. Results show the mean ± SD (n=3).
(E) Developmental arrest at the DN2 cell stage in in vitro differentiated ThymoD p(A)/p(A) using OP9-DL1 stromal cell cultures. Representative FACS plots for DN compartments are shown.
(F) ThymoD and Bcl11b share a common set of target genes in DN2 cells. Venn diagram is shown displaying differentially expressed genes for indicated genotypes.
(G) ThymoD and Bcl11b are linked in a common pathway to induce a T-lineage specific program of gene expression. Data were obtained from three independent RNA-Seq experiments. Table displays a selected group of target genes differentially expressed in sorted ThymoD+/+ and ThymoD p(A)/p(A) DN2 cells. Diagram reveals decreased levels of Bcl11b, Id3, CD3e and Sox13 expression.
(H) ThymoD acts to suppress the expression of genes associated with the developmental progression of alternative cell lineages. Data were obtained from three independent RNA-Seq experiments.
ThymoD Expression Releases the Bcl11b Enhancer from the Nuclear Lamina
Given that the Bcl11b enhancer repositions away from the lamina in developing T cell progenitors, and that ThymoD modulates Bcl11b expression, we considered the possibility that ThymoD transcription acts to release the Bcl11b locus from the lamina to the nuclear interior. To address this possibility, we performed HiC analysis on wild-type and ThymoD p(A)/p(A) DN2 cells. Specifically, adult hematopoietic progenitors were isolated from wild-type and ThymoD p(A)/p(A) hematopoietic progenitors, cultured on OP9-DL1 cells, isolated using cell sorting, formaldehyde fixed and analyzed using HiC. From the HiC reads we generated contact maps and used principal component analysis to segregate the DN2 genomes into A versus B compartments. Upon comparing PC1 values derived from multipotent progenitors versus wild-type DN2 cells we found a large ensemble of genomic regions (2260) that switched compartments during the transition from multipotent progenitors to committed DN2 cells (Figure 4A and Table S1). In contrast, a mere thirty genomic regions were associated in different compartments upon comparing ThymoD+/+ and ThymoD p(A)/p(A) DN2 cells (Figure 4B; Table S1). Conspicuous among these was the Bcl11b intergenic region (Figure 4B). The finding that a rather restricted number of genomic regions flipped PC1 values raised the question as how this compares to changes in compartmentalization during the DN2 to DN3 transition. To explore this possibility, we performed HiC on isolated RAG-deficient thymocytes derived from fetal liver cells and cultured on OP9-DL1 cells. Contact maps were generated from DN3 HiC reads and directly compared to those observed for DN2 cells. We found a total of sixty genomic regions switched compartments during the DN2 to DN3 transition (Table S2 and S3). We next compared changes in the PC1 distribution across chromosomes for wild-type DN2 versus ThymoD p(A)/p(A) DN2, wild-type DN2 versus wild-type DN3 and wild-type DN2 versus multipotent progenitors. The correlation coefficient of interactions for each locus for the paired cell types was calculated and plotted as the mean correlation coefficient for all loci across each chromosome. We found that most of the differences in PC1 values in wild-type versus ThymoD p(A)/p(A) DN2 cells were restricted to chromosome 12 where the Bcl11 locus is located (Figure S4D). These data are consistent with the notion that ThymoD acts locally to orchestrate compartmentalization. As expected the changes in compartmentalization in wild-type DN2 versus ThymoD p(A)/p(A) DN2 cells were also closely associated with differences in long-range genomic interactions that span the Bcl11b locus control region (Figure 4C). In wild-type DN2 cells we observed an elaborate pattern of looping involving the Bcl11b locus control region and the Bcl11b coding region (Figure 4C). In contrast, ThymoD p(A)/p(A) DN2 cells were severely depleted for such interactions. Rather, we observed genomic interactions spanning vast genomic distances and primarily involving neighboring heterochromatic regions (Figure 4C). To validate the changes in compartmentalization we performed immune-3D-FISH using antibodies directed against the lamina and fluorescently labeled probes corresponding to the Bcl11b intergenic region. As expected, we found that in wild-type DN2 cells the Bcl11b intergenic region was predominantly localized at the nuclear interior, whereas in ThymoD p(A)/p(A) DN2 cells the Bcl11b locus was retained for the large majority of cells at or near the nuclear lamina (Figure 4D). Collectively, these observations indicate that ThymoD expression is essential to release the Bcl11b intergenic region from the nuclear lamina.
Figure 4. ThymoD Acts In Cis to Reposition the Bcl11b Enhancer from the Lamina to the Nuclear Interior.

(A) Large ensemble of genomic regions reposition during the transition from multipotent progenitors to differentiated DN2 cells. PC1 values derived from HiC reads are plotted for multipotent progenitors versus differentiated DN2 cells.
(B) ThymoD expression is essential to reposition the Bcl11b locus control region in developing T cell progenitors. Comparison of PC1 values derived from HiC reads generated from ThymoD+/+ versus ThymoD p(A)/p(A) DN2 cells are shown.
(C) ThymoD expression regulates Bcl11b enhancer and promoter compartmentalization. Contact maps derived from HiC reads derived from wild-type and ThymoD p(A)/p(A) DN2 cells are shown. Positive PC1 values are indicated in black whereas negative PC1 values are displayed in gray. Heatmap indicates the ratio of observed versus expected frequencies for genomic interactions as revealed by HiC. Numerical and statistical details are described in the Methods Section.
(D) ThymoD expression is essential to release the Bcl11b super-enhancer from the nuclear lamina. Images represent 3D-FISH analysis using a fosmid probe containing the Bcl11b super-enhancer. The nuclear envelope was visualized using antibodies directed against the lamina and is indicated in red. Green signal indicate Bcl11b super-enhancer. Blue reflects DAPI staining.
(E) ThymoD acts in cis to release the Bcl11b intergenic region from the lamina. Representative images were taken from ThymoD+/p(A) DN2 cells. Bar graph shows localization of Bcl11b in ThymoD p(A)/p(A) (n=226) ThymoD+/p(A) (n=176) and ThymoD p(A)/p(A) DN2 cells (n=85). Images were digitally magnified. Original magnification was ×100. White bar represents 1 μm. p-values are indicated.
Nascent ThymoD Transcription Acts in Cis to Release the Bcl11b Locus Control Region from the Lamina
While the data described above indicate that ThymoD transcription is essential to release the Bcl11b intergenic region from the nuclear lamina it remained to be determined whether ThymoD transcription acts in cis or in trans. To address this question ThymoD+/+ and heterozygous ThymoD+/p(A) DN2 cells were examined for attachment of the Bcl11b intergenic region to the nuclear lamina using immune-3D-FISH. We found that in the vast majority of DN2 +/p(A) cells a single allele of the Bcl11b super-enhancer was sequestered at the nuclear lamina whereas the other allele was positioned in the nuclear interior (Figure 4E). Thus, these data indicate that ThymoD expression acts in cis to modulate the nuclear location of the Bcl11b locus control region. To test whether forced ThymoD2 or ThymoD3 expression in ThymoD +/p(A) DN2 cells has the ability to release the ThymoD p(A) allele in trans, hematopoietic progenitor cells isolated from ThymoD +/p(A) bone marrow were transduced with virus expressing vector alone, ThymoD (exons 1–3) and ThymoD2 (exons 1,2 and 4) and examined for the nuclear positioning of the Bcl11b locus control region (data not shown). We found that the ThymoD p(A) allele remained at the nuclear lamina regardless of ectopic ThymoD or ThymoD2 expression (data not shown). Taken together, these findings demonstrate that (1) nascent ThymoD transcription is required to release the Bcl11b locus control region from the nuclear lamina and (2) ThymoD transcripts generated at wild-type alleles do not have the ability to act in trans and release the other Bcl11b allele from the lamina. We conclude that in developing T cell progenitors ThymoD expression acts in cis to release the Bcl11b locus control region from the nuclear lamina.
ThymoD-Induced Structural Changes in Chromatin Topology Fosters Bcl11b Enhancer-Promoter Communication
To determine how ThymoD transcription modulates local chromatin folding we examined HiC contact reads derived from wild-type and ThymoD p(A)/p(A) DN2 for genomic interactions across the Bcl11b intergenic and genic regions (Figure 5A). In wild-type DN2 cells the most significant interactions across a 7 Mb genomic region involved the Bcl11b enhancer and promoter (Figure 5A). Strikingly, genomic interactions associated with the enhancer and promoter were greatly diminished in DN2 cells derived from ThymoD p(A)/p(A) mice (Figure 5A). Rather we found that ThymoD p(A)/p(A) DN2 cells were highly enriched for loops involving the Bcl11b genic and promoter regions with heterochromatin (Figures 4C and 5A). Thus, ThymoD transcription promotes a pattern of genomic interactions that greatly enrich genomic encounters involving the Bcl11b promoter and enhancer.
Figure 5. ThymoD Transcription Juxtapose the Bcl11b Super-Enhancer and Promoter Regions into a Single Loop Domain.

(A) ThymoD transcription orchestrates cohesin-dependent looping to bring the Bcl11b promoter and enhancer region into a single loop domain. Genomic interactions derived from HiC reads across the Bcl11 intergenic and genic regions are shown for wild-type and ThymoD p(A)/p(A) DN2 cells.
(B) ThymoD p(A)/p(A) DN2 cells are severely depleted for CTCF and SMC3 occupancy across the ThymoD locus. ThymoD+/+, ThymoD+/p(A) and ThymoD p(A)/p(A) DN2 cells were generated from adult hematopoietic progenitors that were cultured in the presence of OP9DL1 and cytokines. Tracks display CTCF and SMC3 occupancy across a genomic region containing the Bcl11b super-enhancer in ThymoD+/+, ThymoD+/p(A)and ThymoD p(A)/p(A) DN2 cells. Note that ChIP-Seq reads were corrected for input reads.
(C) ThymoD p(A)/p(A) DN2 cells are severely depleted of CTCF and SMC3 occupancy across the Bcl11b genic region. Tracks display CTCF and SMC3 occupancy across the Bcl11b genic region in ThymoD+/+, ThymoD+/p(A) and ThymoD p(A)/p(A) DN2 cells. CTCF and SMC3 peaks were called using HOMER and marked as red and black tags, respectively. Note that ChIP-Seq reads were corrected for input reads.
To determine how genomic interactions were affected by ThymoD expression, we examined wild-type and ThymoD p(A)/p(A) DN2 cells for CTCF and cohesin (SMC3) occupancy (Figure 5B). We found that in wild-type DN2 cells CTCF and SMC3 bound to multiple sites that spread across the Bcl11b intergenic and genic regions (Figures 5B and 5C). As expected, the majority of CTCF bound sites overlapped with SMC3 occupancy (Figure 5A). Importantly, both CTCF and SMC3 occupancy across the Bcl11b intergenic region were severely affected in ThymoD p(A)/p(A) DN2 cells (Figure 5B). CTCF and SMC3 occupancy was also affected at the Bcl11b promoter (Figure 5C and S5A).
It could be argued that ThymoD p(A)/p(A) DN2 cells were developmentally distinct from that of wild-type DN2 cells and that the abnormalities observed in CTCF and SMC3 occupancy were caused by a developmental arrest. To address this possibility, we examined CTCF and SMC3 occupancy in ThymoD+/p(A) mice, in which thymocyte development was not perturbed. We found that in ThymoD +/p(A) DN2 cells CTCF and SMC3 occupancy was reduced two-fold across the Bcl11b intergenic region suggesting a direct link between ThymoD transcription and CTCF/SMC3 occupancy (Figure 5B).
As a first approach to determine the mechanism by which ThymoD transcription promotes CTCF/SMC3 occupancy, we examined the ThymoD transcribed region for nucleosome depletion, as well as for the deposition of epigenetic marks (Figures 6A and 6B). We found that ThymoD transcription affected nucleosome depletion and the deposition of epigenetic marks associated with active transcription and the deposition of H3.3 across the Bcl11b promoter, but not genomic regions that flank the Bcl11b regulatory elements (Figure 6A and 6B and Figure S5A).
Figure 6. ThymoD Transcription Promotes the Deposition of Histone Variant H3.3, Activating Histone Marks and Hypomethyation Across the Bcl11b Intergenic Region.

(A) ThymoD expression promotes nucleosome depletion and deposition of epigenetic marks, chromatin accessibility and H3.3 across the Bcl11b intergenic region. Tracks indicate PC1 values derived from HiC reads, ATAC reads, CHiP-Seq reads for CTCF, SMC3, H3K4me1, H3K4me3 and H3.3 for wild-type and ThymoD p(A)/p(A) DN2 cells. Bottom panels indicated RNA-Seq reads across the Bcl11b locus. pAS insertion site is indicated by read arrow. Note that ChIP-Seq reads were corrected for input reads.
(B) ThymoD expression modulates chromatin accessibility and the deposition of epigenetic marks across the ThymoD locus. Tracks indicate ATAC reads, ChIP-Seq reads for CTCF, SMC3, H3K4me1, H3K4me3 and H3.3 for wild-type and ThymoD p(A)/p(A) DN2 cells. RNA-Seq reads across the ThymoD locus for wild-type and ThymoD p(A)/p(A) DN2 cells are indicated. Lower panels shows CpG DNA hyper methylation across the CR2 and CR4 regions in ThymoD poly(A)/poly(A) DN2 cells.
(C) Cohesin occupancy in DN2 cells is closely associated with active transcription start sites. DN2 cells were incubated in the presence of actinomycin D for the indicated time points, formaldehyde fixed and analyzed for SMC3 occupancy using ChIP-Seq. Active transcription start sites were identified using RNA polymerase II ChIP-Seq. Plotted is the −log 10 p-values of the Fisher test of the contingency table (Table S4). The analysis indicates preferential loss of cohesin occupancy at active TSS sites.
(D) SMC3 loading across the ThymoD transcribed region requires nascent transcription. SMC reads were determined across the ThymoD locus (chr12: 108.345–108.405 Mb) in DN2 cells treated in the absence or presence of actinomycin D for indicated times and normalized to the total read number. The mean and standard deviation for each experiment were derived from 20 independent samples using boot strapping for all reads across the genome.
(E) Nascent RNA transcription targets SMC3 to the ThymoD locus. DN2 cells were incubated in the absence or presence of actinomycin D for 15 minutes and analyzed for SMC3 occupancy using ChIP-Seq. Representative track shows a reduction of SMC3 occupancy across the ThymoD locus.
The absence of CTCF occupancy in ThymoD p(A)/p(A) DN2 cells raised the question as to why CTCF failed to bind its cognate sites across the ThymoD transcribed region. Since recent studies have demonstrated that non-coding transcription is closely associated with CpG hypomethylation we considered the possibility that transcription across the ThymoD region acts to recruit members of the Tet protein family (Benner et al., 2015). Hence we examined DNA isolated from wild-type and ThymoD p(A)/p(A) DN2 cells for cytosine DNA methylation using genome-wide bisulfite sequencing (Figure 6B; bottom tracks). Notably, we found that several of differentially methylated CpG residues were associated with CTCF occupancy in wild-type but not in ThymoD p(A)/p(A) DN2 cells (Figure 6B; upper and bottom tracks). Taken together, these data indicate that ThymoD transcription is essential to (i) demethylate CpG residues closely associated with CTCF bound sites, (ii) recruit members of the cohesin complex to the transcribed region and (iii) reposition the Bcl11b enhancer from a repressive to a transcriptionally permissive neighborhood.
Nascent ThymoD Transcription is Essential to Recruit Cohesin to the Bcl11b Locus Control Region
The data described above indicate that ThymoD transcription is associated with modulating chromatin topology to bring the Bcl11b enhancer and promoter elements within close spatial proximity. To determine whether nascent ThymoD transcription is required to recruit members of the cohesin complex to the Bcl11b locus control region wild-type DN2 cells were incubated in the presence of actinomycin D for 15, 30 and 60 minutes. Treated cells were formaldehyde-fixed and immunoprecipitated with antibodies directed against RNA polymerase II and SMC3 followed by genome-wide sequencing. Consistent with previous observations we found that SMC3 occupancy was significantly depleted at sites associated with RNA polymerase II occupancy (Figure 6C; Table S1) (Izumi et al. 2015, Bhardwaj et al 2016). We next evaluated whether in DN2 cells nascent ThymoD transcription directly recruits SMC3. We found that DN2 cells treated with actinomycin D showed a significant reduction of SMC3 occupancy across the transcribed region (Figure 6D and Figure S5B). Notably, a brief 15 minutes incubation with actinomycin D significantly affected SCM3 occupancy (Figure 6E). We conclude that the nascent ThymoD transcription acts in cis to directly promote SMC3 loading across the transcribed region.
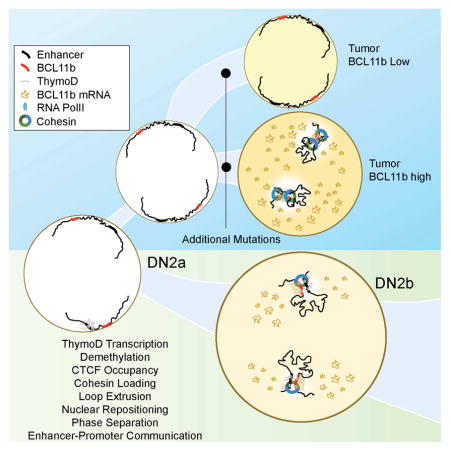
Compartmentalization and Cohesin-Dependent Looping is Reversible in ThymoD-Deficient Lymphomas/Leukemias
As described above ThymoD poly(A)/poly(A) mice readily develop lymphoma/leukemia. To determine whether the Bcl11b intergenic remained attached to the nuclear lamina in ThymoD p(A)/p(A) lymphomas long-term cultures were established from the tumors. Tumors isolated from aged ThymoD poly(A)/poly(A) mice displayed a higher fraction of blasting cells that frequently adopted irregular nuclear morphologies and readily expanded in tissue culture (Figure 7A). Long-term cultures were established either in the absence or presence of OP9-DL1 stromal cells. From these expanding cultures we established independent cell lines representing either lymphomas or leukemias. To determine whether the expression of ThymoD and Bcl11b remained silent in the lymphomas/leukemias, the tumors were examined for ThymoD and Bcl11b expression using RNA-Seq (Figure S6). As expected ThymoD transcription was blocked at the poly(A) insertion in all of the tumors (Figure S6). Intriguingly, three of the tumors expressed anti-sense transcription at relatively high levels in three of the six tumors (Figure 6A). Notably, lymphomas/leukemias that expressed high levels of anti-sense transcription also abundantly expressed Bcl11b (Figure S6). These data indicate that a subset of lymphoid malignancies that develop in ThymoD poly(A)/poly(A) mice display coordinate ThymoD anti-sense and Bcl11b transcription whereas in tumors that failed to induce anti-sense ThymoD transcription, Bcl11b expression remained suppressed.
Figure 7. Compartmentalization and Cohesin-dependent Looping is Reversible in ThymoD-Deficient Lymphomas.

(A) Development of leukemias and lymphomas in ThymoD poly(A)/poly(A) mice. Tumors isolated from aged ThymoD poly(A)/poly(A) mice displayed a higher fraction of blasting cells that frequently adopted irregular nuclear morphologies and readily expanded in tissue culture. DN2 cells derived from wild-type and ThymoD poly(A)/poly(A) DN2 cells as well as several lymphomas and leukemias are shown. Black bar represents 40μm.
(B) ThymoD expression, either sense or anti-sense transcription, promotes SMC3 occupancy and cohesin occupancy, chromatin accessibility and the activation of Bcl11b expression. pAS insertion is indicated by red arrow.
(C) ThymoD transcription is closely associated with compartmentalization. Wild-type and ThymoD p(A)/pA) DN2 cells and ThymoD p(A)/pA) lymphomas were examined for nuclear localization of the Bcl11b intergenic region. The two left images show localization for the Bcl11b super-enhancer at the nuclear lamina in multipotent progenitors. Upper images indicate location of the Bcl11b enhancer in the nuclear interior of ThymoD p(A)/p(A) tumor 3. The bottom images show localization of the Bcl11b enhancer at the nuclear lamina in tumor 5. DAPI staining is shown in blue. Bar graph shows fraction of spatial distances (<500nm) separating the Bcl11b super-enhancer from the nuclear lamina in multipotent progenitor cells (n=175) and WT DN2 cells (n=135). ThymoD p(A)/p(A) DN2 cells (n=120), ThymoD p(A)/p(A) tumor3 (n=206), ThymoD poly(A)/poly(A) tumor5 (n=207). Images were digitally magnified. Original magnification was ×100. White bar represents 1 μm.
To examine whether the activation of ThymoD and Bcl11b expression in a subset of lymphoma cell lines was associated with chromatin accessibility and SMC3 occupancy across the ThymoD region three lymphomas (3, 5 and 6) were examined using ATAC-Seq and SMC3 ChIP-Seq (Figure 7B). As predicted by the analyses described above, lymphomas that were associated with coordinate ThymoD and Bcl11b expression showed widespread ATAC reads across the ThymoD locus and were enriched for SMC occupancy (Figure 7B).
To determine whether the induction of ThymoD and Bcl11b expression as well as the selective increase in SCM3 occupancy was associated with changes in nuclear location, tumors 3 and 5 were examined by 3D-FISH for compartmentalization. We found that the majority of cells derived from tumor 3, which coordinately expressed high levels of ThymoD and Bcl11b, exhibited localization of the Bcl11b intergenic region that was localized away from the nuclear lamina (Figure 7C). In contrast, the localization of ThymoD in tumor 5, which expresses low levels of ThymoD and Bcl11b, was near or on the nuclear lamina (Figure 7C). These data indicate that the insertion of a poly(A) cassette in the ThymoD locus does not irreversibly attach the ThymoD locus to the nuclear lamina. Instead, ThymoD transcription, either sense or anti-sense, promotes SMC3 occupancy and cohesin-dependent looping, changes in compartmentalization and the activation of Bcl11b expression to specify T cell fate.
DISCUSSION
Specifying Immune Cell Fate by Non-Coding Transcription
While it is now well established that genomic regions reposition during developmental progression, it is not understood how such changes in nuclear location are regulated. Here we provide insight into the mechanism that underpins the repositioning of regulatory elements. We identify a non-coding RNA, named ThymoD, which acts to reposition the Bcl11 enhancer away from the lamina to the nuclear interior and sequester the Bcl11b promoter and enhancer region into a single loop domain. How does ThymoD accomplish the repositioning of the Bcl11b enhancer? The phenotype revealed in ThymoD+/p(A) thymocytes strongly suggests that ThymoD acts in cis on nearby gene expression consistent with previous observations that nascent transcription rather than biochemical activities associated with a subset of non-coding RNAs executes their function (Mele and Rinn, 2016; Engreitz et al. 2016). Here we found that the vast majority of DN2+/p(A) cells display one Bcl11b allele located at the nuclear lamina and one in the nuclear interior. Thus, ThymoD acts in cis to release the Bcl11b enhancer from the lamina and to active Bcl11b gene expression. How does ThymoD perform this task? We propose a stepwise mechanism. The induction of Bcl11b expression is initiated and maintained in early T cell progenitors by activating a yet to be identified enhancer associated with the ThymoD locus. Next we suggest that the ThymoD transcribed region directs dioxygenases, Tet proteins, to specific residues that are associated with CTCF occupancy. This explains why in the absence of ThymoD expression CTCF occupancy is perturbed across the transcribed region consistent with previous observations indicating that nascent transcription is closely associated with hypomethylated DNA (Benner et al., 2015). Previous observations have also indicated that mice deficient for Tet2 and Tet3 expression displayed defects in NKT cell development, lymphoproliferative disease and exhibited significantly lower abundance of Bcl11b expression (Tsagaratou et al., 2017). Although still to be proven it is conceivable that the absence of Tet2 and Tet3 expression in NKT cells leads to hypermethylated CpG residues across the ThymoD transcribed region and in a failure to efficiently activate Bcl11b expression plausibly resulting in lympho-proliferative disease and defects in NKT cell development. How the Tet proteins target specific CpG residues spanning the transcribed region is an important question that needs to be addressed but may involve distinct secondary DNA structures that are induced either by sense- or anti-sense transcription. Regardless of the precise mechanism these data link non-coding transcription and Tet targeting into a common pathway.
ThymoD transcription also permits the loading of the cohesin complex in a manner as previously for coding transcription (Izumi et al. 2015; Bhardwaj et al. 2016; Busslinger et al., 2017). Recruitment of the cohesin complex in turn activates an elaborate and coherent pattern of looping, likely involving loop-extrusion, to bring the enhancer and promoter region in a singular loop domain (Nasmyth, 2001). Juxtaposing paired enhancer and promoter regions, separated by vast genomic distances, would yield greatly reduced first-passage times (Lucas et al., 2014). We note that loop extrusion would permit the repositioning of paired enhancers and promoters with great precision even when separated by vast genomic distances. Finally, loop extrusion would sequester the loop domain away from the transcriptionally repressive environment to the euchromatic compartment. We suggest that once localized in the transcriptionally permissive compartment transcriptional regulators associated with T-lineage commitment, including GATA-3, TCF1 and RUNX1, would bind enhancer elements across the loop domain to orchestrate the deposition of activating histone marks as well as the histone variant H3.3 facilitating enhancer-promoter communication that may involve phase separation (Hnisz et al., 2017). Such a sequential order of gene activation is consistent with the notion that ThymoD transcription (DN1 stage) precedes the expression of non-coding RNAs initiated from the enhancer regions (DN2a stage). In sum, we propose a model in which nascent ThymoD transcription dictates enhancer-promoter communication at multiple steps that precede gene activation: (i) demethylation at CpG residues closely associated with CTCF occupancy across the ThymoD genomic region, (ii) activation of cohesin-dependent looping to juxtapose the enhancer and promoter into a single loop domain and to relocalize the enhancer from a heterochromatic to an euchromatic environment and (iii) deposition of activating epigenetic marks across the loop domain once positioned in the euchromatic compartment to facilitate interactions of paired regulatory elements by phase separation.
Mechanisms that Orchestrate Immune Cell Development
Previous studies have established that a subset of genomic regions reposition in the nucleus during the transition from the multipotent progenitor cell stage to committed B-lineage cells (Lin et al. 2012; Miyazaki et al., 2014; van Steensel and Belmont, 2017). Switching the nuclear location of genomic regions is often associated with changes in gene expression (Lin et al., 2012; Kind et al., 2015). Prominent among the genomic regions that switch nuclear location in B cell progenitors are genes encoding for transcriptional regulators that specify B cell fate including the EBF1 and FOXO1 loci (Lin et al. 2012; Mansson et al., 2012). Both loci are associated with the heterochromatic compartment in multipotent progenitors but reposition to the transcriptionally permissive compartment upon developing into committed B-lineage cells (Lin et al. 2012). Here we demonstrate that in developing T cell progenitors, a genomic region associated with the Bcl11b locus that harbors critical regulatory elements, repositions from the nuclear lamina to the transcriptionally permissive nuclear interior. These findings point to a common mechanism that specifies B and T cell fate. In developing B cell progenitors, the EBF1 locus switches from the nuclear lamina to the transcriptionally permissive compartment leading to the induction of EBF1 expression to establish B cell identity. In contrast, in T cell progenitors the Bcl11b enhancer repositions from the lamina to the nuclear interior to activate Bcl11b expression. Why has such a complex mechanism, involving repression, nuclear repositioning and activation, evolved to promote adaptive immune cell fate? We suggest that anchoring regulatory elements to the nuclear lamina permits efficient transcriptional repression across a large genomic region preventing stochastic and premature activation of a lymphoid-specific program of gene expression in hematopoietic progenitors. Only upon reaching a distinct developmental stage, are progenitor cells instructed to release locus control regions or super enhancers from the nuclear lamina to the transcriptionally active compartment. In B cell progenitors, a repositioned EBF1 locus control region in the transcriptionally permissive compartment would associate with B-lineage specific transcription factors such as E2A and FOXO1 to induce a B-lineage specific program of gene expression (Mansson et al., 2014). In T cell progenitors, the Bcl11b locus control region being positioned in the transcriptionally permissive compartment would be able to respond to Notch-signaling and transcriptional regulators that include GATA3, TCF1 and RUNX1 to activate a T-lineage specific transcription signature (Kueh et al. 2015). Thus, specification of adaptive immune cell fate in early lymphoid progenitors is orchestrated by a common mechanism that involves the repositioning of regulatory regions from the lamina to the euchromatic compartment.
Non-Coding Transcription and Tumor Suppression
The activation of anti-sense ThymoD transcription associated with a subset of lymphomas that were derived from ThymoD poly(A)/poly(A) mice was initially puzzling. How is anti-sense ThymoD transcription induced in a subset of lymphomas? Murine T cell lymphomas are frequently associated with mutations that activate the Notch signaling pathway, which activates Bcl111b expression (Weng et al., 2004; Yui and Rothenberg, 2014). Although a detailed characterization of cooperating oncogenic mutations in the ThymoD p(A);p(A) lymphomas will be required, we speculate that the aberrant activation of the Notch signaling cascade leads to the induction of high levels of anti-sense ThymoD transcription, ultimately leading to release of the Bcl11b intergenic region from the lamina. Since both sense- and anti-sense ThymoD transcription is associated with changes in compartmentalization and chromatin folding we suggest that ThymoD transcription rather than its primary nucleotide sequence is the critical determinant that dictates the specificity of Bcl11b enhancer-promoter communication.
Previous studies have demonstrated that somatic mutation of an intergenic region may generate an oncogenic superenhancer (Mansour et al., 2014). Our observations indicate that non-coding transcription may act as a tumor suppressor. Given that non-coding transcription is widespread we suggest that likewise non-coding transcription is a common mechanism to activate the expression of nearby genes that encode for proteins with tumor suppressive function.
Conclusion
It is now well established that the genomes of animal and plant kingdoms are organized into euchromatic and heterochromatic regions. Numerous studies have also identified genomic regions that switch nuclear location during developmental progression. The underlying mechanism that directs changes in compartmentalization, however, remained to be revealed. Here we present data indicating that non-coding transcription dictates remodeling of local chromatin structure, possibly involving loop extrusion, to reposition regulatory elements from the heterochromatic to the euchromatic compartment. We suggest that local remodeling of chromatin topology by non-coding transcription induced loop extrusion is a universal mechanism that permits genomic regions to readily switch compartments.
Enhancers are often separated from promoter regions by vast genomic distances. How enhancer elements select their cognate promoter regions with high precision and specificity has remained to be revealed (Farley et al., 2015). We suggest that developmental and lineage specific patterns of non-coding transcription sequester, by loop extrusion, enhancer and promoter elements into a single loop domain. Non-coding transcription induced loop extrusion explains how enhancers and promoters, even when initially segregated from each other into distinct compartments, find each other with great specificity.
STAR*METHODS
KEY RESOURCES TABLE
The table highlights the genetically modified organisms and strains, cell lines, reagents, software, and source data essential to reproduce results presented in the manuscript. Depending on the nature of the study, this may include standard laboratory materials (i.e., food chow for metabolism studies), but the Table is not meant to be comprehensive list of all materials and resources used (e.g., essential chemicals such as SDS, sucrose, or standard culture media don’t need to be listed in the Table). Items in the Table must also be reported in the Method Details section within the context of their use. The number of primers and RNA sequences that may be listed in the Table is restricted to no more than ten each. If there are more than ten primers or RNA sequences to report, please provide this information as a supplementary document and reference this file (e.g., See Table S1 for XX) in the Key Resources Table.
Please note that ALL references cited in the Key Resources Table must be included in the References list. Please report the information as follows:
- REAGENT or RESOURCE: Provide full descriptive name of the item so that it can be identified and linked with its description in the manuscript (e.g., provide version number for software, host source for antibody, strain name). In the Experimental Models section, please include all models used in the paper and describe each line/strain as: model organism: name used for strain/line in paper: genotype. (i.e., Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J). In the Biological Samples section, please list all samples obtained from commercial sources or biological repositories. Please note that software mentioned in the Methods Details or Data and Software Availability section needs to be also included in the table. See the sample Table at the end of this document for examples of how to report reagents.
- SOURCE: Report the company, manufacturer, or individual that provided the item or where the item can obtained (e.g., stock center or repository). For materials distributed by Addgene, please cite the article describing the plasmid and include “Addgene” as part of the identifier. If an item is from another lab, please include the name of the principal investigator and a citation if it has been previously published. If the material is being reported for the first time in the current paper, please indicate as “this paper.” For software, please provide the company name if it is commercially available or cite the paper in which it has been initially described.
- IDENTIFIER: Include catalog numbers (entered in the column as “Cat#” followed by the number, e.g., Cat#3879S). Where available, please include unique entities such as RRIDs, Model Organism Database numbers, accession numbers, and PDB or CAS IDs. For antibodies, if applicable and available, please also include the lot number or clone identity. For software or data resources, please include the URL where the resource can be downloaded. Please ensure accuracy of the identifiers, as they are essential for generation of hyperlinks to external sources when available. Please see the Elsevier list of Data Repositories with automated bidirectional linking for details. When listing more than one identifier for the same item, use semicolons to separate them (e.g. Cat#3879S; RRID: AB_2255011). If an identifier is not available, please enter “N/A” in the column.
- A NOTE ABOUT RRIDs: We highly recommend using RRIDs as the identifier (in particular for antibodies and organisms, but also for software tools and databases). For more details on how to obtain or generate an RRID for existing or newly generated resources, please visit the RII or search for RRIDs.
Please see the sample Table at the end of this document for examples of how reagents should be cited. To see how the typeset table will appear in the PDF and online, please refer to any of the research articles published in Cell in the August 25, 2016 issue and beyond.
Please use the empty table that follows to organize the information in the sections defined by the subheading, skipping sections not relevant to your study. Please do not add subheadings. To add a row, place the cursor at the end of the row above where you would like to add the row, just outside the right border of the table. Then press the ENTER key to add the row. You do not need to delete empty rows. Each entry must be on a separate row; do not list multiple items in a single table cell.
TABLE FOR AUTHOR TO COMPLETE
Please upload the completed table as a separate document. Please do not add subheadings to the Key Resources Table. If you wish to make an entry that does not fall into one of the subheadings below, please contact your handling editor.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CTCF | EMD Millipore | Cat # 07-729 RRID:AB_441965 |
| Anti-H3K4me1 | Abcam | Cat # ab8895 RRID:AB_306847 |
| Anti-H3K4me3 | Abcam | Cat # ab8580 RRID:AB_306649 |
| Anti-H3.3 | Abcam | ab176840 |
| Anti-RNA polymerase II CTD (8WG16) | Abcam | Cat # ab817 RRID:AB_306327 |
| Anti-SMC3 | Abcam | Cat # ab9263 RRID:AB_307122 |
| Anti-BrdU (IIB5) | Santa Cruz | Cat # sc-32323 AC RRID:AB_626766 |
| Anti-Lamin B1 | Santa Cruz | Cat # sc-6217 RRID:AB_648158 |
| Anti-CD3e (145-2C11) | Biolegend | Cat # 100306 RRID:AB_312671 |
| Anti-CD3e (145-2C11) | eBioscience | Cat # 13-0031-82 RRID:AB_466319 |
| Anti-CD4 (GK1.5) | Biolegend | Cat # 100423 RRID:AB_389302 |
| Anti-CD4 (GK1.5) | Biolegend | Cat # 100408 RRID:AB_312693 |
| Anti-CD8a (53-6.7) | eBioscience | Cat # 11-0081-85 RRID:AB_464916 |
| Anti-CD8a (53-6.7) | Bioleged | Cat # 100725 RRID:AB_493425 |
| Anti-CD11b (M1/70) | eBiosciences | Cat # 11-0112-85 RRID:AB_464936 |
| Anti-CD19 (MB19-1) | eBiosciences | Cat # 11-0191-82 RRID:AB_464965 |
| Anti-B220 (RA3-6B2) | eBiosciences | Cat # 11-0452-82 RRID:AB_465054 |
| Anti-B220 (RA3-6B2) | eBiosciences | Cat # 13-0452-85 RRID:AB_466450 |
| Anti-Gr1 (RB6-8C5) | Biolegend | Cat # 108417 RRID:AB_389309 |
| Anti-Gr1 (RB6-8C5) | eBiosciences | Cat # 13-5931-85 RRID:AB_466801 |
| Anti-NK1.1(PK136) | eBiosciences | Cat # 11-5941-85 RRID:AB_465319 |
| Anti-CD25 (PC61) | Biolegend | Cat # 102016 RRID:AB_312865 |
| Anti-CD27 (LG3A) | Biolegend | Cat # 124211 RRID:AB_1236460 |
| Anti-CD44 (IM7) | Biolegend | Cat # 103020 RRID:AB_493683 |
| Anti-CD45.2 (104) | Biolegend | Cat # 109828 RRID:AB_893350 |
| Anti-CD117 (ack45) | BD Pharmingen | Cat # 553869 RRID:AB_395103 |
| Anti-CD117 (2B8) | eBiosciences | Cat # 13-1171-82 RRID:AB_466569 |
| Anti-CD127 (A7R37) | eBiosciences | Cat # 13-1271-85 RRID:AB_466589 |
| Anti-Flt-3 (A2F10) | eBiosciences | Cat # 13-1351-85 RRID:AB_466600 |
| Anti-TCRb (H57-597) | Biolegend | Cat # 109211 RRID:AB_313434 |
| Anti-TCRgd (eBioGL3) | Biolegend | Cat # 118105 RRID:AB_313829 |
| Anti-Ter119 (TER119) | eBioscience | Cat # 11-5921-82 RRID:AB_465311 |
| Anti-Ter119 (TER119) | eBiosciences | Cat # 13-5921-85 RRID:AB_466798 |
| Anti-Thy1.2 (53-2.1) | eBiosciences | Cat #13-0902-85 RRID:AB_466534 |
| Biological Samples | ||
| Murine thymus | Jackson | Cat # 00056 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Actinomycin D | Thermo FIsher Scientific | Cat # 11805017 |
| Puromycin dihydrochloride hydrate | Fisher Scientific | Cat # AC227420100 |
| Unmethylated lambda DNA | Promega | Cat # D1521 |
| MethoCult M3630 | Stem Cell Technologies | Cat # 3630 |
| Anti-biotin Microbeads | Miltenyi Biotech | Cat # 130-090-485 |
| Complete protease inhibitor cocktail, EDTA-free | Roche | Cat # 4693132001 |
| Ghost dye red 780 | Tonbo bissciences | Cat # 13-0865-T100 |
| Ficol-paque plus | GE health care | Cat #17-1440-02 |
| Formamide | Sigma | Cat # F9037-100ML |
| 5-Bromouridine 5′-triphosphate | Sigma | Cat # B7166-10MG |
| MES sodium salt | Sigma | Cat # M3058-25G |
| SUPERase In™ Rnase inhibitor | Life Technologies | Cat # AM2694 |
| Biotin-14-dATP | Life Technologies | Cat # 19524-016 |
| Qdot 585 streptavidin conjugate | Invitrogen | Cat # Q10111MP |
| N-Lauroylsarcosine sodium salt solution | Sigma | Cat # L7414-10ml |
| 10× T4DNA Ligation Buffer | NEB | Cat # B0202 |
| T4 PNK | NEB | Cat # M0201 |
| T4 RNA Ligase 2, truncated | NEB | Cat # M0242 |
| T4 RNA Ligase 1 (ssRNA Ligase) | NEB | Cat # M0204 |
| 5′ DNA Adenylation Kit | NEB | Cat # E2610 |
| DNA Polymerase I, Large fragment (Klenow) | NEB | Cat # M0210 |
| Klenow 3′-5′ exo minus | NEB | Cat # M0212 |
| MboI | NEB | Cat # R0147 |
| Quick Ligation Kit | NEB | Cat # M2200 |
| Paraformaldehyde Granular | Electron Microscopy Sciences | Cat #19208 |
| Critical Commercial Assays | ||
| Nextera DNA Sample Preparation Kit for ATAC-seq | Illumina | Cat # FC-121-1030 |
| NEBNext Multiplex Oligos for Illumina Set1 and 2 | NEB | Cat # E7335S |
| TruSeq DNA Sample Preparation Kits v2 for WGBS | Illumina | Cat # FC-121-2001 |
| MethylCode™ Bisulfite Conversion Kit | Invitrogen | Cat # MECOV-50 |
| RNeasy Mini Kit | Qiagen | Cat # 74106 |
| RNeasy Micro Kit | Qiagen | Cat # 74004 |
| RNase free Dnase (50 units) | Qiagen | Cat # 79254 |
| Qiagen Miniprep Kit | Qiagen | Cat # 27106 |
| DNA Clean and Concentrator | Zymo Research | Cat # D4014 |
| NEBuider HiFi DNA Assembly Master Mix | NEB | Cat # E2621S |
| dsDNA HS qubit kit | LifeTechnologies | Cat # Q32854 |
| MEGAshortscript T7 Kit | Life technologies | Cat # AM1354 |
| MEGAclear™ Transcription Clean-Up Kit | LifeTechnologies | Cat # AM1908 |
| SuperScriptIII First-Strand Synthesis System | LifeTechnologies | Cat # AM2010 |
| RNA Fragmentation Reagents | Lifetechnologies | Cat # AM8740 |
| Micro Bio-Spin 30 Columns #732-6250 | Biorad | Cat # 732-6250 |
| Ultrafree-MC Centrifugal Filter Units with Microporous Membrane | Millipore | Cat # UFC30HVNB |
| AMPureXP beads | Beckman Coulter | Cat # A63880 |
| ProbeQuant G50 micro-columns | GE Health Care | Cat # 28-9034-08 |
| Nick translation kit | Roche | Cat #11745808910 |
| Deposited Data | ||
| Hi-C, ATAC-Seq, ChIP-Seq, GRO-Seq, RNA-Seq | This manuscript | GSE90958 |
| ATAC-Seq | Bossen et al., 2015 | GSE66978 |
| Hi-C | Yin et al., 2012 | GSE40173 |
| RNA-Seq | Zhang et al., 2012 | GSE31235 |
| RNA-Seq | Longabaugh et al., 2017 | GSE89198 |
| Experimental Models: Cell Lines | ||
| Human: 293T | ATCC | Ca t# ATCC-CRL-3216 |
| Mouse: OP9-DL1 | Schmitt et al, 2002 | N/A |
| Mouse: SCIDadh cells | Carleton et al., 1999 | N/A |
| Experimental Models: Organisms/Strains | ||
| C57Bl/6 Mice | Jackson | Cat # 00064 |
| Recombinant DNA | ||
| Fosmid: BCL11b enhancer | BacPac | Cat # WIBR1-1110C15 |
| pGEM-T Easy Vector System I | Promega | Cat # A1360 |
| Cas9 WT vector (WT Cas9 cut by NcoI/EcoRI from px330 was cloned into the pdcas9 vector by NcoI/EcorI) | This paper | Cat # 44246 |
| pdCas9-humanized | Addgene | N/A |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | N/A |
| pGEM LHA pAS (ThymoD exon3) RHA | This paper | N/A |
| pGEM LHA GFP pAS (CR1-CR2) RHA | This paper | N/A |
| pGEM LHA GFP pAS (ThymoD exon3) RHA | This paper | N/A |
| Sequence-Based Reagents | ||
| sgRNA at CR1-CR2 control region cagctGAATTCTAATACGACTCACTATAGGGGGAAAACACAGACCCTAGTCgttttagagctagaaatagcaagttaaaataaggctagtccGttatcAacttgaaaaagtggcaccgagtcggtgc) | IDT | N/A |
| sgRNA at exon 3 of ThymoD (cagctGAATTCTAATACGACTCACTATAGGGTCTTCAAGGGTGCTATCACAgttttagagctagaaatagcaagttaaaataaggctagtccGttatcAacttgaaaaagtggcaccgagtcggtgc) | IDT | N/A |
| Software and Algorithms | ||
| FlowJo software | Tree Star | N/A |
| GraphPad Prism 7 | graphPad Software | N/A |
| Bowtie | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| HOMER | Heniz et al., 2010 | http://homer.ucsd.edu/homer |
| TopHat | Trapnell et al., 2009 | https://ccb.jhu.edu/software/tophat/index.shtml |
| BSSeeker2 | Guo et al., 2013 | https://github.com/BSSeeker/BSseeker2 |
| Other | ||
| Poly-D-Lysine/Laminin; 12mm dish | Corning | Cat # 08-774-385 |
| Fisherbrand Microscope cover glass | Fisher Scientific | Cat # 12-541AC |
TABLE WITH EXAMPLES FOR AUTHOR REFERENCE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-Snail | Cell Signaling Technology | Cat#3879S; RRID: AB_2255011 |
| Mouse monoclonal anti-Tubulin (clone DM1A) | Sigma-Aldrich | Cat#T9026; RRID: AB_477593 |
| Rabbit polyclonal anti-BMAL1 | This paper | N/A |
| Biological Samples | ||
| Healthy adult BA9 brain tissue | University of Maryland Brain & Tissue Bank; http://medschool.umaryland.edu/btbank/ | Cat#UMB1455 |
| Human hippocampal brain blocks | New York Brain Bank | http://nybb.hs.columbia.edu/ |
| Patient-derived xenografts (PDX) | Children’s Oncology Group Cell Culture and Xenograft Repository | http://cogcell.org/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MK-2206 AKT inhibitor | Selleck Chemicals | S1078; CAS: 1032350-13-2 |
| SB-505124 | Sigma-Aldrich | S4696; CAS: 694433-59-5 (free base) |
| Picrotoxin | Sigma-Aldrich | P1675; CAS: 124-87-8 |
| Human TGF-β | R&D | 240-B; GenPept: P01137 |
| Activated S6K1 | Millipore | Cat#14-486 |
| GST-BMAL1 | Novus | Cat#H00000406-P01 |
| Critical Commercial Assays | ||
| EasyTag EXPRESS 35S Protein Labeling Kit | Perkin-Elmer | NEG772014MC |
| CaspaseGlo 3/7 | Promega | G8090 |
| TruSeq ChIP Sample Prep Kit | Illumina | IP-202-1012 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE63473 |
| B-RAF RBD (apo) structure | This paper | PDB: 5J17 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Experimental Models: Cell Lines | ||
| Hamster: CHO cells | ATCC | CRL-11268 |
| D. melanogaster: Cell line S2: S2-DRSC | Laboratory of Norbert Perrimon | FlyBase: FBtc0000181 |
| Human: Passage 40 H9 ES cells | MSKCC stem cell core facility | N/A |
| Human: HUES 8 hESC line (NIH approval number NIHhESC-09-0021) | HSCI iPS Core | hES Cell Line: HUES-8 |
| Experimental Models: Organisms/Strains | ||
| Streptococcus pyogenes: M1 serotype strain: strain SF370; M1 GAS | ATCC | ATCC:700294 |
| C. elegans: Strain BC4011: srl-1(s2500) II; dpy-18(e364) III; unc-46(e177)rol-3(s1040) V. | Caenorhabditis Genetics Center | WB Strain: BC4011; WormBase: WBVar00241916 |
| D. melanogaster: RNAi of Sxl: y[1] sc[*] v[1]; P{TRiP.HMS00609}attP2 | Bloomington Drosophila Stock Center | BDSC:34393; FlyBase: FBtp0064874 |
| S. cerevisiae: Strain background: W303 | ATCC | ATTC: 208353 |
| Mouse: R6/2: B6CBA-Tg(HDexon1)62Gpb/3J | The Jackson Laboratory | JAX: 006494 |
| Mouse: OXTRfl/fl: B6.129(SJL)-Oxtrtm1.1Wsy/J | The Jackson Laboratory | RRID: IMSR_JAX:008471 |
| Zebrafish: Tg(Shha:GFP)t10: t10Tg | Neumann and Nuesslein-Volhard, 2000 | ZFIN: ZDB-GENO-060207-1 |
| Arabidopsis: 35S::PIF4-YFP, BZR1-CFP | Wang et al., 2012 | N/A |
| Arabidopsis: JYB1021.2: pS24(AT5G58010)::cS24:GFP(-G):NOS #1 | NASC | NASC ID: N70450 |
| Recombinant DNA | ||
| pLVX-Tight-Puro (TetOn) | Clonetech | Cat#632162 |
| Plasmid: GFP-Nito | This paper | N/A |
| cDNA GH111110 | Drosophila Genomics Resource Center | DGRC:5666; FlyBase:FBcl0130415 |
| AAV2/1-hsyn-GCaMP6- WPRE | Chen et al., 2013 | N/A |
| Mouse raptor: pLKO mouse shRNA 1 raptor | Thoreen et al., 2009 | Addgene Plasmid #21339 |
| Sequence-Based Reagents | ||
| siRNA targeting sequence: PIP5K I alpha #1: ACACAGUACUCAGUUGAUA | This paper | N/A |
| Primers for XX, see Table SX | This paper | N/A |
| Primer: GFP/YFP/CFP Forward: GCACGACTTCTTCAAGTCCGCCATGCC | This paper | N/A |
| Morpholino: MO-pax2a GGTCTGCTTTGCAGTGAATATCCAT | Gene Tools | ZFIN: ZDB-MRPHLNO-061106-5 |
| ACTB (hs01060665_g1) | Life Technologies | Cat#4331182 |
| RNA sequence: hnRNPA1_ligand: UAGGGACUUAGGGUUCUCUCUAGGGACUUAGGGUUCUCUCUAGGGA | This paper | N/A |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Other | ||
| Sequence data, analyses, and resources related to the ultra-deep sequencing of the AML31 tumor, relapse, and matched normal. | This paper | http://aml31.genome.wustl.edu |
| Resource website for the AML31 publication | This paper | https://github.com/chrisamiller/aml31SuppSite |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to the Lead Contact, Cornelis Murre (cmurre@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
SCIDadh cell line culture
SCIDadh cells were cultured in IMDM 10% FBS, PSG at 37 °C in 5% CO2.
Mice
ThymoD pA/pA mice were generated using CRISPR-Cas9 engineering in the embryonic core facility at the University of California, San Diego. All mice were bred in specific pathogen-free condition in accordance with the Institutional Animal Care and Use Committee of the University of California, San Diego.
T cell culture
For OP9-DL1 culture, adult bone marrow single cell suspension were incubated with CD3e (145-2C11), B220 (RA3-6B2), CD11b (RB6-8C5), GR1 (RB6-8C5), TER119 (TER119), CD117 (2B8), CD127 (A7R37) and Flt3 (A2F10). Hematopoietic progenitor cells were collected by depleting lineage-positive cells using Automacs. Selected cells were cultured on OP9-DL1 in 6 well plates with αMEM containing 20% FCS, 2% PSG, FLT3L, recombinant IL-7 1-2 ng/ml at 37 °C in 5% CO2.
ThymoD p(A)/p(A) tumor culture
Tumor 3 and tumor 6 were cultured on OP9-DLL1 in MEMa 20% FBS containing 5ng/ml Flt3-L, 5ng/ml IL7, 5ng/ml SCF, PSG at 37 °C in 5% CO2. Tumor 5 was cultured without OP9-DLL1 in MEMa and 20% FBS containing 5ng/ml Flt3-L, 5ng/ml IL7, 5ng/ml SCF, PSG at 37 °C in 5% CO2. All RNAseq data for tumors were obtained from primary cells. SMC3, RNA polII ChIP-seq was done for cultured tumors 3, 5 and primary tumor 6. DNA FISH was done for cultured tumor 3 and tmor 5.
METHOD DETAILS
Generating SCIDadh pA/pA cell line
WT Cas9 was digested using NcoI/EcoRI from pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene) and cloned into pdCas9-humanized vector (Addgene) digested using NcoI/EcoRI. Transformation was done using WT Cas9 and Pcl packaging vector transfected into 293T cells using calcium phosphate precipitation. Virus supernatant was harvested at 48 hours. Spin infection was performed in 1×106 cells/ml concentration at 30°C for 90 minutes. Puromycin (final concentration 10ug/ml) was used for selection. sgRNA was generated by MEGAscriptTM T7 transcription kit and MEGAclearTM transcription clean-up kit (ThermoFisher Scientific). In order to construct a repair template, gBlock gene fragments (Integrated DNA Technologies) carrying bGH poly(A) with 700bps homology arms that flank the gRNA target site were inserted in pGEM-T easy by NEBuilder HiFi DNA Assembly Master Mix (NEB). pAS knock in was conducted in a Neon transfection system (Invitrogen) using 10 μl tips. 1 × 106 of SCIDadh cells were harvested in a 1.5 ml tube and washed with PBS. Cells were suspended in 45μl of buffer R. 3-4μl of sgRNA (~20 ug) and 1-2μl of repair template (10μg/μl) were added in cell suspension to make a total 50 μl solution. 2 × 105 Cells were transfected at one time with run#3 (pulse voltage 1500, Pulse width 20, and Pulse no 1), then transferred to 1 well of 12 well plates with 1.5 ml of IMDM without PSG. The next day cells were transferred into Methocult M3630 (STEMCELL technology). Single colonies were picked and transferred into 96 well plates at day 3 and cultured for 5-6 days. Genotyping was performed for the cells isolated from each well.
sgRNA template sequence at upstream of ThymoD CR1
- cagctGAATTCTAATACGACTCACTATAGGGGGAAAACACAGACCCTAGTCgttttagagctagaaatagcaagttaaaataaggctagtccGttatcAacttgaaaaagtggcaccgagtcggtgc
sgRNA template sequence at exon3 of thymoD (upstream of CR2)
- cagctGAATTCTAATACGACTCACTATAGGGTCTTCAAGGGTGCTATCACAgttttagagctagaaatagcaagttaaaataaggctagtccGttatcAacttgaaaaagtggcaccgagtcggtgc
Primers for genotyping at upstream of ThymoD CR1
| IntCR1-2_F1 | ATGGATGGAGAGGTGGACTG |
|---|---|
| IntCR1-2_R1 | CCCTGAGAGAGCCCTAATCC |
| int5_out | CTCTGTCTGCCAACCCAACT |
Primers for genotyping at exon3 (upstream of CR2)
| ThymoDex3F | GGGCAGACGAAACTGACTGT |
|---|---|
| ThymoDex3R2 | AAGCCCTGCCTTGACTGTAA |
| CR2_ex3_F1 | ACCAGGAGAAGAGTGCTGGA |
Generating ThymoD pA/pA mice
A sgRNA target site (TCTTCAAGGGTGCTATCACA) as described for SCIDadh pA/pA CR2 mutants was computationally identified in ThymoD exon 3 and verified by transfection of sgRNAs into a pro-B cell line (IM3) that stably expressed Streptococcus pyogenes WT Cas9 using the same method for generating SCIDadh mutant cell line. In order to construct a repair template, homology arms that flank the sgRNA target site were isolated from amplified C57Bl/6 genomic DNA. Homology arms flanking a bGH poly(A) addition site were inserted in pGEM-T easy. The inserted poly(A) addition site is located upstream of CR2 region as well as 12kb upstream of the CR1 region (Li et al. 2013). Next a mixture of sgRNA, Cas9 mRNA and repair template were injected in mouse zygotes using standard procedures. The inserted pAS sequence was detected by PCR.
Primer for genotyping
- Set 1 F; 5′-GTGTCTCAAGGCGAGAAAGG-3′
- Set 1 R; 5′-AAGCTCGCTCTGTTTTGAGG-3′
- Set 2 F; 5′-ACTGAACAGCTCTCACCCTC-3′
- Set 2 R; 5′-ACTGTAAAGCCCTGGGTCCT-3′
Flow cytometry
For flow cytometry and cell sorting, single cell suspensions of thymus were prepared and analyzed as follows. Dead cells were eliminated using ghost dye red 780. Cells were stained with CD3e (145-2C11), CD4 (GK1.5; RM4-5), CD8a (53-6.7), CD11b (M1/70), CD19 (MB19-1), B220 (RA3-6B2), Gr1 (RB6-8C5), NK1.1(PK136), CD25 (PC61), CD27 (LG3A), CD44 (IM7), CD45.2 (104), CD117 (ack45), TCRb (H57-597), TCRgd (eBioGL3), Ter119 (TER119), and Thy1.2 (53-2.1) antibodies (Becton Dickinson and e-Biosciences). Antibodies were conjugated to fluorescein isothiocyanate, phycoerythrin, peridinin chlorophyll protein-cyanine 5.5, allophycocyanin, allophycocyanin-indotricarbocyanate, Pacific Blue, eFluor 450, Brilliant Violet 421 or were biotinylated. Biotynylated antibodies were stained using QD588 quantum dots (Life Technologies). Data were collected on a LSRII (BD Biosciences) and analyzed with FlowJo software (TreeStar). Cell sorting was performed on a FACSAriaII (BD).
Wright Giemsa staining
Cytospin was done at 1000rpm for 3min. Cover slips were stained by Wright solution for 3 min. Coverslips were washed into each well in 6well plate 2 times. Giemsa staining (1:10) was done for 7 min and washed into each well in 6well plate 2 times. Images were captured by an Olympus BH2 microscope.
Real-time PCR
For quantitative PCR cells were stored in buffer RLT (Qiagen) with 2-Mercaptoethanol at −80°C. Total RNA was extracted using an RNeasy Mini Kit (Qiagen) with DNase digestion (Qiagen) or alternatively with RNeasy Micro Kit (Qiagen) for small numbers of cells. cDNA was synthesized using a first-strand synthesis kit (Life Technologies) and oligo-dT primers. Conventional PCR was performed for cDNA with SYBR Green Master Mix (Roche). Previously reported primer sets were used for BCL11b and ARP detection (Ikawa et al. 2010).
Primer for qPCR
| ThymoD F | GGGCAGACGAAACTGACTGT |
|---|---|
| ThymoD R | AAGCCCTGCCTTGACTGTAA |
3D-FISH
For 3D-DNA FISH fosmid probes were used. WIBR1-1110C15 was used for the Bcl11b enhancer region and obtained from the BACPAC Resource Center (BPRC) at Children’s Hospital Oakland Research Institute. Fosmid DNA was labeled with fluorochrome by Nick translation. 20ul solution consisted of 1μg of DNA, 4 μl of 5× Alexa fluor mix, 5× nick translatioin mix, 2 μl of 10× buffer was incubated at 15°C for 4 hour, heat killed by adding 1 μl of 0.5M EDTA pH 8 at 65°C for 10 minutes, and purified through ProbeQuant G-50 micro column. 2×105 cells/40 μl were put on the center of the poly-L-Lysine coated coverslip and cultured in 37 °C for 30 minutes in cell culture incubator. Cells were washed with PBS, fixed at room temperature (RT) for 10 minutes with 4% paraformaldehyde in 1× PBS, pH 7.2. Fixed cells were quenched at RT for 10 minutes with 0.1 M Tris-Cl, pH 7.4, washed with PBS and stored in 1xPBS at 4°C for up to 1 month. Cells were permeabilized in 0.1% saponin, 0.1% Triton-X 100, 1× PBS for 10 minutes at RT, incubated 20 minutes at RT with 20% glycerol, 1× PBS, freeze-thawed in liquid nitrogen three times and rinsed in 1× PBS. For immunofluorescence, cells were blocked at 37°C for 30 minutes in 5% BSA, 0.1% triton-X 100, 1× PBS and stained using a primary Lamin B1 (M-20) antibody (sc-6217) obtained from Santa Cruz Biotechnology at 1/150 dilution in the blocking buffer at 37°C for 30 minutes. Primary stained cell were washed for 10 minutes at RT twice in 1× PBS, 0.1% Triton-X 100 at RT with gentle agitation. For second staining, cells were blocked for 30 minutes at 37 °C in 5% BSA, 0.1% triton-X 100, 5 % donkey serum, 1× PBS, followed by secondary staining of donkey anti-goat IgG antibody conjugated to Alexa594 (A11058) (Invitrogen). Primary stained cell were washed for 10 minutes at RT twice in 1× PBS, 0.1% Triton-X 100 at RT with gentle agitation, fixed again for 10 minutes at RT in 2% PFA, 1× PBS and quenched at RT for 10 minutes with 0.1 M Tris-Cl, pH 7.4, washed with PBS and stored in 1xPBS. For DNA-FISH, cells were denatured for 30 minutes in 0.1 M HCl at RT, blocked for 1hr at 37 °C in 3% BSA + 100 μg/ml Rnase A in 1× PBS, and permeabilized for 30 minutes at RT in 0.5% saponin, 0.5 % triton-X 100, 1× PBS. Cells were then washed once in PBS and stored in 2× SSC until hybridization. For hybridization, nuclear DNA was denatured by incubating coverslips for 2 minutes and 30 seconds at 73°C in 2× SSC, 70% formamide solution, followed by an incubation for 1 minute in 2× SSC, 50% formamide solution. The hybridization solution contained 100 ng of labeled fosmid probe, 4 μg of mouse Cot-1 DNA, 1 μg of sheared salmon-sperm DNA dissoleved in 50% formamide, 4× SSC and 20% dextran sulfate. The probes were denatured at 73°C for 5 minutes. Denatured coverslips and probes were sealed and incubated at 37°C for over-night in hybridization oven. On the next day, coverslips were removed and washed once in 2× SSC, 50% formamide solution for 15 minutes and three times in 2× SSC for 5 minutes at 37°C with gentle agitation. Cells were washed once with PBS, excess PBS was removed and coverslips were mounted on slides with Prolong gold anti-fade reagent with DAPI (Invitrogen). Images were acquired on a Deltavision microscope using 100× objective lens. 0.2 μm optical sections were obtained in the DAPI, FITC and Red channels. The distances between the nuclear lamina and the BCL11b intergenic region were measured in 2D with ImageJ software.
In Situ Hi-C
We used in-situ Hi-C with minor modifications to generate a genome-wide contact map essentially consisted of 70 steps as described in the supplementary material (Rao, S.S. et al 2014). Five million cells each of wild-type DN2 and ThymoD−/− DN2 cells were generated using OP9-DL1 stromal cells. For step 7, Complete Protease Inhibitor Cocktail, EDTA-free (Roche) was used. One tablet was dissolved in 1 ml dH2O. 5 μl of 50× Protease Inhibitor was used to 250 ml Hi-C lysis buffer. For step 8 and 9, centrifugation was done at 200× G for 5 minutes. For step 10, nuclei were resuspended in 0.5% SDS diluted in a final concentration of 1× NEB Buffer 2. For step 11, 5 % Triton X-100 was used instead of 10 %. For step 12, digestion efficiency was determined by using 3 μl of sample with 87 μl of 1× tris and 10 μl of proteinase-K. Samples were incubated at 55°C for 30 minutes, at 65°C with shaking for at least 1 hour, and purified with DNA clean & concentrator (Zymo Research). Samples were run on 0.6% agarose gel. For step 27, sheared DNA size should be 300-600 bps. Samples were checked by taking 1μl of sample and run on 2 % agarose gel. For step 54–57 each sequencing library was incubated with USER enzyme for 15 minutes at 37°C after step 56. Final libraries were submitted to paired-end sequencing of 100 bp length on an Illumina HiSeq 2500.
ATAC-Seq
ATAC-seq was performed as previously described (Buenrostro et al. 2013). 50000 cells were used for library preparation. Washed cells were resuspended in lysis buffer. After washing, cells were treated with transposition mix for 30 min at 37 °C. DNA was purified by DNA clean & concentrator (Zymo Research). Library fragments were amplified using 1× NEBnext PCR master mix and 1.25 μM of custom Nextra PCR primers 1 and 2 with following PCR conditions: 72°C for 5 minutes, 98°C for 30 seconds, followed by thermocycling at 98°C for 10 seconds, 63°C for 30 seconds and 72°C for 1 minute. Cycle of PCR amplification was determined by q-PCR with Sybr Green in order to stop amplification prior to saturation. Amplified 100–800 bps of Libraries were selected from 2% agarose gel. The quality of the library was checked with Agilent TapeStation and sequenced on an Illumina Hi-Seq4000.
ChIP-Seq
Chromatin was immunoprecipitated with following antibodies. CTCF antibody (07-729) was obtained from Millipore. H3.3 (ab176840), H3K4me1 (ab8895), H3K4me3 (ab8580), RNAPII 8WG16 (ab817) and SMC3 (ab9263) antibodies were purchased from Abcam. Cells were isolated from OP9-DL1 cultures and stained with FACS antibody and ghost dye to sort DN2 cells. Cells were fixed for 10 min in PBS containing 1% formaldehyde. Formaldehyde was quenched with 0.2 M glycine for 10 min. Lineage negative fixed DN2 cells were sorted by FACS AriaII and washed with PBS. Fixed cells were stored at −80 °C until use. For preparation for cells treated with actinomycin D, lived cells were selected by ficol-paque plus (GE health care) at day 10. Selected live DN2 cells were suspended with MEM containing 20% FCS, 2% PSG, actinomycin D 10 ug/ml, IL7 and FLT3L and cultured without OP9-DLL1 for 1hr. Cells were harvested at 15, 30, and 60 min and immediately spun down and fixed as described above. Nuclei were isolated in cell buffer mix 10 mM of HEPES/KOH, 85 mM of KCL, 1 mM of EDTA, 1 mM of Benzamidine, 1× Protease inhibitor (Roche), 1% of NP-40 for 10–15 min on ice. Nuclei were spun down for 5 min at 3000 rpm and resuspended in lysis buffer with protease inhibitors. Nuclei were sonicated by Biorupter (Diagenode) with 20 cycles of 30 sec on and 30 sec off at high setting. Sonicated chromatin was immunnoprecipitated with antibodies coated on Protein G-Sepharose for over-night at 4 °C. Samples were washed 5 times with LiCl wash buffer, mixing 3 min for each wash on a rotator and washed with TE buffer, mixing 1 min on rotator. Samples were resuspended in 200 μl IP Elution Buffer, incubated with 65 °C thermomixer for 3 hrs, mixed with 2 μl of protenase-K at 50 °C for 1hr. Samples were placed on magnet and transferred to new 1.5ml Eppendorf tube and incubated at 65 °C for over-night in hybridization oven for reverse cross-linking. DNA was purified by ChIP DNA clean & concentrator (Zymo Research). Immunoprecipitated DNA was end-repaired, added with dATP by using Klenow exo-, ligated with adaptors from NEBNext Multiplex Oligos for Illumina (NEB) and purified with SPRI beads. Adaptor ligated DNA was amplified with NEBNext Multiplex Oligos for Illumina (NEB) and size-selected with SPRI beads. The quality of the library was checked with Agilent TapeStation. Libraries were run on Illumina Hi-Seq4000.
RNA-Seq
Total RNA was isolated from DN2 cells sorted by AriaII and treated with TURBO DNase (Ambion). mRNA was purified from total RNA by a Dynabeads mRNA purification kit (Life Technologies). cDNA was generated with a First-Strand Synthesis Kit (Life Technologies) and a random hexamers in presence of actinomycin D. Second-strand synthesis was performed with dUTP instead of dTTP. The double-stranded cDNA was sinicated to a length of 200–400 base pairs with S220 Focused-ultrasonicator (Covaris). Sonicated cDNA was ligated to adaptors. Libraries were prepared by TruSeq Stranded mRNA library prep kit (Illumina). Libraries for DN2 cells were sequenced for 50 cycles on Illumine HiSeq 2500. Total RNA from Tumor samples were isolated from whole enlarged thymus, prepared for library as same as DN2 RNA, and sequenced for 50 cycles on Illumina HiSeq 4000.
GRO-seq
Duplicates of GRO-seq experiments were performed. Nuclei from 5 million cells were isolated by hypotonic lysis. Nuclear run on (NRO) reaction was done for 5 min at 30°C in the presence of BrUTP 750 μM, ATP 750 μM, GTP 750 μM, reduced concentrations of CTP 4.5 μM and 1.65% sarkosyl. Total RNA was purified with Trizol and isopropanol precipitation, DNase treated (TURBO DNase, Ambion), fragmented with 2ul of fragmentation regent (Ambion AM1907). Fragmented RNA was re-buffered by P30 RNase-free spin column (Bio-Rad). RNA fragments were 3′ dephosphorylated with T4 polynucleotide kinase. BrUTP-labeled run-on RNA was immunopurified with anti-BrdUTP-coated agarose beads, washed, and EtOH-precipitated. Run-on RNA was de-capped with tobacco acid pyrophosphatase from FirstChoice RLM-RACE kit (Thermo Fisher Scientific), 5′ phosphorylated with polynucleotide kinase (NEB) and purified with Trizol LS/isopropanol precipitation.3′ adapter ligation was done by ligationg a single-stranded, 3′-blocked, 5″-adenylated 3′ oligonucleotide with mutant (K227Q) truncated RNA ligase 2 (NEB) to the 3′ end of the RNA fragments, followed by annealing a reverse transcription primer complementary to the 3′ adapter to suppress adapter dimer formation, and ligating a hybrid 5′ DNA-RNA oligonucleotide using RNA ligase I and reverse-transcribing with SuperScript III reverse transcriptase (ThermoFisher Scientific). The cDNA was purified with SPRI beads. The libraries were amplified with primers bearing primer landing site compatible with illumine sequencing. The libraries were size-selected with 2% agarose containing SYBR gold to 60–110 bp insert size, followed by the quality of the library was checked with Agilent TapeStation and then sequenced for 50 cycles on Illumina HiSeq 2500.
Whole genome bisulfite sequencing (WGBS)
Library preparation for WGBS was previously described (Benner, C. 2015). DNA was isolated from in-vitro cultured WT DN2 and ThymoDpA/pA DN2 cells. 1 μg of genomic DNA mixed with unmethylated lamda DNA at a concentration of 0.5% of total DNA was sonicated by Biorupter with 20 cycles (30 seconds on and 30 seconds off at low condition). Fragmented DNA was end-repaired using the End-It procedure (Epicentre), and incubated in the presence of Klenow (3′ to 5′ exo minus) for adding an A base to the 3′-end. Next TruSeq adapters were ligated to fragmented DNA and purified using 2% Agarose gel electrophoresis. Bisulfite conversion was performed as described by the manufacturer (MethylCode). Bisulfite-treated DNA was amplified by using a TruSeq PCR primer mixture and Pfu Turbo Cx Polymerase, agarose purified, and sequenced on an Illumina HiSEq 2500 sequencer.
QUANTIFICATION AND STATISTICAL ANALYSIS
Filtering and Normalization of HiC Reads
Reads were first trimmed from the 3′ end of sequences to GATC (MboI restriction enzyme site). Trimmed reads were aligned to mm9 with Bowtie software with parameters --chunkmbs 128 --mm -m1 --best --strata -p4 -S. The remainder of the analysis was performed using Homer (Heinz et al. 2010). Only unique reads and paired end tags with more than 1.5× fragment length on the same chromosome separating them were retained. Sequence reads were checked for GC and nucleotide bias. HiC reads derived from wild-type DN2 and ThymoD−/− DN2 cells were compared to pro-B and multipotent progenitors (Lin et al. 2012). Tag counts for all directories were normalized to the sample with lowest sequencing depth. For multipotent progenitors, pro-B and wild-type DN2 comparisons a map resolution of 50 kb was chosen to ensure that > 80% of loci were associated with at least 1000 genomic contacts. A superRes of 100 kb was used for comparison of wild-type DN2 and ThymoD−/− DN2 cells. A resolution of 40 kb was chosen to ensure that greater than 80% of loci were associated with at least 1000 contacts. Hi-C heatmaps were normalized to output the ratio of observed to expected interactions by assuming each region has an equal chance of interacting with other regions in the genome and that regions are expected to interact depending on their linear distance along the chromosome (Lin et al. 2012). Hi-C heatmaps were visualized using Java Tree View. Circos diagrams were generated using Circos software. Only interactions with p-values <= 0.0001 are shown. Line thickness of Circos diagrams refers to p-values. Darker lines depict lower p-values. In depth explanations of normalization, generation of Hi-C correlation matrices, principal component analysis (PCA) and identifying significant interactions were performed as previously described (Lin et al. 2012).
Analysis for ATAC-Seq
ATAC-Seq data were mapped to reference genome mm9 by Bowtie (version 1.1.1), using following option: --best -m 1, and others as default. The bam file were then processed by Homer to create tag directory with the default setting of Homer makeTagDirectory, using option -tbp 1. For ATAC open region, following options were used: localSize 50000 -size 150-minDist 50 –fragLength 0, and we used the default setting for other peak/region calling.
ChIP-Seq
ChIP-Seq data were mapped to reference genome mm9 by Bowtie (version 1.1.1), using following option: --best -m 1, and others as default. The bam file were then processed by Homer to create tag directory with the default setting of Homer makeTagDirectory, using option -tbp 1. Homer findPeak has been used to identify enriched regions.
Analysis of CTCF direction and significant interaction
p-values associated with CTCF-CTCF interactions were computed as described by HOMER with the following modification. Bins were centered on CTCF bound site in wild-type or ThymoD p(A)/p(A) DN2 cells. CTCF directionality was identified by scanning using FIMO. The following parameter were used --motif CTCF_MOUSE.H10MO.A --text --thresh 0.01. MEME’s mouse DNA-binding protein motif database was used (HOCOMOCOv10_MOUSE_mono_meme_format.meme). For multiple significant CTCF motifs that showed opposite directions within a CTCF binding site interactions originating from that site were identified as “unknown”.
Analysis of SMC3 Occupancy in DN2 Cells Exposed to Actinomycin
DN2 cells were incubated in the presence of actinomycin, formaldehyde fixed and analyzed for SMC3 occupancy using ChIP-Seq as described above. Active transcription start sites were identified using RNA polymerase II ChIP-Seq. Active TSS sites were defined by RNAPII peaks spanning a 2 kb window centered on the TSSs. −log 10 p-values of the Fisher test were calculated for the contingency table. SMC reads were determined across the ThymoD locus (chr12: 108.345–108.405 Mb) in DN2 cells treated in the absence or presence of actinomycin D and normalized to the total number of reads (107 read). The mean and standard deviation for each experiment were derived from 20 independent samples using boot strapping for all reads across the genome.
RNA-Seq
RNAseq data were examined using Omics Pipe applying the RNA-seq count-based differential expression analysis pipeline. Quality control of the raw fastq files was performed using FastQC (Babraham Bioinformatics). Sequencing reads were aligned to the mouse genome (mm10) applying STAR aligner. Read quantification (exons) was performed using htseq-count with UCSC RefSeq annotation. The R BioConductor package DESeq2 was used to calculate size factors in order to normalize library sizes across replicates and calculate means and variances based on a negative binomial distribution model in order to detect differentially expressed genes based on adjusted p-values of < 0.05. Functional enrichment of the differentially expressed genes was done using ToppGene Suite, WebGestalt and Metascape.
Analysis of GRO-seq
GRO-Seq data were mapped to reference genome mm 9 by Bowtie (version 1.1.1), using following option: --best -m 1, and others as default. The bam file are then processed by Homer to create tag directory with the default setting of Homer makeTagDirectory, using option -tbp 1. UCSCfiles were generated with default setting with -strand separate.
Analysis of WGBS
Analysis of bisulfite sequencing was performed using BSSeeker2 (Guo et al., 2013), HOMER (Heinz, S. 2010), and custom awk scripts. Specifically, a bisulfite-sequencing amenable mm9 reference genome was built using the BSSeeker2 script bs_seeker2-build.py with default options. Paired-end sequencing data was aligned to this reference genome using the BSSeeker2 script bs_seeker2-align.py allowing 6 mismatches (-m 6) and a fragment length between 0 and 800 bases (-I 0 -X 800). PCR duplicates were removed from paired end data using picard tools (https://broadinstitute.github.io/picard/). Unpaired reads were saved, aligned individually with the same options, and merged with paired-end data. Methylation levels were called using the BSSeeker2 script bs_seeker2-call_methylation.py with default settings. BSSeeker2 methylation call data was reformatted to the allC format (Lister et al., 2009) and tag directories were created using HOMER’s makeTagDirectory command with the -minCounts 5 option, thus including only those cytosines covered by at least 5 reads. Downstream analysis comparing DNA methylation levels between wild-type and ThymoD pA/pA cells included only those cytosines covered by 5 reads in both data sets.
Supplementary Material
1
2
Table S1. Related to Figures 1, 4, 5 and 6. Reads Obtained for Genome-Wide Analysis are Presented for wild-type, ThymoD-deficient and Actinomycin D treated cells. HIC, ATAC-Seq, GRO-Seq and ChIP-Seq reads are indicated.
Table S2. Related to Figure 4. Numbers of Switched Genomic Regions for Multipotent Progenitors and Thymic Populations. The positions of genomic regions were assigned to A or B compartments based on PC1 values. Genomic regions that switched compartments during developmental progression are shown.
Table S3. Related to Figure 4. Numbers of Switched Genomic Regions Associated with Distinct Chromosomes for Multipotent Progenitors and Thymic Populations. The positions of genomic regions were assigned to A or B compartments based on PC1 values across chromosomes. Genomic regions that switched compartments during developmental progression are shown. Also indicated are changes in compartmentalization for wild-type and ThymoD p(A)/p(A) DN2 cells.
3
- Non-coding transcription directs loop extrusion
- Non-coding transcription dictates compartmentalization
- Non-coding transcription directs enhancer-promoter communication
- Non-coding transcription establishes T cell identity and blocks lymphoid malignancy
Acknowledgments
We thank Alex Bortnick for editing the manuscript. This study was supported by funding from the CCBB (UL1TRR001442), the CIRM (RB5-07025) and the NIH to C.M. (AI102853 and DK107977) and D.W. (AI102853). TI was supported by the Uehara Memorial Foundation.
Footnotes
Supplemental information includes seven figures and three tables and is deposited online with this manuscript.
AUTHORS CONTRIBUTIONS
T.I. and C.M. designed experiments. T.I. performed the majority of the experiments. A.M. performed HiC. V.C. identified ThymoD. Z.H., M.D., K.F. and A.C. performed bioinformatics. P.v.H. characterized ThymoD isoforms. S.F. and D.W. provided cells. T.I. and C.M. wrote the manuscript. C.M. conceived and supervised the study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bain G, Murre C. The role of E-proteins in B and T lymphocyte development. Semin Immunol. 1998;10:143–153. doi: 10.1006/smim.1998.0116. [DOI] [PubMed] [Google Scholar]
- Benner C, Isoda T, Murre C. New roles for DNA cytosine modification, eRNA, anchors, and superanchors in developing B cell progenitors. Proc Natl Acad Sci U S A. 2015;112:12776–127781. doi: 10.1073/pnas.1512995112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj S, Schlackow M, Rabajdova M, Gullerova M. Transcription facilitates sister chromatid cohesion on chromosomal arms. Nucleic Acids Res. 2016;44:6672–6692. doi: 10.1093/nar/gkw252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossen C, Murre CS, Chang AN, Mansson R, Rodewald HR, Murre C. The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat Immunol. 2015;16:775–784. doi: 10.1038/ni.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busslinger GA, Stocsits RR, van der Lelij P, Axelsson E, Tedeschi A, Galjart N, Peters JM. Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature. 2017;544:503–507. doi: 10.1038/nature22063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton M, Ruetsch NR, Berger MA, Rhodes M, Kaptik S, Wiest DL. Signals transduced by CD3e, but not by pre-TCR complexes, are able to induce maturation of an early thymic lymphoma in vitro. J Immunol. 1999;163:2576–2585. [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2017;539:452–455. doi: 10.1038/nature20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC, Zhang J, Protopopov A, Chin L, Dahlberg SE, Neuberg DS, Silverman LB, Winter SS, Hunger SP, Sallan SE, Zha S, Alt FW, Downing JR, Mullighan CG, Look AT. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood. 2011;118:4169–4173. doi: 10.1182/blood-2010-11-318873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Levine MS. Regulatory principles governing tissue specificity of developmental enhancers. Cold Spring Harbor Symposia on Quantitative Biology. 2015;80:27–32. doi: 10.1101/sqb.2015.80.027227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Maillard I, Chakraborti S, Rothenberg EV, Speck NA. Core binding factors are necessary for natural killer cell development and cooperate with Notch signaling during T-cell specification. Blood. 2008;112:480–492. doi: 10.1182/blood-2007-10-120261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, Chen PY, Pellegrini M. BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics. 2013;14:774. doi: 10.1186/1471-2164-14-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Shrivinas K, Young RA, Chakraborty AK, Sharp PA. A phase-separation model for transcriptional control. Cell. 2017;169:13–23. doi: 10.1016/j.cell.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa T, Kawamoto H, Goldrath AW, Murre C. E proteins and Notch signaling cooperate to promote T cell lineage specification and commitment. J Exp Med. 2006;203:1329–1342. doi: 10.1084/jem.20060268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikawa T, Hirose S, Masuda K, Kakugawa K, Satoh R, Shibano-Satoh A, Kominami R, Katsura Y, Kawamoto H. An essential developmental checkpoint for production of the T cell lineage. Science. 2010;329:93–96. doi: 10.1126/science.1188995. [DOI] [PubMed] [Google Scholar]
- Izumi K, Nakato R, Zhang A, Edmonson AC, Katou Y, Shirahige K, Krantz ID. Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nature Gen. 2015;47:338–347. doi: 10.1038/ng.3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura K, Ohi H, Kubota T, Okazuka K, Yoshikai Y, Wakabayashi Y, Aoyagi Y, Mishima Y, Kominami R. Haploinsufficiency of Bcl11b for suppression of lymphomagenesis and thymocyte development. Biochem Biophys Res Commun. 2007;355:538–542. doi: 10.1016/j.bbrc.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Kind J, Pagie L, de Vries SS, Nahidiazar L, Dey SS, Bienko M, Zhan Y, Lajoie B, de Graaf CA, Amendola M, Fudenberg G, Imakaev M, Mirny LA, Jalink K, Dekker J, van Oudenaarden A, van Steensel B. Genome-wdie maps of nuclear lamina interactions in single human cells. Cell. 2015;163:134–147. doi: 10.1016/j.cell.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see) Nat Rev Immunol. 2014;14:377–391. doi: 10.1038/nri3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh HY, Yui MA, Ng KK, Pease SS, Zhang JA, Damle SS, Freedman G, Siu S, Bernstein ID, Elowitz MB, Rothenberg EV. Asynchronous combinatorial action of four regulatory factors activates Bcl11b for T cell commitment. Nat Immunol. 2016;8:956–965. doi: 10.1038/ni.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Leid M, Rothenberg EV. An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science. 2010;329:89–93. doi: 10.1126/science.1188989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC, Lu D, Campos L, Goulding D, Ng BL, Dougan G, Huntly B, Gottgens B, Jenkins NA, Copeland NG, Colucci F, Liu P. Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science. 2010;329:85–89. doi: 10.1126/science.1188063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Zhang JA, Dose M, Kueh HY, Mosadeghi R, Gounari F, Rothenberg EV. A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood. 2013;122:902–911. doi: 10.1182/blood-2012-08-447839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Benner C, Mansson R, Heinz S, Miyazaki K, Miyazaki M, Chandra V, Bossen C, Glass CK, Murre C. Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nat Immunol. 2012;13:1196–1204. doi: 10.1038/ni.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q, Edsall ML, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Li P, Burke S. Critical roles of Bcl11b in T-cell development and maintenance of T-cell identity. Immunol Rev. 2010;238:138–149. doi: 10.1111/j.1600-065X.2010.00953.x. [DOI] [PubMed] [Google Scholar]
- Longabaugh WJR, Zeng W, Zhang JA, Hosokawa H, Jansen CS, Li L, Romero-Wolf M, Liu P, Kueh HY, Mortazavi A, Rothenberg EV. Bcl11b and combinatorial resolution of cell fate in the T cell gene regulatory network. Proc Natl Acad Sci USA. 2017;114:5800–5807. doi: 10.1073/pnas.1610617114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas JS, Zhang Y, Dudko OK, Murre C. 3D Trajectories adopted by coding and regulatory DNA elements: First-passage times for genomic interactions. Cell. 2014;158:339–352. doi: 10.1016/j.cell.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour MR, Sanda T, Young RA, Look AT. An oncogenic super-enhancer formed through somatic mutation of a non-coding intergenic element. Science. 2014;346:1373–1377. doi: 10.1126/science.1259037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansson R, Welinder E, Åhsberg J, Lin YC, Benner C, Glass CK, Lucas JS, Sigvardsson M, Murre C. Positive intergenic cirtcuitry, involvign EBF1 and FOXO1, orchestrates B cell fate. Proc Natl Acad Sci USA. 2012;109:21028–21033. doi: 10.1073/pnas.1211427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele M, Rinn JL. “Cat’s Cradling” the 3D Genome by the Act of LncRNA Transcription. Molecular Cell. 2016;62:657–664. doi: 10.1016/j.molcel.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Miyazaki K, Miyazaki M, Murre C. The establishment of B versus T cell identity. Trends Immunology. 2014;35:840–853. doi: 10.1016/j.it.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Miyazaki K, Chen K, Jin Y, Turner J, Moore AJ, Saito R, Yoshida K, Ogawa S, Rodewald HR, Lin YC, Kawamoto H, Murre C. The E-Id proteins axis specifies adaptive lymphoid cell identity and suppressed thymic innate lymphoid cell development. Immunity. 2017;46:818–834. doi: 10.1016/j.immuni.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito T, Tanaka H, Naoe Y, Taniuchi I. Transcriptional control of T-cell development. Int Immunol. 2011;23:661–668. doi: 10.1093/intimm/dxr078. [DOI] [PubMed] [Google Scholar]
- Nasmyth K. Disseminating the genome: joining, resolving and separating siste chromatids during mitosis and meiosis. Annu Rev Genet. 2001;35:673–745. doi: 10.1146/annurev.genet.35.102401.091334. [DOI] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Lieberman-Aiden E. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt TM, Zuniga-Pflucker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. 2002;17:749–756. doi: 10.1016/s1074-7613(02)00474-0. [DOI] [PubMed] [Google Scholar]
- Schmitt S, Prestel M, Paro R. Intergenic transcription through a polycomb group response element counteracts silencing. Genes & Devel. 2005;19:697–708. doi: 10.1101/gad.326205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsagaratou A, González-Avalos E, Rautio S, Scott-Browne JP, Togher S, Pastor WA, Rothenberg EV, Chavez L, Lähdesmäki H, Rao A. TET proteins regulate the lineage specification and TCR-mediated expansion of iNKT cells. Nat Immunol. 2017;18:45–53. doi: 10.1038/ni.3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Belmont AS. Lamina-associated domains: Links with chromosome architecture, heterochromatin and gene repression. Cell. 2017;169:7801–791. doi: 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber BN, Chi AW, Chavez A, Yashito-Ohtani Y, Yang Q, Shestova O, Bhandoola A. A critical role for TCF-1 in T-lineage specification and differentiation. Nature. 2011;476:63–68. doi: 10.1038/nature10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of Notch1 in human T cell acute lymphoblastoid leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Janenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui MA, Rothenberg EV. Developmental gene networks: a triathlon on the course to T cell identity. Nat Rev Immunol. 2014;14:529–545. doi: 10.1038/nri3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JA, Mortazavi A, Williams BA, Wold BJ, Rothenberg EV. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell. 2012;149:467–482. doi: 10.1016/j.cell.2012.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2
Table S1. Related to Figures 1, 4, 5 and 6. Reads Obtained for Genome-Wide Analysis are Presented for wild-type, ThymoD-deficient and Actinomycin D treated cells. HIC, ATAC-Seq, GRO-Seq and ChIP-Seq reads are indicated.
Table S2. Related to Figure 4. Numbers of Switched Genomic Regions for Multipotent Progenitors and Thymic Populations. The positions of genomic regions were assigned to A or B compartments based on PC1 values. Genomic regions that switched compartments during developmental progression are shown.
Table S3. Related to Figure 4. Numbers of Switched Genomic Regions Associated with Distinct Chromosomes for Multipotent Progenitors and Thymic Populations. The positions of genomic regions were assigned to A or B compartments based on PC1 values across chromosomes. Genomic regions that switched compartments during developmental progression are shown. Also indicated are changes in compartmentalization for wild-type and ThymoD p(A)/p(A) DN2 cells.
3