The RNA modification landscape in human disease (original) (raw)
Abstract
RNA modifications have been historically considered as fine-tuning chemo-structural features of infrastructural RNAs, such as rRNAs, tRNAs, and snoRNAs. This view has changed dramatically in recent years, to a large extent as a result of systematic efforts to map and quantify various RNA modifications in a transcriptome-wide manner, revealing that RNA modifications are reversible, dynamically regulated, far more widespread than originally thought, and involved in major biological processes, including cell differentiation, sex determination, and stress responses. Here we summarize the state of knowledge and provide a catalog of RNA modifications and their links to neurological disorders, cancers, and other diseases. With the advent of direct RNA-sequencing technologies, we expect that this catalog will help prioritize those RNA modifications for transcriptome-wide maps.
Keywords: RNA modification, epitranscriptome, disease, detection methods, direct RNA sequencing
INTRODUCTION
Advances in genomic sequencing technologies have revolutionized our understanding of the mammalian genome and its transcriptional output. It is now evident that most of the genome does not code for protein but is transcribed to produce, in addition to messenger RNAs, a vast pool of intronic, intergenic, and antisense RNAs (Djebali et al. 2012). These non-protein-coding RNAs, most of which have yet to be biologically characterized, are likely to fulfill a wide variety of roles in cell and developmental biology, including the guidance of epigenetic processes (Mattick 2010, 2011; Mercer and Mattick 2013; Morris and Mattick 2014).
While the triplet code of the open reading frame is well understood for mRNAs, the language used by noncoding RNAs to execute their biological functions remains elusive. Dynamic regulation of RNA expression patterns, localization, structure, splicing, stability, and interactions with RNA-binding proteins will intricately dictate this language. In this already complex scenario, RNA modifications, for which more than 100 different types have been described (http://modomics.genesilico.pl/sequences/; http://mods.rna.albany.edu) (Cantara et al. 2011; Machnicka et al. 2013), overlay the RNA sequence information, expanding its lexicon (Hussain and Bashir 2015).
RNA modifications were first detected in highly abundant “infrastructural” RNAs, such as rRNAs and tRNAs, followed by snoRNAs and snRNAs, and have been generally viewed as irreversible decorations important for RNA structural stability and/or catalytic function (Karijolich and Yu 2010). However, the RNA modification field was greatly stimulated by the discovery that at least some RNA modifications are reversible (Jia et al. 2011; Liu et al. 2016), leading to the birth of the term “epitranscriptome” (He 2010). Comparative analyses of these modification sites across closely related species have shown that these dynamic, reversible modifications are evolutionarily conserved (Schwartz et al. 2013; Batista et al. 2014; Li and Mason 2014; Dominissini et al. 2016).
The functional and evolutionary relevance of the epitranscriptome is yet unknown, but it may represent the crossroads of gene–environment interactions for physiological adaptation and cognition (Mattick 2010; Hussain and Bashir 2015). There is a progressive expansion of enzymes that impart RNA editing and the extent of RNA editing in the brain during cognitive evolution (Mattick and Mehler 2008; Behm and Öhman 2016). A clear example of this expansion is the family of adenosine deaminases acting on RNA (ADARs), responsible for the editing of adenosine to inosine, thus increasing gene product diversity, particularly in primates (Paz-Yaacov et al. 2010). Comparative analysis of ADARs suggests that these enzymes evolved from adenosine deaminases acting on tRNAs (ADATs)—present in both Bacteria and Eukarya—after the split of protozoa and metazoa (Grice and Degnan 2015). Thus, A-to-I editing of tRNAs is likely ancestral to editing of other RNAs, achieving its maximal diversity in vertebrates, via the appearance of ADAR3, whose expression is largely restricted to brain (Chen et al. 2000), and the expansion of the ABOBEC family (which catalyze C/meC deamination to U/T) in mammals, with strong positive selection in the primates (Sawyer et al. 2004).
Recent studies have provided detailed maps of the location and abundance of a handful of RNA modifications (Dominissini et al. 2012; Meyer et al. 2012; Khoddami and Cairns 2013; Carlile et al. 2014; Schwartz et al. 2014a; Delatte et al. 2016), mostly obtained by coupling antibody immunoprecipitation or chemical treatments to next-generation sequencing. Through these approaches, _N_6-methyladenosine (m6A) modification has been identified as an important factor in the determination of mammalian cell fate transition and embryonic stem cell differentiation (Batista et al. 2014; Wang et al. 2014). It is also involved in the regulation of circadian rhythms in hypothalamic mouse brain (Fustin et al. 2013), sex determination in flies (Haussmann et al. 2016; Lence et al. 2016), and maternal mRNA clearance in zebrafish (Zhao et al. 2017). Transcriptome-wide maps have also been obtained for _N_1-methyladenosine (m1A) (Dominissini et al. 2016), _N_6,2′-_O_-dimethyladenosine (m6Am) (Linder et al. 2015; Mauer et al. 2016), 5-methylcytosine (m5C) and pseudouridine (Y), revealing their involvement in biological processes such as stress responses (Carlile et al. 2014), protein synthesis quality control (Tuorto et al. 2012; Hussain et al. 2013; Blanco et al. 2014), and mRNA stability (Mauer et al. 2016), among others. There are several excellent reviews on these few relatively well-characterized RNA modifications (Klungland and Dahl 2014; Li and Mason 2014; Frye et al. 2016; Gilbert et al. 2016; Li et al. 2016; Schwartz 2016; Zhao et al. 2016).
In addition to mapping RNA modifications in a genome-wide fashion, several studies have attempted to characterize the biological function of RNA modifications by comparing wild-type cells to those that lack a specific RNA modification enzyme (Zinshteyn and Gilbert 2013; Nedialkova and Leidel 2015). Although most RNA modifications do not appear to be essential for viability in fungi—but may play important roles in fitness—they have been shown to be critical for maintaining protein homeostasis (Nedialkova and Leidel 2015; Klassen et al. 2016), proper cellular signaling (Zinshteyn and Gilbert 2013), and translation fidelity (Patil et al. 2012; Agris et al. 2017).
RNA modifications have been historically considered to be relatively static fine-tuners of the RNA structure and function. However, in the last few years, it has become evident that the epitranscriptomic layer is not only dynamic and reversible (Jia et al. 2011; Zheng et al. 2013; Wang and He 2014; Liu et al. 2016)—catalyzed by RNA modification “erasers” (Meyer and Jaffrey 2017)—but also that the activity of RNA modifications can be regulated by a wide variety of factors, including environmental conditions (Chan et al. 2010; Dedon and Begley 2014; Alings et al. 2015; Han et al. 2015).
The past two decades have witnessed considerable progress in the identification of novel RNA modifications (Grosjean 2015). Unfortunately, the distribution of most remains uncharacterized. Here we provide an overview of current methodologies that have been used to date to map RNA modifications, and consider novel technologies such as direct RNA sequencing, especially useful in the case of RNA modifications for which no other genome-wide method exists. To help decide which RNA modifications to prioritize, we also provide an overview of RNA modifications shown to be linked to human disease, including their distribution and the enzymes involved.
HUMAN RNA MODIFICATIONS AND THEIR IMPLICATIONS IN DISEASE
A comprehensive catalog of RNA modifications and their association to human disease is provided in Table 1 (with the complete list in Supplemental Table S1). Mutations in approximately half of the currently known RNA modification enzymes have been linked to human diseases, including cancer, cardiovascular diseases, genetic birth defects, metabolic diseases, neurological disorders, and mitochondrial-related defects (Fig. 1). From the more than 100 different associations between mutations in RNA modification enzymes and human disease (Supplemental Table S1), we find that neurological diseases are largely overrepresented, in agreement with the observed enrichment of several RNA modifications in neuronal tissues (Paul and Bass 1998; Chi and Delgado-Olguin 2013) and in neuronal dysfunction (Najmabadi et al. 2011; Abbasi-Moheb et al. 2012; Davarniya et al. 2015; Lence et al. 2016).
TABLE 1.
RNA modifications linked to human disease
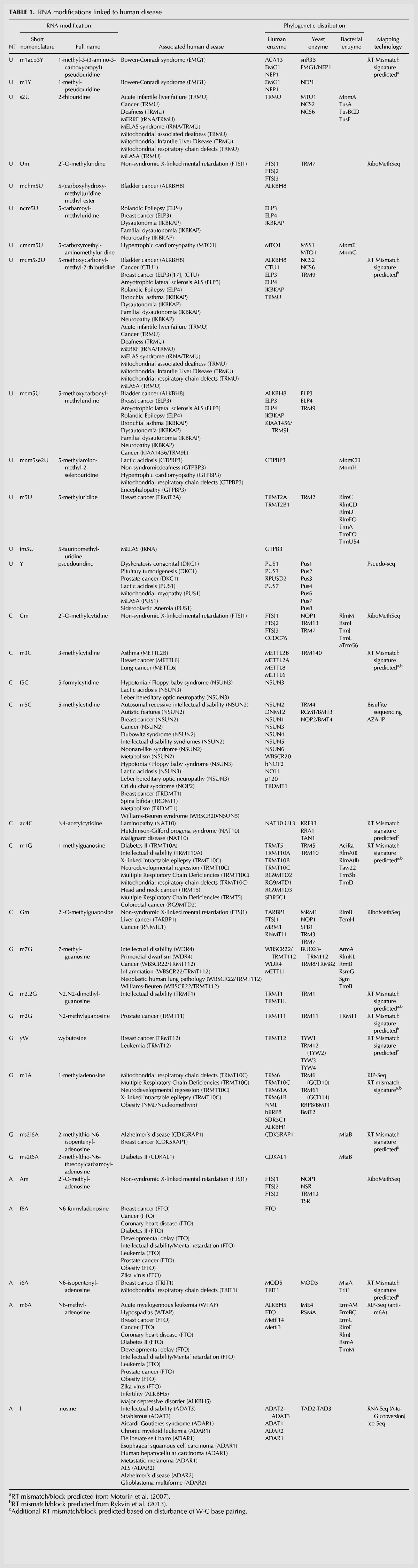
FIGURE 1.

RNA modifications and their links to human disease. The set of known RNA modifications classified by their reference nucleotide, highlighting those that have been associated to human diseases (red), as well as those for which a transcriptome-wide detection method has been established (circled in green).
From the battery of known RNA modifications, those present in rRNAs and tRNAs largely dominate the landscape (Supplemental Table S1). Consequently, previous studies characterizing the associations between RNA modifications and human diseases have been mainly focused on modifications occurring in tRNA molecules (Sarin and Leidel 2014; Torres et al. 2014; Schaffrath and Leidel 2017). However, pseudouridine, originally thought to be exclusive to these infrastructural RNAs, has been detected in several mRNAs (Carlile et al. 2014; Schwartz et al. 2014a), suggesting that additional modifications typically thought to be restricted to “classical” RNA molecules may actually occur more widely.
The mechanisms whereby a lack of modification may lead to disease is a field of active debate. It has been postulated that specific RNA modifications may be essential to tune the proteomic outcome under stress conditions, and that the lack of regulation may affect the ability of the cell to responsively tune its proteome (Patil et al. 2012; Deng et al. 2015). On the other hand, the lack of specific RNA modifications may affect global and/or local translation rates, and consequently cause increased protein aggregation (Nedialkova and Leidel 2015). Finally, it has also been proposed that RNA modifications transduce information that connect the cell's metabolic state to its translational output, and therefore, that their dysregulation may cause an imbalance between metabolic rates and protein synthesis (Helm and Alfonzo 2014). Future work will be needed to disentangle the causal relationship between RNA modification dysregulation and human disease.
Neurological diseases
Defects in RNA metabolism, including RNA synthesis, processing, function, and degradation, have been found to be associated with motor neuron disorders (Lemmens et al. 2010). In this regard, several RNA methyltransferase-encoding genes have been linked to intellectual disability, supporting the relevance of RNA modifications in the development of cognitive functions (Bednářová et al. 2017). These include FTSJ1, identified in X-linked nonsyndromic intellectual disability in which mental retardation is the sole clinical feature (Freude et al. 2004); TRMT1, which has been identified as the cause of autosomal-recessive intellectual disability (ARID) (Najmabadi et al. 2011; Davarniya et al. 2015); and the m5C methyltransferase NSUN2, which has been associated with defects in memory and learning in Drosophila and NSUN2-deficient mouse models (Abbasi-Moheb et al. 2012; Blanco et al. 2014). Mechanistically, it has been shown that lack of NSUN2 in mice leads to fragmentation of tRNAs, which may trigger apoptosis in the brain (Blanco et al. 2014). However, it is still unclear to which degree this mechanism may actually contribute to the intellectual disability phenotypes observed in human.
Defects in demethylation of RNAs have been also linked to neurological defects. Deletion of the FTO gene in mice, which is one of the two enzymes responsible for reversing or “erasing” m6A modifications (Zhao et al. 2016), results in an impairment of dopamine receptor control of neuronal activity and behavioral responses (Hess et al. 2013). ALKBH5, also responsible for m6A demethylation, has been linked to major depressive disorders (Du et al. 2015), suggesting that m6A may be playing an important regulatory role in the function of the mammalian brain.
A-to-I editing defects have also been associated with neurological diseases (Hideyama and Kwak 2011; Hideyama et al. 2012; Gaisler-Salomon et al. 2014; Tomaselli et al. 2015), such as amyotrophic lateral sclerosis (ALS), the most common adult-onset motor neuron disease (Hideyama and Kwak 2011; Hideyama et al. 2012). More specifically, the glutamate receptor 2 (GluA2) mRNA, which is constitutively edited in some of its nucleotides, has been found to be unedited in motor neurons in individuals with sporadic ALS (Hideyama and Kwak 2011). On the other hand, ADAR2 expression levels were found to be down-regulated in ALS individuals, further supporting the importance of editing in proper motor neuronal functioning (Hideyama et al. 2012). In addition, ADAR2 knockout mice show increased cell death rates in their motor neurons (Sasaki et al. 2015), in agreement with the results observed in ALS individuals, indicating a pivotal role of A-to-I editing in proper neuronal functioning and brain development in mammals.
Cancer
Dysregulation and mutations in several RNA modification enzymes have been associated with various types of cancers, including breast cancer, bladder cancer, and leukemia, among others (Supplemental Table S1). For example, the enzymes responsible for mcm5s2 modification, ELP3 and CTU1/2, have been found to be up-regulated in breast cancer and to sustain metastasis (Delaunay et al. 2016). Similarly, the methyltransferase NSUN2 has been found to be overexpressed in breast cancer and its expression levels have been shown to correlate with cancer development and progression (Yi et al. 2016). In contrast, the tRNA methyltransferase TRM9L/KIAA1456 has been found to be down-regulated in breast cancer cells, as well as in other forms of cancer (Begley et al. 2013). Lastly, TRMT12 was found to be overexpressed in 87% of breast tumors (Rodriguez et al. 2007). Taken together, these point to a role of RNA modifications in cancer, although it is yet to be shown whether these enzymes could be used as possible targets for cancer treatment or as biomarkers of disease prognosis.
Genetic defects
Most serious genetic birth defects are caused by mutations in protein-coding genes, but the regulation of gene expression may also cause deviation from normal development. Numerous genetic birth defects have been associated with mutations in RNA modification enzymes, such as the Cri du chat syndrome (NOP2/NOL1/p120/NSUN1) (Wu et al. 2005), the Dubowitz syndrome (NSUN2) (Martinez et al. 2012), the Noonan-like syndrome (NSUN2) (Fahiminiya et al. 2014), or the William–Beuren syndrome (WBSCR20/WBSCR22/NSUN5) (Doll and Grzeschik 2001). Furthermore, mutations in RNA modification enzymes have also been shown to cause developmental defects, such as Hutchinson–Gilford progeria syndrome (NAT10) (Larrieu et al. 2014), and primordial dwarfism (WDR4) (Shaheen et al. 2015). In addition, mutations in RNA modification enzymes can cause the spinal cord to expose outside the body like spina bifida (TRDMT1) (Franke et al. 2009) and infant death (EMG1) (Armistead et al. 2009).
Note of caution
Here we provide an updated comprehensive catalog of RNA modifications and their associations with human disease, which we expect will provide useful starting points to prioritize the study of additional RNA modifications. However, caution must be taken when interpreting the available data. For example, the fat mass and obesity-associated (FTO) gene obtained its name in 2007 after a strong association between a single-nucleotide polymorphism (SNP) in the FTO locus and obesity had been identified in multiple populations. Ever since, FTO has been widely cited as an example of how an RNA modification dysregulation can be linked to human disease. However, this SNP was recently shown to be unrelated to FTO function (Claussnitzer et al. 2015). Instead, this intronic SNP disrupts a conserved motif for the ARID5B repressor, which in turn leads to a de-repression of a potent pre-adipocyte enhancer, causing increased expression of two nearby genes, IRX3 and IRX5, involved in early adipocyte differentiation (Claussnitzer et al. 2015).
In a similar fashion, a variant of CDKAL1 was found to be associated to type 2 diabetes in various ethnic groups (Benrahma et al. 2014; Lasram et al. 2015), but a different study on Caucasian UK residents found evidence against a role of deregulated expression of CDKAL1-v1 in susceptibility to type II diabetes (Locke et al. 2015), emphasizing the fact that caution should be taken in inferring causality from an association between disease-risk genotypes and expression levels. These examples highlight the importance of understanding the underlying mechanisms driving the association between RNA modification enzyme mutations and human diseases, as well as the need to validate these associations.
DETECTING RNA MODIFICATIONS: PAST, PRESENT, AND FUTURE EFFORTS
Classical approaches to detect modified ribonucleosides have relied on thin-layer chromatography, capillary electrophoresis, or related techniques. In all cases, the sample is reduced to nucleotides, which are then separated based on their physicochemical properties. To increase sensitivity, 32P-radioactive labeling can be combined with two-dimensional TLC separation on cellulose, enabling the detection of femtomole quantities of modified nucleotides (Keith 1995). However, these methods are labor-intensive, require the use of radioactive labeling, and are semiquantitative at best (Reddy et al. 1981; Zhao and Yu 2004; Hengesbach et al. 2008).
More recent approaches have used liquid chromatography–tandem mass spectrometry (LC-MS/MS) methodologies, which can provide accurate quantitation of multiple RNA modifications across conditions and cell types (Chan et al. 2010; Addepalli and Limbach 2011; Yan et al. 2013; Su et al. 2014) and have been successfully applied to a wide variety of species (Chan et al. 2010; Patil et al. 2012; Begley et al. 2013). Selective enzymatic digestions of individual RNAs with a battery of RNases, coupled to LC–MS/MS techniques, has been shown to be extremely useful for comparative RNA modification analysis across species and conditions (Li and Limbach 2012). In addition, the use of isotopically-labeled compounds (13C,15N,18O) has lowered the detection methods to the femtomole scale, allowing the detection of small differences of RNA modifications between RNA samples (Nikcevic et al. 2011; Li and Limbach 2012).
A key requirement to obtain meaningful results from LC–MS/MS approaches, however, is the isolation of specific RNA species, free from contamination with other RNAs. Thus, LC–MS/MS analyses of RNA modifications have been mostly focused on rRNA or tRNA molecules, due to their high abundance of modifications and simplicity of isolation (Chan et al. 2010; Su et al. 2014). Among the few attempts to identify RNA modifications beyond tRNAs and rRNAs (Yan et al. 2013), several were detected in other RNA pools, suggesting that some may have broader distribution. Unfortunately, a major limitation of LC–MS/MS approaches is that the information of both the transcript that carried the modified nucleosides, as well as their location within the sequence, remains unknown.
Accurate transcriptome-wide mapping of modified nucleosides is now possible due to advances in next-generation sequencing technologies (Saletore et al. 2012; Helm and Motorin 2017; Novoa et al. 2017). These fall into three different categories: (i) immunoprecipitation of fragmented RNAs using modification-specific antibodies, followed by sequencing of the enriched RNA fragments (RIP-seq), which has been used for mapping m6A (Dominissini et al. 2012; Meyer et al. 2012), hm5C (Delatte et al. 2016) and m1A (Hauenschild et al. 2015; Dominissini et al. 2016); (ii) chemical treatment of RNA prior to sequencing, which exploits the differential reactivity of modified bases, such as using sodium bisulfite for detection of m5C (Squires et al. 2012) or CMC [_N_-cyclohexyl-_N_9-(2-morpholinoethyl)-carbodiimidemetho-p-toluenesulphonate] for detection of pseudouridine (Carlile et al. 2014; Schwartz et al. 2014a) (Chem-seq); and (iii) nonrandom mismatch signatures in RNA sequencing data produced during conversion of RNA to cDNA by reverse transcriptase during library preparation (Fig. 2; Hauenschild et al. 2015). To date, these technologies and sequencing adaptations have produced transcriptome-wide maps for six RNA modifications including 5-methylcytidine (m5C), _N_6-methyladenosine (m6A), _N_6-2′-_O-_dimethyladenosine (m6Am), pseudouridine (Ψ), 1-methyladenosine (m1A), 5-hydroxymethylcytidine (hm5C), in addition to adenosine to inosine (A-to-I) editing, which can be detected using traditional RNA-seq protocols (Ramaswami et al. 2013; Shafik et al. 2016).
FIGURE 2.

Current genome-wide detection methods used to identify RNA modifications. (A) In the left panel, antibody-based methods (RIP-seq) show how RNA-modification enriched fragments are selected using pool-down, and compared to a total fragmented sample (input), which is used for normalization, obtaining genome-wide maps with peak resolution. (B) In the middle panel, RNA samples are pretreated with chemical reagents (Chem-seq), which inhibit the reverse transcription reaction beyond the chemically modified position. (C) In the right panel, mismatch signature-based methods, which are based on the increased mismatch rates that occur upon reverse transcription at certain RNA-modified positions, are depicted.
RIP-seq
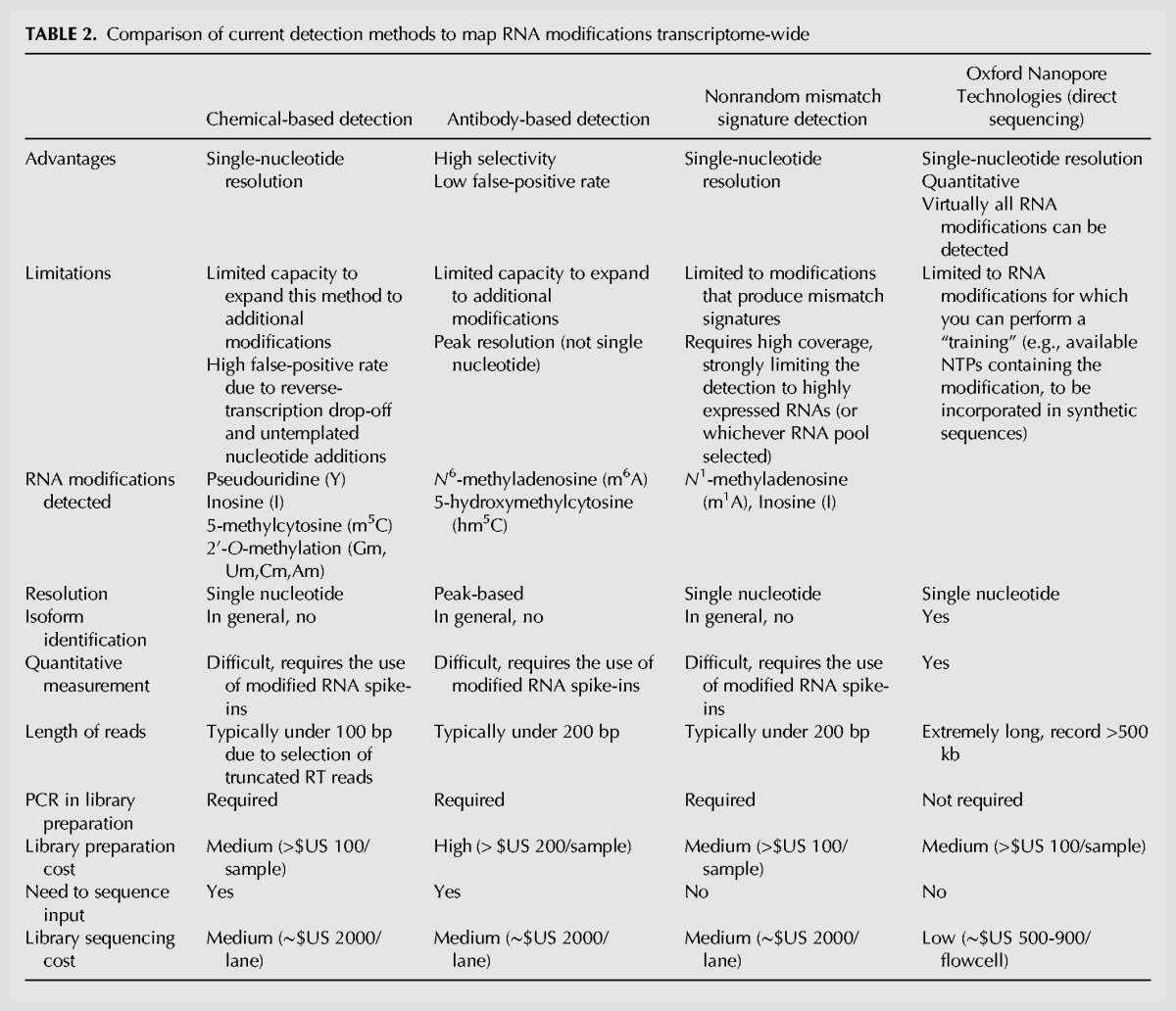
Building on the principles of chromatin immunoprecipitation-sequencing (ChIP-seq), antibody-based detection methods have been successfully applied to detect RNA modifications in a transcriptome-wide fashion, where read densities of immunoprecipitated modified RNA are compared to an untreated input (Fig. 2A; Dominissini et al. 2012; Batista et al. 2014). These methods have proven to be highly sensitive, but they are limited by the available repertoire of commercial antibodies (i.e., at present only those against m6A, m1A, m5C, and hm5C) (Table 2). They have had the disadvantage of lacking single-nucleotide resolution, although this limitation can be overcome by slight modification of the protocol (Linder et al. 2015).
TABLE 2.
Comparison of current detection methods to map RNA modifications transcriptome-wide
Chem-seq
Chemical-based detection methods rely on the use of chemical reagents that selectively react with specific modified RNA nucleotides (Fig. 2B). Upon reverse transcription (RT), chemically modified positions induce RT drop-off, leading to the accumulation of reads ending at the same position. These chemically modified positions can be then precisely located through the identification of increased reverse transcription termination sites (RTTS). Successful application of this technique is exemplified by Pseudo-seq (Carlile et al. 2014; Schwartz et al. 2014a), where CMC-modified pseudouridines block reverse transcription. Although chemical-based detection methods have the strength of producing single-nucleotide resolution RNA modification maps, RT drop-off can be caused by many factors, such as increased RNA secondary structures (Aviran and Pachter 2014), the presence of binding of proteins (Konig et al. 2010), or other RNA modifications (Table 2; Motorin et al. 2007; Ryvkin et al. 2013). In addition, RT enzymes have the undesirable ability of adding nontemplated nucleotides, thus generating an additional source of false positives (Chen and Patton 2001).
Several successful chemical-detection methods do not induce RT drop-off, but instead change the pairing ability of the modified position. In the case of RNA editing, treatment with glyoxal protects guanosines but not inosines (the product of adenosine deamination, which behaves like guanosine) from RNase T1 activity prior to reverse transcription (Cattenoz et al. 2013). Similarly, in the case of bisulphite sequencing, unmethylated cytosine is sulfonated by sodium bisulphite and is subsequently deaminated to uridine, while methylated cytosine is refractory, remaining as cytosine (Schaefer et al. 2009). Upon conversion to cDNA, unmethylated cytosine will be read by the sequencer as thymine. Unfortunately, due to the incomplete conversion of C-to-U (in the case of bisulfite sequencing) or glyoxal protection of guanosines, these methods suffer from high false-positive rates. Thus, to correct for false positives, these methods require deep sequencing of RNAs both from a wild-type in combination with the knockout/knockdown of the enzyme of interest.
A hybrid method combining chemical modification and immunoprecipitation is 5-azacytidine-mediated RNA immunoprecipitation (Aza-IP), a mechanism-based technique that exploits the covalent bond formed between an RNA methyltransferase and the cytidine analog 5-azacytidine to selectively recover m5C modified RNA targets by immunoprecipitation (Khoddami and Cairns 2013).
Mismatch signature analysis
Lastly, mismatch-signature–based analyses have been used to produce transcriptome-wide maps of RNA modifications with single-nucleotide resolution (Hauenschild et al. 2015). These methods rely on the interference of certain RNA modifications in Watson–Crick (W–C) base-pairing, generating nonrandom mismatch patterns at modified positions during enzymatic RT readthrough (Fig. 2C). Unfortunately, multiple RNA modifications disturb W–C base-pairing and generate increased mismatch rates, hindering the proper identification of the specific underlying modification. To deconvolute these signatures and identify the underlying modifications, previous efforts have used bioinformatic approaches to classify the nonrandom mismatch signatures, using mismatch patterns observed at known tRNA modification sites to train the algorithm (Ryvkin et al. 2013). In our hands, however, tRNA modification signatures are not representative of mismatch patterns observed in other RNA classes and locations, perhaps due to the rich modification environment of tRNA molecules.
Overall, current transcriptome-wide mapping methods have provided highly valuable information to broaden our understanding of the epitranscriptome, but are constrained by the limited repertoire of commercial antibodies (e.g., those against m6A and hm5C) and the lack of selective chemical reactivities of uncharacterized RNA modifications (Table 2).
Future approaches: direct RNA sequencing
A major limitation of current genome-wide sequencing methods is that they are based on sequence-by-synthesis (SBS) technologies, and consequently, are blind to DNA and RNA modifications (with the exception of A-to-I editing, which causes an A-to-G mismatch). In the case of DNA modifications, this information is lost in the amplification step, e.g., m5C will be read as a C, and a G will be placed in this position. In the case of RNA modifications, the loss occurs during reverse transcription, whereby RNA is converted back to cDNA in order to be sequenced. These processes strip all edited bases and epigenetic information from the molecules, and occasionally introduce substantial artifacts (Chen and Patton 2001). In addition, standard RT enzymes are sensitive to RNA length and may terminate early when encountering stable RNA structures during extension (Aviran and Pachter 2014).
Third generation sequencing (TGS) appeared only a few years ago, and has emerged as a promising alternative to genome-wide map RNA modifications. TGS technologies distance themselves from second generation sequencing (SGS) technologies for their capability of generating very long reads (>100 kb) (Laver et al. 2015; Rhoads and Au 2015). First single-molecule sequencing attempts were performed by the now defunct Helicos Biosciences, which used a single-molecule SBS strategy. Its ability to identify modified and nonstandard RNA bases is unknown, but would likely be subject to similar constraints as cDNA-based technologies, given that a polymerase is nonetheless required for sequencing.
The first alternative SBS approach for detecting RNA modifications was offered by Pacific Bioscience's single-molecule real-time sequencing (SMRT) platform, which was proven successful in detecting the differences between m6A-modified and unmodified synthetic sequences of RNA, but was too labor-intensive and expensive for subsequent development (Saletore et al. 2012). Another promising TGS technology has been developed by Oxford Nanopore Technologies (ONT). This platform is based on direct measurement of disruptions in the current as the DNA or RNA molecules pass through a porous bacterial transmembrane protein (Loman and Watson 2015). These changes in current intensity can then be used to identify the transiting nucleotides, including modified RNA and DNA nucleotides (Fig. 3A). Initially limited to DNA sequencing, ONT published their first results of direct RNA sequencing at the end of 2016 (Garalde et al. 2016). Several months later, a direct RNA-sequencing (DRS) kit finally became available to the general public in April 2017 (Fig. 3B). In the first step of the DRS library preparation protocol, a double-stranded DNA adapter with a poly(T) or sequence-specific complementary single-stranded overhang is ligated to the 3′ end of template RNA molecules. Next, an optional reverse-transcription step can be performed to linearize and stabilize the template RNAs. Finally, a proprietary sequencing adapter is ligated to the double-stranded DNA adapter before being loaded into a flow cell for sequencing. It is important to note that only the RNA strand will be sequenced (in 3′–5′ orientation). Remarkably, the low sample manipulation required for the library preparation (Fig. 3B) vastly diminishes the biases typically introduced during SBS library preparation, such as those introduced by fragmentation, PCR amplification, or immunoprecipitation.
FIGURE 3.
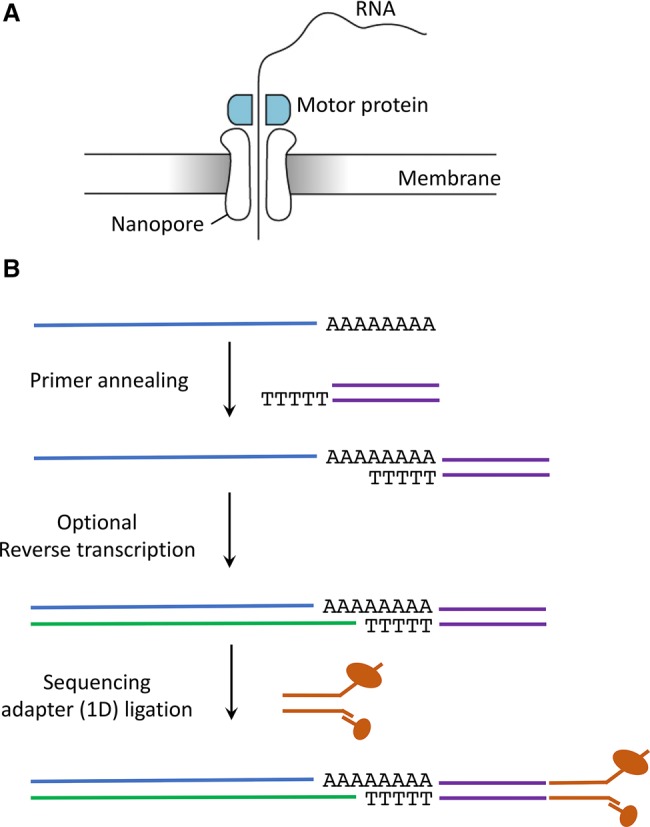
Direct RNA sequencing library preparation steps using Oxford Nanopore Technologies. (A) Schematic representation of a nanopore embedded in the membrane of the flowcell. (B) Overview of the main library preparation steps in ONT direct RNA sequencing.
The simplest way to convert ionic current traces into base-called nucleotides is to run local “live” base-calling, where samples are base-called on-the-fly as the molecules exit individual pores using ONT's proprietary algorithm Albacore (which combines a recurrent neural network and a hidden Markov model). Base-calling can also be performed a posteriori with Albacore on a personal computer, a high performance computing server, or ONT's cloud-based analysis service known as Metrichor (https://metrichor.com). A third option is to use one of the multiple open-source base-calling algorithms, which use various machine- or deep-learning algorithms, including hidden Markov models (David et al. 2017; Simpson et al. 2017) and recurrent neural networks (Boža et al. 2017; Stoiber and Brown 2017). Unfortunately, these algorithms are typically trained to predict exclusively four bases (A, C, G, T), and thus cannot directly identify DNA- or RNA-modified nucleotides. There have nonetheless been recent reports describing computational models capable of detecting modified DNA bases, by training models from biological control data and by observing conspicuous alterations of ionic current at specific positions (Stoiber et al. 2016; McIntyre et al. 2017; Rand et al. 2017; Simpson et al. 2017). With respect to DRS, these strategies have recently been applied to characterize the epitranscriptome, namely the identification of m6A (Garalde et al. 2016) and conserved 16S ribosomal RNA base modifications and a 7-methylguanosine modification associated with antibiotic resistance (Smith et al. 2017). It is likely that, following the release of the direct RNA-sequencing kit, additional algorithms to detect and predict RNA modifications from DRS data will become available.
For many years, a major limitation of ONT technologies has been its relatively low base-calling accuracy. However, in the last three years, its base-calling accuracy has increased from 70%–88% (using R7 pore technologies and HMM-based algorithms) to 90%–98%, due to a more efficient protein nanopore (currently R9.5), homopolymer-aware RNN base-calling, and a paired-end consensus read strategy. Nonetheless, base-calling errors can be corrected a posteriori either by determining a consensus sequence, probabilistic refinement (Jain et al. 2015), or via a process known as “polishing” (Loman et al. 2015; Sarkozy et al. 2017), where reads are aligned to a reference genome or transcriptome to guide error correction by revisiting the raw signal. However, initial base-calling attempts using ONT to sequence 16s rRNA from E. coli only using DRS only yielded an accuracy of 87% (Smith et al. 2017). The authors found that these errors were mainly due to deletion errors occurring in G-rich regions, which are abundant in noncoding infrastructural RNAs such as 16s rRNA. It is likely that the highly modified nature of rRNA molecules may be in fact a confounder for proper RNA base-calling. Newer algorithms, previously trained with known modified RNA nucleotides, will likely produce higher base-calling identities in RNA molecules.
A second major limitation of direct RNA-sequencing technologies is the yield of each individual sequencing run. Although the throughput of ONT sequencing has greatly increased in the last years for (c)DNA sequences, achieving yields of 3–15 billion bases (Gb) per run in a standard R9.4 MinION FLO-MIN106 flow cell (Lu et al. 2016; Jain et al. 2017), the yields obtained from direct RNA sequencing are still far from these values. More specifically, the expected number of cDNA reads using a high-quality FLO-MIN106 flow cell ranges between 6–10 million reads, whereas the expected number of reads from direct RNA sequencing is only 1 million (https://nanoporetech.com/rna).
Despite these limitations, the possibility of detecting RNA modifications in each individual RNA molecule opens new avenues to explore the cross talk and dependencies that may exist between multiple RNA modifications within the same RNA molecule. Current indirect SBS-based methods are unable to decipher whether two RNA modifications present in a given mRNA sequence actually coexist in the same RNA molecule, or if instead, they are exclusively present in different molecules. Furthermore, compared with SBS-based methods, ONT offers the possibility to identify in which RNA transcript isoform the modification is found, and thus may be able to provide quantitative stoichiometric measurements of modified RNA nucleotides at each position in an isoform-specific manner.
DISCUSSION
During the past few decades we have learned how vital epigenetic processes are for learning and memory formation (Day et al. 2013). RNA modifications form an additional language, much less characterized, which is capable of overwriting and redefining the hard-wired transcriptome, extending and diversifying the function of transcripts, thus adding an unchartered layer of regulation affecting genome function.
One of the major surprises in the last decades was the observation that developmental complexity is not correlated with the number of protein-coding genes. One of the answers to this enigma came with the discovery of long noncoding RNAs, whose number increases with developmental complexity (Mattick 2011). Thus, the appearance of multitudes of noncoding RNAs created additional layers of gene expression and genetic information, which provides the regulatory power and plasticity required for the developmental and cognitive capacity of mammals and, in particular, primates (Qureshi et al. 2010; Mattick 2011).
In a similar fashion, RNA modifications may not be simple fine-tuners of RNA function, such as mRNA half-lives (Batista et al. 2014; Schwartz et al. 2014b) or translation efficiency (Wang et al. 2015), but also provide additional capacity for increasing developmental and cognitive complexity. RNAs undergo an enormous amount of editing, especially in primates and especially in the brain (Paul and Bass 1998). A large variety of studies have shown that the activity ADARs (adenosine deaminases acting on RNAs), responsible for A-to-I editing, are highly expressed in nervous systems (Picardi et al. 2015), and markedly increased during primate evolution (Paz-Yaacov et al. 2010). These modifications do not only affect protein-coding genes, but also noncoding transcripts, and thus may be central to learning and plasticity in brain function (Lence et al. 2016; Nainar et al. 2016).
Moreover, there is now evidence that transgenerational inheritance can be mediated by RNAs (Chen et al. 2016; Sharma et al. 2016), thus raising the possibility that RNA is not just the underlying engine of cell biology, developmental biology, and cognition, but perhaps also of evolution itself. Interestingly, the identified molecules involved in transgenerational inheritance were tRNA-derived fragments, which are likely highly modified. Whether these RNAs display different RNA modifications to execute or regulate their function is still an open question, hopefully to be answered in the following years. Once we are capable of systematically mapping RNA modifications in a genome-wide fashion, we may be able to uncover the potential roles of RNA modifications in human development, as well as the effects of their dysregulation in disease.
In recent years, TGS technologies have made possible the sequencing of very long reads from single RNA/DNA molecules. Despite being in their infancy, they have already demonstrated to be powerful technologies capable of overcoming challenges that could not be solved by SGS, such as providing quick in situ diagnoses of virus outbreaks (Quick et al. 2016, 2017), or obtaining genome-wide structural variant information from patient genomes (Cretu Stancu et al. 2017). While the use of TGS to detect RNA modifications is still not fully benchmarked, we expect that in the near future TGS will provide us with single-molecule genome-wide maps of RNA modifications, allowing us to investigate the dependencies between different modified sites, as well as those between different RNA modification types (e.g., m6A and pseudouridine), in a genome-wide fashion.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
Supplemental Material
ACKNOWLEDGMENTS
N.J. is supported by a UNSW International PhD fellowship. M.A.S. is partially supported by a Cancer Council NSW project grant (RG 14-18). E.M.N. was supported by a long-term postdoctoral fellowship from the Human Frontier Science Program (LT000307/2013-L), and is currently supported by a Discovery Early Career Researcher Award (DE170100506) from the Australian Research Council. This work was supported by National Health and Medical Research Council funds (Project Grant APP1070631 to J.S.M.).
Footnotes
REFERENCES
- Abbasi-Moheb L, Mertel S, Gonsior M, Nouri-Vahid L, Kahrizi K, Cirak S, Wieczorek D, Motazacker MM, Esmaeeli-Nieh S, Cremer K, et al. 2012. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am J Hum Genet 90: 847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addepalli B, Limbach PA. 2011. Mass spectrometry-based quantification of pseudouridine in RNA. J Am Soc Mass Spectrom 22: 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agris PF, Narendran A, Sarachan K, Väre VYP, Eruysal E. 2017. The importance of being modified: the role of RNA modifications in translational fidelity. Enzymes 41: 1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alings F, Sarin LP, Fufezan C, Drexler HC, Leidel SA. 2015. An evolutionary approach uncovers a diverse response of tRNA 2-thiolation to elevated temperatures in yeast. RNA 21: 202–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armistead J, Khatkar S, Meyer B, Mark BL, Patel N, Coghlan G, Lamont RE, Liu S, Wiechert J, Cattini PA, et al. 2009. Mutation of a gene essential for ribosome biogenesis, EMG1, causes Bowen-Conradi syndrome. Am J Hum Genet 84: 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviran S, Pachter L. 2014. Rational experiment design for sequencing-based RNA structure mapping. RNA 20: 1864–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. 2014. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15: 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednářová A, Hanna M, Durham I, VanCleave T, England A, Chaudhuri A, Krishnan N. 2017. Lost in translation: defects in transfer RNA modifications and neurological disorders. Front Mol Neurosci 10: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley U, Sosa MS, Avivar-Valderas A, Patil A, Endres L, Estrada Y, Chan CT, Su D, Dedon PC, Aguirre-Ghiso JA, et al. 2013. A human tRNA methyltransferase 9-like protein prevents tumour growth by regulating LIN9 and HIF1-α. EMBO Mol Med 5: 366–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm M, Öhman M. 2016. RNA editing: a contributor to neuronal dynamics in the mammalian brain. Trends Genet 32: 165–175. [DOI] [PubMed] [Google Scholar]
- Benrahma H, Charoute H, Lasram K, Boulouiz R, Atig RK, Fakiri M, Rouba H, Abdelhak S, Barakat A. 2014. Association analysis of IGF2BP2, KCNJ11, and CDKAL1 polymorphisms with type 2 diabetes mellitus in a Moroccan population: a case-control study and meta-analysis. Biochem Genet 52: 430–442. [DOI] [PubMed] [Google Scholar]
- Blanco S, Dietmann S, Flores JV, Hussain S, Kutter C, Humphreys P, Lukk M, Lombard P, Treps L, Popis M, et al. 2014. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J 33: 2020–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boža V, Brejová B, Vinař T. 2017. DeepNano: deep recurrent neural networks for base calling in MinION nanopore reads. PLoS One 12: e0178751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF. 2011. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res 39: D195–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. 2014. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515: 143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattenoz PB, Taft RJ, Westhof E, Mattick JS. 2013. Transcriptome-wide identification of A > I RNA editing sites by inosine specific cleavage. RNA 19: 257–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CT, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. 2010. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet 6: e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Patton JT. 2001. Reverse transcriptase adds nontemplated nucleotides to cDNAs during 5′-RACE and primer extension. Biotechniques 30: 574–580, 582. [DOI] [PubMed] [Google Scholar]
- Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. 2000. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6: 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Yan M, Cao Z, Li X, Zhang Y, Shi J, Feng GH, Peng H, Zhang X, Zhang Y, et al. 2016. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 351: 397–400. [DOI] [PubMed] [Google Scholar]
- Chi L, Delgado-Olguin P. 2013. Expression of NOL1/NOP2/sun domain (Nsun) RNA methyltransferase family genes in early mouse embryogenesis. Gene Expr Patterns 13: 319–327. [DOI] [PubMed] [Google Scholar]
- Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, et al. 2015. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med 373: 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretu Stancu M, van Roosmalen MJ, Renkens I, Nieboer M, Middelkamp S, de Ligt J, Pregno G, Giachino D, Mandrile G, Espejo Valle-Inclan J, et al. 2017. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. bioRxiv. 10.1101/129379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davarniya B, Hu H, Kahrizi K, Musante L, Fattahi Z, Hosseini M, Maqsoud F, Farajollahi R, Wienker TF, Ropers HH, et al. 2015. The role of a novel TRMT1 gene mutation and rare GRM1 gene defect in intellectual disability in two Azeri families. PLoS One 10: e0129631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David M, Dursi LJ, Yao D, Boutros PC, Simpson JT. 2017. Nanocall: an open source basecaller for Oxford Nanopore sequencing data. Bioinformatics 33: 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JJ, Childs D, Guzman-Karlsson MC, Kibe M, Moulden J, Song E, Tahir A, Sweatt JD. 2013. DNA methylation regulates associative reward learning. Nat Neurosci 16: 1445–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedon PC, Begley TJ. 2014. A system of RNA modifications and biased codon use controls cellular stress response at the level of translation. Chem Res Toxicol 27: 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, Deplus R, Calonne E, Hassabi B, Putmans P, Awe S, et al. 2016. RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 351: 282–285. [DOI] [PubMed] [Google Scholar]
- Delaunay S, Rapino F, Tharun L, Zhou Z, Heukamp L, Termathe M, Shostak K, Klevernic I, Florin A, Desmecht H, et al. 2016. Elp3 links tRNA modification to IRES-dependent translation of LEF1 to sustain metastasis in breast cancer. J Exp Med 213: 2503–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Babu IR, Su D, Yin S, Begley TJ, Dedon PC. 2015. Trm9-catalyzed tRNA modifications regulate global protein expression by codon-biased translation. PLoS Genet 11: e1005706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. 2012. Landscape of transcription in human cells. Nature 489: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll A, Grzeschik KH. 2001. Characterization of two novel genes, WBSCR20 and WBSCR22, deleted in Williams-Beuren syndrome. Cytogenet Cell Genet 95: 20–27. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201–206. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, et al. 2016. The dynamic _N_1-methyladenosine methylome in eukaryotic messenger RNA. Nature 530: 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Rao S, Wu L, Ye N, Liu Z, Hu H, Xiu J, Shen Y, Xu Q. 2015. An association study of the m6A genes with major depressive disorder in Chinese Han population. J Affect Disord 183: 279–286. [DOI] [PubMed] [Google Scholar]
- Fahiminiya S, Almuriekhi M, Nawaz Z, Staffa A, Lepage P, Ali R, Hashim L, Schwartzentruber J, Abu Khadija K, Zaineddin S, et al. 2014. Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clin Genet 86: 134–141. [DOI] [PubMed] [Google Scholar]
- Franke B, Vermeulen SH, Steegers-Theunissen RP, Coenen MJ, Schijvenaars MM, Scheffer H, den Heijer M, Blom HJ. 2009. An association study of 45 folate-related genes in spina bifida: Involvement of cubilin (CUBN) and tRNA aspartic acid methyltransferase 1 (TRDMT1). Birth Defects Res A Clin Mol Teratol 85: 216–226. [DOI] [PubMed] [Google Scholar]
- Freude K, Hoffmann K, Jensen LR, Delatycki MB, des Portes V, Moser B, Hamel B, van Bokhoven H, Moraine C, Fryns JP, et al. 2004. Mutations in the FTSJ1 gene coding for a novel S-adenosylmethionine-binding protein cause nonsyndromic X-linked mental retardation. Am J Hum Genet 75: 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Jaffrey SR, Pan T, Rechavi G, Suzuki T. 2016. RNA modifications: what have we learned and where are we headed? Nat Rev Genet 17: 365–372. [DOI] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, et al. 2013. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155: 793–806. [DOI] [PubMed] [Google Scholar]
- Gaisler-Salomon I, Kravitz E, Feiler Y, Safran M, Biegon A, Amariglio N, Rechavi G. 2014. Hippocampus-specific deficiency in RNA editing of GluA2 in Alzheimer's disease. Neurobiol Aging 35: 1785–1791. [DOI] [PubMed] [Google Scholar]
- Garalde DR, Snell EA, Jachimowicz D, Heron AJ, Bruce M, Lloyd J, Warland A, Pantic N, Admassu T, Ciccone J, et al. 2016. Highly parallel direct RNA sequencing on an array of nanopores. bioRxiv. 10.1101/068809. [DOI] [PubMed] [Google Scholar]
- Gilbert WV, Bell TA, Schaening C. 2016. Messenger RNA modifications: form, distribution, and function. Science 352: 1408–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice LF, Degnan BM. 2015. The origin of the ADAR gene family and animal RNA editing. BMC Evol Biol 15: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H. 2015. RNA modification: the Golden Period 1995–2015. RNA 21: 625–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Kon Y, Phizicky EM. 2015. Functional importance of Ψ38 and Ψ39 in distinct tRNAs, amplified for tRNAGln(UUG) by unexpected temperature sensitivity of the s2U modification in yeast. RNA 21: 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauenschild R, Tserovski L, Schmid K, Thuring K, Winz ML, Sharma S, Entian KD, Wacheul L, Lafontaine DL, Anderson J, et al. 2015. The reverse transcription signature of _N_-1-methyladenosine in RNA-Seq is sequence dependent. Nucleic Acids Res 43: 9950–9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG, Soller M. 2016. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 540: 301–304. [DOI] [PubMed] [Google Scholar]
- He C. 2010. Grand challenge commentary: RNA epigenetics? Nat Chem Biol 6: 863–865. [DOI] [PubMed] [Google Scholar]
- Helm M, Alfonzo JD. 2014. Posttranscriptional RNA modifications: playing metabolic games in a cell's chemical Legoland. Chem Biol 21: 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M, Motorin Y. 2017. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet 18: 275–291. [DOI] [PubMed] [Google Scholar]
- Hengesbach M, Meusburger M, Lyko F, Helm M. 2008. Use of DNAzymes for site-specific analysis of ribonucleotide modifications. RNA 14: 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess ME, Hess S, Meyer KD, Verhagen LA, Koch L, Bronneke HS, Dietrich MO, Jordan SD, Saletore Y, Elemento O, et al. 2013. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci 16: 1042–1048. [DOI] [PubMed] [Google Scholar]
- Hideyama T, Kwak S. 2011. When does ALS start? ADAR2–GluA2 hypothesis for the etiology of sporadic ALS. Front Mol Neurosci 4: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideyama T, Yamashita T, Aizawa H, Tsuji S, Kakita A, Takahashi H, Kwak S. 2012. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol Dis 45: 1121–1128. [DOI] [PubMed] [Google Scholar]
- Hussain S, Bashir ZI. 2015. The epitranscriptome in modulating spatiotemporal RNA translation in neuronal post-synaptic function. Front Cell Neurosci 9: 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S, Aleksic J, Blanco S, Dietmann S, Frye M. 2013. Characterizing 5-methylcytosine in the mammalian epitranscriptome. Genome Biol 14: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M. 2015. Improved data analysis for the MinION nanopore sequencer. Nat Methods 12: 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Koren S, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S, et al. 2017. Nanopore sequencing and assembly of a human genome with ultra-long reads. bioRxiv. 10.1101/128835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. 2011. _N_6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7: 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karijolich J, Yu YT. 2010. Spliceosomal snRNA modifications and their function. RNA Biol 7: 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith G. 1995. Mobilities of modified ribonucleotides on two-dimensional cellulose thin-layer chromatography. Biochimie 77: 142–144. [DOI] [PubMed] [Google Scholar]
- Khoddami V, Cairns BR. 2013. Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat Biotechnol 31: 458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen R, Ciftci A, Funk J, Bruch A, Butter F, Schaffrath R. 2016. tRNA anticodon loop modifications ensure protein homeostasis and cell morphogenesis in yeast. Nucleic Acids Res 44: 10946–10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klungland A, Dahl JA. 2014. Dynamic RNA modifications in disease. Curr Opin Genet Dev 26: 47–52. [DOI] [PubMed] [Google Scholar]
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. 2010. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17: 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrieu D, Britton S, Demir M, Rodriguez R, Jackson SP. 2014. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science 344: 527–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasram K, Ben Halim N, Benrahma H, Mediene-Benchekor S, Arfa I, Hsouna S, Kefi R, Jamoussi H, Ben Ammar S, Bahri S, et al. 2015. Contribution of CDKAL1 rs7756992 and IGF2BP2 rs4402960 polymorphisms in type 2 diabetes, diabetic complications, obesity risk and hypertension in the Tunisian population. J Diabetes 7: 102–113. [DOI] [PubMed] [Google Scholar]
- Laver T, Harrison J, O'Neill PA, Moore K, Farbos A, Paszkiewicz K, Studholme DJ. 2015. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol Detect Quantif 3: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens R, Moore MJ, Al-Chalabi A, Brown RH Jr, Robberecht W. 2010. RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci 33: 249–258. [DOI] [PubMed] [Google Scholar]
- Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, Kreim N, Andrade-Navarro MA, Poeck B, Helm M, et al. 2016. m6A modulates neuronal functions and sex determination in Drosophila. Nature 540: 242–247. [DOI] [PubMed] [Google Scholar]
- Li S, Limbach PA. 2012. Method for comparative analysis of ribonucleic acids using isotope labeling and mass spectrometry. Anal Chem 84: 8607–8613. [DOI] [PubMed] [Google Scholar]
- Li S, Mason CE. 2014. The pivotal regulatory landscape of RNA modifications. Annu Rev Genomics Hum Genet 15: 127–150. [DOI] [PubMed] [Google Scholar]
- Li X, Xiong X, Yi C. 2016. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat Methods 14: 23–31. [DOI] [PubMed] [Google Scholar]
- Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. 2015. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12: 767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Clark W, Luo G, Wang X, Fu Y, Wei J, Wang X, Hao Z, Dai Q, Zheng G, et al. 2016. ALKBH1-mediated tRNA demethylation regulates translation. Cell 167: 1897. [DOI] [PubMed] [Google Scholar]
- Locke JM, Wei FY, Tomizawa K, Weedon MN, Harries LW. 2015. A cautionary tale: the non-causal association between type 2 diabetes risk SNP, rs7756992, and levels of non-coding RNA, CDKAL1-v1. Diabetologia 58: 745–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Watson M. 2015. Successful test launch for nanopore sequencing. Nat Methods 12: 303–304. [DOI] [PubMed] [Google Scholar]
- Loman NJ, Quick J, Simpson JT. 2015. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12: 733–735. [DOI] [PubMed] [Google Scholar]
- Lu H, Giordano F, Ning Z. 2016. Oxford nanopore MinION sequencing and genome assembly. Genomics Proteomics Bioinformatics 14: 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. 2013. MODOMICS: a database of RNA modification pathways--2013 update. Nucleic Acids Res 41: D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabriel S, Frye M, Al-Gazali L, Gleeson JG. 2012. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J Med Genet 49: 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS. 2010. RNA as the substrate for epigenome-environment interactions: RNA guidance of epigenetic processes and the expansion of RNA editing in animals underpins development, phenotypic plasticity, learning, and cognition. Bioessays 32: 548–552. [DOI] [PubMed] [Google Scholar]
- Mattick JS. 2011. The central role of RNA in human development and cognition. FEBS Lett 585: 1600–1616. [DOI] [PubMed] [Google Scholar]
- Mattick JS, Mehler MF. 2008. RNA editing, DNA recoding and the evolution of human cognition. Trends Neurosci 31: 227–233. [DOI] [PubMed] [Google Scholar]
- Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, et al. 2016. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 541: 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre ABR, Alexander N, Burton AS, Castro-Wallace S, Chiu CY, John KK, Stahl SE, Li S, Mason CE. 2017. Nanopore detection of bacterial DNA base modifications. bioRxiv. 10.1101/127100 [DOI] [Google Scholar]
- Mercer TR, Mattick JS. 2013. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol 20: 300–307. [DOI] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. 2017. Rethinking m6A readers, writers, and erasers. Annu Rev Cell Dev Biol 33: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KV, Mattick JS. 2014. The rise of regulatory RNA. Nat Rev Genet 15: 423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, Muller S, Behm-Ansmant I, Branlant C. 2007. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol 425: 21–53. [DOI] [PubMed] [Google Scholar]
- Nainar S, Marshall PR, Tyler CR, Spitale RC, Bredy TW. 2016. Evolving insights into RNA modifications and their functional diversity in the brain. Nat Neurosci 19: 1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, et al. 2011. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478: 57–63. [DOI] [PubMed] [Google Scholar]
- Nedialkova DD, Leidel SA. 2015. Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161: 1606–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikcevic I, Wyrzykiewicz TK, Limbach PA. 2011. Detecting low-level synthesis impurities in modified phosphorothioate oligonucleotides using liquid chromatography - high resolution mass spectrometry. Int J Mass Spectrom 304: 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa EM, Mason CE, Mattick JS. 2017. Charting the unknown epitranscriptome. Nat Rev Mol Cell Biol 18: 339–340. [DOI] [PubMed] [Google Scholar]
- Patil A, Chan CT, Dyavaiah M, Rooney JP, Dedon PC, Begley TJ. 2012. Translational infidelity-induced protein stress results from a deficiency in Trm9-catalyzed tRNA modifications. RNA Biol 9: 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul MS, Bass BL. 1998. Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J 17: 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Yaacov N, Levanon EY, Nevo E, Kinar Y, Harmelin A, Jacob-Hirsch J, Amariglio N, Eisenberg E, Rechavi G. 2010. Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc Natl Acad Sci 107: 12174–12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, Manzari C, Mastropasqua F, Aiello I, D'Erchia AM, Pesole G. 2015. Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci Rep 5: 14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, Bore JA, Koundouno R, Dudas G, Mikhail A, et al. 2016. Real-time, portable genome sequencing for Ebola surveillance. Nature 530: 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J, Grubaugh ND, Pullan ST, Claro IM, Smith AD, Gangavarapu K, Oliveira G, Robles-Sikisaka R, Rogers TF, Beutler NA, et al. 2017. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 12: 1261–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi IA, Mattick JS, Mehler MF. 2010. Long non-coding RNAs in nervous system function and disease. Brain Res 1338: 20–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O'Connell MA, Li JB. 2013. Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10: 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M, Paten B. 2017. Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods 14: 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy R, Henning D, Epstein P, Busch H. 1981. Primary and secondary structure of U2 snRNA. Nucleic Acids Res 9: 5645–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads A, Au KF. 2015. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13: 278–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez V, Chen Y, Elkahloun A, Dutra A, Pak E, Chandrasekharappa S. 2007. Chromosome 8 BAC array comparative genomic hybridization and expression analysis identify amplification and overexpression of TRMT12 in breast cancer. Genes Chromosomes Cancer 46: 694–707. [DOI] [PubMed] [Google Scholar]
- Ryvkin P, Leung YY, Silverman IM, Childress M, Valladares O, Dragomir I, Gregory BD, Wang LS. 2013. HAMR: high-throughput annotation of modified ribonucleotides. RNA 19: 1684–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saletore Y, Meyer K, Korlach J, Vilfan ID, Jaffrey S, Mason CE. 2012. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol 13: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarin LP, Leidel SA. 2014. Modify or die? - RNA modification defects in metazoans. RNA Biol 11: 1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkozy P, Jobbágy Á, Antal P. 2017. Calling homopolymer stretches from raw nanopore reads by analyzing k-mer dwell times. In EMBEC & NBC 2017: joint conference of the European Medical and Biological Engineering Conference (EMBEC) and the Nordic-Baltic Conference on Biomedical Engineering and Medical Physics (NBC), Tampere, Finland, June 2017 (ed. Eskola H, et al.), pp. 241–244. Springer Singapore, Singapore. [Google Scholar]
- Sasaki S, Yamashita T, Kwak S. 2015. Autophagy in spinal motor neurons of conditional ADAR2-knockout mice: an implication for a role of calcium in increased autophagy flux in ALS. Neurosci Lett 598: 79–84. [DOI] [PubMed] [Google Scholar]
- Sawyer SL, Emerman M, Malik HS. 2004. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol 2: E275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M, Pollex T, Hanna K, Lyko F. 2009. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res 37: e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffrath R, Leidel SA. 2017. Wobble uridine modifications—a reason to live, a reason to die?! RNA Biol 23: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S. 2016. Cracking the epitranscriptome. RNA 22: 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, Tabach Y, Mikkelsen TS, Satija R, Ruvkun G, et al. 2013. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155: 1409–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, Leon-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES, et al. 2014a. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159: 148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. 2014b. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8: 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafik A, Schumann U, Evers M, Sibbritt T, Preiss T. 2016. The emerging epitranscriptomics of long noncoding RNAs. Biochim Biophys Acta 1859: 59–70. [DOI] [PubMed] [Google Scholar]
- Shaheen R, Abdel-Salam GM, Guy MP, Alomar R, Abdel-Hamid MS, Afifi HH, Ismail SI, Emam BA, Phizicky EM, Alkuraya FS. 2015. Mutation in WDR4 impairs tRNA m7G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol 16: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, Belleannee C, Kucukural A, Serra RW, Sun F, et al. 2016. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 351: 391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W. 2017. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods 14: 407–410. [DOI] [PubMed] [Google Scholar]
- Smith AM, Jain M, Mulroney L, Garalde DR, Akeson M. 2017. Reading canonical and modified nucleotides in 16S ribosomal RNA using nanopore direct RNA sequencing. bioRxiv. 10.1101/132274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, Preiss T. 2012. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res 40: 5023–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoiber M, Brown J. 2017. BasecRAWller: streaming nanopore basecalling directly from raw signal. bioRxiv. 10.1101/133058. [DOI] [Google Scholar]
- Stoiber MH, Quick J, Egan R, Lee JE, Celniker SE, Neely R, Loman N, Pennacchio L, Brown JB. 2016. De novo identification of DNA modifications enabled by genome-guided nanopore signal processing. bioRxiv. 10.1101/094672. [DOI] [Google Scholar]
- Su D, Chan CT, Gu C, Lim KS, Chionh YH, McBee ME, Russell BS, Babu IR, Begley TJ, Dedon PC. 2014. Quantitative analysis of ribonucleoside modifications in tRNA by HPLC-coupled mass spectrometry. Nat Protoc 9: 828–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli S, Galeano F, Alon S, Raho S, Galardi S, Polito VA, Presutti C, Vincenti S, Eisenberg E, Locatelli F, et al. 2015. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol 16: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AG, Batlle E, Ribas de Pouplana L. 2014. Role of tRNA modifications in human diseases. Trends Mol Med 20: 306–314. [DOI] [PubMed] [Google Scholar]
- Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G, Lyko F. 2012. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol 19: 900–905. [DOI] [PubMed] [Google Scholar]
- Wang X, He C. 2014. Dynamic RNA modifications in posttranscriptional regulation. Mol Cell 56: 5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang ZL, Zhao JC. 2014. _N_6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16: 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. 2015. _N_6-methyladenosine modulates messenger RNA translation efficiency. Cell 161: 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Niebuhr E, Yang H, Hansen L. 2005. Determination of the ‘critical region’ for cat-like cry of Cri-du-chat syndrome and analysis of candidate genes by quantitative PCR. Eur J Hum Genet 13: 475–485. [DOI] [PubMed] [Google Scholar]
- Yan M, Wang Y, Hu Y, Feng Y, Dai C, Wu J, Wu D, Zhang F, Zhai Q. 2013. A high-throughput quantitative approach reveals more small RNA modifications in mouse liver and their correlation with diabetes. Anal Chem 85: 12173–12181. [DOI] [PubMed] [Google Scholar]
- Yi J, Gao R, Chen Y, Yang Z, Han P, Zhang H, Dou Y, Liu W, Wang W, Du G, et al. 2016. Overexpression of NSUN2 by DNA hypomethylation is associated with metastatic progression in human breast cancer. Oncotarget 8: 20751–20765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Yu YT. 2004. Detection and quantitation of RNA base modifications. RNA 10: 996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Roundtree IA, He C. 2016. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18: 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, He C. 2017. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542: 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinshteyn B, Gilbert WV. 2013. Loss of a conserved tRNA anticodon modification perturbs cellular signaling. PLoS Genet 9: e1003675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material