Chemosensitivity prediction by transcriptional profiling (original) (raw)
Abstract
In an effort to develop a genomics-based approach to the prediction of drug response, we have developed an algorithm for classification of cell line chemosensitivity based on gene expression profiles alone. Using oligonucleotide microarrays, the expression levels of 6,817 genes were measured in a panel of 60 human cancer cell lines (the NCI-60) for which the chemosensitivity profiles of thousands of chemical compounds have been determined. We sought to determine whether the gene expression signatures of untreated cells were sufficient for the prediction of chemosensitivity. Gene expression-based classifiers of sensitivity or resistance for 232 compounds were generated and then evaluated on independent sets of data. The classifiers were designed to be independent of the cells' tissue of origin. The accuracy of chemosensitivity prediction was considerably better than would be expected by chance. Eighty-eight of 232 expression-based classifiers performed accurately (with P < 0.05) on an independent test set, whereas only 12 of the 232 would be expected to do so by chance. These results suggest that at least for a subset of compounds genomic approaches to chemosensitivity prediction are feasible.
A long-term goal of pharmacogenomics research is the accurate prediction of patient response to drugs, as it would facilitate the individualization of patient treatment. Such an approach is particularly needed in cancer therapy, where commonly used agents are ineffective in many patients, and where side effects are common, given the nonspecific mechanism of action of most chemotherapeutic drugs. Previous efforts to use genetic information to predict drug sensitivity primarily have focused on individual genes that have broad effects, such as multidrug resistance genes mdr1 and mrp1 (1). Here we describe a predictive methodology that seeks to tap more complex genetic contributions to drug sensitivity. The recent development of DNA microarrays, which permit the simultaneous measurement of the expression levels of thousands of genes, raises the possibility of an unbiased, genomewide approach to the genetic basis of drug response.
Prediction of chemosensitivity in the clinic is particularly challenging because drug responses reflect not only properties intrinsic to the target cell, but also host metabolic properties. By modeling this approach in cultured cells, we limited our study to cell-intrinsic properties that are exposed in culture. A panel of 60 such cancer cell lines has been used extensively by the National Cancer Institute's Developmental Therapeutics Program, and the merits and limitations of their use as screening tools for drug development have been described (2–5). These cell lines have been analyzed for their sensitivity to a broad range of chemical compounds and thus offer an extensive database for the testing of our methodology.
We investigated the feasibility of chemosensitivity prediction by using oligonucleotide microarrays to measure the expression levels of 6,817 genes in each of the 60 cell lines in the NCI-60 panel. The data can be found at www.genome.wi.mit.edu/MPR/NC160/NC160.html. We then asked whether patterns of gene expression were sufficient to predict sensitivity or resistance of the cell lines to 232 chemical compounds. To maintain statistical rigor, the data set was divided into two groups—a training set, which was used to develop a gene expression-based chemosensitivity classifier, and a test set, on which we evaluated the accuracy of the classifier. When compared with random prediction, a significant number of the expression-based classifiers performed accurately, indicating that the response of cancer cell lines to drugs is indeed predictable.
Materials and Methods
Compound Selection.
The 60 cell lines were previously assayed for their sensitivity to a variety of compounds as a part of the Developmental Therapeutics Program at the National Cancer Institute, as described (refs. 2 and 3; see also: http://dtp.nci.nih.gov). Briefly, each cell line was exposed to each compound for 48 h, and growth inhibition was assessed by the sulforhodamine B assay for cellular protein. The concentration of compound required for 50% growth inhibition was scored as the GI50. For each compound, log10(GI50) values were normalized across the 60 cell lines. Cell lines with log10(GI50) at least 0.8 SDs above the mean were defined as resistant to the compound, whereas those with log10(GI50) at least 0.8 SDs below the mean were defined as sensitive. Cell lines with log10(GI50) within 0.8 SDs of the mean were considered to be intermediate and were eliminated from analysis. Prediction analysis was performed for compounds that had a minimum of 30 sensitive and resistant lines, with at least 10 each sensitive and resistant. To avoid choosing drug compounds with narrow dynamic ranges of drug responses, which are essentially sensitive or resistant to most of the 60 cell lines, we also required that the 1.6-SD window around the mean GI50 correspond to at least 1 order of magnitude in raw GI50 values. Of 5,084 compounds evaluated, 232 met these criteria. Importantly, gene expression data were not used in any way in compound selection.
Training and Test Set Selection.
For each selected compound, a set of training cell lines was chosen in the following manner. Within each tissue type (e.g., breast cancer; see Results), the most sensitive and most resistant cell line were chosen. If a tissue type lacked either sensitive or resistant cell lines according to the criteria above, it was not used in training. All sensitive or resistant cell lines not selected for training were reserved as a test set for final evaluation of the classifier.
Gene Expression Data.
RNA was isolated as described (6). Poly(A) selected RNA (1.5 μg) from each cell line was used to prepare biotinylated cRNA targets as described (7); details are provided at www.genome.wi.mit.edu/MPR. Targets were hybridized to Affymetrix (Santa Clara, CA) high-density Hu6800 arrays, washed, stained with phycoerythrin-conjugated streptavidin (Molecular Probes), and signal-amplified with biotinylated anti-streptavidin antibody (Vector Laboratories). Expression values (average difference units) were calculated by using Affymetrix genechip software. An expression level of 100 units was assigned to measurements <100.
An earlier version of the gene expression data was generated by hybridizing the biotinylated targets to an earlier-generation, low-density Affymetrix HU6800 four-chip set (HU6800 subA, subB, subC, subD). These data were analyzed by Butte et al. (13), using relevance networks, and are available at http://www.genome.wi.mit.edu/MPR. However, all analyses described in this article were performed on the data from the newer, higher density arrays.
Weighted Voting Classification.
We used a weighted voting scheme to classify each cell line as sensitive or resistant on the basis of gene expression data. In this scheme, a set of marker genes “vote” on the class of each cell line (8). For each compound being classified, genes were excluded if they varied by less than 5-fold and 500 units across training cell lines, and by less than 2-fold across each pair of training cell lines of a single tissue type. The remaining genes on the microarray were ranked according to the correlation between their expression level and the sensitivity and resistance profile of the training cell lines. We used a measure of correlation, P(g,c), as described (8). Let [μ1(g), σ1(g)] and [μ2(g), σ2(g)] denote the means and SDs of the expression levels of gene g for the samples in class 1 and class 2, respectively. Let P(g,c) = [μ1(g) − μ2(g)]/[ σ1(g) + σ2(g)], which reflects the difference between the class means relative to the variance within the classes. Large values of P(g,c) indicate strong correlation between gene expression and class distinction, whereas the sign of P(g,c) indicates whether higher expression correlates with class 1 or class 2. The vote for each gene can be expressed as the weighted difference between the normalized log expression in the cell line to be classified and the average of the sensitive and resistance class mean expression levels, where weighting is determined by the correlation P(g,c) from the training set. The class of the cell line is determined by the sum of votes for all marker genes used in a classifier. In previous work (8), classification was subjected to a confidence (prediction strength) threshold; no such threshold was used here.
Optimizing Classifiers by Cross-Validation.
Classifiers with 1–200 marker genes were used for training set cross-validation to determine the number of marker genes that best classify each compound. For each classifier, cross-validation was performed with the entire training set: one cell line was removed, the classifier was trained on the remaining cell lines and then tested for its ability to classify the withheld cell line. This procedure was repeated for each cell line in the training set. Cross-validation accuracy rates are available at www.genome.wi.mit.edu/MPR/NC160/NC160.html.
Evaluating Classifier Accuracy.
The model that was most accurate in cross-validation was chosen as the optimized classifier for that compound. In the case of multiple models that scored identically, the model with the larger number of genes was chosen. The training set-optimized classifier for each compound was then used to classify test cell lines. Performance was measured as the average of the accuracy of classifying sensitive cell lines and the accuracy of classifying resistant cell lines. As a control, 1,000 iterations of a simulation were run to classify the same 232 test sets by random coin flip. The distributions from observed and random results were compared by using the Kolmogorov–Smirnov test (9), which is a test for whether two sets of data are drawn from different distributions (see www.genome.wi.mit.edu/MPR/NC160/NC160.html for details).
For computing the significance of individual classifier performance, we computed the probability of the observed prediction accuracy occurring by chance if such predictions were the result of a fair coin flip. Consider a compound with n cell lines in the test set, and a classifier that predicts j of the n cell lines correctly. Because training introduces no class bias, the probability of doing at least this well by chance, Pr(j correct predictions), is the same as Pr(≥j heads of n fair coin flips), which can be represented as
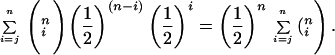
Results
Our classification scheme is outlined in Fig. 1. We approached chemosensitivity prediction as a binary classification problem, and thus for each compound, two classes of cell lines were defined: sensitive and resistant. The majority of the 5,084 compounds demonstrated relatively uniform growth inhibitory activities (GI50) across the 60 cancer cell lines, but we restricted our analysis to compounds that included a balance of sensitive and resistant lines (see Materials and Methods and Fig. 2). A total of 232 compounds met these criteria (see www.genome.wi.mit.edu/MPR/NC160/NC160.html for complete list of compounds and cell lines).
Figure 1.

General scheme for classification of compound sensitivity in cell lines by using gene expression data.
Figure 2.

Example of compound (NSC 749; Azaguanine) with bimodal distribution of growth inhibition. For each compound, log(GI50) values were normalized across the 60 cell lines, and cell lines with log(GI50) within 0.8 SDs of the mean are eliminated from analysis; remaining cell lines were defined as sensitive or resistant to the compound. Compounds with at least 30 cell lines outside the 1.6-SD window, and for which the window represents at least 1 order of magnitude in raw GI50 data were analyzed further. A total of 232 compounds met these criteria.
For each of the 232 compounds, the sensitive and resistant cell lines were divided into a training set and a test set, again by using only drug sensitivity data to make these assignments. One approach would be to select a set of cell lines at random for training and use the remaining lines as a test set. The problem with this approach is that the cell lines in the NCI-60 panel are derived from nine broad categories of tissue of origin (lung, breast, colon, kidney, bone marrow, melanocyte, central nervous system, prostate, and ovary). Sensitivity to some drugs correlates with tissue of origin, and thus one runs the risk of developing classifiers that simply classify according to tissue type, rather than according to drug sensitivity per se. To circumvent this problem, we designed “tissue-aware” training sets. Each training set included one sensitive and one resistant cell line from each of multiple tissue types. A tissue type was used in training only if it included both sensitive and resistant cell lines for the compound, and thus the 232 training sets contained variable numbers of cell lines (6 to 18). For each compound, the remaining cell lines (16 to 35) were reserved as a test set that was used to independently evaluate prediction accuracy. All reported prediction accuracies are for test set samples only.
To create a gene expression database, RNA was extracted from the 60 cell lines before any drug treatment. These RNAs were then analyzed on oligonucleotide microarrays containing probes for 6,817 known human genes. The genes were not selected to be particularly informative for the present experiments, but rather they represent the named human genes identified in GenBank at the time the array was designed. The expression levels of the 6,817 genes in each of the 60 cell lines are available at www.genome.wi.mit.edu/MPR/NC160/NC160.html.
To build and train classifiers, we used both drug sensitivity data and gene expression data. The GI50 profile of each training set was used as a template for marker gene selection. Each gene was ranked according to the correlation in the training set between its expression level and the sensitivity-resistance class distinction (see Materials and Methods). Classification (sensitive vs. resistant) was performed by using a weighted voting algorithm, in which correlated genes “vote” on whether a cell line is predicted to be sensitive or resistant (8). The vote for each gene is a function of its expression in the cell line to be classified and the degree to which its expression is correlated with sensitivity or resistance in the training set (see Materials and Methods). Classifiers with up to 200 correlated genes were tested through cross-validation by holding back one cell line, training on the remaining lines, predicting the class of the withheld line, and repeating this cycle for each cell line in the training set. For each compound, the classifier model that was most accurate in training set cross-validation was selected as the optimized classifier for that compound, and it was evaluated without further modification on the independent test set. Each optimized classifier contained between five and 200 genes, with an average of 68 genes per classifier (all classifier genes and weights are available at www.genome.wi.mit.edu/MPR/NC160/NC160.html). This process of cross-validation diminishes the problem of overfitting during selection of the optimal classifier, a particular problem when dealing with small number of cases and large numbers of variables.
Each classifier, optimized on a training set, was evaluated on a test set of cell lines that had not participated in training. The distribution of accuracies from expression-based classification was compared with the distribution obtained from random classification of the same 232 test sets (Fig. 3). The difference between the two distributions is highly significant, as indicated by the Kolmogorov–Smirnov test (P ≤ 10−24) (9, 10), with the expression-based distribution clearly skewed toward higher accuracy.
Figure 3.

Distribution of classification accuracies for 232 compounds. Percent accuracy for each compound is the average accuracy for classification of sensitive and resistant test cell lines. The control distribution represents results obtained from random classification (1,000 iterations) of the 232 test sets.
The significance of each classifier's performance was assessed by determining the probability of obtaining the observed accuracy rate by chance if each classification was the result of a fair coin toss (see Materials and Methods). A total of 88 of 232 (38%) expression-based classifiers performed accurately with a significance of P ≤ 0.05, whereas only 12 such classifiers (5% of 232) would be expected to do so by chance. The statistically significant classifiers had a median accuracy of 75% (range 64% to 92%). This result indicates that for a substantial subset of compounds gene expression data were sufficient for accurate prediction of chemosensitivity.
The compounds whose chemosensitivity was highly predictable spanned multiple structural categories, the majority functioning through unknown mechanisms of action. We observed no obvious connection between mechanism of drug action and classifier accuracy. No obvious relationship was seen between prediction accuracy and number of genes used or number of cell lines used for training (data not shown).
In addition to yielding accurate predictors of chemosensitivity, the gene expression data generated herein provide potential insights into mechanisms of drug resistance. In general, the gene expression correlates of drug sensitivity were complex, and their biological significance not easily interpretable (all lists of genes and weights are available at www.genome.wi.mit.edu/MPR/NC160/NC160.html). Our method required variable expression across multiple pairs of training cell lines, which explains some notable absences, such as mdr1, whose expression level surpassed our detection threshold (100 average difference units) in only three cell lines. However, anecdotal relationships between correlated marker genes and known mechanisms of drug action suggest that marker genes may provide insights into mechanisms of drug action—or of sensitivity or resistance—for compounds with unknown mechanism of action.
For example, the 120-gene classifier for cytochalasin D (NSC 209835) classified 20 cell lines with accuracy of 80% (significant at a threshold of P < 0.0013). The marker genes for the cytochalasin D classifier included 29 genes (24%) related to the cytoskeleton or extracellular matrix (ECM). This set is enriched relative to the ≈5% known cytoskeletal/ECM genes on the entire array (data not shown). The top 30 cytochalasin D marker genes are shown in Fig. 4, along with the expression level of each gene across the 20 cell lines (a classifier built on only 30 genes similarly yields 80% accuracy). Cytochalasin D binds to actin and induces dimers that interfere with polymerization, thus disrupting cytoskeletal integrity (11), but it has not been previously suspected that the expression pattern of cytoskeletal genes in untreated cells would be predictive of cytochalasin D sensitivity. Interestingly, an excess of cytoskeletal/ECM genes also was observed for a number of other classifiers, including ones for compounds that are not thought to act through cytoskeletal components. For example, the 100-gene classifier for the antifolate, NSC 633713 is highly accurate (87.5% accuracy; significant at a threshold of P < 0.0003) and includes 21 (21%) cytoskeletal/ECM genes. It is possible that cytoskeletal signatures may reflect cellular components that influence sensitivity to a variety of compounds rather than functioning as direct targets of compound activity.
Figure 4.

Top 30 classifier genes for cytochalasin D (NSC-209835). The red and blue matrix represents the normalized expression patterns for each gene across the cell lines (brightest red indicates highest relative expression, darkest blue indicate, lowest relative expression). (Top) The sensitive and resistant cell lines are shown. Tissue of origin for each cell line is indicated as follows: L, lung (nonsmall cell); C, colon; B, breast; O, ovarian; E, leukemia; R, renal; M, melanoma; P, prostate; N, central nervous system. Lines used as training sets are shown in bold. The list at right shows the weighting factor [measure of correlation; weights were computed by using negative log(GI50) values and thus a positive value correlates with sensitivity], the GenBank accession number, and the gene name. Genes whose products are known to have cytoskeletal and/or extracellular matrix functions are shown in bold.
Discussion
Implicit in the goal of personalized medicine is the notion that an individual patient's response to drugs should be predictable. However, experimental data supporting the genetic basis of differential drug response are limited. We report here a systematic approach for gene expression-based prediction of chemosensitivity. We have applied this methodology to the prediction of cytotoxicity for 232 compounds in 60 cell lines by using the gene expression profiles of untreated cells. The NCI-60 panel has been used extensively in drug evaluation efforts at the National Cancer Institute, and more recently, it has been studied at the gene expression level by using an alternative approach to gene expression profiling (cDNA microarrays) (6, 12). Those and other studies (13) clearly demonstrate that biological correlates of gene expression are identifiable. In the present study, we explored whether such gene-drug correlates are sufficiently robust to permit development of a chemosensitivity classifier built exclusively on gene expression data.
A particular challenge in such an effort is the small size of the data set. DNA microarrays allow for the measurement of thousands of genes, yet most experiments contain relatively few samples. The NCI-60 panel contains a total of 60 cell lines, but only 2–9 cell lines represent each tissue type (e.g., kidney, colon). When analyzing small data sets, one runs the risk of overfitting a model to the data. This can result in overestimating the classifier's accuracy. We addressed this problem in two ways. First, we used a leave-one-out cross-validation procedure to build the prediction models. Second, we divided the data set into two parts: a training set on which chemosensitivity predictors were developed, and a test set, on which they were evaluated. One disadvantage to this approach is that it further reduces the size of the data set used to generate the model, and therefore accuracy can be potentially compromised. A particular goal of this study was thus to determine whether a data set of only 60 diverse cell lines would be sufficiently large to generate accurate, statistically significant chemosensitivity classifiers.
Given the above limitations, the observed accuracies are quite remarkable. Classification accuracy was far greater than one would expect by chance alone, with approximately one-third of the evaluated compounds being predictable with statistical significance (P < 0.05). These results suggest that, for at least some compounds, chemosensitivity is predictable by using only the gene expression patterns of untreated cells. The results further suggest that the identification of such patterns is feasible in data sets of only modest size.
The training sets were specifically designed to identify gene expression correlates of chemosensitivity within a tissue type, so as to reduce the confounding problem of chemosensitivity-tissue type correlations. However, such correlations may not be entirely avoided by the method. The selection of extreme cell lines within a given tissue type (i.e., those with the highest and lowest GI50s) for the training of the classifier leaves open the possibility that the training samples are atypical in their lack of chemosensitivity-tissue type correlation. For example, the classification of cytochalasin D sensitivity (Fig. 4) is in part correlated with tissue type in that the ovarian cancer cell lines tend to be resistant, whereas the central nervous system (CNS) cell lines are sensitive. Notably, neither ovarian nor CNS cell lines were used to train the classifier.
For some compounds, gene-based classification was no more accurate than random classification. There are several possible explanations for this. First, we measured the expression level of only 6,817 genes, estimated to represent roughly one-fifth of the human genome (14). It is possible that if the entire genome were analyzed, the number of compounds with predictable chemosensitivity would increase. It is also conceivable that alternative gene selection or machine learning algorithms would be more successful. Second, we limited ourselves to a binary classification scheme, whereas a multiclass or continuous definition of sensitivity may be more appropriate for some compounds. It is likely that larger data set would be required for such efforts. Finally, for some compounds, chemosensitivity may be governed by mechanisms that are not readily revealed at the transcriptional level, such as posttranscriptional regulation, posttranslational modification, proteasome function, or protein–protein interactions. The ability to increase prediction accuracy by capturing such information by using proteomic approaches, for example, remains to be determined.
To achieve the goal of personalized medicine, chemosensitivity prediction must be extended beyond cell line models to include the analysis of primary patient material, and the prediction of intermediate levels of chemosensitivity that were not addressed in our experiments. Although few clinical studies have been reported to date, early indications are that clinically relevant gene expression patterns can be extracted from tumor samples (8, 12, 15, 16). However, the current study demonstrates the potential for screening samples for genetic determinants of drug sensitivity and, thus, suggests that the goal of individualizing patient treatment plans based on genetic features of a tumor may indeed be feasible.
Acknowledgments
We thank current and former members of the Whitehead/Massachusetts Institute of Technology Center for Genome Research for helpful discussions and reviewers for comments on the manuscript. We are especially grateful to Nathan Siemers for scientific input and Julian Fowler for the web-page implementation. This work was supported in part by Affymetrix, Millennium Pharmaceuticals, and Bristol-Myers Squibb (to E.S.L.), and by grants from the National Cancer Institute and National Cancer Institute Intramural Breast Cancer Think Tank (to U.S., J.N.W., J.K.L., and W.O.R.).
Abbreviation
GI50
50% growth inhibition
References
- 1.Sonneveld P. J Intern Med. 2000;247:521–534. [PubMed] [Google Scholar]
- 2.Grever M R, Schepartz S A, Chabner B A. Semin Oncol. 1992;19:622–638. [PubMed] [Google Scholar]
- 3.Stinson S F, Alley M C, Kopp W C, Fiebig H H, Mullendore L A, Pittman A F, Kenney S, Keller J, Boyd M R. Anticancer Res. 1992;12:1035–1053. [PubMed] [Google Scholar]
- 4.Monks A, Scudiero D A, Johnson G S, Paull K D, Sausville E A. Anticancer Drug Des. 1997;12:533–541. [PubMed] [Google Scholar]
- 5.Weinstein J N, Myers T G, O'Connor P M, Friend S H, Fornace A J, Jr, Kohn K W, Fojo T, Bates S E, Rubinstein L V, Anderson N L, et al. Science. 1997;275:343–349. doi: 10.1126/science.275.5298.343. [DOI] [PubMed] [Google Scholar]
- 6.Scherf U, Ross D T, Waltham M, Smith L H, Lee J K, Tanabe L, Kohn K W, Reinhold W C, Myers T G, Andrews D T, et al. Nat Gent. 2000;24:236–244. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- 7.Lockhart D, Dong H, Byrne M, Follettie M, Gallo M, Chee M, Mittmann M, Wang C, Kobayashi M, Horton H, El B. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 8.Golub T R, Slonim D K, Tamayo P, Huard C, Gaasenbeek M, Mesirov J P, Coller H, Loh M L, Downing J R, Caligiuri M A, et al. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 9.Press W H, Teukolsky S A, Vetterling W T, Flannery B P. Numerical Recipes in C: The Art of Scientific Computing. 2nd Ed. Cambridge, U.K.: Cambridge Univ. Press; 1992. [Google Scholar]
- 10.Zar J H. Biostatistical Analysis. Upper Saddle, NJ: Prentice–Hall; 1999. [Google Scholar]
- 11.Goddette D W, Frieden C. J Biol Chem. 1986;261:15974–15980. [PubMed] [Google Scholar]
- 12.Ross D T, Scherf U, Eisen M B, Perou C M, Rees C, Spellman P, Iyer V, Jeffrey S S, Van de Rijn M, Waltham M, et al. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- 13.Butte A, Tamayo P, Slonim D, Golub T R, Kohane I S. Proc Natl Acad Sci USA. 2000;97:12182–12186. doi: 10.1073/pnas.220392197. . (First Published October 10, 2000; 10.1073/pnas.220392197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.International Human Genome Sequencing Consortium. Nature (London) 2001;409:860–921. [Google Scholar]
- 15.Alizadeh A A, Eisen M B, Davis R E, Ma C, Lossos I S, Rosenwald A, Boldrick J C, Sabet H, Tran T, Yu X, et al. Nature (London) 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 16.Bittner B, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, et al. Nature (London) 2000;406:536–540. doi: 10.1038/35020115. [DOI] [PubMed] [Google Scholar]