Eradication of spontaneous malignancy by local immunotherapy (original) (raw)
. Author manuscript; available in PMC: 2018 Jun 12.
Published in final edited form as: Sci Transl Med. 2018 Jan 31;10(426):eaan4488. doi: 10.1126/scitranslmed.aan4488
Abstract
It has recently become apparent that the immune system can cure cancer. In some of these strategies the antigen targets are pre-identified and therapies are custom-made against these targets. In others, antibodies are used to remove the brakes of the immune system, allowing pre- existing T cells to attack cancer cells. We have employed another non-customized approach called in situ vaccination. Here, immune enhancing agents are injected locally into one site of tumor, thereby triggering a T cell immune response locally that then attacks cancer throughout the body. We have employed a screening strategy in which the same syngeneic tumor is implanted at two separate sites in the body. One tumor is then injected with the test agents and the resulting immune response is detected by the regression of the distant, untreated tumor. Using this assay the combination of unmethylated CG-enriched oligodeoxynucleotide (CpG) -a TLR9 ligand- and anti-OX40 antibody provided the most impressive results. TLRs are components of the innate immune system that recognize molecular patterns on pathogens. Low doses of CpG injected into a tumor induce the expression of OX40 on CD4+ T cells in the microenvironment in mouse or human tumors. An agonistic anti-OX40 antibody can then trigger a T cell immune response, which is specific to the antigens of the injected tumor. Remarkably, this combination of a TLR ligand and an anti-OX40 antibody can cure multiple types of cancer and prevent spontaneous genetically-driven cancers.
Introduction
T cells that recognize tumor antigens are present in the tumor microenvironment and their activity is modulated through stimulatory and inhibitory receptors. Once cancer is well established, the balance between these inputs is tipped toward immunosuppression (1, 2). The inhibitory signals on T cells are delivered through molecules such as CTLA4 and PD1 by interaction with their respective ligands expressed on cancer cells and/or antigen presenting cells. But these same tumor-reactive T cells express stimulatory receptors including members of the TNFR superfamily. Therefore, many attempts are being made to relieve the negative checkpoints on the anti-tumor immune response and/or to stimulate the activation pathways of the tumor- infiltrating T effector cells.
Here, we conducted a preclinical screen to identify candidate immunostimulatory agents that could trigger a systemic anti-tumor T cell immune response when injected locally into one site of tumor. We found that TLR9 ligands induce the expression of OX40 on CD4 T cells in the tumor microenvironment. OX40 is a costimulatory molecule belonging to the TNFR superfamily, it is expressed on both activated T effector and T regulatory cells. OX40 signaling can promote T effector cell activation as well as inhibit T regulatory cell function.
The addition of an agonistic anti-OX40 antibody then can provide a synergistic stimulus to elicit an anti-tumor immune response that cures distant sites of established tumors. This combination of TLR9 ligand and anti-OX40 antibody can even treat spontaneous breast cancers, overcoming the effect of a powerful oncogene. This in situ vaccine maneuver is safe because it employs low doses of the immunoenhancing agents and practical because the therapy can be applied to many forms of cancer without prior knowledge of their unique tumor antigens.
Results
In-situ vaccination with a TLR 9 ligand induces the expression of OX40 on intra-tumoral CD4 T cells
Toll Like Receptors (TLRs) are known to signal the activation of a variety of cells of the innate and adaptive immune system. To exploit this for cancer therapy, we implanted a tumor subcutaneously into syngeneic mice and after the tumor had become established we injected a CpG oligonucleotide –a ligand for TLR9- into the tumor nodule. We then analyzed the intratumoral T cells for their expression of inhibitory and activation markers. Prior to treatment we observed that OX40 was expressed on CD4 cells in the tumor microenvironment (Fig. 1a, upper panel) and this was restricted mainly to the T-regulatory cells as has been previously reported (3–5) (Fig. 1b, upper panel). Further subsetting of the CD4 T cells demonstrated that OX40 is also expressed to a lesser degree on T effector/memory cells (Fig. 1b, upper panel) as has also been previously reported (6). After intratumoral injection of CpG there was up- regulation of OX40 on CD4 T cells (Fig. 1a, middle panel) mostly among the effector CD4 cells that greatly outnumber the Tregs (Figs. 1a and b, lower panels). This inductive effect was specific to the activating receptor OX40 and did not occur for inhibitory T cell checkpoint targets such as CTLA4 and PD1 (Fig. S1a). Moreover, this OX40 upregulation on CD4 cells also occurred in a patient with follicular lymphoma that had been treated with low dose radiation and intratumoral injection of CpG (Fig. 1c) and in tumor-infiltrating cell populations from lymphoma patients’ samples that were exposed to CpG in vitro (Fig. 1d, Fig. 1e, Fig S2). In these human cases the enhancement of OX40 expression was observed on both effector T cells and in T regulatory cells (Fig. 1d). All of these changes occurred only in the tumor that was injected with CpG and not in the tumor at the untreated site (Fig. S1b).
Fig. 1. CpG induces the expression of OX40 on CD4 T cells.
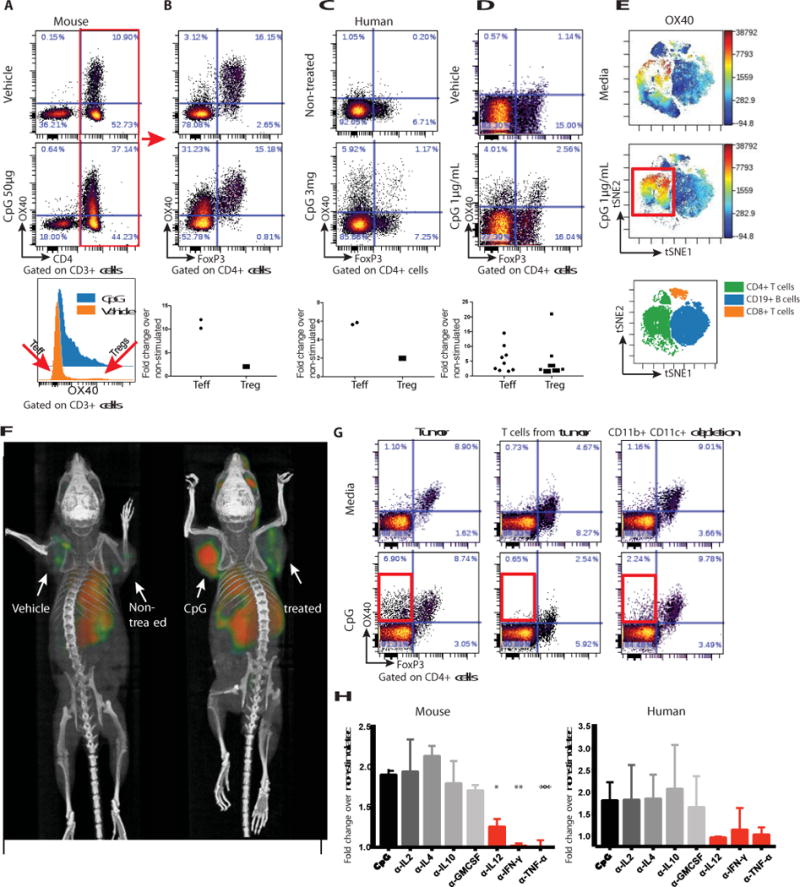
(A) A20 tumor-bearing mice were treated either with vehicle (top) or CpG (middle). 48h later tumors were excised and a single cell suspension was stained and analyzed by flow cytometry. (B) OX40 expression within the CD3+CD4+ subset was separately analyzed for FoxP3 negative (Teff) and positive (Treg) subsets. Fold changes of OX40 positive cells were calculated according to their frequencies in the vehicle vs CpG treatment, n=2. (C) Fine needle aspirates from a CpG injected and non-injected tumors of a follicular lymphoma patient were obtained 22h after treatment. FACS plots of OX40 expression within the CD4+ subset following a 24h rest in media top-non treated tumor, bottom-CpG treated tumor. Top-non-treated lesion, bottom-CpG treated site n=2. (D) Single cell suspensions from biopsy specimens of human lymphoma (5 mantle cell lymphomas, 5 follicular lymphomas) were exposed in vitro to CpG for 48h and analyzed for OX40 expression as in B. (E) CpG-stimulated human lymphoma infiltrating CD4+ T cells, CD8+ T cells and CD19+ B cells were gated, and visualized in t-SNE space using Cytobank software. The viSNE map (bottom) green blue and orange, shows the location of each CD4+, CD19+ and CD8+ cell population respectively. Cells in the viSNE maps were colored according to intensity of OX40 expression. CpG upregulation of OX40 expression on a subset of CD4+ T cells is highlighted by a red box. (F) BALB/c mice were implanted subcutaneously with A20 lymphoma cells (5 × 106) on both the right and left shoulders. When tumors reached between 0.7-1cm in the largest diameter (typically on day 8-9 post inoculation) PBS or CpG (50μg) were injected into one tumor site (left tumor). 16h later 64Cu-DOTA-OX40 was administered intravenously via the tail vein. PET imaging of mice was performed 40h post in- situ treatment. Left image: vehicle treated, right image: CpG treated. These images are representative of 6 mice/group. (G) Fresh A20 tumors were excised from animals (typically 5-6 days after inoculation) and either whole tumors (left), T cells purified from the tumor (middle) or whole tumor depleted of CD11b and CD11c expressing cells (right) were treated for 48h with media (top) or CpG (bottom) and were analyzed for their expression of OX40 by flow cytometry. (H) Left: A20 tumors were excised as in F, Right: Single cell suspensions from biopsy specimens of human follicular lymphoma. Tumors were treated for 48h with media, CpG with or without 1μg/mL of antibodies to IL-2, IL-4, IL-10, GM-CSF, IL-12, IFN-γ, or TNFα and were analyzed for their expression of OX40 by flow cytometry. α-IL-12 *P=0.0144, α-IFN-γ**P=0.0032, α-TNFα**P=0.008 unpaired t-test, either depleting-antibody vs CpG alone.
In vivo imaging of CpG- induced OX-40 Expression
The enhancement of OX40 expression by intratumoral injection of CpG could be visualized in mice by whole body small animal PET (positron emission tomography) imaging after tail-vein administration of an anti-OX40 antibody labeled with 64Cu (Fig. 1f). Remarkably, the systemically injected antibody revealed that OX40 was induced in the microenvironment of the injected tumor, as opposed to a second non-injected tumor site in the same animal. This result indicates that the effect of CpG at this low dose to upregulate OX40 expression is predominately local.
Mechanism of OX40 upregulation on T cells
Purified tumor-infiltrating T cells do not upregulate OX40 when exposed to CpG in vitro (Fig. 1g). The T cells within whole tumor cell populations similarly fail to upregulate OX40 after depletion of macrophages and dendritic cells (Fig. 1g). From these results, we conclude that myeloid-derived cells communicate the CpG signal to T cells. Therefore, we tested for the role of several cytokines in this cellular crosstalk. In human and mice tumors, antibody neutralization of IL-12, interferon (IFN)-γ, and TNF-α each prevented the CpG-induced upregulation of OX40 on T cells in these tumor cell populations (Fig. 1h). In contrast, neutralization of IL-2, IL-4, IL-10 and GMCSF had no effect (Fig. S3).
In situ vaccination with a TLR ligand and anti-OX-40 antibody induces T cell immune responses that cure established cancers
Based on the results above we hypothesized that an agonistic anti-OX40 antibody could augment CpG treatment and help to induce anti-tumor immune responses. To test this hypothesis, we implanted mice with A20 B cell lymphoma tumors at two different sites in the body, allowed the tumors to become established and then injected a TLR agonist together with a checkpoint antibody into only one tumor site (Fig. 2a). The animals were then monitored for tumor growth at both the injected and the distant sites (Fig. 2b). The tumors of vehicle-treated mice grew progressively at both sites. CpG caused complete regression of tumors at the local injected site but had only a slight delay in growth of the distant non-treated tumor. The anti-OX40 antibody alone induced a slight delay in growth of both the treated and non-treated tumors. However, the combination of CpG and anti-OX40 resulted in complete regression of both injected and non- injected tumors. Consistent with the time needed to induce an adaptive T cell response, the kinetics of regression at the two sites was different, with the distant site following the local site by several days (Fig. S1c). Tumor regressions in response to the combined treatment were long- lasting and led to cure of most of the mice (Fig. 2b, lower panel).
Fig. 2. in situ vaccination of CpG in combination with anti-OX40 antibody cures established local and distant tumors.
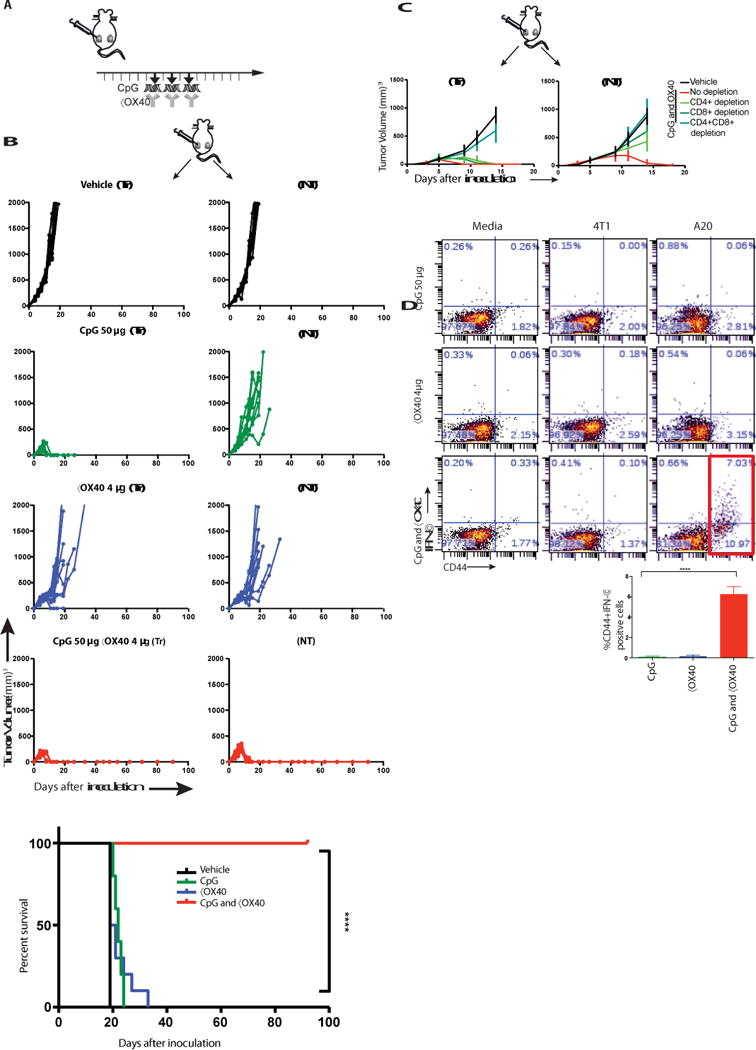
(A) Treatment schema. BALB/c mice were implanted subcutaneously with A20 lymphoma cells (5 × 106) on both the right and left sides of the abdomen. When tumors reached between 0.5- 0.7cm in the largest diameter (typically on day 4-5 post inoculation), αOX40 (4μg) and CpG (50μg) were injected into one tumor site every other day for a total of three doses. Tumors sizes were serially measured by caliper. (B) Tumor growth curves. Left column: treated tumors (Tr), right column: non-treated tumors (NT). Top to bottom: Vehicle, CpG, αX40, CpG and αOX40. Survival plots of the treated mice, (n = 10 mice per group). Shown is one representative experiment out of 9. (C) Effect of CD4/CD8 depletion. Mice were implanted with bilateral tumors and one tumor was injected with CpG and αOX40 antibody according to the schema in A. CD4 (0.5 mg) or CD8 (0.1 mg) depleting antibodies were injected i.p. on days 6, 8, 12, and 15 (n = 10 mice per group). (D) CD8 T cell immune response. Splenocytes from the indicated groups obtained on day 7 post-treatment were co-cultured with either media or with 1 × 106 irradiated 4T1 (unrelated control tumor) or with A20 (homologous tumor) cells for 24h. Intracellular IFN-γ was measured in CD8+ T cells by flow cytometry as a percentage of CD44hi (memory CD8) T cells shown in dot plots and in bar graph summarizing data from three experiments (n = 9 mice per group).
The systemic anti-tumor response required the presence of both CD4+ and CD8+ T cells as mice treated with the corresponding depleting antibodies were unable to control tumor growth (Fig. 2c). Indeed, CD8+ T cells derived from mice treated with both CpG and anti-OX40 antibody responded to tumor cells in vitro as measured by IFN-γ production (Fig. 2d). CD4+ T cells from mice treated by the combination also responded to tumors in vitro, but with a lesser magnitude (Fig. S4). Immediately after CpG and anti-OX40 injection the proportion of the CD4 effector/memory T cell subset increased at the treated site. Twenty-four hours later this subset increased in the spleen and five days later the same occurred at the distant, non-treated site (Fig. S5).
Distant tumors occasionally did recur in mice treated with the effective combination (3/90 mice), and interestingly, these late recurring tumors were sensitive to re-treatment by anti-OX40 and CpG (Fig. S6). An alternative TLR agonist, Resiquimod, (R848) a ligand for TLR7/8, in combination with anti-OX40 induced a similar systemic anti-tumor immune response (Fig. S7a). Anti-OX40 antibody was especially effective compared to other immune checkpoint antibodies, such as anti-PD1, anti-PDL1, (Fig. S7b) that delayed tumor growth in the non-treated site but were not curative.
In-situ vaccination with CpG and anti-OX40 was effective not only against lymphoma, but also against tumors of a variety of histologic types, such as breast carcinoma (4T1), colon cancer (CT26) and melanoma (B16F10) (Fig. S8a-c). In all these tumor models the systemic therapeutic effects were induced by extremely low doses of both the CpG (typically 50μg), and the anti- OX40 antibody (typically 8μg) or even lower (Fig. S9). However, the TLR agonist worked best when it was injected directly into the tumor, consistent with its action to up-regulate the OX40 target in the T cells of the tumor microenvironment. Similar systemic effects were obtained when the OX40 antibody was given systemically, rather than into the tumor, but at higher doses (Fig. S10).
In-situ vaccination protects animals genetically prone to spontaneous breast cancers
Female FVB/N-Tg(MMTV-PyVT) 634 Mul/J mice (also known as PyVT/PyMT) develop highly invasive mammary ductal carcinomas that give rise to a high frequency of lung metastases (7). By 6-7 weeks of age all female carriers develop the first palpable mammary tumor (8), and eventually tumors develop in all of their 10 mammary fat pads. This provided an opportunity for therapeutic intervention in a spontaneous tumor model where the site of tumor development is known, and is accessible for in-situ vaccination.
Young mice were observed and as their first tumor reached 50-75mm3 we injected it with CpG and anti-OX40 antibody (Fig. 3a). In some cases, a second tumor was present at the beginning of therapy, and in these mice with coincident tumors, treatment at a single tumor site with CpG and anti-OX40 led to significant retardation of growth of the contralateral tumor (Fig. 3b) establishing the combination as a therapy for established and disseminating tumors. The injected tumors and the non-injected tumors regressed and, remarkably, the treated mice were protected against the occurrence of independently arising tumors in their other mammary glands (Fig. 3c). The treated mice had significantly lower eventual total tumor burdens (Fig. 3c-d) and developed far fewer lung metastases (Fig. 3e). This in situ vaccination with CpG and anti-OX40 not only caused tumor regression and reduced tumor incidence but also had a major effect on the survival of these cancer-prone mice (Fig. 3f). Following CpG and anti-OX40 treatment these mice developed anti-tumor CD8 T cells in their spleens as indicated by their ability to produce IFNγ when exposed in vitro to autologous tumor cells from the non-injected tumor site (Fig. 3g). These results establish that the anti-tumor immune response was elicited against tumor antigens shared by all the independently arising tumors in these mice, rather than antigens unique to the injected tumor, and accounted for the impressive therapeutic effects seen.
Fig. 3. In-situ vaccination with CpG and anti-OX40 is therapeutic in a spontaneous tumor model.
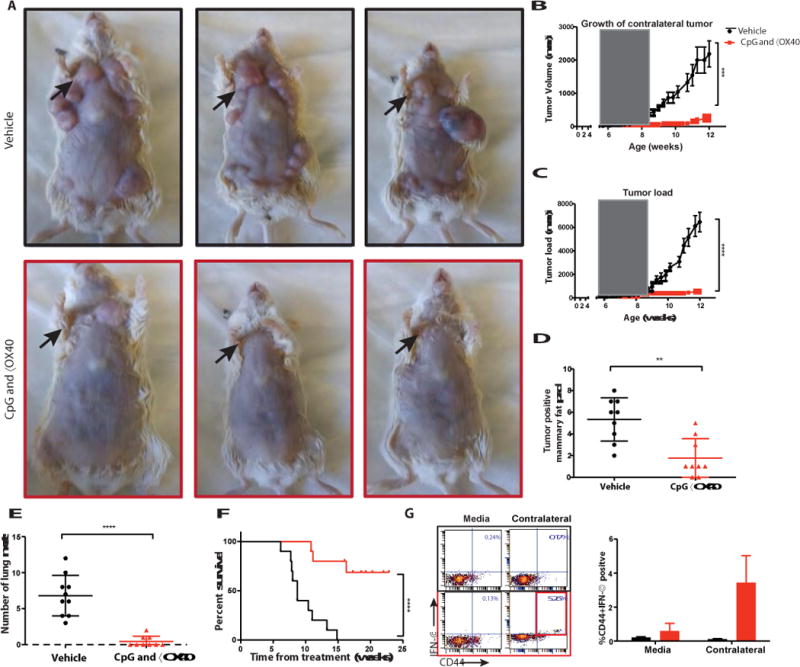
(A) MMTV-PyMT transgenic female mice were injected into the first arising tumor (black arrow) with either vehicle (top) or with CpG and anti-OX40 (bottom); pictures were taken on day 80. (B) CpG and αOX40 decrease the tumor size of a non-treated contralateral tumor. Growth curves representing the volume of a contralateral (untreated) tumor in mice that had 2 palpable tumors at the beginning of treatment. Mice treated by in-situ vaccination (red, n=6) or vehicle (black, n=6) ***P=0.0008, unpaired t-test. (C) CpG and αOX40 decrease the total tumor load. Growth curves represent the sum of the volume of 10 tumors from the different fat pads of each mouse, measured with calipers (n=10 mice per group), the window of treatment is indicated by the grey square, ****P < 0.0001, unpaired t test. (D) Time-matched quantification of the number of tumor positive mammary fat pads. **P=0.011, unpaired t-test (n=9 mice per group). (E) Mice were sacrificed at the age of 80 days, lungs were excised and analyzed ex-vivo for the number of metastases ****P < 0.0001, unpaired t-test (n=10 vehicle group, n=9 CpG and αOX40). (F) Survival plots of the treated mice ****P < 0.0001. Data are means ± s.e.m (n=10 mice per group). (G) CD8 T cell immune response. Splenocytes from the indicated groups obtained on days 7-15 post-treatment were co-cultured for 24h with either media or with 1 × 106 irradiated tumor cells taken from an independent contralateral site on the body. Intracellular IFN- γ was measured in CD8+ T cells by flow cytometry as shown in dot plots and in bar graph summarizing data as a percentage of CD44hi (memory CD8) T cells (n = 3 mice per group).
Therapeutic effect of in-situ vaccination is antigen-specific and triggered at the site of local injection
The results of cross-protection against independently arising tumors in the spontaneous breast cancer model raises the question of antigen specificity. We approached this question using two different tumors that are antigenically distinct. Mice cured by in-situ vaccination of the A20 lymphoma were immune to re-challenge with the same tumor (A20), but not to a different tumor (CT26) (Fig. S11). Conversely, mice cured of the CT26 tumor were immune to re-challenge with CT26 but not with A20. Therefore, these two tumors are antigenically distinct.
To further demonstrate the specificity of the anti-tumor response we implanted tumors into mice at 3 different body sites, two with the A20, and one with CT26 (Fig. 4a). One A20 tumor site was then injected with CpG and anti-OX40 antibody. Both A20 tumors, the injected one and the non-injected one, regressed but the unrelated CT26 tumor continued to grow (Fig. 4a). In a reciprocal experiment, we injected mice with two CT26 tumors and one A20 tumor and treated one CT26 tumor. Once again, only the homologous distant tumors, in this case CT26, regressed but not the unrelated A20 tumor (Fig. 4b). This result confirmed that the immune response induced by the therapy was tumor-specific. Furthermore, it demonstrated that in-situ vaccination with these low doses of agents works by triggering an immune response in the microenvironment of the injected site, rather than by diffusion of the injected agents to systemic sites.
Fig. 4. Immunizing effects of intratumoral CpG and anti-OX40 are local and tumor- specific.
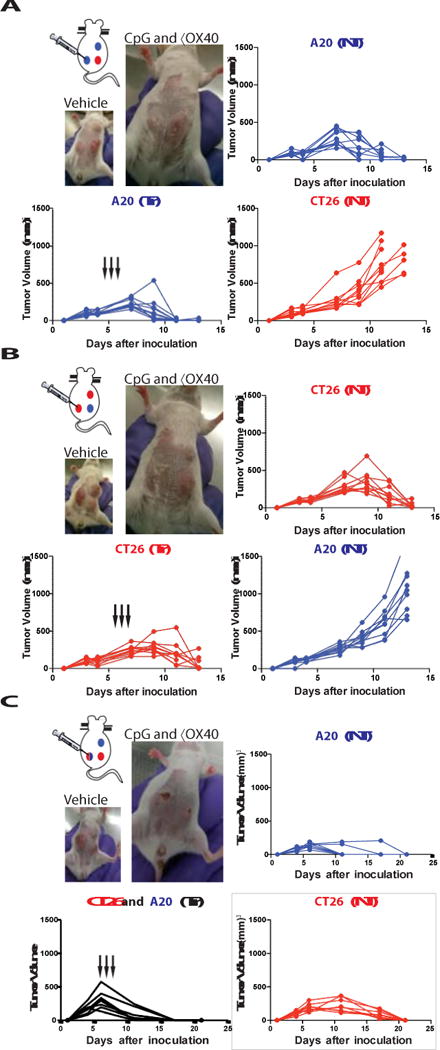
(A) Three tumor model: each mouse was challenged with 3 tumors, 2 of them A20-lymphoma (blue) and one CT26-colon cancer (red). Mice were treated at the indicated times (black arrows). Tumor growth curves of the treated tumor (bottom left), the homologous non-treated A20 tumor (top right) and the heterologous CT26 tumor (bottom right). Photo of a representative mouse at day 11 after tumor challenge from the vehicle treated group (top) and from the group with A20 tumors treated with intratumoral CpG and αOX40 (n = 10 mice per group). (B) Reciprocal three tumor model with two CT26 tumors and one A20 tumor. Treatment was given to one CT26 tumor and growth curves are shown for the treated CT26 tumor site (bottom right), the non- treated homologous CT26 tumor site (top right) and the heterologous A20 tumor (bottom right). Photo of a representative mouse from this experiment (n = 10 mice per group). (C) Mixed three tumor model, each mouse was challenged with: one A20 (blue, top right abdomen) one CT26 (red, bottom right abdomen) and one mixture A20 and CT26 tumor cells (blue and red gradient, left abdomen). Mice were treated only in the mixed tumor at the indicated times (black arrows). Tumor growth curves of the treated tumor (bottom left), the non-treated A20 tumor (top right) and the non-treated CT26 tumor (bottom right). Photo of a representative mouse at day 11 after tumor challenge from the vehicle treated group (top) and on day 17 from the intratumoral CpG and αOX40 (n = 8 mice per group).
Naturally arising tumors can show intra-tumoral antigenic heterogeneity. In order to test whether CpG and anti-OX40 treatment can trigger an immune response against multiple different tumor antigens at the same time, we injected mice with a mixture of A20 and CT26 tumor cells at one site, treated that site with local CpG and anti-OX40 antibody and monitored two additional sites of tumor containing each of the single tumor cells, A20 and CT26, respectively. In situ vaccination of the mixed tumor site simultaneous induced immune responses protective of each of the respective other two pure tumor sites (Fig. 4c). These results demonstrate the power of in situ vaccination to simultaneously immunize against a panoply of different tumor antigens.
Requirement for Fc competency of the anti-OX40 antibody
OX40 is expressed on both intratumoral FoxP3+ regulatory T cells (Tregs) and on activated T effector cells (Teff) (Fig. 1b). The immuno-enhancing activity of the anti-OX40 antibody could therefore be mediated by inhibition/depletion of Tregs or by stimulation of Teff cells, or by a combination of both. We tested the anti-OX40 Treg depletion hypothesis by replacing it with an antibody against folate receptor 4 (FR4), a Treg depleting agent (9) (Fig. S12a). Tregs were partially depleted (43% reduction) by the anti-FR4 in combination with CpG, but no distant therapeutic occurred (Fig. 5a). We investigated this question further using mice genetically engineered to express diphtheria toxin under the FoxP3 promoter (10, 11). Injection of diphtheria toxin led to complete Treg depletion in these mice (Fig. S12b). But when combined with intratumoral CpG no distant therapeutic effect was observed (Fig. 5b). As others have shown, stimulation of Tregs through OX40 can impair their function (4, 12, 13) which we confirm here (Fig. S13a-b). Therefore, we conclude that Treg stimulatory impairment but not depletion is involved in the mechanism of therapeutic synergy with CpG. To dissect the mechanism of these potent therapeutic effects we compared two different forms of the anti- OX40 antibody that differ in their ability to bind to CD16, the activating Fc receptor on NK cells and macrophages. When used in combination with CpG the native, Fc-competent version of anti- OX40 antibody induced systemic anti-tumor immunity, whereas the Fc-mutant version did not (Fig. 5a). We repeated the in situ vaccination experiment in mice deficient in the Fc common γ chain, a component of the activating Fc γ Receptors I, III and IV (14). Once again, in the absence of Fc receptor interaction, this time at the level of the host, the effect of in situ vaccination with CpG and anti-OX40 antibody was lost (Fig. S14). These results could implicate ADCC function of the antibody or alternatively an Fc-dependent agonistic action of the anti- OX40 antibody (15, 16).
Fig. 5. A competent Fc is required for the anti-tumor immune response.
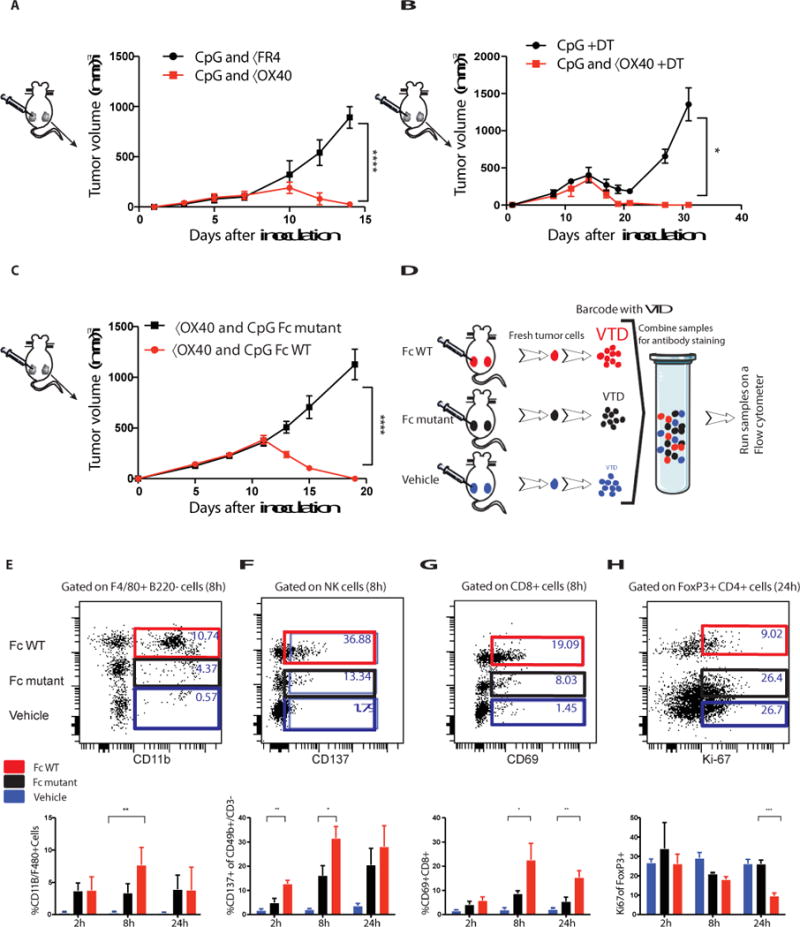
(A, B) Effect of Treg depletion. (A) Tumors were implanted according to the schema in A. Mice were treated with either CpG and anti-FR4 antibody (15μg) or CpG and anti-OX40 as described in A and the NT was measured over time ****P<0.0001 unpaired t test n=10 mice per group. (B) DEREG mice were implanted with B16F10 (0.05 × 106) melanoma cells on both the right and left sides of the abdomen. Diphtheria toxin (DT, 1μg) was injected IP on days 1, 2, 7, 14. CpG or CpG and anti-OX40 were given on days 7, 9 and 11. The NT was measured over time. *P=0.0495 unpaired t test n=4 mice per group. (C) A20 cells were inoculated and treated as described in Fig 2a, tumor volumes were measured following treatment of CpG with either αOX40 rat IgG1 (red) or αOX40 rat IgG1 Fc mutant (black) ****P < 0.0001, unpaired t test (n=10 mice per group). (D) Tumors from control and treated mice were excised at the indicated times after a single treatment, and the cell populations from the different groups were differentially labeled (barcoded) with two different levels of violet tracking dye (VTD) and mixed together, stained, and analyzed as a single sample. (C-F n=3 mice per group). (E-H) Dot plots for single time point and bar graphs for replicates of multiple time points. (E) Number of F4/80 CD11b+ myeloid cells. **P=0.009 8h FcWT vs Vehicle. (F) CD137 expression on NK cells, **P=0.0035 2h, *P=0.0343 8h unpaired t-test FcWT vs Fc mutant. (G) CD69 expression on CD8+ T cells, *P=0.025 8h **P=0.0064 24h, unpaired t-test FcWT vs Fc mutant. (H) Treg cell proliferation ***P=0.0003 24h, unpaired t-test FcWT vs Fc mutant.
Therefore, we examined immune cells in the tumor microenvironment during the early phases of treatment with intratumoral CpG and compared the changes induced with the Fc-competent to those induced by the Fc-mutant version of the anti-OX40 antibody. Early after in situ vaccination, within 24 hours, the tumor infiltrating cell populations from animals treated with Fc-competent or Fc-mutant antibodies were bar coded (17), then pooled and co-stained by a panel of antibodies to identify subsets of immune cells and their activation states (Fig. 5b). The cell populations derived from the different treatment groups were then separately identified by their bar codes. In response to the anti-OX40 antibody with the native Fc, there was an increase in myeloid cell infiltration (Fig. 5c), a cell population important in the cross talk between CpG and the T cells (see above, Fig. 1f-g). NK cells showed an Fc-dependent upregulation of their CD137 activation marker (Fig. 5d). In addition, the Fc-competent but not the Fc-mutant antibody induced activation of a population of CD8 T cells, as indicated by increased CD69 expression (Fig. 5e). Tregs were inhibited in their proliferation by comparison to those exposed to the antibody with the mutated Fc region (Fig. 5f). Neither Tregs nor Teff cells were killed by the Fc- competent antibody (Fig. S15). These early cellular changes occurred only in the tumor microenvironment of the treated site and were not evident at other sites throughout the mouse (Fig. S1b).
These results imply that anti-OX40 antibodies, in conjunction with TLR ligands, can induce therapeutic systemic anti-tumor immune responses by a combination of NK cell activation, Treg inhibition and Teff cell activation, all at the treated tumor site.
Discussion
We have developed a practical strategy for immunotherapy of cancer. It takes advantage of the pre-existing T cell immune repertoire within the tumor microenvironment. The combination of a TLR agonist and an activating antibody against OX40 amplify these anti-tumor T cells and induce their action throughout the body against tumor at non-treated sites. This in situ vaccination does not require knowledge of the tumor antigens. Potential drawbacks include reliance on adequate immune infiltrates and the availability of a suitable injectable site of tumor. After screening a series of immune activators and checkpoint antibodies, we identified the combination of CpG oligonucleotide (TLR9 ligand) and anti-OX40 antibody to be the most potent form of in situ vaccination in multiple mouse models. TLR 7/8 agonists could substitute for CpG, but checkpoint antibodies against PD1, PDL1 or CTLA4 could not substitute for anti- OX40.
The synergistic therapeutic effect between locally injected CpG and anti-OX40 antibodies is explained by the fact that CpG induced the expression of the OX40 target on CD4+ T cells in the tumor microenvironment. CpG also induced OX40 in CD4+ T cells in the tumor microenvironment of human lymphoma tumors and therefore our results are likely to translate to human cancer.
It has been reported that local intratumoral administration of CpG together with systemic antibody against IL-10R leads to rejection of the injected tumor and distant metastases (6, 18). This combination was shown to deflect M2 to M1 macrophages in the tumor microenvironment (19). Therefore, we examined the requirement for induction of OX40 expression on CD4 T cells by CpG in our system. We found that it was dependent on cytokines secreted by myeloid cells, including IL-12, IFN-γ, or TNFα but not IL-2, IL-4, IL-10 nor GMCSF.
The therapeutic effect at the distant sites was specific for antigens expressed by the tumor at the injected site that were shared with the tumor cells at the distant sites. This result established not only the tumor specificity of the immunization but also proved that it was the local effect of the injected agents in the tumor microenvironment rather than their systemic delivery that triggered the systemic anti-tumor immune response.
Autoimmune toxicities are a common complication of systemically administered immune checkpoint antibodies (20–24). By contrast, direct-injection of the antibodies into the tumor at very low doses can avoid these side effects (25, 26). In our experiments, in-situ injection of microgram quantities of immune stimulants and checkpoint antibodies proved to be sufficient to induce the required local immune modulation, resulting in a systemic anti-tumor immune response.
A major challenge in tumor immunotherapy lies in breaking tumor immune tolerance. In a previous report, we showed that depletion of tumor-specific Tregs by the addition of anti CTLA4 antibody was associated with enhanced anti-tumor efficacy (27). But here we find that activating antibody against OX40 is sufficient. It is known that OX40 is expressed on both Treg and Teff cells in the tumor microenvironment and as we now realize, OX40 can be further induced on CD4+ T cells in response to CpG. Modulating both Teff and Tregs is essential to obtain therapeutic effect (28–30). Antibodies to OX40 co-stimulate effector T cells (31–37) and they also inhibit the function of regulatory T cells (13, 27, 38–40).
Having demonstrated the potent therapeutic efficacy of in situ immunotherapy in several different transplanted tumor types, we assessed this form of therapy in a spontaneous arising tumor. The MMTV-PyMT mouse model recapitulates several of the characteristics of virulent human breast cancer, among them: showing histology similarity, having loss of estrogen and progesterone receptors and overexpressing ErbB2/Neu and cyclin D1 (7, 41, 42). Although the tumors within a mouse arise independently in different mammary glands, they all share the expression of the PyMT antigen (43). Injection of CpG and anti-OX40 antibody into the first tumor to occur in each mouse resulted in reduced tumor load in the other mammary fat pads and prevented lung metastases. These results demonstrate the potency of the in situ vaccine maneuver in a situation spontaneous cancer driven by a strong oncogene, suggesting the possibility of a direct application to human cancer. By analogy to the genetically prone mice we can imagine administering an in situ vaccine at the site of the primary tumor prior to surgery in patients at high risk for the occurrence of metastatic disease and/or in patients genetically prone to develop second primary cancers, such as those with inherited mutation in the BRCA genes. The CpG used here, SD-101, is currently being tested in patients as a single agent and in combination with other therapeutic modalities (NCT02927964, NCT02266147, NCT01745354, NCT02254772, NCT02521870). Anti-OX40 antibody is also currently being studied in phase I clinical trials (NCT02559024, NCT01644968, NCT02221960, NCT02318394, NCT02274155, NCT01862900, NCT01303705, NCT02205333). The results from our current preclinical studies provide strong rationale for combining CpG with agonistic anti-OX40 antibodies in a therapeutic format of in situ vaccination in patients with lymphoma and solid tumors. As we have shown, CpG and anti-OX40 antibodies work locally at very low doses that should provide the advantage of avoiding toxicities that occur with their systemic administration.
Materials and Methods
Study Design
Our objective was to develop a new immunotherapy for cancer by using the tumor itself as a source of antigen, of immune reactive cells and as a site for injecting immune activating agents- in situ vaccination. Our general strategy was to implant the same syngeneic tumor at two separate sites in the body of mice. One tumor was then injected with the test agents and tumor size was measured in both the treated and non-treated sites. Using this assay for abscopal therapeutic effects we identified the combination of unmethylated CG-enriched oligodeoxynucleotide (CpG) -a TLR9 ligand- and an agonistic antibody against OX40 as the most promising immunostimulatory regimen.
Since transplanted syngeneic tumor models lack certain aspects of naturally occurring tumors, we also studied the effects of our combination in a spontaneous model of breast cancer. This model is driven by the Polyoma middle T oncogene under the control of the MMTV promoter and the female mice develop independently arising breast cancers in all of their mammary glands between 5-14 weeks of age.
We observed the mice and when their first breast cancer tumor arose, we injected it with our combination of CpG and anti-OX40 antibody. Each mouse was then monitored for the regression of simultaneously present second tumors, the occurrence of newly arising tumors, for metastatic disease in their lungs and for their survival. The data were analyzed by Kaplan Meier curves with events scored as time to reach 2 cm in the largest diameter at which time the mouse was sacrificed. The data were analyzed using the Log-Rank test.
We studied the mechanism of the therapy, by examining the requirement for T cells and their subsets, including Treg cells and by the requirement for Fc function of the anti-OX40 antibody. These requirements were tested by depleting specific T cells and by substituting a Fc mutant for the native anti-OX40 antibody.
In all therapy experiments, to ensure similar tumor sizes in all treatment groups, mice were randomized only after tumors were established. To ensure statistical power experimental groups were typically composed of 10 animals each. For each experiment mice numbers, statistical tests and numbers of experimental replicates are described in the figure legends. Data include all outliers. Investigators were not blinded during evaluation of the in vivo experiments. Raw data for all therapy experiments are provided in the Supplementary Materials (Table S1).
Reagents
CpG SD101 was provided by Dynavax Technologies. Anti-mouse CD8a, clone 2.43; and anti- mouse CD4, clone GK1.5, antibodies were purchased from BioXcell. Anti-OX40 (CD134) mAb (rat IgG1, clone OX86; European Collection of Cell Cultures), isotype control rat hybridoma, SFR8-B6 (ATCC HB-152) were produced as ascites in SCID mice by Bionexus. Fc silent Anti- OX40 (CD134) mAb (Absolute Antibody).
The following monoclonal antibodies (mAbs) were used for flow cytometry: CD4-PerCP Cy5.5, CD3-PerCP Cy5.5 or BV786, CD4-BV650, CD8a-FITC or APC H7, CD44-APC, IFN-gamma- PE, B220-PerCP Cy5.5, CD49b-PE CY7, CD69- BV605, CD137-PE and ICOS (CD278)-PE Cy7, FoxP3-PE and Ki-67-BV711. These antibodies and their isotype controls were purchased from either BD Biosciences, Biolegend or eBioscience.
Cell lines and mice
A20 B cell lymphoma, B16-F10 melanoma, and CT26 colon carcinoma line were obtained from ATCC (Manassas, VA), 4T1-Luc breast carcinoma cell line was a gift from the S. Strober laboratory and the C. Contag laboratory (both at Stanford University). Tumor cells were cultured in complete medium (RPMI 1640 DMEM- for B16-F10; Cellgro) containing 10% fetal bovine serum (FBS;HyClone), 100 U/mL penicillin, 100μg/mL streptomycin, and 50μM 2-ME (Gibco). Cell lines were routinely tested for mycoplasma contamination.
Six to eight-week-old female BALB/c and C57BL/6 were purchased from Charles River (http://www.criver.com). FVB/N-Tg(MMTV-PyVT)634Mul/J male FVB/NJ females, C57BL/6- Tg(Foxp3-DTR/EGFP)23.2Spar also known as DEREG mice were purchased from JAX Laboratories (http://jaxmice.jax.org/). Mice were housed in the Laboratory Animal Facility of the Stanford University Medical Center (Stanford, CA). All experiments were approved by the Stanford administrative panel on laboratory animal care and conducted in accordance with Stanford University animal facility guidelines.
Tumor inoculation and animal studies
A20, CT26, 4T1 and B16-F10 tumor cells (5 × 106, 0.5 × 106, 0.01 × 106 and 0.05 × 106 respectively) were injected subcutaneously at sites on both right and left of the abdomen. When tumors size reached 0.5-0.7 cm in the largest diameter, mice were randomized to the experimental groups. CpG and anti-OX40 were injected into the tumor only on the right side of the animals in a volume of 50μL. Tumors size was monitored on both sides of the animals with a digital caliper (Mitutoyo) every 2 to 3 days and expressed as volume (length × width × height). Mice were sacrificed when tumor size reached 1.5cm in the largest diameter as per guidelines.
All mice that developed tumors on both sides of the abdomen were included in the experiments. The investigator was not blinded to the group allocation during the experiment and/or when assessing the outcome.
4T1 tumor challenged mice were analyzed for lung metastasis by injecting India ink through the trachea after euthanasia. Lungs were then excised, washed once in water and fixed in Fekete’s solution (100 ml 70% alcohol, 10 mL formalin, and 5 mL glacial acetic acid) at room temperature. Surface metastases subsequently appeared as white nodules at the surface of black lungs and were counted under a microscope.
DEREG mice were implanted with B16-F10 tumor cells as described above. Diphtheria toxin (Sigma-Aldrich) DT, 1μg was injected IP on days 1, 2, 7, 14 following tumor implantations. CpG or CpG and anti-OX40 were given on days 7, 9 and 11 following tumor implantations.
Flow cytometry
Cells were surface stained in phosphate-buffered saline [PBS], 1% BSA, and 0.01% sodium azide, fixed in 2% paraformaldehyde, and analyzed by flow cytometry on an FACSCalibur or LSRII (BD Biosciences). Data were stored and analyzed using Cytobank (www.cytobank.org).
Multiplex flow cytometry- Fluorescent cell barcoding
Excised tumors from mice treated with an Fc-competent OX40 antibody, an Fc-silent OX40 antibody (Absolute Antibody). or saline were mechanically processed into single cell suspensions and barcoded using 3 different concentrations of CellTrace Violet Proliferation reagent (Life Techologies) ranging from 5 – 0.1μM. Once barcoded, equal numbers of cells from each group were combined and stained with LIVE/DEAD Fixable Green Dead Cell Stain (Life Technologies) followed by antibodies against surface antigens to determine populations of interest such as tumor (B220), T cells (CD3, CD4, CD8) and NK cells (CD56) as well as those to look at activation (CD69, CD137 and ICOS). Cells were then fixed and permeabilized (eBioscience, Inc.) stained for FoxP3 (Tregs) and Ki-67 (proliferation) and were analyzed by flow cytometry.
IFNγ production assay
Single cell suspensions were made from spleens of treated mice (on day 7 post-treatment), red cells were lysed with ammonium chloride, potassium buffer (Quality Biological). Splenocytes were then co-cultured with 1×106 irradiated A20 or 4T1 cells for 24 hours at 37oC and 5% CO2 in the presence of 0.5 μg anti-mouse CD28mAb (BD PharMingen). Monensin (Golgistop; BD Biosciences) was added for the last 5-6 hours. Intracellular IFNγ expression was assessed using BD Cytofix/Cytoperm Plus Kit per manufacturer’s instructions.
Depletion of CD4 and CD8 T cells
Anti-CD4 (GK1.5 clone- rat IgG2b) and anti-CD8 (2.43 clone- rat IgG2b) mAbs (BioXcell) were injected 2 days and 1 day before therapy, on the day therapy was begun, and at 5, 8, and 19 days after beginning of therapy, at a dose of 0.5 or 0.1 mg per injection for CD4 and CD8 respectively. The depletion conditions were validated by flow cytometry of blood showing specific depletion of more than 95% of each respective cell subset.
In-vitro assessment of OX40 expression
Fresh tumor cells were excised from mice, processed into single-cell suspensions and incubated for 48h with 1μg/mL CpG. Cells were then stained for the surface antigens CD3, CD4, CD8 and OX40. They were then fixed and permeabilized using reagents from eBioscience, followed by FoxP3staining.
T cells isolation, and depletion of T cells and CD11b and CD11c expressing cells from tumors used Miltenyi Biotec kits.
Tetramer staining
PE-conjugated H-2Ld tetramer to peptide SPSYVYHQF (MuLV env gp70 423–431) was purchased from Proimmune, PE-conjugated H-2Ld tetramer to peptide IASNENMETMESSTLE (influenza nucleoprotein 365-380) was a gift from the M. Davis lab (Stanford University). Antibodies were used at 5 μg/ml, tetramer staining was performed in FACS buffer for 10 min at room temperature and followed by a surface staining on ice for 20 min.
Activation and suppression assay
T cell activation assay
C57/BL6 splenocytes were isolated, VTD labeled and incubated in the presence of anti-CD3 antibody (0.05μg/mL) for 72h with or without anti-OX40 antibodies. T cell activation and proliferation were determined by VTD dilution and the expression of the activation marker CD69.
Treg suppression assay
To determine the impact of OX40 antibodies on Treg activity, violet tracking dye (VTD) labeled splenic cells were co-cultured with OX40-KO Tregs (OX40 WT splenocytes:OX40 KO Treg = 1:1). Cells were co-cultured in 96 well plate in the presence of anti-CD3 and anti-CD28 beads (ThermoFisher) for 96h with or without anti-OX40 antibody (1μg/mL). Proliferation of the WT labeled Teff cells was measured by flow cytometry and calculated by VTD dilution. Tregs were isolated using kits from Miltenyi Biotec.
PET imaging
PET imaging of mice was performed using the microPET/CT hybrid scanner (Inveon, Siemens). PET images were reconstructed using 2 iterations of 3-dimensional ordered subsets expectation maximization algorithm (12 subsets) and 18 iterations of the accelerated version of 3D-MAP (i.e., FASTMAP) – matrix size of 128 × 128 × 159. CT images were acquired just before each PET scan to enable attenuation correction of the PET data set and provided an anatomic reference for the PET image. Mice were anesthetized using isoflurane gas (2.0%–3.0% for induction and 2.0%–2.5% for maintenance). 64Cu-DOTA-OX40 (80-110μCi radiochemical purity of 99% as determined by TLC and SA, specific activity is 185 MBq/mg) was administered intravenously via the tail vein 16 hours after CpG and vehicle intratumoral injections. Static PET scans (10 min) were acquired 16 hours after intravenous administration of 64Cu-DOTA-OX40 (40 hours post intra-tumoral injections). Once reconstructed using a three-dimensional ordered subsets expectation maximization (3DOSEM) algorithm, PET images were co-registered with CT images to generate figures using the Inveon Research Workplace (IRW) image analysis software (version 4.0; Siemens).
Upregulation of OX40 on CD4 T cells infiltrating Human B cell Tumors
Tumor samples from patients with Follicular and Mantle Cell B cell lymphoma who were part of ongoing clinical trials (Stanford IRB protocols IRB-31224, IRB-36750 and IRB-5089) were available for in vitro analysis.
Single-cell suspensions were incubated for either 24 or 48 hours in RPMI media containing 5% fetal bovine serum (HyClone), 100 U/mL penicillin, 100μg/mL streptomycin, and then stained for T cell surface antigens including CD3, CD4, CD8 and OX40. They were also fixed and permeabilized using reagents from eBioscience and then stained for FoxP3. For in vitro stimulation studies CpG at a concentration of 1ug/ml was added to the media.
For response of tumor infiltrating cells to CpG in vivo a sample was obtained from a site of tumor that had been injected with CpG (3mg) 24h prior and compared to a site of tumor that had not been injected. Both of these sites shared as part of the clinical protocol an exposure to low dose radiation (2Gy on each of two successive days). Single cell suspensions were rested in media with no further exposure to CpG for 24h prior to analysis of OX40 expression by flow cytometry.
Statistical analysis
Prism software (GraphPad) was used to analyze tumor growth and to determine statistical significance of differences between groups by applying an unpaired Student’s t-test. P values <0.05 were considered significant. The Kaplan-Meier method was employed for survival analysis, P values were calculated using the log-rank test (Mantel-Cox).
Supplementary Material
Supplemental
Fig. S1. In-situ vaccination with a TLR 9 ligand induces the local expression of OX40 but not of PD-1 or CTLA-4.
Fig. S2. CpG induces the expression of OX40 on CD4 T cells.
Fig. S3. Cytokines are playing a role in the CpG T cell crosstalk.
Fig. S4. Intracellular IFNγ production of CD4+ cells.
Fig. S5. Frequency of T cell subsets.
Fig. S6. Tumor recurrence is sensitive to treatment with anti-OX40 and CpG.
Fig. S7. Resiquimod (R848) in combination with anti-OX40 and anti-PD1/PD-L1 in combination with CpG.
Fig. S8. In-situ vaccination with CpG and anti-OX40 is effective against breast carcinoma, colon cancer and melanoma.
Fig. S9. Dose de-escalation of CpG and αOX40 antibody.
Fig. S10. Systemic administration of anti-αOX40 antibody.
Fig. S11. Long-term memory in cured mice.
Fig. S12. Confirmation of Treg depletion from the tumor.
Fig. S13. Anti- OX40 antibody stimulates Teff cells and inhibits function of Tregs.
Fig. S14. Requirement for Fc competency of the anti-OX40 Antibody.
Fig. S15. Neither Tregs nor Teff are depleted by anti-OX40 antibody.
Table S1
Table S1. Primary data
One Sentence Summary.
In-situ vaccination with low doses of TLR ligands and anti-OX40 antibodies can cure widespread cancers in pre-clinical models.
Acknowledgments
Funding: Supported by grants from the NIH R01 CA188005, and the Leukemia and Lymphoma Society, TAP- 120921, and by the Boaz and Varda Dotan and the Phil N. Allen foundations.
Footnotes
Author contributions: I.SB, D.C, I.SL and A.TM designed and performed all experiments, I.SB, SL, S.SB and RL wrote the paper.
Materials availability: CpG SD101 is available from Dynavax under a material transfer agreement.
Competing interest: Dr Gambhir is founder and equity holder of CellSIght Inc. that develops and translates multimodality strategies for imaging cell trafficking/transplantation. Other authors declare no potential conflicts of interest.
References and Notes
- 1.Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, Lu Z, Fein S, Schoenberger S, Levitsky HI. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of CD40. Nature medicine. 1999;5:780–787. doi: 10.1038/10503. [DOI] [PubMed] [Google Scholar]
- 2.Staveley-O’Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, Pardoll D, Levitsky H. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc Natl Acad Sci U S A. 1998;95:1178–1183. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and antigen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J Immunol. 2007;179:1427–1430. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 4.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Li XC. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Burocchi A, Pittoni P, Gorzanelli A, Colombo MP, Piconese S. Intratumor OX40 stimulation inhibits IRF1 expression and IL-10 production by Treg cells while enhancing CD40L expression by effector memory T cells. Eur J Immunol. 2011;41:3615–3626. doi: 10.1002/eji.201141700. [DOI] [PubMed] [Google Scholar]
- 7.Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ, Pollard JW. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. The American journal of pathology. 2003;163:2113–2126. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi T, Hirota K, Nagahama K, Ohkawa K, Takahashi T, Nomura T, Sakaguchi S. Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity. 2007;27:145–159. doi: 10.1016/j.immuni.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 10.Lahl K, Sparwasser T. In vivo depletion of FoxP3+ Tregs using the DEREG mouse model. Methods Mol Biol. 2011;707:157–172. doi: 10.1007/978-1-61737-979-6_10. [DOI] [PubMed] [Google Scholar]
- 11.Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. The Journal of experimental medicine. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Voo KS, Bover L, Harline ML, Vien LT, Facchinetti V, Arima K, Kwak LW, Liu YJ. Antibodies targeting human OX40 expand effector T cells and block inducible and natural regulatory T cell function. J Immunol. 2013;191:3641–3650. doi: 10.4049/jimmunol.1202752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol Cell Biol. 2014;92:475–480. doi: 10.1038/icb.2014.26. [DOI] [PubMed] [Google Scholar]
- 14.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nature reviews Immunology. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 15.Li F, Ravetch JV. Inhibitory Fcgamma receptor engagement drives adjuvant and anti- tumor activities of agonistic CD40 antibodies. Science. 2011;333:1030–1034. doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang D, Goldberg MV, Chiu ML. Fc Engineering Approaches to Enhance the Agonism and Effector Functions of an Anti-OX40 Antibody. The Journal of biological chemistry. 2016;291:27134–27146. doi: 10.1074/jbc.M116.757773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krutzik PO, Nolan GP. Fluorescent cell barcoding in flow cytometry allows high- throughput drug screening and signaling profiling. Nature methods. 2006;3:361–368. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- 18.Vicari AP, Chiodoni C, Vaure C, Ait-Yahia S, Dercamp C, Matsos F, Reynard O, Taverne C, Merle P, Colombo MP, O’Garra A, Trinchieri G, Caux C. Reversal of tumor- induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti- interleukin 10 receptor antibody. The Journal of experimental medicine. 2002;196:541–549. doi: 10.1084/jem.20020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65:3437–3446. doi: 10.1158/0008-5472.CAN-04-4262. [DOI] [PubMed] [Google Scholar]
- 20.Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, Powderly JD, Heist RS, Carvajal RD, Jackman DM, Sequist LV, Smith DC, Leming P, Carbone DP, Pinder-Schenck MC, Topalian SL, Hodi FS, Sosman JA, Sznol M, McDermott DF, Pardoll DM, Sankar V, Ahlers CM, Salvati M, Wigginton JM, Hellmann MD, Kollia GD, Gupta AK, Brahmer JR. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33:2004–2012. doi: 10.1200/JCO.2014.58.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DC, Brahmer JR, Carvajal RD, Hammers HJ, Puzanov I, Hodi FS, Kluger HM, Topalian SL, Pardoll DM, Wigginton JM, Kollia GD, Gupta A, McDonald D, Sankar V, Sosman JA, Atkins MB. Survival, Durable Response, and Long-Term Safety in Patients With Previously Treated Advanced Renal Cell Carcinoma Receiving Nivolumab. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33:2013–2020. doi: 10.1200/JCO.2014.58.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez-Rodriguez E, Rodriguez-Abreu D, I.-B. Spanish Group for Cancer Immune Checkpoint Inhibitors: Review and Management of Endocrine Adverse Events. The oncologist. 2016 doi: 10.1634/theoncologist.2015-0509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kottschade L, Brys A, Peikert T, Ryder M, Raffals L, Brewer J, Mosca P, Markovic S, Midwest Melanoma P. A multidisciplinary approach to toxicity management of modern immune checkpoint inhibitors in cancer therapy. Melanoma research. 2016 doi: 10.1097/CMR.0000000000000273. [DOI] [PubMed] [Google Scholar]
- 24.Sgambato A, Casaluce F, Sacco PC, Palazzolo G, Maione P, Rossi A, Ciardiello F, Gridelli C. Anti PD-1 and PDL-1 Immunotherapy in the Treatment of Advanced Non- Small Cell Lung Cancer (NSCLC): A Review on Toxicity Profile and its Management. Current drug safety. 2016;11:62–68. doi: 10.2174/1574886311207040289. [DOI] [PubMed] [Google Scholar]
- 25.Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:5381–5389. doi: 10.1158/1078-0432.CCR-12-0781. [DOI] [PubMed] [Google Scholar]
- 26.Marabelle A, Kohrt H, Levy R. Intratumoral anti-CTLA-4 therapy: enhancing efficacy while avoiding toxicity. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:5261–5263. doi: 10.1158/1078-0432.CCR-13-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, Luong R, Rosenblum MD, Steinman L, Levitsky HI, Tse V, Levy R. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. The Journal of clinical investigation. 2013;123:2447–2463. doi: 10.1172/JCI64859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. The Journal of experimental medicine. 2008;205:825–839. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.James Ziai GF, Zhang Liping, Kozlowski Cleopatra, Gilbert Houston, Smith Jacquelyn, Kowanetz Marcin, Kim Jeong M, Huseni Mahrukh. Prevalence analysis of OX40- positive cell populations in solid tumors. Journal for ImmunoTherapy of Cancer. 2015;3 [Google Scholar]
- 30.Mallett S, Fossum S, Barclay AN. Characterization of the MRC OX40 antigen of activated CD4 positive T lymphocytes–a molecule related to nerve growth factor receptor. EMBO J. 1990;9:1063–1068. doi: 10.1002/j.1460-2075.1990.tb08211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co- stimulatory molecule OX40. Nature reviews Immunology. 2004;4:420–431. doi: 10.1038/nri1371. [DOI] [PubMed] [Google Scholar]
- 32.Triplett TA, Tucker CG, Triplett KC, Alderman Z, Sun L, Ling LE, Akporiaye ET, Weinberg AD. STAT3 Signaling Is Required for Optimal Regression of Large Established Tumors in Mice Treated with Anti-OX40 and TGFbeta Receptor Blockade. Cancer immunology research. 2015;3:526–535. doi: 10.1158/2326-6066.CIR-14-0187. [DOI] [PubMed] [Google Scholar]
- 33.Murphy KA, Lechner MG, Popescu FE, Bedi J, Decker SA, Hu P, Erickson JR, O’Sullivan MG, Swier L, Salazar AM, Olin MR, Epstein AL, Ohlfest JR. An in vivo immunotherapy screen of costimulatory molecules identifies Fc-OX40L as a potent reagent for the treatment of established murine gliomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:4657–4668. doi: 10.1158/1078-0432.CCR-12-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murata S, Ladle BH, Kim PS, Lutz ER, Wolpoe ME, Ivie SE, Smith HM, Armstrong TD, Emens LA, Jaffee EM, Reilly RT. OX40 costimulation synergizes with GM-CSF whole-cell vaccination to overcome established CD8+ T cell tolerance to an endogenous tumor antigen. J Immunol. 2006;176:974–983. doi: 10.4049/jimmunol.176.2.974. [DOI] [PubMed] [Google Scholar]
- 35.Curti A, Parenza M, Colombo MP. Autologous and MHC class I-negative allogeneic tumor cells secreting IL-12 together cure disseminated A20 lymphoma. Blood. 2003;101:568–575. doi: 10.1182/blood-2002-03-0991. [DOI] [PubMed] [Google Scholar]
- 36.Song A, Song J, Tang X, Croft M. Cooperation between CD4 and CD8 T cells for anti- tumor activity is enhanced by OX40 signals. Eur J Immunol. 2007;37:1224–1232. doi: 10.1002/eji.200636957. [DOI] [PubMed] [Google Scholar]
- 37.Hirschhorn-Cymerman D, Budhu S, Kitano S, Liu C, Zhao F, Zhong H, Lesokhin AM, Avogadri-Connors F, Yuan J, Li Y, Houghton AN, Merghoub T, Wolchok JD. Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. The Journal of experimental medicine. 2012;209:2113–2126. doi: 10.1084/jem.20120532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res. 2008;68:5206–5215. doi: 10.1158/0008-5472.CAN-07-6484. [DOI] [PubMed] [Google Scholar]
- 39.Kitamura N, Murata S, Ueki T, Mekata E, Reilly RT, Jaffee EM, Tani T. OX40 costimulation can abrogate Foxp3+ regulatory T cell-mediated suppression of antitumor immunity. International journal of cancer. 2009;125:630–638. doi: 10.1002/ijc.24435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105:2845–2851. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- 41.Lapidus RG, Nass SJ, Davidson NE. The loss of estrogen and progesterone receptor gene expression in human breast cancer. Journal of mammary gland biology and neoplasia. 1998;3:85–94. doi: 10.1023/a:1018778403001. [DOI] [PubMed] [Google Scholar]
- 42.Menard S, Tagliabue E, Campiglio M, Pupa SM. Role of HER2 gene overexpression in breast carcinoma. Journal of cellular physiology. 2000;182:150–162. doi: 10.1002/(SICI)1097-4652(200002)182:2<150::AID-JCP3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 43.Maglione JE, Moghanaki D, Young LJ, Manner CK, Ellies LG, Joseph SO, Nicholson B, Cardiff RD, MacLeod CL. Transgenic Polyoma middle-T mice model premalignant mammary disease. Cancer Res. 2001;61:8298–8305. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental
Fig. S1. In-situ vaccination with a TLR 9 ligand induces the local expression of OX40 but not of PD-1 or CTLA-4.
Fig. S2. CpG induces the expression of OX40 on CD4 T cells.
Fig. S3. Cytokines are playing a role in the CpG T cell crosstalk.
Fig. S4. Intracellular IFNγ production of CD4+ cells.
Fig. S5. Frequency of T cell subsets.
Fig. S6. Tumor recurrence is sensitive to treatment with anti-OX40 and CpG.
Fig. S7. Resiquimod (R848) in combination with anti-OX40 and anti-PD1/PD-L1 in combination with CpG.
Fig. S8. In-situ vaccination with CpG and anti-OX40 is effective against breast carcinoma, colon cancer and melanoma.
Fig. S9. Dose de-escalation of CpG and αOX40 antibody.
Fig. S10. Systemic administration of anti-αOX40 antibody.
Fig. S11. Long-term memory in cured mice.
Fig. S12. Confirmation of Treg depletion from the tumor.
Fig. S13. Anti- OX40 antibody stimulates Teff cells and inhibits function of Tregs.
Fig. S14. Requirement for Fc competency of the anti-OX40 Antibody.
Fig. S15. Neither Tregs nor Teff are depleted by anti-OX40 antibody.
Table S1
Table S1. Primary data