Terminal spreading depolarization and electrical silence in death of human cerebral cortex (original) (raw)
Abstract
Objective
Restoring the circulation is the primary goal in emergency treatment of cerebral ischemia. However, better understanding of how the brain responds to energy depletion could help predict the time available for resuscitation until irreversible damage and advance development of interventions that prolong this span. Experimentally, injury to central neurons begins only with anoxic depolarization. This potentially reversible, spreading wave typically starts 2 to 5 minutes after the onset of severe ischemia, marking the onset of a toxic intraneuronal change that eventually results in irreversible injury.
Methods
To investigate this in the human brain, we performed recordings with either subdural electrode strips (n = 4) or intraparenchymal electrode arrays (n = 5) in patients with devastating brain injury that resulted in activation of a Do Not Resuscitate–Comfort Care order followed by terminal extubation.
Results
Withdrawal of life‐sustaining therapies produced a decline in brain tissue partial pressure of oxygen (ptiO2) and circulatory arrest. Silencing of spontaneous electrical activity developed simultaneously across regional electrode arrays in 8 patients. This silencing, termed “nonspreading depression,” developed during the steep falling phase of ptiO2 (intraparenchymal sensor, n = 6) at 11 (interquartile range [IQR] = 7–14) mmHg. Terminal spreading depolarizations started to propagate between electrodes 3.9 (IQR = 2.6–6.3) minutes after onset of the final drop in perfusion and 13 to 266 seconds after nonspreading depression. In 1 patient, terminal spreading depolarization induced the initial electrocerebral silence in a spreading depression pattern; circulatory arrest developed thereafter.
Interpretation
These results provide fundamental insight into the neurobiology of dying and have important implications for survivable cerebral ischemic insults. Ann Neurol 2018;83:295–310
The brain is the most vulnerable organ of the body to hypoxia and ischemia. Neocortical pyramids of layers III, IV, and V, hippocampal CA1 pyramidal cells, striatal cells, and cerebellar Purkinje cells display the highest vulnerability.1, 2 Massive, irreversible injury of these cells develops within <10 minutes when circulation completely ceases, such as after cardiac arrest.3, 4 The pathophysiological cascades resulting in this injury have been extensively investigated in various animal species for more than half a century. However, in humans, scalp electroencephalographic (EEG) recordings have only conveyed a vague hint at what truly happens in the brain when the circulation ceases.5 Accordingly, interpretations and conclusions vary widely in the clinical literature, including views deviating significantly from the observations in animals. For example, notions are at times encountered that brain death occurs when the EEG becomes silent. Other notions recognize that neurons of the human cortex can remain polarized for several minutes during electrical silence associated with acute, complete ischemia, and that failure of membrane pumps thereafter would indicate the tissue's death.6 However, animal studies demonstrate that such failure only initiates the cytotoxic processes leading to death and only occurs as a consequence of a terminal depolarization wave, a phenomenon that has not yet been documented in the human brain.
In animals, complete global ischemia causes complete electrical inactivity (ie, isoelectricity) of affected gray matter within about 20 to 40 seconds.7 This cerebral silencing is regarded as an austerity program to curb neuronal energy usage by the shutdown of nonessential cell functions well before the neuronal adenosine triphosphate (ATP) pool is depleted and prospects of tissue recovery vanish.4, 8 Accordingly, the electrical inactivity corresponds to a hyperpolarization of neurons,9 which may be mediated by several mechanisms, as reviewed recently.10 A hallmark of electrical silencing after onset of severe ischemia is its simultaneous development in affected tissue, which has been termed “nonspreading depression.”11 Despite this austerity program, sodium and calcium always leak into cells and potassium leaks out. These movements are driven by the release of Gibbs free energy. Eventually, ATP is depleted and the Na,K‐ATPase fails to restore the leaking ions. When a threshold level of failure, usually reached 1 to 5 minutes after nonspreading depression, has rendered the ischemic tissue isoelectric, neurons then undergo abrupt, nonlinear rundown in the ion gradients across the cellular membranes. This depolarization develops en masse from either one or multiple foci and spreads in the tissue as a self‐propagating wave.7, 9, 12, 13, 14 Initially termed “anoxic” or “asphyxial” depolarization,15 this spreading depolarization (SD) initiates a plethora of toxic changes, including intracellular calcium and sodium loading, steep rise in extracellular glutamate, and cytotoxic edema, as reviewed recently.16, 17, 18 Importantly, cellular injury does not develop during the initial period of nonspreading depression, but only begins with a delay after SD ignition.10
Knowledge of analogous pathologic processes in the human brain would inform not only research on treatment strategies of cardiac arrest and stroke that may complement efforts to reestablish circulation, but also the debate on organ donation after cardiocirculatory death (DCD), where death is declared between 2 and 10 minutes following the cessation of circulatory function.19, 20, 21 Therefore, here we analyzed the sequence of pathophysiologic events in patients during abrupt hypoxia–ischemia after withdrawal of life‐sustaining treatments. Patients underwent neuromonitoring with intracranial electrodes during intensive care treatment following aneurysmal subarachnoid hemorrhage (aSAH) and clip ligation of the aneurysm, decompressive hemicraniectomy for malignant hemispheric stroke (MHS), or traumatic brain injury (TBI). Following poor clinical course and family discussion, neuromonitoring was continued through the dying process after activation of a Do Not Resuscitate–Comfort Care (DNR‐CC) order.
Materials and Methods
Patients undergoing invasive neuromonitoring were enrolled at Charité–Universitätsmedizin Berlin (n = 4) and the University of Cincinnati Medical Center (n = 5) in research protocols approved by the local ethics committees; written informed consent was obtained from the patients' legally authorized representatives. Research was conducted in accordance with the Declaration of Helsinki.
In 3 patients with aSAH, a linear electrode strip (6 platinum/iridium [Pt/Ir] contacts spaced at 10mm; Wyler, Ad‐Tech Medical Instrument Corporation, Oak Creek, WI) was placed on the cortical surface of the vascular territory of the aneurysm‐carrying vessel following surgery to secure the aneurysm (Fig 1B).22, 23 In 1 patient with MHS, the electrode strip was placed on perfused, noninfarcted peri‐infarct tissue after decompressive hemicraniectomy.24, 25 In 5 patients with TBI, an intraparenchymal depth electrode array (8 Pt/Ir contacts spaced at 2.2mm; Spencer, Ad‐Tech Medical) was placed in the frontal lobe through a multilumen bolt after admission to neurocritical care.26 In Patient 5 of Tables 1 and 2, the electrode array was implanted on the left side with no lesions present, contralateral to subdural hematoma (Fig 2A). By contrast, the array was located in the right hemisphere in 3 other patients with extensive bifrontal hemorrhagic contusions and another one with perforating gunshot wound from right temporoparietal through left occipital regions. Subdermal platinum needles placed at the ipsilateral mastoid were used as reference.
Figure 1.
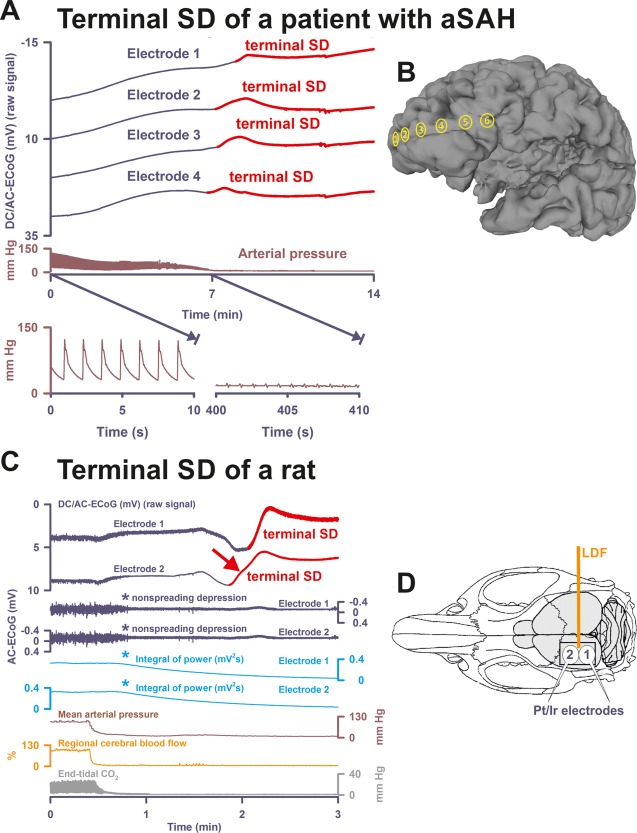
Terminal spreading depolarization (SD) during death in the human brain in comparison to terminal SD during death in the rat brain. (A) In Patient 1, the first direct current (DC) change after terminal extubation was a very slow, homogeneous DC positivity (not shown; cf Table 1) that began before the circulatory arrest. Terminal SD then started at electrode 4 (transition from dark blue to red DC traces) after the arterial pressure had reached its minimum (brown trace). Systemic arterial pressure was recorded in the radial artery. The 2 insets show the arterial pressure fluctuations during the initial period and during the arrest of the systemic circulation. Note that there is still evidence of cardiac cycles in the right inset, but the cardiac output is too low to maintain a sufficient systemic pressure (electromechanical dissociation). This increasingly pulseless electrical activity of the heart disappeared 30 seconds thereafter. Spontaneous brain activity had ceased in response to the barbiturate thiopental several hours before the circulatory arrest. Therefore, no nonspreading depression of activity occurred (not shown). (B) Projection of the electrodes of a typical subdural, collinear recording strip on a human brain. (C) Dying process after circulatory arrest in a rat. (D) Original cranial window experiment using 2 subdural Pt/Ir plate electrodes for use in humans (DC/AC‐ECoG) and a laser‐Doppler flowmetry (LDF) probe (regional cerebral blood flow [rCBF]). Note the steep falls in arterial pressure and rCBF that mark the moment of circulatory arrest. In animals, the typical sequence of nonspreading depression followed by terminal SD during global cerebral anoxia–ischemia has been extensively documented since its first description in 1947.7, 11, 12, 13, 14 Note the similarity between the patterns of the terminal SD in the rodent experiment and in the patient (red arrows in A and C). In both cases, the terminal SD propagated from one electrode to the next corresponding with previous evidence from experiments in animals in vivo and in brain slices.7, 12, 13, 14 In contrast to the spread of the terminal SD, the nonspreading depression (cf asterisks) is seen in the rodent recordings as a silencing of the spontaneous electrical activity (alternating current [AC]–electrocorticography [ECoG]: 0.5–45Hz) that develops simultaneously across the array of the regional 2 electrodes (cf Figs 3B, 4B, 5A, 5C, and 6 for nonspreading depression in patients). In the clinic, it has become conventional to review the raw signal alongside a leaky integral of the total power of the bandpass‐filtered (AC‐ECoG) data and measure depression duration based on the latter.16, 23 Therefore, the integral of power is shown here to display the pattern of nonspreading depression when this type of data presentation is chosen (light blue traces, cf Patient 3 in Fig 4A and Patient 7 in Fig 6). The experiment was performed in a male Wistar rat (300g, 12 weeks old, supplied by Charles River, Sulzfeld, Germany) anesthetized with 100mg/kg body weight thiopental–sodium intraperitoneally (Trapanal; BYK Pharmaceuticals, Konstanz, Germany), tracheotomized, and artificially ventilated (Effenberger Rodent Respirator; Effenberger Med.‐Techn. Gerätebau, Pfaffing/Attel, Germany; approved by the Office for Occupational Safety and Health and Technical Security Berlin [G0152/11]). Sudden circulatory arrest was induced in the rat by injection of 10ml of air into the heart via the femoral vein. This explains why mean arterial pressure rapidly dropped in the rat in contrast to the patient, in whom the circulatory arrest developed gradually after extubation. The Supplementary Table provides the statistical description of 5 rats with a previously healthy brain in which death resulted from sudden circulatory arrest after air injection into the femoral vein and heart under thiopental anesthesia. aSAH = aneurysmal subarachnoid hemorrhage.
Table 1.
Summary of Demographic, Treatment, and SD‐Related Data of the Patients
| aSAH, Subdural Electrode Strip | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | Age, yr; Sex | Lesions | WFNS Grade | Fisher Grade | Location of Aneurysm | Intervention | Location of Electrode | Condition Resulting in Case Fatality | Day of Death | SDs before Terminal Events, No. | Recording Time, h |
| 1 | 52; F | Infarcts: R hem, L O; midline shift: 10mm; brain herniation | 3 | 3 | ACoA | Aneurysm clipping, EVD | R Fr cortex | Acute cerebral ischemia, brain herniation | 6 | 13 | 56.4 |
| 2 | 56; F | ICH R T; infarcts: R hem, L Fr, L O; midline shift: 17mm | 5 | 3 | MCA | Aneurysm clipping, EVD | R Fr cortex | Acute cerebral ischemia, large ICH, brain herniation | 2 | 5 | 27.7 |
| 3 | 78; F | ICH: L Fr, R Fr | 4 | 3 | ACoA | Aneurysm coiling and clipping, EVD | R Fr cortex | Liver cirrhosis progressing to hepatorenal failure | 12 | 13 | 210.9 |
| MHS, Subdural Electrode Strip | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Age, yr; Sex | Lesions | Infarct Etiology | Intervention | Location of Electrode | Condition Resulting in Case Fatality | Day of Death | SDs before Terminal Events, No. | Recording Time, h |
| 4 | 63; M | Infarcts: R MCA territory, L posterior MCA territory, R PICA, R and L cerebellum | Suspicion of cardiac embolism | Decompressive hemicraniectomy | R ACA territory | Multiple large brain infarcts | 5 | 0 | 75.3 |
| TBI, Depth Electrode | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | Age, yr; Sex | Lesions | History of Injury | Initial GCS | Intervention | Location of Electrode | Condition Resulting in Case Fatality | Day of Death | SDs before Terminal Events, No. | Recording Time, h |
| 5 | 47; M | tSAH, SDH, skull vault and base fractures, multiple foci of petechial shear | Vehicle struck by train | 1T1 (3T) | None | L Fr cortex | Severe blunt TBI | 3 | 0 | 22.3 |
| 6 | 71; M | tSAH; SDH; contusions: L Fr, R Fr; skull fractures: L Fr, L T | Pedestrian struck by vehicle | 3T5 (9T) | None | R Fr cortex | Severe blunt TBI | 2 | 0 | 40.8 |
| 7 | 57; M | tSAH; DAI; contusions: L Fr, R Fr, L T, R T | Fall down stairs | 2T5 (8T) | None | R Fr cortex | Severe blunt TBI | 5 | 5 | 27.3 |
| 8 | 29; M | tSAH, callosal hemorrhage, contusions | Gunshot wound | 1T5 (7T) | None | R Fr cortex | Severe penetrating TBI | 9 | 0 | 30.2 |
| 9 | 68; M | Massive contusions, cerebral edema | Struck by cow, fall to concrete | 3T5 (9T) | Craniotomy, evacuation of EDH | R Fr cortex | Severe blunt TBI | 5 | 8 | 115.0 |
Table 2.
Characteristics of the End‐of‐Life DC‐Electrocorticography
| No. | Slow Initial Homogeneous DC Change, mV (IQR) | Depression Type | Delay from Nonspreading Depression to Terminal SD, s | Initial DC Component of the Terminal SD, cf Figs 2 and 3 | Largest TOAD of Terminal SD, s | Late and Terminal DC Component, mV, NUP, cf Figs 2 and 3 | ||
|---|---|---|---|---|---|---|---|---|
| Largest Amplitude, mV | Median Amplitude, mV | Duration, s | ||||||
| 1 | 7.5 (7.0 to 8.2) | Barbiturate‐induced depression | — | −2.8 | −2.6 (−2.3 to −2.7) | 154 (122 to 176) | 42 | −7.3 (−6.2 to −8.9) |
| 2 | 20.8 (20.4 to 21.7) | Nonspreading depression | 53 | −4.4 | −1.4 (−1.3 to −2.9) | 82 (60 to 100) | 324 | −19.2 (−15.5 to −24.0) |
| 3 | 6.6 (6.3 to 8.2) | Spreading and nonspreading depression | — | −7.6 | −5.4 (−4.1 to −6.2) | 65 (64 to 113) | 383 | −8.3 (−6.7 to −9.6) |
| 4 | 2.8 (2.7 to 3.2) | Nonspreading depression | 13 | −4.6 | −3.7 (−3.4 to −4.0) | 322 (318 to 326) | 10 | −1.9 (−0.1 to −4.9) |
| 5 | 2.5 (1.3 to 2.8) | Nonspreading depression | 73 | −6.3 | −3.2 (−1.7 to −3.7) | 183 (153 to 225) | 80 | −18.9 (−16.5 to −21.6) |
| 6 | 9.2 (6.7 to 10.1) | Nonspreading depression | 266 | −0.5 | −0.3 (−0.2 to −0.4) | 97 (84 to 102) | — | −20.1 (−14.4 to −21.3) |
| 7 | −8.0 (−5.6 to −9.0) | Nonspreading depression | 106 | −0.7 | −0.3 (−0.2 to −0.5) | 94 (66 to 113) | 15 | −2.2 (1.0 to −4.3) |
| 8 | 0 | Nonspreading depression | 78 | −0.6 | −0.6 (−0.5 to −0.6) | 398 (360 to 416) | 5 | −5.8 (−5.4 to −6.0) |
| 9 | 5.2 (5.1 to 5.4) | Nonspreading depression | Not recorded |
Figure 2.
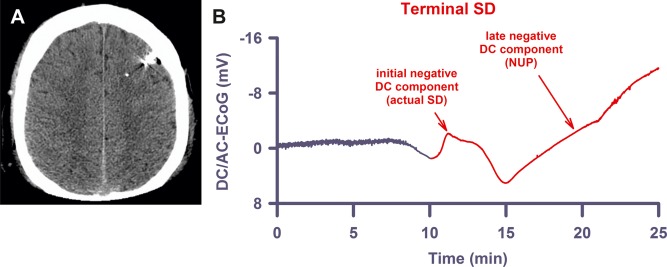
Similar to the terminal spreading depolarization (SD) in subdural recordings, the terminal SD in recordings with depth electrodes shows an initial and a late negative direct current (DC) component. (A) The computed tomogram of Patient 5 displays the intraparenchymal electrode array in the left frontal hemisphere with no lesions present. (B) The late component, the so‐called negative ultraslow potential (NUP), is similar to the negative DC shifts of prolonged SD, but specifically refers to a negative potential component generated by progressive recruitment of neurons into cell death in the wake of SDs (terminal SD at electrode 3 of Patient 5).16 AC = alternating current; ECoG = electrocorticography.
Continuous electrocorticography (ECoG) and multimodal monitoring were performed following the recently published recommendations of the Co‐Operative Studies on Brain Injury Depolarizations (COSBID) while patients were in neurocritical care.16 For aSAH patients and the MHS patient, electrodes were connected to a BrainAmp amplifier (BrainProducts, Munich, Germany) and data were acquired with BrainVision Recorder software (BrainProducts). For TBI patients, recordings were made with the Component Neuromonitoring System (Moberg ICU Solutions, Ambler, PA). In both groups, amplifiers were direct current (DC)‐coupled (0–100Hz). In 2 patients, a subdural optoelectrode strip allowed the simultaneous measurement of ECoG and regional cerebral blood flow (rCBF) using laser‐Doppler flowmetry (LDF; Perimed, Järfälla, Sweden).22, 27, 28 In 2 other patients, rCBF was monitored with a parenchymal thermal diffusion probe (Hemedex, Cambridge, MA).29 Tissue partial pressure of oxygen (ptiO2) was measured using an intraparenchymal sensor in 6 patients (Licox CC1P1; Integra Lifesciences Corporation, Plainsboro, NJ).22, 29, 30, 31 Intracranial pressure (ICP) was monitored via ventricular drainage catheter (extraventricular drainage [EVD]) or ICP transducer (Codman, San Jose, CA or Camino Laboratories, San Diego, CA). The systemic arterial pressure was continuously recorded via radial artery catheter.
Data Analysis
All data in text and figures are given as median, 1st quartile, and 3rd quartile. No patients were excluded from the analysis. The data were analyzed in an unblinded fashion with LabChart‐7 software (ADInstruments, Bella Vista, Australia).
The first 24‐hour period after the initial insult was denoted as “day 0.” SDs were identified by the consecutive occurrence of slow potential changes (<0.05Hz) of neighboring ECoG channels. In electrically active tissue, SD typically causes spreading depression of spontaneous activity in the alternating current (AC)‐ECoG range (0.5–45Hz).23 The duration of the depression period was scored beginning at the initial decrease in the integral of power and ending at the start of the recovery phase. Depression durations were scored to measure the total depression duration per recording day (TDDD), following the recently published recommendations of COSBID.16 The peak TDDD (PTDDD) was defined as the maximal value among all recording days. SDs in electrically active tissue received the epithet “spreading depression.” By contrast, SDs measured in a zone of electrically inactive tissue were denoted with the adjective “isoelectric.”16
Results
Tables 1 and 2 summarize the characteristics of 9 patients in whom multimodal monitoring was performed after devastating aSAH, MHS, or TBI and through the dying process after withdrawal of life‐sustaining therapies.
Illustrative Cases
Patient 1
Patient 1 was a 52‐year‐old woman with aSAH. Until day 3, she had 13 SDs (2 spreading depressions and 11 isoelectric SDs) with a PTDDD of 487.9 minutes that occurred on day 3. On the same day, a computed tomography (CT) scan showed multiple infarcts in the territories of the right middle cerebral artery (MCA) and both posterior cerebral arteries. On the following day, a control scan revealed increasing lesions, 10mm midline shift, and beginning brain herniation. From the beginning, she required intravenous noradrenaline at a rate of 0.08 µg/kg/min to maintain adequate cerebral perfusion pressure (CPP) under sedation with propofol. Stress cardiomyopathy was ruled out using echocardiography, electrocardiography, and cardiac biomarkers. However, noradrenaline had to be further increased to 0.26 µg/kg/min on day 4 when she developed _Staphylococcus aureus_–caused pneumonia and midazolam was added to the sedative regimen to control progressive increase in ICP. Thiopental was added for the same reason 12 hours before she died on day 6. Thiopental resulted in cerebral isoelectricity, and intravenous noradrenaline had to be increased to a rate of 0.43 µg/kg/min. Eventually, a DNR‐CC order was activated after family discussion and the patient was terminally extubated. Withdrawal of the life‐sustaining treatment resulted in complete circulatory arrest after 17 minutes. Figure 1A shows the time course of the systemic arterial pressure including the circulatory arrest and subsequent terminal SD that started at electrode 4 and then spread to the other electrodes. For comparison, Figure 1C displays a typical terminal SD of a rodent brain after injection of air into the heart via the femoral vein (experimental setup in Fig 1D); circulatory arrest is followed by nonspreading depression and then terminal SD. The Supplementary Table provides the statistical data of 5 thiopental‐anesthetized rats with previously healthy brains in which death resulted from sudden circulatory arrest by air injection into the femoral vein.
Patient 2
Patient 2 was a 56‐year‐old woman with aSAH. ICP increased up to 25mmHg despite EVD, artificial ventilation, and analgosedation. She became hemodynamically unstable and required intravenous noradrenaline at a rate of 0.56 µg/kg/min. ECoG and LDF displayed 5 SDs, which induced spreading depressions of activity, spreading ischemias of up to 190‐second duration (Fig 3A), and spreading suppressions of low‐frequency vascular fluctuations.22 Spreading ischemia concurred with disturbed autoregulation similar to a previous report.29 Accordingly, ptiO2 seemed to follow the fluctuations of mean arterial pressure (MAP). The PTDDD was 102.3 minutes and occurred on day 1.
Figure 3.
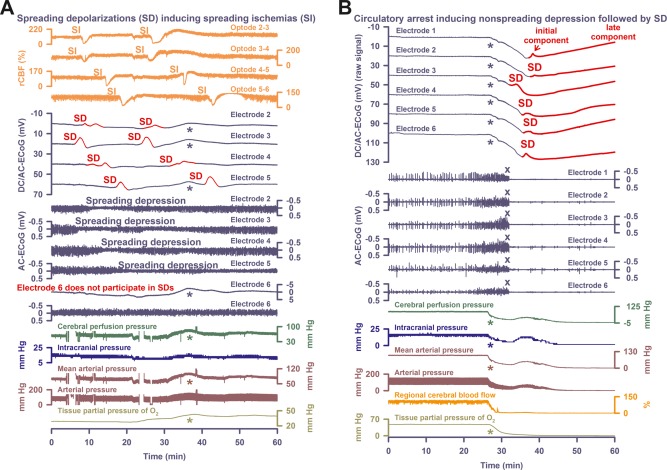
Comparison of spreading depolarization (SD)‐induced spreading depression versus nonspreading depression followed by terminal SD in death. (A) In Patient 2, electrocorticography (ECoG) and laser‐Doppler flowmetry displayed 5 SDs, which induced spreading ischemias (SIs) of up to 190‐second duration, spreading depressions of activity, and spreading suppressions of low‐frequency vascular fluctuations.22 Spreading ischemia concurred with disturbed autoregulation. Accordingly, tissue partial pressure of oxygen (ptiO2) seemed to follow the fluctuations of mean arterial pressure (MAP; cf asterisks). Oxygen is known to interfere with platinum. Accordingly, the fluctuations of ptiO2 are reflected in the direct current (DC) recordings (cf dark blue asterisks). (B) Discontinuation of the life‐sustaining measures caused progressive decline in MAP, regional cerebral blood flow (rCBF), and ptiO2. Nonspreading depression (cf X's) rendered the brain isoelectric within 338 seconds after onset of the decline in MAP or, respectively, 220 seconds after rCBF had fallen to 20% of baseline. A very slow, homogeneous DC positivity (cf Table 2) started simultaneously with the drop in ptiO2 (cf asterisks). Superimposed on this DC positivity, the terminal SD started in electrode 3, 53 seconds after complete cessation of spontaneous activity (transition from dark blue to red DC traces). From electrode 3, terminal SD spread to the other electrodes. Note that the terminal SD shows an initial and a late negative DC component (cf Fig 2B and Table 2). AC = alternating current.
In addition to a large intracerebral hemorrhage (ICH) in the right temporal lobe, magnetic resonance imaging (MRI) on day 1 revealed large infarcts in the territories of both anterior cerebral arteries, the right MCA, and both posterior cerebral arteries, as well as lacunar infarcts in right thalamus and pons. On day 2, a family discussion was held and a DNR‐CC order was activated. The patient was terminally extubated. Figure 3B shows the nonspreading depression of activity during the falling phase of ptiO2. A very slow, homogeneous DC positivity started simultaneously with the drop in ptiO2. Superimposed on this DC positivity, the terminal SD then started at electrode 3.
Patient 3
Patient 3, a 78‐year‐old woman, showed large bifrontal ICH in addition to aSAH. She had 12 SDs on days 2 and 3, and 1 on day 10. One SD was a spreading convulsion.32 The others were classified as spreading depressions. The PTDDD was 144.1 minutes and occurred on day 2. Her past medical history was significant for liver cirrhosis of unknown etiology. During her clinical course, she developed a hepatorenal syndrome with progressive failure of liver, kidneys, lungs, and circulation. A family discussion was held and a DNR‐CC order was activated followed by terminal extubation on day 11. As shown in Figure 4A, the ECoG showed a deviation from the 2 previous cases because the terminal SD started in electrode 3 at 7 minutes before the circulatory arrest. In this moment, spontaneous brain activity was still present. Accordingly, the terminal SD induced spreading depression at electrode 3. From electrode 3, the SD spread to the other electrodes. Finally, nonspreading depression rendered the remaining spontaneous brain activity isoelectric while the terminal SD was still propagating. Presumably, the last SD originated from the rim of the previous ICH. The circulatory arrest then converted this SD into a terminal one.
Figure 4.
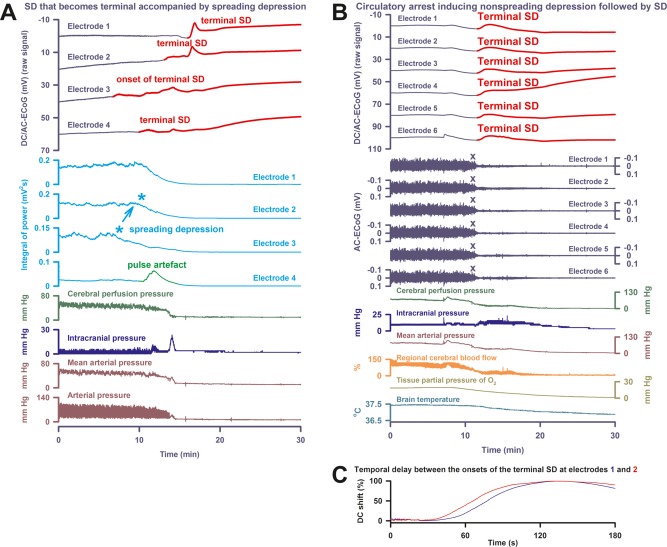
Electrocerebral silence in death can develop in a spreading or nonspreading pattern. (A) In Patient 3, the first direct current (DC) change after discontinuation of the life‐sustaining measures was again a very slow, homogeneous DC positivity (cf Table 2). However, electrocorticography (ECoG) showed a deviation from the 2 previous cases, because terminal spreading depolarization (SD; transition from dark blue to red DC traces) started in electrode 3 at 7 minutes before the circulatory arrest. In this moment, spontaneous brain activity was still present. Accordingly, the terminal SD induced spreading depression at electrode 3 (cf light blue asterisks). From electrode 3, SD spread to the other electrodes. During the SD‐induced spreading depression, a pulse artifact, similar to previous observations,32 developed at electrode 4. Finally, nonspreading depression rendered the remaining spontaneous brain activity isoelectric while the terminal SD was still propagating. (B) Patient 4 suffered from malignant hemispheric stroke. Discontinuation of life‐sustaining measures caused progressive decline in mean arterial pressure, regional cerebral blood flow, and tissue partial pressure of oxygen (ptiO2) followed by nonspreading depression (cf X's) and terminal SD. First DC change was a very slow, homogeneous DC positivity. Similar to Patient 2, it started together with the decline in ptiO2. Note the fall in brain temperature after the circulatory arrest. (C) Patient 4's longest time of terminal SD arrival difference between 2 neighboring electrodes is displayed. AC = alternating current.
Patient 4
Patient 4, a 63‐year‐old man with previous myocardial infarction, hypertension, and diabetes mellitus, suffered from a CT‐proven infarct of the complete right MCA territory. Decompressive hemicraniectomy was performed 37 hours after onset of the left‐sided hemiparesis. On day 4, MRI additionally revealed infarcts in the vascular territories of the left MCA and right posterior inferior cerebellar artery as well as both cerebellar hemispheres. No SDs were recorded before the terminal one. A family discussion was held and a DNR‐CC order was activated followed by terminal extubation on day 5. Figure 4B shows the nonspreading depression of activity during the falling phase of ptiO2. A very slow, homogeneous DC positivity started simultaneously with the drop in ptiO2. Superimposed on this DC positivity, the terminal SD then started at electrode 3.
Patient 5
Patient 5 was a 47‐year‐old male occupant of a car struck by a train with reported field Glasgow Coma Scale (GCS) = 3. The patient was intubated for airway protection and arrived at the trauma center with presentation GCS = 3T. Bilateral chest tubes were placed for traumatic pneumothoraces. Once stabilized, the patient had a postresuscitation GCS = 1T3. CT of the head demonstrated skull vault fractures, diffuse traumatic SAH, subdural hematoma, and diffuse petechial shear injuries. He was admitted to neurocritical care, and an ICP monitor was placed with elevated ICP noted. A family discussion was held on day 2, a DNR‐CC order was activated, and the patient was terminally extubated. Similar to Patients 2 and 4, nonspreading depression occurred while ptiO2 showed the final drop. The terminal SD started 73 seconds after nonspreading depression had rendered the ECoG isoelectric (Fig 5A, B).
Figure 5.
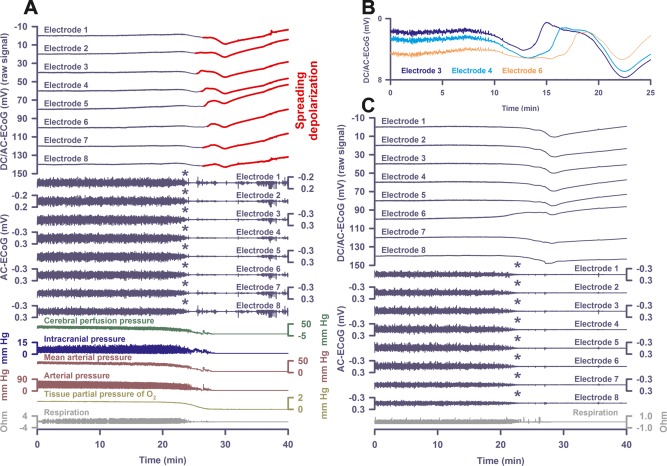
Nonspreading depression followed by terminal spreading depolarization (SD) recorded with intraparenchymal electrode arrays. (A) In Patient 5, similar to Patients 2 and 4, nonspreading depression occurred while tissue partial pressure of oxygen (ptiO2) showed the final drop (cf asterisks). When the electrocorticography (ECoG) became isoelectric, mean arterial pressure had fallen to 18mmHg and ptiO2 to 0.6mmHg. The first direct current (DC) change was a very slow, homogeneous DC positivity. Similar to Patient 2 and 4, it started together with the decline in ptiO2. The terminal SD started superimposed on the homogeneous DC positivity 73 seconds after nonspreading depression had rendered the ECoG isoelectric. (B) shows an overlay of electrodes 3, 4, and 6 from recordings in A, illustrating the time of SD arrival difference of the terminal SD. (C) The end‐of‐life recordings in Patient 6 showed nonspreading depression (cf asterisks) similar to A. Also, the diffuse changes of the DC potential are similar to A. Although the part of the curve where the terminal SD starts may be derived from the comparison with A, occurrence of the terminal SD is more speculative in C, because amplitudes are smaller and spread between electrodes was less obvious. AC = alternating current.
Patient 6
Patient 6 was a 71‐year‐old male pedestrian struck by a vehicle with reported field GCS = 14. The patient was intubated for airway compromise, and arrived at the trauma center with presentation GCS = 3T5. His primary survey revealed polytrauma with gross limb deformities and signs of craniofacial injury. A Focused Assessment with Sonography for Trauma examination was negative; CT of the head revealed diffuse traumatic SAH and a small falcine subdural hematoma. He was found to have coagulopathy by admission laboratory evaluation. After resuscitation and correction of coagulopathy, the patient was admitted to neurocritical care and an ICP monitor was placed. Repeat cranial imaging demonstrated worsening bifrontal contusions and an enlarging left temporal contusion. The patient's neurologic examination declined to GCS = 3T despite aggressive medical management for refractory ICP. A family discussion was held on day 2, at which time a DNR‐CC order was activated. The patient was terminally extubated. The end‐of‐life recordings are shown in Figure 5C.
Patient 7
Patient 7 was a 57‐year‐old male who was found at the base of a stairway. The patient neurologically deteriorated in the field, with decline in the GCS from 13 to 8, was intubated in the emergency department, and was admitted to the neuroscience intensive care unit. CT scans showed hemorrhagic contusions, subdural hematoma, and traumatic SAH. ICP, ptiO2, and rCBF probes, and intraparenchymal depth electrodes were placed in the right frontal lobe and continuous scalp DC/AC‐EEG was monitored using Ag/AgCl‐electrodes. Five SDs were observed on day 2, and elevated ICP at >40mmHg became intractable despite administration of muscle relaxants, pentobarbital, and 3% saline. Per discussions with family, a DNR‐CC order was placed on day 5 and the patient was terminally extubated. Approximately 10 minutes thereafter, a slow, homogeneous DC shift was observed on intraparenchymal DC/AC‐ECoG and scalp DC/AC‐EEG electrodes in synchrony with the decline in oxygen saturation and PtiO2, a final ICP spike, and declining cerebral perfusion and respiratory arrest. Nonspreading depression was observed across intraparenchymal and scalp electrodes during the falling phase of ptiO2. Asystole for 20 seconds began 3 minutes later, only 13 seconds before the start of terminal SD (Fig 6).
Figure 6.
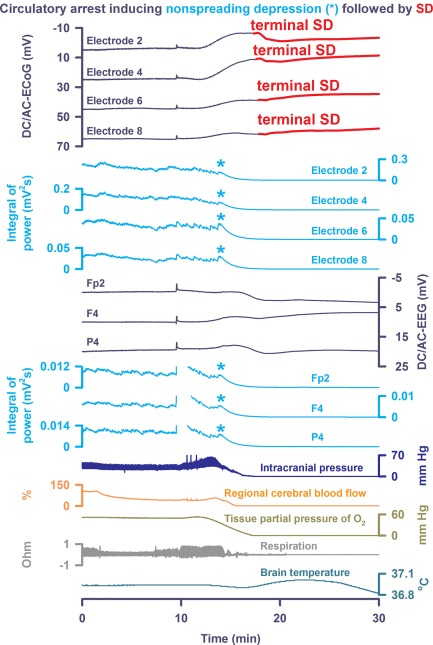
Scalp electroencephalographic (EEG) recordings indicated that nonspreading depression occurs simultaneously not only at intracranial electrodes, but also throughout the hemisphere. In Patient 7, approximately 10 minutes after terminal extubation, a slow, homogeneous direct current (DC) shift was observed on intraparenchymal DC/alternating current (AC)–electrocorticography (ECoG) and scalp DC/AC‐EEG electrodes in synchrony with the decline in oxygen saturation (not shown) and tissue partial pressure of oxygen (ptiO2), a final intracranial pressure spike, and declining cerebral perfusion. Nonspreading depression (cf light blue asterisks) was then observed across intraparenchymal and scalp electrodes during the fall of ptiO2. Asystole for 20 seconds began 3 minutes later (not shown), only 13 seconds before the start of terminal SD. Note the correlate of the terminal spreading depolarization (SD) in the scalp DC/AC‐EEG. Brain temperature was recorded in this case. Note the transient rise in brain temperature during terminal SD followed by decline thereafter.
Nonspreading Depression and Terminal SD in Human Intracranial Recordings following Circulatory Arrest
In general, terminal events were initiated with circulatory arrest after withdrawal of life‐sustaining treatment. The time period from withdrawal of life‐sustaining treatment to complete circulatory arrest was 23 (interquartile range [IQR] = 19–30) minutes (range = 16–101 minutes). In Case 9, the recordings ended with the circulatory arrest, whereas they continued for another 33 (IQR = 21–40) minutes in the remaining 8 patients. Following circulatory arrest, brain temperature dropped at a rate of 0.044 (IQR = 0.032–0.052) °C/min (n = 4; see Figs 4B, 6).
Circulatory arrest caused a simultaneous nonspreading depression of activity across the electrode array in 8 of 9 patients. In 3 patients, scalp EEG recordings confirmed that nonspreading depression occurred simultaneously not only at intracranial electrodes, but also throughout the hemisphere (see Fig 6). At the time of nonspreading depression onset, brain ptiO2 values were 11 (IQR = 7–14) mmHg (n = 6), MAP was 25 (IQR = 23–37) mmHg (n = 5), CPP was 22 (IQR = 19–28) mmHg (n = 5), ICP was 11 (IQR = 6–25) mmHg (n = 7), and rCBF was 16% (IQR = 6–29%) compared to baseline (100%; n = 4). Respiration simultaneously ceased with the nonspreading depression in 3 of 3 cases (see Figs 5, 6). Nonspreading depression was followed by negative DC shifts of terminal SD after delays ranging 13 to 266 seconds (see Table 2; Figs 3B, 4, 5, 6). Patient 8 was an outlier, because an ICP increase to 100mmHg in parallel with a decline in ptiO2 to 6mmHg caused nonspreading depression at a CPP of ∼25mmHg followed by terminal SD >3 hours before extubation and arrest of the systemic circulation. In the 1 case not showing nonspreading depression, electrocerebral silence was induced prior to death by barbiturate medication (Patient 1). However, the patient displayed a terminal SD in the wake of the circulatory arrest (see Fig 1). Patient 9 showed nonspreading depression, but recordings ended before the terminal SD.
After onset of the final drops in perfusion (marked by asterisks in Figure 3B), terminal SDs abruptly started with a delay of 3.9 (IQR = 2.6–6.3) minutes. When terminal SD reached the last electrode, brain ptiO2 values were 2 (IQR = 1–6) mmHg (n = 5), MAP was 15 (IQR = 13–37) mmHg (n = 7), CPP was 13 (IQR = 8–21) mmHg (n = 7), ICP was 9 (IQR = 1–10) mmHg (n = 7), rCBF was 3% (IQR = 2–21%; n = 3), and respiration had ceased (n = 3). Increasingly pulseless electrical activity of the heart could continue for 5.1 (IQR = 0.3–10.6) minutes beyond the onset of terminal SD (n = 6). As explained above, the sequence of events was more complex in Patient 3, because terminal SD started before the final drop in perfusion. The circulatory arrest then developed as the SD spread. As a result, the terminal pattern of electrical silencing was a mixture of spreading and nonspreading depression (see Fig 4A).
DC shifts of terminal SD were usually superimposed on a very slow DC drift that occurred simultaneously on all electrodes. These slow drifts are likely caused by multiple sensitivities of Pt/Ir electrodes and/or generated at the blood–brain barrier (BBB; cf discussion), as they coincided with changes in cerebral variables such as ptiO2 (see Figs 3B, 4B, 5, 6). Simultaneous slow DC drifts were also observed at all electrodes in 3 patients with scalp EEG recordings (see Fig 6).
In animals, the SD extreme in ischemic tissue is characterized by prolonged depolarization durations, in addition to a slow baseline variation termed the negative ultraslow potential (NUP). The NUP is initiated by SDs and similar to the negative DC shifts of prolonged SD, but specifically refers to a negative potential component generated by progressive recruitment of neurons into cell death in the wake of SDs.16 These two potential components of the terminal SD are illustrated, for example, in Figure 2B (depth electrode) and Figure 3B (subdural electrode). The amplitude of the initial component of the terminal SD, representing the actual SD process, was −2.0 (IQR = −1.5 to −2.8) mV (n = 8). The amplitude of the subsequent NUP was −7.8 (IQR = −6.4, −9.3) mV. It should, however, be noted that the recordings usually ended before the NUP had reached its full negativity (see Fig 2B).
In all 4 subdural electrode strip recordings, the spreading nature of the terminal DC shift was evidenced by differences in the time of SD arrival among the electrodes (see Table 2, Figs 1A, 3B, 4A, 4C). With depth electrodes, the spread of DC shifts was evidenced in 2 of 4 cases (see Figs 5A, 5B, 6). In 2 other cases, unequivocal delay between different electrodes was not seen (see Fig 5C).
Discussion
In patients with acute brain injury, subdural ECoG has shown that persistent states of electrical silence in cortex are in many cases induced by SD, because the initial depression occurs in a spreading pattern.22, 23, 24, 33, 34 Thereafter, SDs can repetitively invade the already silent tissue but cannot induce further depression, and are thus termed “isoelectric” SDs.16, 35, 36 However, it was unknown whether a new, abruptly developing hypoxic–ischemic insult in the human brain causes electrocortical silence in the pattern of nonspreading depression, as in animals. It was further unknown whether such a pattern is followed by the depolarization, known as anoxic or terminal SD.15 In patients with malignant hemispheric stroke after MCA occlusion, electrophysiological monitoring begins hours after the initial insult,24, 25 precluding observation of nonspreading depression and anoxic SD in the ischemic core region near the time of ischemic onset. However, a different situation exists in the case of moribund patients when a medical decision is reached to withdraw life‐sustaining therapies, because intracranial electrodes are already in place when circulatory arrest acutely induces severe hypoxia–ischemia. Thus, here we performed continuous patient monitoring following DNR‐CC orders to investigate mechanisms of acute cerebral ischemia and the relative timing of circulatory and cerebral events in the dying process.
Nonspreading Depression of Activity
We found electrophysiological evidence for the occurrence of nonspreading depression in 8 patients undergoing global cerebral ischemia. Subdural electrode strips covered the cortical surface at 6 adjacent locations over a distance of 5cm, whereas intraparenchymal depth arrays were recorded from a single location but extended through cortex as well as white matter. Combining these two approaches, it was possible to demonstrate that the depression of activity following global arrest of the circulation does not spread in either the horizontal or the vertical plane. Thus, the term nonspreading depression accurately describes this phenomenon. Nonspreading depression typically preceded the terminal SD by several tens of seconds, similar to previous observations in animals.7 It has been proposed that the underlying mechanism of nonspreading depression is related to a neuronal or glial oxygen sensor that shuts down neuronal activity once it senses a steep decline in ptiO2.4 Accordingly, we found here in patients with simultaneous recordings of ECoG and ptiO2 that nonspreading depression occurred during a steep decline in ptiO2.
In the case of focal ischemia, the initial SD developing in the electrically silent hypoxic–ischemic zone also propagates through ischemic border zones into normally perfused and spontaneously active tissue against the gradients of oxygen, glucose, and perfusion. Notably, this wave and all SD waves, whether in well‐nourished or severely ischemic tissue, maintain their essential defining features such as the spreading nature, degree of ionic changes, and cytotoxic edema, among others.10, 17 Nonetheless, several characteristics and consequences of SD change depending on local tissue conditions, including duration/persistence of depolarization, cellular toxicity, hemodynamic response, and pharmacology. These variations are described as the SD continuum.10, 17 A critical aspect of the continuum is that SD induces a spreading depression of spontaneous electrical activity in tissue that is healthy or only mildly ischemic, as complete neuronal depolarization precludes the generation of action potentials and postsynaptic potentials. Thus, whether electrical silence of cortical tissue develops in a nonspreading or spreading pattern distinguishes whether the changes are induced by a newly developing insult of severe hypoxia–ischemia, or by SD as a secondary process, respectively.
Terminal SD
The human recordings with subdural electrode strips did not suggest gross differences with animal recordings concerning nature, timing, or propagation of the terminal SD, but a fraction of the time of SD arrival differences between neighboring electrodes was relatively short. This could reflect a multifocal origin, as others have noted that if conditions for SD are very favorable, it may spontaneously originate from several foci practically simultaneously, so that spread is only evident with detailed spatiotemporal mapping provided by neuroimaging techniques.13, 15
In 1 of the patients, terminal SD started before arrest of the systemic circulation. Possibly, this SD originated at the rim of a CT‐proven ICH. The electrophysiological pattern during the dying process is interesting in this patient, because it is analogous to experiments in rabbits by Aristides Leão in which he interrupted the circulation after first triggering an SD that propagated through the tissue with accompanying spreading depression. As a consequence of the circulatory arrest, the negative DC shift became prolonged.7 Our observation here suggests that this constellation can also occur in humans and further strengthens Leão's notion that “in the spreading depression, a change of the same nature as one, resulting from prolonged interruption of the circulation, occurs in the cerebral cortex. The electrical sign of this change is the negative slow voltage variation.”7
Failure of the energy‐dependent recovery under continued oxygen and glucose depletion is the most obvious discriminator between SD in ischemic and normal tissue.37 Accordingly, the negative DC shift of SD becomes one of indefinite duration under anoxia–severe ischemia. However, anoxia‐triggered SD is fully reversible without any signs of cellular damage, if the oxidative substrate supply is reestablished before the so‐called commitment point, defined as the time when neurons start dying under persistent depolarization.3, 4, 38 The delay between SD onset and the commitment point, which can range from a few minutes to >10 minutes, depends critically on the absolute local level of perfusion and temperature, and differs between different types of neurons.2, 39, 40 Terminal SD, as used presently, describes the particular cases of SD when it is confirmed that the commitment point is reached and/or that repolarization never occurs, as in death. Importantly, the reversibility of persistent depolarization and the concept of the commitment point indicate that the onset of the brain's terminal SD does not mark the onset of brain death, but only of the toxic changes eventually leading to death.
Limitations
In patients, the current gold standard for the recording of DC potentials is subdural Pt/Ir plate electrode strips on the brain surface.16 Pt/Ir‐based intraparenchymal depth arrays can also be used for clinical monitoring.41 The advantage of intraparenchymal depth arrays, in contrast to subdural electrode strips, is that they can be placed at the bedside in the intensive care unit. However, a disadvantage is that the positioning of electrode contacts of an intraparenchymal array relative to cortex in gyri and sulci is less certain. For instance, the long SD arrival differences for Patient 5 may reflect an electrode array location along and in close proximity to a sulcal cortex band, where the DC potential is generated. However, often only 1 or 2 contacts are located in gyral cortex, with the others in deeper white matter where SD does not occur. The unfavorable geometry may explain why intraparenchymal depth arrays unequivocally displayed the propagation of the terminal SD in only 2 of 4 patients. In addition, the tissue in proximity to the electrode array had no lesions present in Patient 5, and in animals, terminal SD is the more pronounced the more the tissue is intact until the dying process begins because more cells can be recruited into the depolarization.
Both subdural and intraparenchymal Pt/Ir electrodes show a good safety profile in humans,27, 42 but it should be noted that electrode polarization of platinum causes significant distortion and underestimation of the true amplitude of the negative DC shift.43, 44 Moreover, platinum displays a strong catalytic capacity for many chemical reactions in biological media. Thus, recordings from intracranial platinum electrodes reflect not only the DC potential of SD, but likely also the signals of ptiO2 and pH.22, 30, 45, 46 Such contamination may explain the large, homogeneous DC potential deflections in both gray and white matter in the end‐of‐life recordings. These overlapped with the terminal SD. They seemed to start with the final drop in ptiO2, which in addition is characterized by progressive acidification.46, 47 This timing suggests that they resulted, at least partially, from interferences of ptiO2 and pH with platinum. However, a complementary hypothesis posits that the homogeneous DC potential deflections were in part of biological rather than solely of artificial origin, because the DC potential generated at the BBB is strongly influenced by tissue partial pressure of CO2 (ptiCO2) and O2.48, 49, 50 PtiO2 decreases whereas ptiCO2 increases after terminal extubation, and the appearance of the DC potential deflections would accord with a DC potential generated at the BBB, because they were typically characterized by a homogeneous laminar profile throughout cortex and white matter, in contrast to the zonal gradients observed during functional activation, electrographic seizure activity, or SD.50 In addition, appearance of the homogenous DC potential deflections in the scalp recordings supported their generation at the BBB.49, 50 Although ptiCO2 recordings were not performed here, they would be interesting to study in relation to ICP and SD, especially considering that SDs are known to abolish cerebrovascular reactivity to ptiCO2, which should blunt ptiCO2's effect on ICP.51
TBI patients with intraparenchymal Pt/Ir electrodes and aSAH patients with subdural electrodes showed typical differences such as a preponderance of women in aSAH and of men in TBI, and a higher proportion of patients with SDs after aSAH than after TBI.18 Additionally, the lower number of captured SDs in the TBI patients was presumably due to restricted sampling of intraparenchymal arrays.52 No information was available regarding whether the patients suffered from migraine aura during their lifetime. Migraine with aura could predispose to an earlier onset of terminal SD.53 However, fundamental differences of terminal SD between patients with and without migraine with aura during their lifetime or between patients with and without previous brain injuries are unlikely, because all properly investigated animal species with a somewhat more complex central nervous system, including mammals, reptiles, and insects, display terminal SD during the dying process.15, 54, 55
Conclusion
In this observational study, we found that nonspreading depression and terminal SD occur in patients in the acutely injured brain undergoing global cerebral ischemia in the dying process. These results confirm that the human brain responds to acute severe cerebral ischemia by the same sequence of pathological processes as originally described by Aristides Leão in animals with previously healthy brains.7 In slight deviation from the condition of systemic circulatory arrest reported here, Leão had discovered this in a model of global cerebral ischemia in which he produced sudden ischemia of the rabbit cortex by clamping temporarily the common carotid arteries with the basilar artery occluded at the level of the pons, and the external carotid arteries tied just above their origin. Knowledge of these pathologic mechanisms informs treatment strategies that complement the reestablishment of the systemic circulation, as neuroprotective interventions could be directed at the depolarization wave with the aim of prolonging the survival time. Potential treatment approaches targeting SDs have been discussed elsewhere.10, 11, 56, 57, 58
Because the mere absence of spontaneous EEG activity is ambiguous concerning the onset of cytotoxic cellular processes leading to cortical death, a noninvasive clinical method to diagnose the onset of terminal SD, as well as its reversal upon return of the circulation, may have practical value for recovery prognosis and as an indication/endpoint for resuscitation efforts. It has been shown that the negative DC shifts of SD can be detected with scalp DC‐EEG,33, 59 although important ambiguities remain and further research is needed to develop a reliable technique. In the case of abrupt ischemia, recordings might be particularly complicated by large BBB‐generated DC potentials due to ptiO2 and ptiCO2 changes.49, 50 Nonetheless, results presented here should have immediate relevance to the biological understanding of human brain death,60 which should at least require the occurrence of terminal SD in cerebral gray matter. Our results suggest that this occurs with a certain biological variability after circulatory arrest and cessation of spontaneous brain activity, which may have implications for ethical debates concerning DCD. There is presently no indicator—experimental or clinical—to determine when terminal SD becomes irreversible (ie, when restoration of function is no longer possible), yet the depolarized state after circulatory arrest will not reverse spontaneously in the absence of intervention.
Author Contributions
Study concept and design: J.P.D. and J.A.H. Data acquisition and analysis: all authors. Drafting manuscript and figures: J.P.D., J.A.H.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Additional supporting information can be found in the online version of this article.
Supporting Information 1
Acknowledgment
Supported by grants of the Federal Ministry of Education and Research Center for Stroke Research Berlin (01 EO 0801, J.P.D.), German Research Association (DR 323/5‐1, J.P.D., J.W.; DFG WO 1704/1‐1, J.W.), U.S. Army Congressionally Directed Medical Research Programs Psychological Health and Traumatic Brain Injury Research Program (contract No. W81XWH‐08‐2‐0016, J.A.H.), and Mayfield Education and Research Foundation (J.A.H.).
References
- 1.García JL, Anderson ML. Circulatory disorders and their effect on the brain In: Davis RL, Robertson DM, eds. Textbook of neuropathology. Baltimore, MD: Williams & Wilkins, 1997:715–822. [Google Scholar]
- 2.Pulsinelli WA. Selective neuronal vulnerability: morphological and molecular characteristics. Prog Brain Res 1985;63:29–37. [DOI] [PubMed] [Google Scholar]
- 3.Ayad M, Verity MA, Rubinstein EH. Lidocaine delays cortical ischemic depolarization: relationship to electrophysiologic recovery and neuropathology. J Neurosurg Anesthesiol 1994;6:98–110. [DOI] [PubMed] [Google Scholar]
- 4.Somjen GG. Irreversible hypoxic (ischemic) neuron injury In: Somjen GG, ed. Ions in the brain. New York, NY: Oxford University Press, 2004:338–372. [Google Scholar]
- 5.Norton L, Gibson RM, Gofton T, et al. Electroencephalographic recordings during withdrawal of life‐sustaining therapy until 30 minutes after declaration of death. Can J Neurol Sci 2017;44:139–145. [DOI] [PubMed] [Google Scholar]
- 6.Göbel K, Meuth SG. Blut‐Hirn‐Schranke, Liquor cerebrospinalis, Hirndurchblutung und Hirnstoffwechsel In: Pape HC, Kurtz A, Silbernagl S, eds. Physiologie. 7th ed. Stuttgart: Thieme, 2014:949–963. [Google Scholar]
- 7.Leão AAP. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol 1947;10:409–414. [DOI] [PubMed] [Google Scholar]
- 8.Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci U S A 1996;93:9493–9498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Revah O, Lasser‐Katz E, Fleidervish IA, Gutnick MJ. The earliest neuronal responses to hypoxia in the neocortical circuit are glutamate‐dependent. Neurobiol Dis 2016;95:158–167. [DOI] [PubMed] [Google Scholar]
- 10.Dreier JP, Reiffurth C. The stroke‐migraine depolarization continuum. Neuron 2015;86:902–922. [DOI] [PubMed] [Google Scholar]
- 11.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 2011;17:439–447. [DOI] [PubMed] [Google Scholar]
- 12.Farkas E, Pratt R, Sengpiel F, Obrenovitch TP. Direct, live imaging of cortical spreading depression and anoxic depolarisation using a fluorescent, voltage‐sensitive dye. J Cereb Blood Flow Metab 2008;28:251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jarvis CR, Anderson TR, Andrew RD. Anoxic depolarization mediates acute damage independent of glutamate in neocortical brain slices. Cereb Cortex 2001;11:249–259. [DOI] [PubMed] [Google Scholar]
- 14.Bogdanov VB, Middleton NA, Theriot JJ, et al. Susceptibility of primary sensory cortex to spreading depolarizations. J Neurosci 2016;36:4733–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall WH. Spreading cortical depression of Leao. Physiol Rev 1959;39:239–279. [DOI] [PubMed] [Google Scholar]
- 16.Dreier JP, Fabricius M, Ayata C, et al. Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: review and recommendations of the COSBID research group. J Cereb Blood Flow Metab 2017;37:1595–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartings JA, Shuttleworth CW, Kirov SA, et al. The continuum of spreading depolarizations in acute cortical lesion development: examining Leao's legacy. J Cereb Blood Flow Metab 2017;37:1571–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dreier JP, Lemale CL, Kola V, et al. Spreading depolarization is not an epiphenomenon but the principal mechanism of the cytotoxic edema in various gray matter structures of the brain during stroke. Neuropharmacology doi: 10.1016/j.neuropharm.2017.09.027 (in press). [DOI] [PubMed] [Google Scholar]
- 19.Reich DJ, Mulligan DC, Abt PL, et al. ASTS recommended practice guidelines for controlled donation after cardiac death organ procurement and transplantation. Am J Transplant 2009;9:2004–2011. [DOI] [PubMed] [Google Scholar]
- 20.Shemie SD, Baker AJ, Knoll G, et al. National recommendations for donation after cardiocirculatory death in Canada: donation after cardiocirculatory death in Canada. CMAJ 2006;175:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhanani S, Hornby L, Ward R, Shemie S. Variability in the determination of death after cardiac arrest: a review of guidelines and statements. J Intensive Care Med 2012;27:238–252. [DOI] [PubMed] [Google Scholar]
- 22.Dreier JP, Major S, Manning A, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 2009;132(pt 7):1866–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dreier JP, Woitzik J, Fabricius M, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 2006;129(pt 12):3224–3237. [DOI] [PubMed] [Google Scholar]
- 24.Dohmen C, Sakowitz OW, Fabricius M, et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol 2008;63:720–728. [DOI] [PubMed] [Google Scholar]
- 25.Woitzik J, Hecht N, Pinczolits A, et al. Propagation of cortical spreading depolarization in the human cortex after malignant stroke. Neurology 2013;80:1095–1102. [DOI] [PubMed] [Google Scholar]
- 26.Waziri A, Claassen J, Stuart RM, et al. Intracortical electroencephalography in acute brain injury. Ann Neurol 2009;66:366–377. [DOI] [PubMed] [Google Scholar]
- 27.Drenckhahn C, Windler C, Major S, et al. Complications in aneurysmal subarachnoid hemorrhage patients with and without subdural electrode strip for electrocorticography. J Clin Neurophysiol 2016;33:250–259. [DOI] [PubMed] [Google Scholar]
- 28.Oliveira‐Ferreira AI, Milakara D, Alam M, et al. Experimental and preliminary clinical evidence of an ischemic zone with prolonged negative DC shifts surrounded by a normally perfused tissue belt with persistent electrocorticographic depression. J Cereb Blood Flow Metab 2010;30:1504–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinzman JM, Andaluz N, Shutter LA, et al. Inverse neurovascular coupling to cortical spreading depolarizations in severe brain trauma. Brain 2014;137(pt 11):2960–2972. [DOI] [PubMed] [Google Scholar]
- 30.Bosche B, Graf R, Ernestus RI, et al. Recurrent spreading depolarizations after SAH decrease oxygen availability in human cerebral cortex. Ann Neurol 2010;67:607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winkler MK, Dengler N, Hecht N, et al. Oxygen availability and spreading depolarizations provide complementary prognostic information in neuromonitoring of aneurysmal subarachnoid hemorrhage patients. J Cereb Blood Flow Metab 2017;37:1841–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dreier JP, Major S, Pannek HW, et al. Spreading convulsions, spreading depolarization and epileptogenesis in human cerebral cortex. Brain 2012;135(pt 1):259–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drenckhahn C, Winkler MKL, Major S, et al. Correlates of spreading depolarizations in human scalp electroencephalography. Brain 2012;135(pt 3):853–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hartings JA, Watanabe T, Bullock MR, et al. Spreading depolarizations have prolonged direct current shifts and are associated with poor outcome in brain trauma. Brain 2011;134:1529–1540. [DOI] [PubMed] [Google Scholar]
- 35.Fabricius M, Fuhr S, Bhatia R, et al. Cortical spreading depression and peri‐infarct depolarization in acutely injured human cerebral cortex. Brain 2006;129(pt 3):778–790. [DOI] [PubMed] [Google Scholar]
- 36.Hartings JA, Bullock MR, Okonkwo DO, et al. Spreading depolarisations and outcome after traumatic brain injury: a prospective observational study. Lancet Neurol 2011;10:1058–1064. [DOI] [PubMed] [Google Scholar]
- 37.Milakara D, Grozea C, Dahlem M, et al. Simulation of spreading depolarization trajectories in cerebral cortex: correlation of velocity and susceptibility in patients with aneurysmal subarachnoid hemorrhage. Neuroimage Clin 2017;16:524–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nozari A, Dilekoz E, Sukhotinsky I, et al. Microemboli may link spreading depression, migraine aura, and patent foramen ovale. Ann Neurol 2010;67:221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dreier JP, Victorov IV, Petzold GC, et al. Electrochemical failure of the brain cortex is more deleterious when it is accompanied by low perfusion. Stroke 2013;44:490–496. [DOI] [PubMed] [Google Scholar]
- 40.Heiss WD, Rosner G. Functional recovery of cortical neurons as related to degree and duration of ischemia. Ann Neurol 1983;14:294–301. [DOI] [PubMed] [Google Scholar]
- 41.Jeffcote T, Hinzman JM, Jewell SL, et al. Detection of spreading depolarization with intraparenchymal electrodes in the injured human brain. Neurocrit Care 2014;20:21–31. [DOI] [PubMed] [Google Scholar]
- 42.Stuart RM, Schmidt M, Kurtz P, et al. Intracranial multimodal monitoring for acute brain injury: a single institution review of current practices. Neurocrit Care 2010;12:188–198. [DOI] [PubMed] [Google Scholar]
- 43.Tallgren P, Vanhatalo S, Kaila K, Voipio J. Evaluation of commercially available electrodes and gels for recording of slow EEG potentials. Clin Neurophysiol 2005;116:799–806. [DOI] [PubMed] [Google Scholar]
- 44.Li C, Narayan RK, Wu PM, et al. Evaluation of microelectrode materials for direct‐current electrocorticography. J Neural Eng 2016;13:016008. [DOI] [PubMed] [Google Scholar]
- 45.Windmuller O, Lindauer U, Foddis M, et al. Ion changes in spreading ischaemia induce rat middle cerebral artery constriction in the absence of NO. Brain 2005;128(pt 9):2042–2051. [DOI] [PubMed] [Google Scholar]
- 46.Mutch WA, Hansen AJ. Extracellular pH changes during spreading depression and cerebral ischemia: mechanisms of brain pH regulation. J Cereb Blood Flow Metab 1984;4:17–27. [DOI] [PubMed] [Google Scholar]
- 47.Taylor DL, Obrenovitch TP, Symon L. Changes in extracellular acid‐base homeostasis in cerebral ischemia. Neurochem Res 1996;21:1013–1021. [DOI] [PubMed] [Google Scholar]
- 48.Kang EJ, Major S, Jorks D, et al. Blood‐brain barrier opening to large molecules does not imply blood‐brain barrier opening to small ions. Neurobiol Dis 2013;52:204–218. [DOI] [PubMed] [Google Scholar]
- 49.Voipio J, Tallgren P, Heinonen E, et al. Millivolt‐scale DC shifts in the human scalp EEG: evidence for a nonneuronal generator. J Neurophysiol 2003;89:2208–2214. [DOI] [PubMed] [Google Scholar]
- 50.Lehmenkühler A, Richter F, Poppelmann T. Hypoxia‐ and hypercapnia‐induced DC potential shifts in rat at the scalp and the skull are opposite in polarity to those at the cerebral cortex. Neurosci Lett 1999;270:67–70. [DOI] [PubMed] [Google Scholar]
- 51.Wahl M, Lauritzen M, Schilling L. Change of cerebrovascular reactivity after cortical spreading depression in cats and rats. Brain Res 1987;411:72–80. [DOI] [PubMed] [Google Scholar]
- 52.Hartings JA, Li C, Hinzman JM, et al. Direct current electrocorticography for clinical neuromonitoring of spreading depolarizations. J Cereb Blood Flow Metab 2017;37:1857–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eikermann‐Haerter K, Hyun Lee J, Yuzawa I, et al. Migraine mutations increase stroke vulnerability by facilitating ischemic depolarizations. Circulation 2012;125:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Evans JJ, Xiao C, Robertson RM. AMP‐activated protein kinase protects against anoxia in Drosophila melanogaster. Comp Biochem Physiol A Mol Integr Physiol 2017;214:30–39. [DOI] [PubMed] [Google Scholar]
- 55.Okada YC, Huang JC, Rice ME, et al. Origin of the apparent tissue conductivity in the molecular and granular layers of the in vitro turtle cerebellum and the interpretation of current source‐density analysis. J Neurophysiol 1994;72:742–753. [DOI] [PubMed] [Google Scholar]
- 56.Ayata C, Lauritzen M. Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 2015;95:953–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 2014;15:379–393. [DOI] [PubMed] [Google Scholar]
- 58.Lauritzen M, Strong AJ. ‘Spreading depression of Leao’ and its emerging relevance to acute brain injury in humans. J Cereb Blood Flow Metab 2017;37:1553–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hartings JA, Wilson JA, Hinzman JM, et al. Spreading depression in continuous electroencephalography of brain trauma. Ann Neurol 2014;76:681–694. [DOI] [PubMed] [Google Scholar]
- 60.Shemie SD, Hornby L, Baker A, et al. International guideline development for the determination of death. Intensive Care Med 2014;40:788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information can be found in the online version of this article.
Supporting Information 1