Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer (original) (raw)
. Author manuscript; available in PMC: 2018 Jul 9.
Published in final edited form as: Nat Immunol. 2017 Jun 19;18(8):940–950. doi: 10.1038/ni.3775
Abstract
Therapies that boost the anti-tumor responses of cytotoxic T lymphocytes (CTLs) have shown promise; however, clinical responses to the immunotherapeutic agents currently available vary considerably, and the molecular basis of this is unclear. We performed transcriptomic profiling of tumor-infiltrating CTLs from treatment-naive patients with lung cancer to define the molecular features associated with the robustness of anti-tumor immune responses. We observed considerable heterogeneity in the expression of molecules associated with activation of the T cell antigen receptor (TCR) and of immunological-checkpoint molecules such as 4-1BB, PD-1 and TIM-3. Tumors with a high density of CTLs showed enrichment for transcripts linked to tissue-resident memory cells (TRM cells), such as CD103, and CTLs from CD103hi tumors displayed features of enhanced cytotoxicity. A greater density of TRM cells in tumors was predictive of a better survival outcome in lung cancer, and this effect was independent of that conferred by CTL density. Here we define the ‘molecular fingerprint’ of tumor-infiltrating CTLs and identify potentially new targets for immunotherapy.
Immunotherapy is rapidly gaining its place as a standard treatment for solid tumors1,2, including lung cancer3. Nonetheless, only ~30% of patients benefit from this approach4. Much remains to be learned about how immunotherapy works and how to choose the right treatment or combination of treatments for a particular patient. Understanding the mechanisms and molecular basis of effective anti-tumor immune responses will be essential for the development of novel immunotherapeutic agents for those patients who do not respond to the immunotherapies now available.
Immunotherapy is thought to enhance the antitumor responses of cytotoxic T lymphocytes (CTLs); i.e., CD8+ T cells that infiltrate into the tumor5. Indeed, a high density of tumor-infiltrating lymphocytes (TILs) is predictive of a good prognosis in a wide range of cancers6,7. However, it remains unclear why the degree of infiltration by TILs varies substantially even between people with the same cancer. It is also unknown whether there are merely quantitative differences in TILs or whether qualitative differences also exist in TILs from tumors with a high TIL density that might contribute to the superior outcome seen in patients with such tumors. Understanding of the TIL transcriptome and the molecular basis of TIL heterogeneity could not only lead to novel biomarkers for patient stratification for therapy but also identify novel immunological pathways to be targeted by future immunotherapeutic strategies.
So far, transcriptional studies of CD8+ T cells from patients with cancer have analyzed cells in peripheral blood or metastatic sites8–11. The precise state of the activation, differentiation and function of CD8+ T cells within primary tumors is poorly understood; however, this must be a key reference point from which to begin elucidating the biology of immunological attack at the time of diagnosis, during tumor progression and after intervention with immunotherapy. To fully characterize the molecular nature of immune responses at the tumor site, we have taken an unbiased approach to define the global transcriptional profile of purified CD8+ TILs from well- characterized cohorts of patients with two epithelial cancers: non– small-cell lung cancer (NSCLC), and head-and-neck squamous-cell cancer (HNSCC).
RESULTS
Major transcriptional changes characterize CD8+ TILs
To identify the core transcriptional signature of CD8+ TILs, we used RNA-based next-generation sequencing (RNA-Seq) to analyze purified populations of CD8+ T cells present in tumor samples (CD8+ TILs) from patients (n = 36) with treatment-naive early-stage NSCLC (Supplementary Fig. 1a and Supplementary Tables 1 and 2). We also generated matched transcriptional profiles of CD8+ T cells isolated from the adjacent non-tumor lung tissue (CD8+ N-TILs) to discriminate features linked to lung-tissue residence from those related to tumor infiltration. To assess conservation of the transcriptional program of CD8+ TILs in a related solid tumor of epithelial origin, we used a similar data set generated from patients (n = 41) with HNSCC from both human papilloma virus–positive (virus-driven) subtypes and human papilloma virus–negative subtypes.
We identified a large number of transcripts (n = 1,403) that were expressed differentially by CD8+ TILs relative to their expression by CD8+ N-TILs (Fig. 1a and Supplementary Table 3), which suggested major changes in the transcriptional landscape of CD8+ TILs in lung tumor tissue. The expression of such lung-cancer ‘CD8+ TIL–associated transcripts’ did not differ according to histological subtype (Supplementary Fig. 1b). Principal-component analysis and hierarchical clustering also showed that CD8+ TILs from both subtypes of lung cancer mostly clustered together, distinct from the CD8+ N-TILs (Fig. 1b and Supplementary Fig. 1c,d). Notably, that set of lung-cancer ‘CD8+ TIL–associated transcripts’ was expressed similarly by CD8+ TILs in both subtypes of HNSCC (Fig. 1a and Supplementary Fig. 1b), which also clustered together with CD8+ TILs from lung cancer (Fig. 1b and Supplementary Fig. 1c,d); this indicated a conserved TIL transcriptome for these two tumor types.
Figure 1.
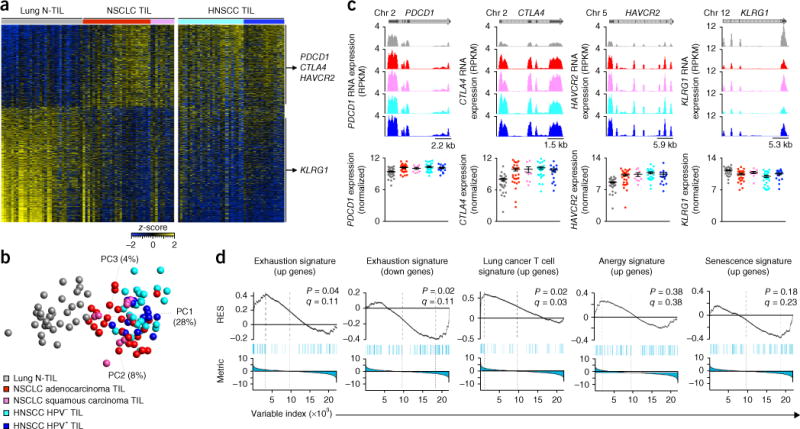
Core transcriptional profile of CD8+ TILs. (a) RNA-Seq analysis of genes (one per row) expressed differentially by lung CD8+ N-TILs (left; n = 32 donors) versus NSCLC CD8+ TILs (middle and right; n = 36 donors) (pairwise comparison; change in expression of 1.5-fold with an adjusted P value of <0.05 (DESeq2 analysis; Benjamini-Hochberg test)), presented as row-wise _z_-scores of normalized read counts in CD8+ TILs from donors with NSCLC adenocarcinoma (red) or squamous carcinoma (pink) or HNSCC negative (light blue) or positive (dark blue) for human papilloma virus (HNSCC TIL; n = 41 donors); each column represents an individual sample; right margin, genes encoding exhaustion-associated molecules (vertical lines group genes upregulated (top) or downregulated (bottom) in NSCLC CD8+ TILs relative to their expression in lung CD8+ N-TILs). (b) Principal-component analysis of CD8+ T cell core transcriptomes (symbols) in N-TILs and TILs as in a (key); numbers along perimeter indicate principal components (PC1–PC3), and numbers in parentheses indicate percent variance for each. HPV, human papilloma virus. (c) RNA-Seq analysis of genes encoding exhaustion-associated molecules (as in a) in N-TILs and TILs (key in b), presented as reads per kilobase per million (RPKM) mapped as University of California Santa Cruz genome browser tracks (top) or as a summary of the results (bottom; log2 normalized counts). Each symbol (bottom) represents an individual sample; small horizontal lines indicate the mean (± s.e.m.). Above plots, position of exons (including untranslated regions) (dark grey) and introns (light grey) in each gene, as well as the chromosome (Chr) on which the gene is present. (d) GSEA of various gene sets (above plots) in the transcriptome of CD8+ TILs versus that of CD8+ N-TILs from donors with NSCLC, presented as the running enrichment score (RES) for the gene set as the analysis ‘walks down’ the ranked list of genes (reflective of the degree to which the gene set is over-represented at the top or bottom of the ranked list of genes) (top), the position of the gene-set members (blue vertical lines) in the ranked list of genes (middle), and the value of the ranking metric (bottom). P values, Kolmogorov-Smirnov test. Data are from one experiment with n = 32 donors (lung N-TILs), n = 36 donors (NSCLC TILs) and n = 41 donors (HNSCC TILs).
Features associated with inhibited function, anergy and senescence of T cells have been described for TILs12–14. Gene-set–enrichment analysis (GSEA) revealed significantly higher expression of genes encoding molecules linked to the so-called ‘exhaustion stage’, such as PDCD1 (which encodes the immunological-checkpoint molecule PD-1), CTLA4 (which encodes the immunomodulatory receptor CTLA-4 (CD152)) and HAVCR2 (which encodes the cell-surface marker TIM-3) in CD8+ TILs than in CD8+ N-TILs, while CD8+ TILs did not have higher expression of genes encoding molecules associated with T cell anergy and senescence (Fig. 1c,d). Our data set also showed higher expression of T cell–associated genes derived from The Cancer Genome Atlas (TCGA) of lung cancer15 in CD8+ TILs than in CD8+ N-TILs (Fig. 1d). Together these findings suggested that our strategy for ‘micro-scaled’ RNA-Seq analysis of freshly purified ex vivo CD8+ TILs and CD8+ N-TILs reliably identified transcripts previously linked to TILs.
Cell-cycle and TCR-activation pathways in CD8+ TILs
To gain broad insight into the functional relevance of the CD8+ TIL transcriptional program, we performed gene-pathway analysis. Notably, we observed significant enrichment in TILs for transcripts from overlapping sets of genes encoding products involved in cell-cycle control, mitosis, DNA replication and signaling via the pathways of the tumor suppressor p53, the cyclin-dependent kinase ATM and the kinase PLK, relative to the abundance of those transcripts in N-TILs (Fig. 2a–c and Supplementary Table 4); this indicated that the TIL populations (tumors) showed enrichment for proliferating CD8+ T cells. Furthermore, we observed enrichment in CD8+ TILs, relative to their abundance in N-TILs, for transcripts encoding components of canonical pathways involved in antigen-specific T cell activation, especially the pathway mediated by 4-1BB (encoded by TNFRSF9; called ‘_4-1BB_’ here) and the CD27 co-stimulatory pathway, which are activated following the engagement of TCRs and their co-stimulation by antigen-presenting cells, respectively16,17 (Fig. 2a,d). The higher expression of 4-1BB in CD8+ TILs than in N-TILs was confirmed at the protein level by flow cytometry (Fig. 2e). Together these data suggested that the engagement TCRs and their co-stimulation, presumably provided by antigen-presenting cells expressing tumor-associated antigens (TAAs), were probably involved in the antigen-specific activation and proliferation of CD8+ TILs, which indicated that the tumor milieu sustained the clonal expansion of presumed TAA-specific CD8+ T cells. That suggestion was further supported by analysis of the TCR repertoire, which indicated significantly greater clonal expansion of CD8+ TILs than of N-TILs (Fig. 2f and Supplementary Table 5).
Figure 2.
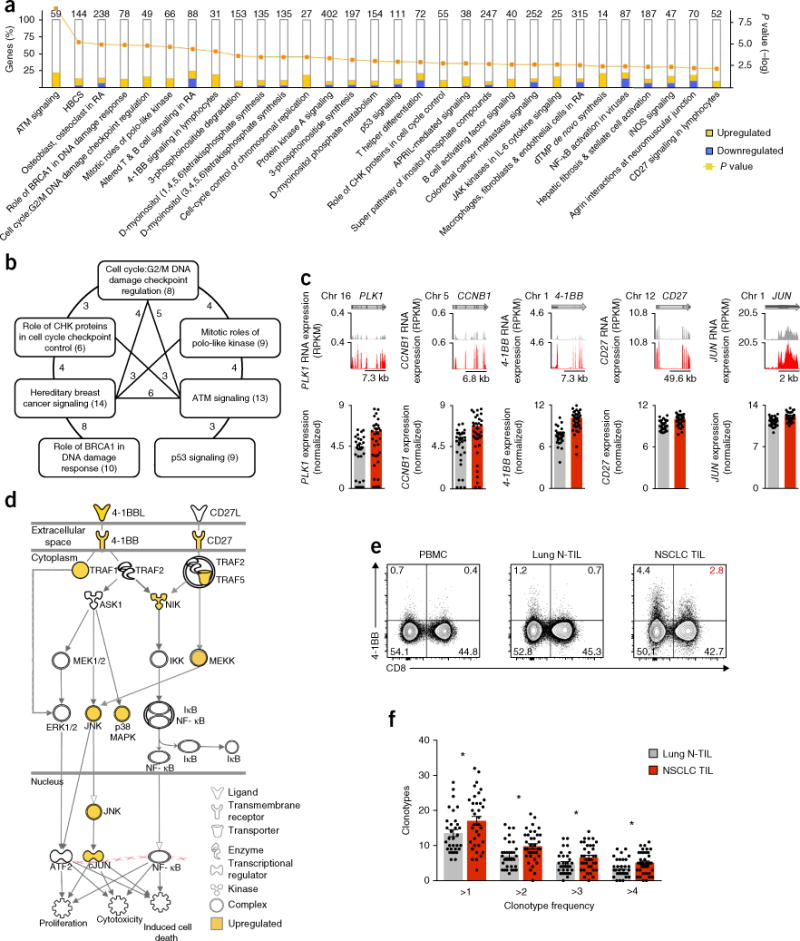
Pathways for which CD8+ TILs show enrichment. (a) Analysis of canonical pathways from the Ingenuity pathway analysis database (horizontal axis; bars in plot) for which CD8+ TILs show enrichment, presented as the frequency of differentially expressed genes encoding components of each pathway that are upregulated or downregulated (key) in CD8+ TILs relative to their expression in CD8+ N-TILs (left vertical axis), and adjusted P values (right vertical axis; line; Fisher’s exact test); numbers above bars indicate total genes in each pathway. HBCS, hereditary breast cancer signaling; BRCA, tumor suppressor; RA, rheumatoid arthritis; CHK, checkpoint kinase; APRIL, proliferation-inducing ligand; dTMP, deoxythymidine monophosphate; NF-κB, transcription factor; iNOS, inducible nitric oxide synthase. (b) Overlap of genes encoding components of the cell-cycle and proliferation pathways in CD8+ TILs and in CD8+ N-TILs: numbers in parentheses indicate total genes in each pathway; numbers along lines indicate total genes shared by the pathways connected by the line. (c) RNA-Seq analysis of PLK1 (encoding the serine-threonine kinase PLK1), CCNB1 (encoding cyclin B1), 4-1BB, CD27 and JUN (encoding the transcription factor c-Jun) in lung N-TILs and NSCLC TILs (key in f) (presented as in Fig. 1c). (d) Ingenuity pathway analysis of genes upregulated in CD8+ TILs relative to their expression in N-TILs (yellow), encoding components of the canonical 4-1BB and CD27 signaling pathways (shape indicates function (key)) in lymphocytes. (e) Flow-cytometry analysis of the surface expression of 4-1BB and CD8 on live and singlet-gated CD45+CD3+ T cells obtained from peripheral blood mononuclear cells (PBMC), lung N-TILs and NSCLC TILs (above plots) from the same patient. Numbers in quadrants indicate percent cells in each throughout; red indicates percent cells among TILs throughout. (f) Quantification of clonotypes (average values) among CD8+ N-TILs and NSCLC CD8+ TILs (key) according to their frequency in each donor (horizontal axis), derived from RNA-Seq analysis of genes encoding TCR β-chains. Each symbol (c,f) represents an individual sample; small horizontal lines indicate the mean (± s.e.m.). *P < 0.05 (unpaired Student’s two-tailed _t_-test). Data are from one experiment (a–d,f) or are representative of six experiments (e).
Heterogeneity in targets of immunotherapy
The considerable success of immunological-checkpoint blockers, such as agents directed against PD-1 and CTLA-4, in humans and in model organisms4,18 suggests that CD8+ TILs with features of TCR engagement and strong co-stimulation probably mount robust anti-tumor immune responses. However, the response to such treatment is highly variable and is limited to a minority of patients. We reasoned that the molecular profile of CD8+ TILs might explain the inter-person variability in the response to agents directed against PD1 or CTLA-4 and might also reveal alternative immune-system-evasion mechanisms beyond PD-1- and CTLA-4-based pathways. Therefore, we assessed the expression of a spectrum of potential targets of immunotherapy to determine the extent of molecular heterogeneity in CD8+ TILs. We observed substantial variability in the expression of transcripts encoding PD-1 and other potential targets of immunotherapy by CD8+ TILs from patients with lung cancer (Fig. 3a,b) or HNSCC (Supplementary Fig. 2). We confirmed PD-1 expression at the protein level and showed that the abundance of PDCD1 transcripts correlated with the average number of PD-1-expressing cells in the tumors (Supplementary Fig. 3a,b). We also found varying combinations of expression of co-inhibitory molecules; for example, CD8+ TILs from some patients with lung cancer had upregulation of transcripts encoding four targets of immunotherapy (PD-1, TIM-3, LAG-3 and CTLA-4) relative to the expression of those transcripts by other patients, while some patients showed upregulation of expression of three or two molecules or even a single molecule (Fig. 3a,b). The high molecular resolution and breadth of our data suggested that baseline transcriptional profiling of tumor-infiltrating CD8+ T cells might guide the selection of appropriate immunotherapies for each patient and the development of biomarkers that can be used to predict the clinical response to checkpoint blockade with monotherapy or combination therapies.
Figure 3.
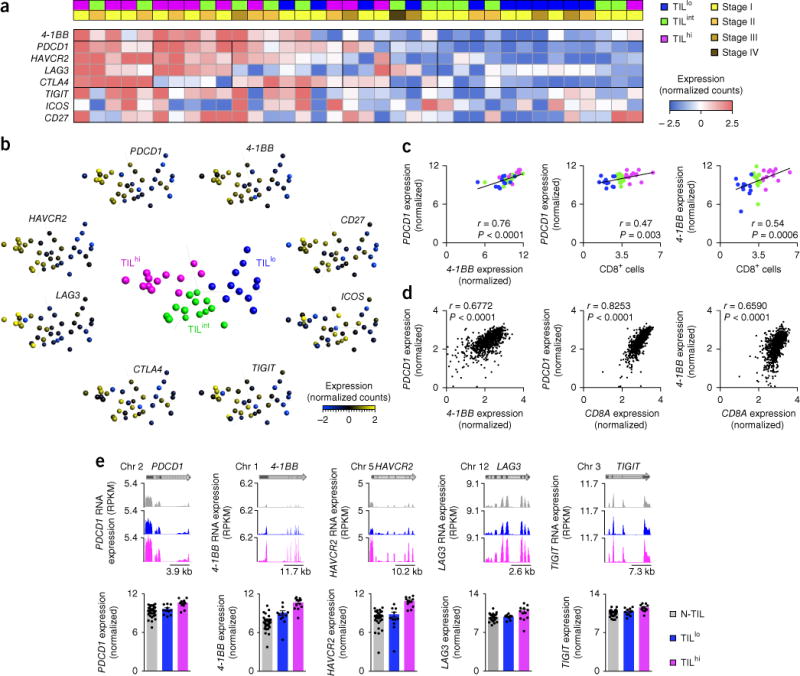
Heterogeneity among targets of immunotherapy. (a) RNA-Seq analysis (row-wise normalized counts; bottom key) of various transcripts (left margin; one per row) in CD8+ TILs from patients with NSCLC (one per column); above, CD8+ TIL density (top row; top left key) and tumor stage (bottom row; top right key) for each patient. (b) Principal-component analysis of CD8+ TILs from patients with NSCLC with TILlo, TILint or TILhi tumors (middle plot), and expression (key) of the transcripts in a in CD8+ TILs (plots along perimeter). Each symbol represents an individual patient. (c) Correlation of the expression of PDCD1 transcripts and that of 4-1BB transcripts (log2 normalized counts) in NSCLC CD8+ TILs (left), and correlation of the expression of PDCD1 transcripts (middle) or 4-1BB transcripts (right) and the number of tumor-infiltrating CD8+ cells (quantified by immunohistochemistry). Each symbol represents an individual patient (colors match those in b, middle). (d) Correlation of the expression of PDCD1 transcripts and that of 4-1BB transcripts (left), and correlation of the expression of PDCD1 transcripts (middle) or 4-1BB transcripts (right) and that of CD8A transcripts in the TCGA lung cancer RNA-Seq data set (extreme outliers are not presented here). Each symbol represents an individual patient (n = 1,013). (e) RNA-Seq analysis of PDCD1, 4-1BB, HAVCR2, LAG3 and TIGIT in N-TILS and TILs from TILhi or TILlo tumors (key) (presented as in Fig. 1c). r values (c,d) indicate the Spearman correlation coefficient; P values (c,d), Spearman correlation. Data are from one experiment.
PDCD1 expression correlates with TIL density
The considerable heterogeneity observed in the abundance of PDCD1 transcripts led us to investigate factors linked to PDCD1 expression in CD8+ TILs. Despite the perceived role of PD-1 as a negative regulatory immunological checkpoint, it serves as a marker for clonally expanded, antigen-specific T cells capable of lysing autologous tumor cells19,20. Furthermore, we found a strong positive correlation between the expression of PDCD1 and that of 4-1BB (Fig. 3c), which is expressed following engagement of the TCR and is thus a marker of antigen-specific T cells16,17,21. The heterogeneity in the expression of these surrogate markers for antigen specificity suggested that not all tumors contained similar numbers of tumor-reactive CD8+ TILs. Hence, we sought to determine which factors might influence the enrichment for PDCD1- or _4-1BB_-expressing CD8+ TILs; i.e., cells we presumed to be TAA specific. We found no correlation between the abundance of PDCD1 or 4-1BB transcripts and clinical or pathological characteristics such as patient age, sex, histological subtype, stage of disease, performance status (the ability to perform activities of daily living without assistance) or smoking status (Supplementary Fig. 3c). However, there was a positive correlation between the abundance of each of those transcripts and the average number of CD8+ TILs that infiltrated each tumor sample (Fig. 3c). A similar correlation was also observed between the abundance of each of those transcripts and CD8A transcripts (encoding the co-receptor CD8α) in lung-tumor samples from the TCGA RNA-Seq data set (Fig. 3d).
In addition to their higher expression of PDCD1 and 4-1BB, tumors with a high density of TILs (‘TILhi’ tumors; tumors were classified as TILhi, TILint and TILlo on the basis of the average number of CD8+ T cells that infiltrated the tumors; Supplementary Fig. 4) also had higher expression of transcripts encoding several other targets of immunotherapy, such as TIM-3, LAG-3 or TIGIT, than that of TILlo tumors (Fig. 3e). Published studies have linked PD-1 and 4-1BB to both exhaustion22 and antigen-specific TCR activation19,20, but the positive correlation of their expression with TIL density indicated that their higher expression probably reflected enrichment for activated TAA-specific CD8+ T cells.
TILhi tumors show enrichment for CD8+ TRM cells
Patients with a high density of TILs in tumors have a better survival outcome that that of patients with a low density of TILs6. It is not known if beyond the numerical changes in T cells, there are qualitative differences between these groups in tumor-infiltrating CD8+ T cells. Defining such features might provide insight into the mechanisms that govern the magnitude and specificity of anti-tumor CD8+ T cell responses.
We found 109 transcripts whose expression differed significantly between TILhi tumors and TILlo tumors (Fig. 4a and Supplementary Table 6). As expected, transcripts encoding products involved in TCR activation (4-1BB and PDCD1) were upregulated in TILhi tumors (Fig. 4a), consistent with enrichment for presumed TAA-specific CD8+ T cells, although such specificity would require further experimental confirmation. Several other transcripts encoding products associated with the tissue retention of lymphocytes and tissue-resident memory T cells (TRM cells) were expressed differentially by TILhi tumors relative to their expression by TILlo tumors (Supplementary Table 6 and Supplementary Fig. 5a). For example, ITGAE encodes the αE subunit of the integrin αEβ7 (CD103), which binds the adhesion molecule E-cadherin expressed by epithelial cells in barrier tissues23,24. TILhi tumors had higher expression of the gene encoding this marker of TRM cells (CD103) than did TILlo tumors (Fig. 4a,b), and its expression positively correlated with the average number of CD8+ cells within tumors (Fig. 4c). That finding was also confirmed in the TCGA lung-cancer data set (Fig. 4c). We confirmed CD103 expression in CD8+ TILs at the protein level by immunohistochemistry and flow cytometry (Fig. 4d,e). Surface molecules linked to TRM cells25,26, such as CD69 and CD49a (ITGA1), were co-expressed with CD103, and surface molecules linked to effector memory cells (KLRG1) and central memory cells (CCR7 and CD62L) had lower expression on CD103+CD8+ TILs than on CD103−CD8+ TILs (Fig. 4f and Supplementary Fig. 5b), which suggested that the former population represented TRM cells. We also observed co-expression of PD-1 and 4-1BB in 6% of CD103+CD8+ TILs and 4% of CD103+CD8+ TILs, respectively, in a representative patient sample (Fig. 4e).
Figure 4.
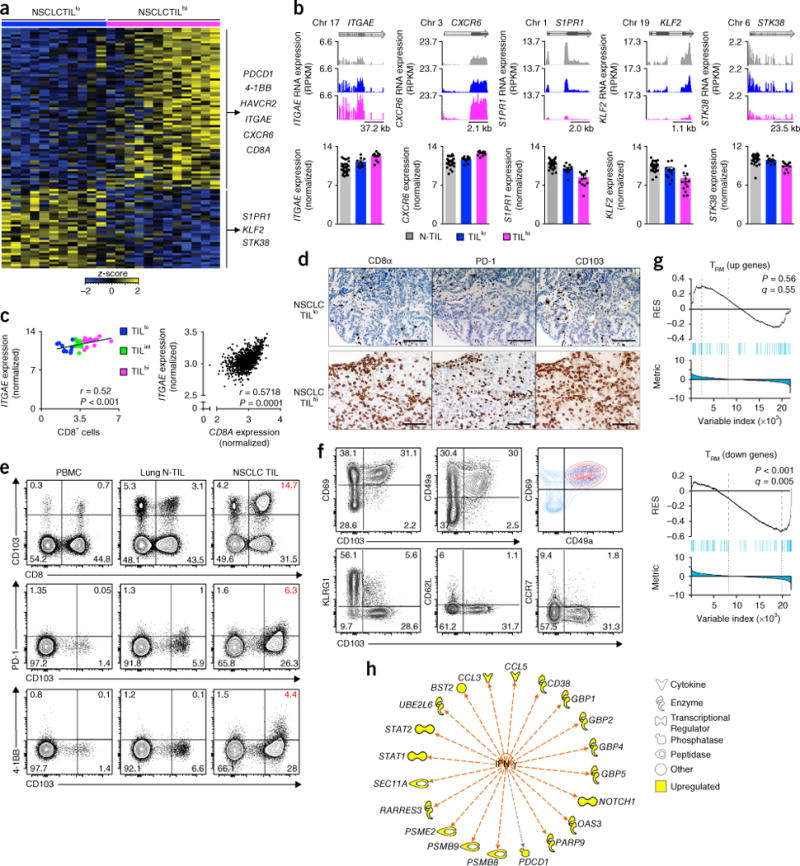
Tissue-residency features of TILhi tumors. (a) RNA-Seq analysis of genes (one per row) expressed differentially (P values as in Fig. 1a) by NSCLC CD8+ TILs from TILlo tumors versus those from TILhi tumors (presented as in Fig. 1a); right margin, genes encoding exhaustion- and TRM cell–associated molecules. (b) RNA-Seq analysis of ITGAE, CXCR6, S1PR1, KLF2 and STK38 (presented as in Fig. 1c). Each symbol (bottom) represents an individual sample; small horizontal lines indicate the mean (± s.e.m.). (c) Correlation of the expression of ITGAE transcripts (log2 normalized counts) in NSCLC CD8+ TILs (key) and the number of tumor-infiltrating CD8+ cells (quantified by immunohistochemistry) (left), and of the expression of ITGAE transcripts and that of CD8A transcripts in the TCGA lung cancer RNA-Seq data set (right; extreme outliers not presented here) (r values and P values as in Fig. 3c,d). Each symbol represents an individual patient (n = 36 (left) or n = 1,013 (right)). (d) Immunohistochemistry microscopy of CD8α, PD-1 and CD103 (above images) in TILlo and TILhi NSCLC tumors (left margin). Scale bars, 100 μm. (e) Flow-cytometry analysis of the surface expression of CD8 and CD103 (top), PD-1 and CD103 (middle) and 4-1BB and CD103 (bottom) on live and singlet-gated T cells obtained from peripheral blood mononuclear cells, lung N-TILs and NSCLC TILs (above plots) from the same patient. (f) Flow-cytometry analysis of the expression of CD69 or CD49a versus that of CD103 (top row, left and middle), and of KLRG1, CD62L or CCR7 versus that of CD103 (bottom row) in live and singlet-gated CD45+CD3+CD8+ T cells; top right, overlay of CD103+CD8+ TILs (red) with CD103−CD8+ TILs (blue). (g) GSEA of TRM cell signature genes upregulated (top) or downregulated (bottom) in the transcriptome of CD8+ TILs from NSCLC TILhi tumors relative to their expression in other TILs and N-TILs (presented as in Fig. 1d). (h) Ingenuity pathway analysis of transcripts (perimeter) upregulated in NSCLC TILhi tumors that are regulated by interferon-γ (orange arrows) and encode products with various functions (key); gray arrow indicates an unpredicted effect of IFN-γ. Data are from one experiment (a–c,g,h) or are representative of ten experiments (d) or six experiments (e,f).
Another transcript with higher expression in TILhi tumors than in TILlo tumors was CXCR6 (which encodes the chemokine receptor CXCR6) (Fig. 4a,b); not only is CXCR6 expression linked to TRM cells25 but also CXCR6 is important for the localization and function of tissue-residing T cells27,28. S1PR1 transcripts (encoding the G protein–coupled receptor S1P1) and KLF2 transcripts (encoding the transcription factor KLF2), which are known to be downregulated in TRM cells23, were also diminished in TILhi tumors relative to their abundance in TILlo tumors (Fig. 4b). Downregulation of S1PR1 is necessary for the egress of T cells from the lymph nodes and their subsequent retention in tissues, as T cells with high expression of S1P1 are retained in the lymph nodes and also easily exit tissues due to the higher concentration of its ligand (S1P) in the lymph nodes and blood. S1PR1 is a target of KLF2; downregulation of KLF2 has been shown to result in lower expression of S1PR1, and the products of both of these genes together have an important role in the establishment and retention of TRM cells in tissues29. GSEA also revealed that TILhi tumors exhibited low expression of genes typically downregulated among a core set of TRM cell signature genes, such as SIPR5, STK38 and FAM65B23,25 (Fig. 4g). Pathway analysis of the genes expressed differentially by TILhi tumors relative to their expression by TILlo tumors showed higher expression (by TILhi tumors than by TILlo tumors) of genes encoding products involved in the canonical interferon pathway (Supplementary Fig. 5c). Interferon-γ (IFN-γ) was also predicted by Ingenuity pathway analysis to be an upstream regulator of the genes expressed differentially by TILhi tumors relative to their expression by TILlo tumors (Fig. 4h). Because IFN-γ produced by TRM cells has been shown to recruit circulating T cells to potentiate robust immune responses in tissues30,31, we inferred that the interferon response signature seen in TILhi tumors might have been the result of the activation of TRM cells by TAAs (tumor-specific TRM activity). Overall, these results demonstrated that TILhi tumors showed enrichment for TRM cells.
CD103 density is predictive of survival in lung cancer
We next assessed the transcriptome of CD8+ TILs from tumors enriched for TRM cells (CD103hi tumors) for features that would support a robust anti-tumor immune response. Ingenuity pathway analysis of the genes expressed differentially by CD8+ TILs from CD103hi tumors relative to their expression by such cells from CD103lo tumors (classified on the basis of the expression of ITGAE transcripts (encoding CD103) in CD8+ TILs; Supplementary Fig. 6a,b and Supplementary Table 7) indicated cell proliferation and cytotoxicity as the key activated functions (Supplementary Table 8). Consistent with that analysis, the expression of several transcripts encoding products linked to cell cycle and proliferation32 was substantially upregulated in CD8+ TILs from CD103hi tumors relative to their expression in such cells from CD103lo tumors (Fig. 5a,b). We confirmed by flow cytometry that CD103+CD8+ TILs expressed the cell-proliferation marker Ki67 (Fig. 5c). The expression of several transcripts encoding products linked to the cytotoxic function of CD8+ T cells (IFNG, GZMA, GZMB, SEMA7A, KLRB1, CCL3, STAT1, RAB27A, IL21R and FKBP1A)32 was also significantly upregulated in CD8+ TILs from CD103hi tumors relative to their expression in such cells from CD103lo tumors (Fig. 5d,e and Supplementary Table 7). We confirmed at the protein level that CD103+CD8+ TILs expressed molecules linked to cytotoxicity, such as granzyme B, granzyme A, perforin and CD107a, and produced IFN-γ (Fig. 5f and Supplementary Fig. 7a,b), and demonstrated that CD103+CD8+ TILs were the main producers, among CD8+ TILs, of both granzyme A and granzyme B (Supplementary Fig. 7c). However, we found no significant difference between CD103+CD8+ TILs and CD103−CD8+ TILs from each patient in a comparison of the proportion of cells expressing granzyme A, granzyme B, IFN-γ and CD107a; however, perforin had lower expression in CD103+CD8+ TILs (Supplementary Fig. 7d). To address the question of whether CD8+ TILs from CD103hi tumors had greater effector potential, we compared the mean fluorescence intensity of those molecules and the frequency of cells expressing them in CD103hi tumors relative to that in CD103lo tumors (Fig. 5f and Supplementary Fig. 7e). Notably, we found that CD8+ TILs from CD103hi tumors had significantly higher expression of granzyme B than that of CD103lo tumors (Fig. 5f). These results suggested that tumors rich in TRM cells (CD103hi tumors) harbored CD8+ T cells that actively proliferated in the tumor milieu and displayed enhanced production of cytotoxic molecules, all hallmarks of robust anti-tumor immunity.
Figure 5.
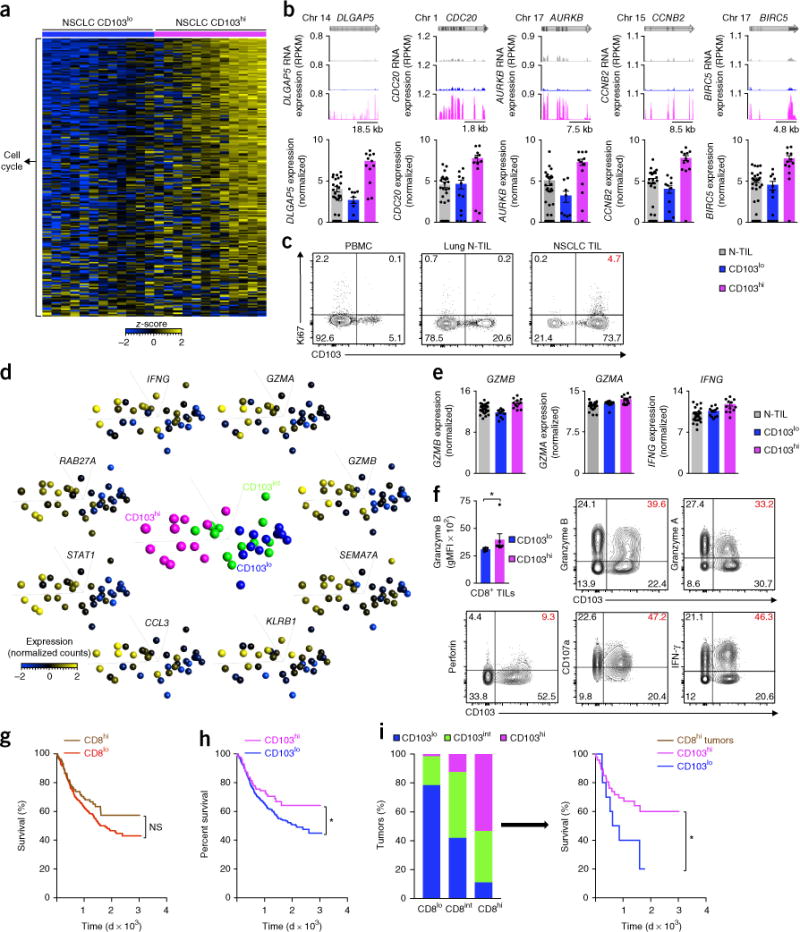
CD103 density predicts survival in lung cancer. (a) RNA-Seq analysis of the expression of genes (one per row) encoding products related to cell cycle and proliferation, by NSCLC CD8+ TILs from CD103lo or CD103hi tumors (presented as in Fig. 1a). (b) RNA-Seq analysis of DLGAP5, CDC20, AURKB, CCNB2A and BIRC5, all encoding products linked to cell cycle and proliferation (presented as in Fig. 1c). Each symbol (bottom) represents an individual sample; small horizontal lines indicate the mean (± s.e.m.). (c) Flow-cytometry analysis of the expression of Ki67 and CD103 in live and singlet-gated CD45+CD3+CD8+ T cells obtained from peripheral blood mononuclear cells, lung N-TILs and NSCLC TILs (above plots) from the same patient. (d) Principal-component analysis of CD8+ TILs from patients with NSCLC with CD103lo, CD103int or CD103hi tumors (middle plot), and expression (key) of the transcripts encoding cytotoxicity-related products in CD8+ TILs (plots along perimeter). Each symbol represents an individual patient. (e) Expression of GZMB, GZMA and IFNG transcripts (log2 normalized counts) in cells as in b (key). (f) Expression of granzyme B (geometric mean fluorescence intensity (gMFI)) in CD8+ TILs from CD103lo tumors (n = 5) or CD103hi tumors (n = 7) (top left), and flow-cytometry analysis of the expression of granzyme B, granzyme A, perforin, CD107a (LAMP-1) or IFN-γ versus that of CD103 in live and singlet-gated CD45+CD3+CD8+ T cells obtained from NSCLC TILs. *P = 0.0025 (Mann-Whitney test). (g,h) Survival of patients (n = 689) with lung cancer, with a low density (CD8lo) or high density (CD8hi) of CD8+ cells (key) in tumors (g) or a low density (CD103lo) or high density (CD103hi) of CD103+ cells (key) in tumors (h), presented as Kaplan–Meier curves. NS, P = 0.086 (g), and *P = 0.043 (h) (log-rank test). (i) Frequency of CD103hi, CD103int or CD103lo tumors (key) among those pre-classified on the basis of CD8 density (horizontal axis) (left), and survival of patients with lung cancer with CD8hi tumors sub-classified according to the density of CD103-expressing cells (key) (right), presented as Kaplan–Meier curves. *P = 0.036 (log-rank test). Each symbol (e,f) represents an individual sample (e) or patient (f); small horizontal lines indicate the mean (± s.e.m.). Data are from one experiment (a,b,d,e,g–i) or are representative of six experiments (c) or twelve experiments (f).
On the basis of the findings reported above, we hypothesized that a high density of CD103+ TILs in tumors (tumors enriched for TRM cells) might also confer a survival advantage beyond that previously found to be associated with the density of CD8+ TILs6,7. In an independent, large cohort of patients (n = 689) with predominantly early-stage lung cancer (83% stage I–IIIA; Supplementary Table 9) monitored from 2007 to 2016, we assessed retrospectively the survival outcome of patients whose tumors were classified on the basis of the density of cells expressing CD8 or CD103 (Supplementary Table 9). A higher density of CD8+ TILs was associated with a 28% reduction in mortality (P = 0.077 (Cox proportional-hazards model)) and a trend toward improved survival (Fig. 5g). Notably, patients with lung cancer who had CD103hi tumors had significantly lower mortality (34% lower risk of mortality; P = 0.045 (Cox proportional-hazards model)) and a better survival outcome than that of patients with CD103lo tumors (Fig. 5h). That finding was also observed in the TCGA data set for lung cancer (Supplementary Fig. 7f). To better understand the dependence on the density of CD103 and CD8 in tumors, we determined the status of the density of cells expressing CD103 (CD103hi, CD103int or CD103lo) in tumors pre-classified on the basis of the density of cells expressing CD8. As expected, the proportion of CD103hi tumors was higher among CD8hi tumors than among CD8lo tumors; however, there was some discordance, as tumors with CD103lo or CD103int status were also observed among CD8hi tumors (Fig. 5i). Notably, even in the subgroup of patients with lung cancer who had a high density of CD8+ TILs (CD8hi tumors), patients who had tumors with a higher density of cells expressing CD103 (CD103hi tumors) had significantly lower mortality (60% lower risk of mortality; P = 0.043 (Cox proportional-hazards model)) than that of patients with CD103lo tumors and survived significantly longer than did patients with CD103lo tumors (Fig. 5i). Our results suggested that patients with a robust intra-tumoral TRM cell response had better long-term survival outcomes and that this effect was above that conferred by the density of CD8+ TILs.
Molecules linked to tumor immune response
Tumors with CD8hi and CD103hi TIL status had higher expression of transcripts encoding molecules shown to be effective targets of immunotherapy, such as PDCD1, TIM3 and LAG3, and CD8hi status and CD103hi status were both independently linked to better anti-tumor immunity and survival outcome. Therefore, we reasoned that other molecules encoded by the list of genes upregulated in tumors with CD8hi and CD103hi TIL status might also have important functions in modulating the magnitude and specificity of anti-tumor immune responses (Fig. 6a and Supplementary Table 7). An example of this was CD39 (encoded by ENTPD1), a cell-surface ectonucleotidase that dephosphorylates ATP to AMP (Fig. 6b,c). We found that the expression of CD39 protein was much higher in CD103+CD8+ TILs than in CD103−CD8+ TILs (Fig. 6d). High concentrations of ATP in the tumor microenvironment can have toxic effects on cells via signaling through the purinergic receptor P2RX7 (refs. 33,34). Given that CD8+ TILs from CD103hi tumors and those from CD103lo tumors exhibited similar expression of transcripts encoding P2RX7 (Fig. 6c), we speculated that the greater abundance of CD39 probably ‘preferentially’ protected TRM cells (CD103+CD8+ TILs) from ATP-induced cell death. Notably, however, adenosine produced by CD39 might also suppress the function of natural killer T cells, natural killer cells and probably CD8+ T cells35,36. CD38 is another ectonucleotidase and type II trans-membrane glycoprotein with various functions, including regulation of adenosine signaling, adhesion and transduction of activation and proliferation signals37,38. Expression of CD38 protein was also higher in CD103+CD8+ TILs than in CD103−CD8+ TILs (Fig. 6d). Given that purinergic receptors can be targeted therapeutically, it might be pertinent to determine how CD39 and CD38 modulate ATP and purinergic signaling pathways to influence the development and function of anti-tumor TRM cells (CD103+CD8+ TILs).
Figure 6.
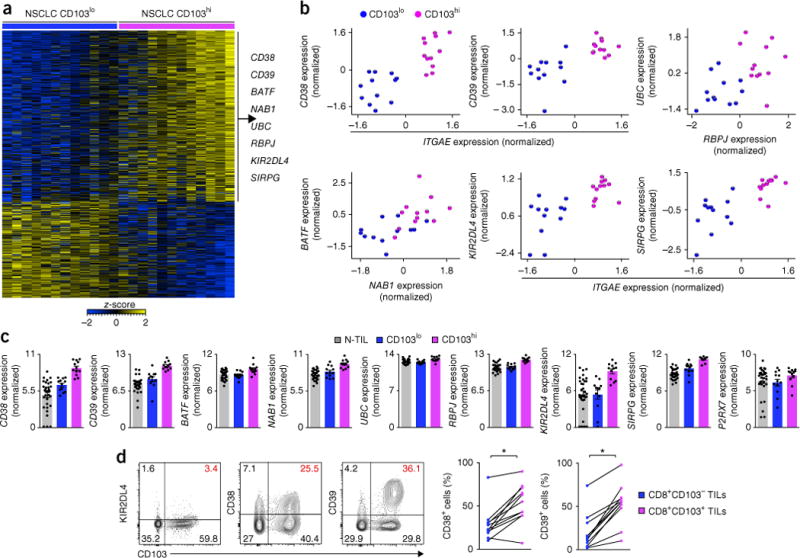
Molecules newly linked to the tumor immune response. (a) RNA-Seq analysis of genes (one per row) expressed differentially by NSCLC CD8+ TILs from CD103lo tumors versus those from CD103hi tumors (presented as in Fig. 1a). (b) RNA-Seq analysis (_z_-scores of normalized counts) of various transcripts (horizontal axes) plotted against that of other transcripts (vertical axes) in NSCLC CD8+ TILs from CD103hi or CD103lo tumors (key in c). (c) Expression of the transcripts in b (log2 normalized counts) in N-TILs or in NSCLC CD8+ TILs from CD103hi or CD103lo tumors (key). (d) Flow-cytometry analysis of the expression of KIR2DL4, CD38 or CD39 versus that of CD103 in live and singlet-gated CD45+CD3+CD8+ T cells obtained from NSCLC TILs (left), and frequency of CD38+ cells or CD39+ cells among CD8+CD103− TILs or CD8+CD103+ TILs (key). *P = 0.0006, CD38+ cells, or P < 0.0001, CD39+ cells (paired Student’s two-tailed _t_-test). Each symbol (b–d) represents an individual patient (b) or sample (c,d); small horizontal lines (c) indicate the mean (± s.e.m.); diagonal lines (d) connect data from the same patient (n = 11 donors). Data are from one experiment (a–c) or are representative of six experiments (d, left) or eleven experiments (d, right).
CD8+ TILs from CD103hi tumors had higher expression of several transcripts encoding components of the Notch signaling pathway (NOTCH, RBPJ, DTX2, UBC and UBB), relative to their expression in CD8+ TILs from CD103lo tumors (Fig. 6b,c), suggestive of an important role for this pathway in boosting TRM cell responses in lung cancer; this speculation is supported by a report showing that the Notch pathway supports the development of TRM cells in the lungs39. CD8+ TILs from CD103hi tumors had higher expression of transcripts encoding two transcription factors (BATF and NAB1) potentially linked to CD4+ T cell–mediated help of CD8+ T cells, relative to their expression in CD8+ TILs from CD103lo tumors (Fig. 6b,c).
Other examples of transcripts upregulated in CD8+ TILs from CD103hi tumors included KIR2DL4, which encodes the killer-cell immunoglobulin-like receptor KIR2DL4 with activating and inhibitory functions40; expression of KIR2DL4 protein was confirmed in CD103+CD8+ TILs (Fig. 6d). HLA-G, a non-classical major histocompatibility complex class I molecule, has been shown to engage KIR2DL4 and increase the production of cytokines and chemokines by natural killer cells41. Although the expression of HLA-G is highly restricted, several reports have shown that its expression is increased in tumor tissue, especially in lung cancer42, so we speculated that HLA-G expressed in tumors might also convey activation signals via the receptor KIR2DL4 to CTLs. SIRPG encodes SIRPG, a member of the immunoglobulin superfamily of signal-regulatory proteins that interact with the ubiquitously expressed signal-regulatory protein CD47 (ref. 43). Notably, SIRPG is the only member of that family that is expressed on T cells, and its interaction with CD47 expressed on antigen-presenting cells has been shown to enhance T cell proliferation and IFN-γ production44. Given the higher expression of SIRPG transcripts in CD8+ TILs from CD103hi tumors than in those from CD103lo tumors (Fig. 6b,c), we speculated that SIRPG might also serve as an important co-stimulatory molecule and that its function could be exploited to enhance the anti-tumor function of CTLs. Several candidate molecules described here have not been fully assessed for their potential as immunotherapeutic targets in cancer; the importance of these molecules in boosting anti-tumor immune responses should be verified in further functional studies.
DISCUSSION
We have taken an unbiased discovery-based approach to identify transcripts for which CD8+ TILs showed enrichment and those that were linked to robust anti-tumor immune responses and good outcomes. Published transcriptional studies of anti-tumor CD8+ T cells from patients with cancer have been largely restricted to the analysis of whole tumor tissue or of CD8+ T cells from peripheral blood or metastatic sites8–11. Our study surveyed over 100 transcriptomes from purified CD8+ TILs and N-TILs derived from tumor tissue and the best control tissue available: the adjacent uninvolved lung. Bioinformatics analysis of those data sets revealed a core CD8+ TIL transcriptional profile comprising ~1,400 genes that was shared by various tumor subtypes and was distinct from that of N-TILs. This profile suggested extensive molecular reprogramming within the tumor microenvironment and enrichment for presumably TAA-specific cells that were actively proliferating following TCR engagement and co-stimulation, all hallmarks of effective anti-tumor immunity.
Despite our use of purified CD8+ TIL populations for our analyses, we observed substantial heterogeneity among patients in their expression of genes encoding molecules involved in the cell cycle, TCR activation, co-stimulation and inhibition. Such underlying molecular heterogeneity in the anti-tumor CTL response might partly explain the variability in clinical responses to the immunological-checkpoint blockers currently available. We propose that baseline transcriptional profiling of purified tumor-infiltrating CTLs might enable the rational selection of immunotherapies. Our strategy of purifying relevant immune-cell populations from relatively small tumor samples and performing ‘micro-scaled’ RNA-Seq assays to generate high-resolution genome-wide data can be readily applied to any accessible tumor type. This approach can thus be used to develop biomarkers of the response to immunotherapy and to discover novel targets for immunotherapy.
Another unique aspect of our study was our evaluation of CD8+ TIL transcriptomes relative to TIL density (a feature linked to outcome). This analysis revealed various features linked to robust anti-tumor immune responses, such as TIL density; the most striking of these was tissue residence. CD8+ TILs with enrichment for TRM cells (CD103hi) had features of enhanced cytotoxicity and proliferation, which suggested that patients whose tumors had a high density of TRM cell markers, such as CD103, had a more-robust anti-tumor immune response and that this feature in the tumor might independently influence clinical outcome. In a large, independent cohort of patients with lung cancer, we showed that a higher density of cells expressing CD103 was predictive of a better survival outcome. Most notably, we confirmed that this effect was independent of that conferred by the density of CD8+ TILs; this finding was biologically relevant and has not been addressed by published studies, to our knowledge45–47. Thus, our study has not only revealed a close link among TIL density, TRM cell features and enhanced survival but has also shed light on the global molecular features that endow CD8+ TILs from TRM cell–rich tumors with robust anti-tumor properties. We speculate that the generation of a robust anti-tumor TRM cell response should be an important goal of vaccination approaches targeting neo antigens or shared tumor antigens.
Since patients with lung cancer who had a high density of CD8+ or CD103+ TILs had a better survival outcome, our comparison of the transcriptional profiles of CD8+ TILs from tumors with either a high density or a low density of cells expressing CD8 or CD103 would probably highlight features linked to the generation of robust anti-tumor immunity. The list of transcripts expressed differentially included those encoding molecules such as PD-1, TIM-3, CTLA-4, LAG-3, CD27, CD8 and OX40, which are effective targets of cancer immunotherapy in humans or in model organisms. Other molecules in that list might also have an important role in modulating the magnitude and specificity of anti-tumor immune response. For example, several promising molecules that we identified, such as CD38, CD39, BATF, NAB1, KIR2DL4, SIPRG and components of Notch signaling, deserve further investigation as immunotherapeutic targets in cancer. BATF has been shown to regulate the metabolism and survival of CD8+ T cells and to diminish the inhibited phenotype of CD8+ T cells48,49. In a model of infection with lymphocytic choriomeningitis virus, the expression of BATF in CD8+ T cells, induced by the cytokine IL-21 derived from CD4+ T cells, was shown to be essential for maintaining the effector response of CTLs, and overexpression of BATF restored the effector function of CD8+ T cells that had not received help from CD4+ T cells49. NAB1 is a transcription factor whose mouse homolog (NAB2) is induced in CD8+ T cells that have received help from CD4+ T cells and is needed to prevent activation-induced cell death of those ‘helped’ CD8+ T cells50. Thus, we hypothesize that NAB1, which has high sequence homology to NAB2, might also have a similar role in preventing the apoptosis of tumor-infiltrating CTLs and that its increased expression might identify tumors in which CD8+ TILs have received help from CD4+ T cells. Further functional experiments will be needed to verify the role of these molecules.
Our study has revealed the transcriptional program of CD8+ TILs at the tumor site and has identified the inter-patient heterogeneity that presumably underlies the variability in clinical responses to checkpoint blockade. It has provided insight into the molecular mechanisms that govern robust anti-tumor CTL responses and lends support to the proposal that anti-tumor vaccines should be designed to enable the generation of CD8+ TRM cells for durable immunity. The ability to perform ‘micro-scaled’ RNA-Seq analysis of purified CD8+ TILs from patients’ tumors allowed us to identify gene-expression programs that might inform personalized immunotherapeutic treatment strategies and thereby provide a useful tool for translational application.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Patient characteristics and sample processing
The Southampton and South West Hampshire Research Ethics Committee and the Ethics Committee of La Jolla Institute approved the study, and written informed consent was obtained from all subjects. Newly diagnosed, untreated patients with NSCLC and HNSCC (Supplementary Table 1) referred to Southampton University Hospitals NHS Foundation Trust and Poole Hospital NHS Foundation trust, UK, between 2014 and 2016 were prospectively recruited. Freshly resected tumor tissue and matched adjacent non-tumor lung tissue (in the case of patients with NSCLC) was obtained following surgical resection. T cells were isolated from tumor (TILs) or adjacent uninvolved lung (N-TILs) using a combination of mechanical and enzymatic dissociation. In brief, tumor or lung tissue was cut into small fragments and incubated at 37 °C for 15 min in an orbital shaker with 2 ml RPMI-1640 medium (Fisher Scientific) containing 0.15 WU/ml Liberase DL (Roche) and 800 units/ml DNase I (Sigma-Aldrich). Dispersed cells were then passed through a 70-μm filter and centrifuged and were re-suspended in MACS buffer (phosphate-buffered saline containing 2 mM EDTA and 0.5% bovine serum albumin) for sorting or analysis by flow cytometry. For isolating and phenotyping of CD8+ T cells from tumor or lung tissue, dispersed cells were first incubated with FcR block (Miltenyi Biotec), then were stained with a mixture of the following fluorescence- conjugated antibodies (each at the concentration recommended by the manufacturer): anti-CD45-FITC (HI30; BioLegend), anti-CD4-PE (RPA-T4; BD Biosciences), anti-CD3-PE-Cy7 (SK7; BioLegend), anti-CD8α-PerCP-Cy5.5 (cSK1; BD Biosciences), anti-HLA-DR-APC (L243; BD Biosciences), anti-CD14-APC-H7 (MϕP9; BD Biosciences), anti-CD19-PerCP-Cy5.5 (clone HIB19; BioLegend) and anti-CD20-PerCP-Cy5.5 (clone 2H7; BioLegend). Stained samples were analyzed with a BD FACSAria (BD Biosciences) and FlowJo software (Treestar), and CD8+ T cells were sorted into ice-cold TRIzol LS reagent (Ambion)51,52. Phenotypic analysis of CD8+ TILs for TRM markers was performed by staining with anti-CD69-BV605 (FN50; BioLegend), anti-CD49a-PE (TS2/7; BioLegend), anti-KLRG1-APC (SA231A2; BioLegend), anti-CD62L-BV510 (DREG-56; BioLegend), anti-CCR7-AF700 (TS2/7; BioLegend) (each at the concentration recommended by the manufacturer). Flow-cytometry analysis of CD8+CD103+ T cells and intra-cellular assessment of Ki67 were carried out with the following antibodies (each at the concentration recommended by the manufacturer): anti-CD45-FITC (HI30; BioLegend), anti-Ki67-PE (Ki67; BioLegend), anti-CD3-APC-Cy7 (SK7; BioLegend), anti-CD8α-PerCP-Cy5.5 (SK1; BD Biosciences), anti-CD103-APC (Ber-ACT8; BioLegend), anti-PD-1-PE-Cy7 (eBioJ105; eBioscience), anti-4-1BB-Pacific blue (4B4-1; BioLegend). The True-Nuclear Transcription Factor Buffer set (BioLegend) was used for the intracellular staining of Ki67. Flow-cytometry analysis of novel molecules and intracellular assessment of cytotoxic molecules were performed using the following antibodies (each at the concentration recommended by the manufacturer): anti-granzyme A-APC (CB9; BioLegend), anti-granzyme B-PE (REA226; Miltenyi Biotec), anti-Perforin-PE or −BV421 (B-D48; BioLegend), anti-KIR2DL4-PE (mAb33; BD BioLegend), anti-CD38-APC-Cy7 (HB-7; BioLegend), anti-CD39-PE (A1; BioLegend). For cytokine and CD107a assays, CD8+ TILs were stimulated ex vivo with 20 nM PMA (phorbol 12-myristate 13-acetate) and 1 μM ionomycin for 4 h, and 5 μg/ml brefeldin was added during the final 2 h of stimulation. Anti-CD107a-PE (H4A3; BioLegend; at the concentration recommended by the manufacturer) was added to the PMA-and-ionomycin stimulation mixture for the final 2 h. Intracellular assessment of interferon-γ was performed using anti-IFNG-BV-421 (4S.B3; BioLegend; at the concentration recommended by the manufacturer) at the end of stimulation. Assays were performed in at least six patients and representative plots are presented. Stained samples were analyzed using a BD FACSCanto II (BD Biosciences). Dead cells were excluded using a LIVE/DEAD Fixable Aqua dead cell stain kit (Life Technologies) or DAPI (4,6-diamidino-2-phenylindole).
Histology and immunohistochemistry
Immunohistochemistry was performed on formalin-fixed paraffin-embedded ] tumor sections with anti-cytokeratin (AE1/AE3; Dako; pre-diluted ‘ready to use’), anti-CD8α (C8/144B; Dako; 1:100), anti-CD103 (EPR4166(2); Abcam; 1:500), anti-PD-1 (NAT105; Abcam; 1:100) and anti-granzyme B (GrB-7; Dako; 1:50). Assays were performed in at least ten patients and representative staining is presented. TILs were quantified using a Zeiss AxioCam MRc5 microscope (Zeiss) and Zeiss Axiovision software (version 4.8.1.0; Zeiss). An average of ten high-powered fields (×400) across representative areas of each tumor were counted to account for intratumoral heterogeneity; these were averaged to generate an intratumoral TIL score. Tumors with an average quantification of CD8+ cells in the top one-third or bottom one-third percentile were classified as TILhi or TILlo, respectively; the lowest quantification of CD8+ cells in the TILhi tumors was at least twofold greater than the highest quantification of CD8+ cells in the TILlo tumors (Supplementary Fig. 4). For overall survival analyses (Fig. 5e–g), tumor-tissue microarrays from patients with NSCLC were stained with anti-CD8α (C8/144B; Dako; 1:100) or anti-CD103 (EPR4166(2); Abcam; 1:500) and were viewed under low-powered magnification (2.5× objective) to determine the density of CD8+ or CD103+ cells, as described previously53.
Survival data and analysis
In an independent large cohort of patients with predominantly early-stage NSCLC (n = 689; Supplementary Table 9) followed up from January 2007 to June 2016 (minimum follow up, 3.4 years), we retrospectively analyzed survival according to density of CD8+ or CD103+ TILs. The primary endpoint was overall survival, and survival time was measured from the date of diagnosis until the date of death or date last seen alive. Kaplan–Meier plots (with log-rank tests to determine significance of overall survival; P values in Fig. 5g–i) and unadjusted Cox proportional hazards model (to determine relative risk of death) were used to analyze the survival data, as described previously54. Patients were excluded from analysis if survival was <30 d, to exclude the possibility of surgery-related mortality. Survival analysis based on the expression of ITGAE transcripts (encoding CD103) in tumor samples from patients with lung adenocarcinoma in the TCGA was derived from the OncoLnc tool (http://www.oncolnc.org).
RNA sequencing
Total RNA was purified using a miRNAeasy micro kit (Qiagen) and was quantified as described previously55 (on average, ~4,230 CD8+ T cells per sample were processed for RNA-Seq analysis). Purified total RNA was amplified according to the smart-seq2 protocol55. cDNA was purified using AMPure XP beads (1:1.1 ratio, Beckman Coulter). From this step, 1 ng of cDNA was used to prepare a standard Nextera XT sequencing library (Nextera XT DNA sample preparation kit and index kit, Illumina). Samples were sequenced using HiSeq2500 (Illumina) to obtain 50-bp single-end reads. Quality-control steps were included to determine total RNA quality and quantity, optimal number of PCR pre-amplification cycles, and cDNA fragment size56. Samples that failed quality control were eliminated from further downstream steps.
RNA-Seq analysis
RNA-Seq data were mapped against the hg19 reference using TopHat57 (v1.4.1.,–library-type fr-secondstrand-C) and the RefSeq gene annotation downloaded from the University of California Santa Cruz Genome Bioinformatics site. Sequencing read coverage per gene was counted using HTSeq-count (-m union -s yes -t exon -i gene_id, http://www-huber.embl.de/users/anders/HTSeq/). To identify genes expressed differentially by various patient groups, we performed negative binomial tests for paired and unpaired comparisons by employing the Bioconductor package DESeq2 disabling the default options for independent filtering and Cooks cutoff58. We considered genes to be expressed differentially by any comparison when the DESeq2 analysis resulted in a Benjamini-Hochberg–adjusted P value of <0.05. The Qlucore Omics Explorer 3.2 software package was used for visualization and representation (heat maps, principal component analysis) of RNA-Seq data52. Unsupervised hierarchical clustering of samples based on the expression of genes (n = 1,000) with the highest variance, which accounted for 20% of the total variance, was performed using DESeq package functions and custom scripts on R (Supplementary Fig. 1c). TCR sequences were retrieved from CD8+ T cell RNA-Seq data sets, and the frequency of TCRβ chain clonotypes was determined using default parameters of the MiXCR package59 (Supplementary Table 5). The CD103 status of TILs was determined on the basis of the transcript abundance of ITGAE (CD103) in CD8+ TILs. Tumors with CD8+ TIL expression of ITGAE transcripts in the top one-third or bottom one-third percentile were classified as CD103hi or CD103lo, respectively (Supplementary Fig. 6).
Knowledge-based network generation and pathway analysis
The biological relevance of differentially expressed genes identified by DESeq2 analysis was further investigated using the Ingenuity Pathways Analysis platform. The enrichment of canonical pathways (pre-defined, well-described metabolic and signaling pathways curated from literature reviews) among differentially expressed genes was assessed, with significance determined by right-tailed Fisher’s exact test (P < 0.05). For network analysis, differentially expressed genes were progressively linked together on the basis of a measure of their interconnection, derived from previously characterized functional interactions.
GSEA
The Qlucore Omics Explorer 3.2 software package was used for GSEA. GSEA was used to further assess significant enrichment of specific biological pathways or signatures in one group relative to that in another group. GSEA determines whether an a priori defined ‘set’ of genes (such as a signature) show significant cumulative changes in gene expression between phenotypic subgroups60. In brief, all genes are ranked on the basis of their differential expression in one group versus their expression in another group. Next, a running enrichment score (RES) is calculated for a given gene set on the basis of how often its members appear at the top or bottom of the ranked differential list. 1,000 random permutations of the phenotypic subgroups are used to establish a null distribution of RES against which a normalized running enrichment score (NES) and false-discovery-rate-corrected q values are calculated using Kolmogorov-Smirnov statistic. GSEA was run with a focused group of gene signatures encoding products related to exhaustion22, lung-cancer-associated T cell signature15, anergy61, senescence62 and tissue residency25. These gene signatures (Figs. 1d and 4g and Supplementary Table 10) were selected to test the null hypothesis that CD8+ T cell groups did not show significant enrichment for different CD8+ T cell phenotypes.
Statistical analysis
Comparison between two groups was assessed with two-tailed unpaired or paired Student’s _t_-test (Figs. 2f and 6d, Supplementary Figs. 5b and 7c,d) or Mann-Whitney test (Fig. 5f and Supplementary Fig. 7e) or Kolmogorov-Smirnov test (Supplementary Fig. 3c) using GraphPad Prism 6. Spearman correlation coefficient (r value) was calculated to assess the significance of correlation of the expression of any two transcripts of interest.
Data availability
Sequencing data are deposited into the Gene Expression Omnibus (accession code GSE90730). Source data files for Figures 5 and 6 and Supplementary Figures 5 and 7 are available online.
Supplementary Material
Supp Table1
Supp Table2
Supp Table6
Supp Table7
Supp figures 1-7
Supp table10
Supp table3
Supp table4
Supp table5
Supp table8
Supp table9
Acknowledgments
We thank M. Chamberlain, K. Amer, C. Fixmer and B. Johnson for assistance with recruitment of study subjects and processing of samples; A. Easton for help with the assignment of scores to TILs; Z. Fu and J. Greenbaum for help with the processing and analysis of sequencing data; and H. Cheroutre, M. Kronenberg and J. Moore for reviewing the manuscript and providing insight. Supported by the Wessex Clinical Research Network and the National Institute of Health Research, UK (sample collection), Cancer Research UK (O.W., E.V.K., C.H.O.; and C11512/A20256, for CD103 pathology analysis), the William K. Bowes Jr Foundation (P.V. and A.-P.G.) and the Faculty of Medicine of the University of Southampton (P.V., T.S.-E. and C.H.O.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
A.-P.G., P.S.F., T.S.-E., P.V. and C.H.O. conceived of the work and designed, performed and analyzed experiments; A.-P.G., D.S-C. and D.S. performed micro-scaled RNA-Seq experiments and analysis under the supervision of G.S., P.V. and C.H.O.; A.-P.G. performed data analysis and wrote the manuscript under the supervision of P.V. and C.H.O.; J.C., O.W., E.M.G.-M. and T.M. performed the cell isolations and immunohistochemistry data analysis under the supervision of G.J.T. and C.H.O.; and S.J.C., A.A., E.W. and E.V.K. assisted in patient recruitment, obtaining consent and sample collection.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat Rev Clin Oncol. 2014;11:24–37. doi: 10.1038/nrclinonc.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garon EB, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 4.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galon J, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 7.Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125:3335–3337. doi: 10.1172/JCI83871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baitsch L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bindea G, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 10.Collins N, et al. Transcriptional hallmarks of tumor infiltrating lymphocyte responses to melanoma. Blood. 2013;122:3491. [Google Scholar]
- 11.Tirosh I, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown IE, Blank C, Kline J, Kacha AK, Gajewski TF. Homeostatic proliferation as an isolated variable reverses CD8+ T cell anergy and promotes tumor rejection. J Immunol. 2006;177:4521–4529. doi: 10.4049/jimmunol.177.7.4521. [DOI] [PubMed] [Google Scholar]
- 14.Montes CL, et al. Tumor-induced senescent T cells with suppressor function: a potential form of tumor immune evasion. Cancer Res. 2008;68:870–879. doi: 10.1158/0008-5472.CAN-07-2282. [DOI] [PubMed] [Google Scholar]
- 15.Johnston RJ, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell. 2014;26:923–937. doi: 10.1016/j.ccell.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 16.Curran MA, et al. Systemic 4-1BB activation induces a novel T cell phenotype driven by high expression of Eomesodermin. J Exp Med. 2013;210:743–755. doi: 10.1084/jem.20121190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willoughby JE, et al. Differential impact of CD27 and 4-1BB costimulation on effector and memory CD8 T cell generation following peptide immunization. J Immunol. 2014;193:244–251. doi: 10.4049/jimmunol.1301217. [DOI] [PubMed] [Google Scholar]
- 18.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gros A, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124:2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fourcade J, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207:2175–2186. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolfl M, et al. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood. 2007;110:201–210. doi: 10.1182/blood-2006-11-056168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Mackay LK, et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- 24.Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013;31:137–161. doi: 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- 25.Mackay LK, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:459–463. doi: 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- 26.Sathaliyawala T, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. 2013;38:187–197. doi: 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee LN, et al. CXCR6 is a marker for protective antigen-specific cells in the lungs after intranasal immunization against Mycobacterium tuberculosis. Infect Immun. 2011;79:3328–3337. doi: 10.1128/IAI.01133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tse SW, Radtke AJ, Espinosa DA, Cockburn IA, Zavala F. The chemokine receptor CXCR6 is required for the maintenance of liver memory CD8+ T cells specific for infectious pathogens. J Infect Dis. 2014;210:1508–1516. doi: 10.1093/infdis/jiu281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skon CN, et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14:1285–1293. doi: 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ariotti S, et al. T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science. 2014;346:101–105. doi: 10.1126/science.1254803. [DOI] [PubMed] [Google Scholar]
- 31.Schenkel JM, et al. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 2014;346:98–101. doi: 10.1126/science.1254536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Best JA, et al. Transcriptional insights into the CD8+ T cell response to infection and memory T cell formation. Nat Immunol. 2013;14:404–412. doi: 10.1038/ni.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bono MR, Fernández D, Flores-Santibáñez F, Rosemblatt M, Sauma D. CD73 and CD39 ectonucleotidases in T cell differentiation: beyond immunosuppression. FEBS Lett. 2015;589:3454–3460. doi: 10.1016/j.febslet.2015.07.027. [DOI] [PubMed] [Google Scholar]
- 34.Kuhny M, Hochdörfer T, Ayata CK, Idzko M, Huber M. CD39 is a negative regulator of P2X7-mediated inflammatory cell death in mast cells. Cell Comm Signal. 2014;12:40. doi: 10.1186/s12964-014-0040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol. 2016;7:109. doi: 10.3389/fimmu.2016.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta PK, et al. CD39 expression identifies terminally exhausted CD8+ T cells. PLoS Pathog. 2015;11:e1005177. doi: 10.1371/journal.ppat.1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandoval-Montes C, Santos-Argumedo L. CD38 is expressed selectively during the activation of a subset of mature T cells with reduced proliferation but improved potential to produce cytokines. J Leukoc Biol. 2005;77:513–521. doi: 10.1189/jlb.0404262. [DOI] [PubMed] [Google Scholar]
- 38.Muñoz P, et al. Antigen-induced clustering of surface CD38 and recruitment of intracellular CD38 to the immunologic synapse. Blood. 2008;111:3653–3664. doi: 10.1182/blood-2007-07-101600. [DOI] [PubMed] [Google Scholar]
- 39.Hombrink P, et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat Immunol. 2016;17:1467–1478. doi: 10.1038/ni.3589. [DOI] [PubMed] [Google Scholar]
- 40.Faure M, Long EO. KIR2DL4 (CD158d), an NK cell-activating receptor with inhibitory potential. J Immunol. 2002;168:6208–6214. doi: 10.4049/jimmunol.168.12.6208. [DOI] [PubMed] [Google Scholar]
- 41.Rajagopalan S, Long EO. KIR2DL4 (CD158d): an activation receptor for HLA-G. Front Immunol. 2012;3:258. doi: 10.3389/fimmu.2012.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wiśniewski A, et al. Genetic polymorphisms and expression of HLA-G and its receptors, KIR2DL4 and LILRB1, in non-small cell lung cancer. Tissue Antigens. 2015;85:466–475. doi: 10.1111/tan.12561. [DOI] [PubMed] [Google Scholar]
- 43.Brooke G, Holbrook JD, Brown MH, Barclay AN. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J Immunol. 2004;173:2562–2570. doi: 10.4049/jimmunol.173.4.2562. [DOI] [PubMed] [Google Scholar]
- 44.Piccio L, et al. Adhesion of human T cells to antigen-presenting cells through SIRPbeta2-CD47 interaction costimulates T-cell proliferation. Blood. 2005;105:2421–2427. doi: 10.1182/blood-2004-07-2823. [DOI] [PubMed] [Google Scholar]
- 45.Wang ZQ, et al. CD103 and intratumoral immune response in breast cancer. Clinical Cancer Res. 2016;22:6290–6297. doi: 10.1158/1078-0432.CCR-16-0732. [DOI] [PubMed] [Google Scholar]
- 46.Webb JR, Milne K, Nelson BH. Location, location, location: CD103 demarcates intraepithelial, prognostically favorable CD8+ tumor-infiltrating lymphocytes in ovarian cancer. OncoImmunology. 2014;3:e27668. doi: 10.4161/onci.27668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Djenidi F, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol. 2015;194:3475–3486. doi: 10.4049/jimmunol.1402711. [DOI] [PubMed] [Google Scholar]
- 48.Kurachi M, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol. 2014;15:373–383. doi: 10.1038/ni.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xin G, et al. A critical role of IL-21-induced BATF in sustaining CD8-T-cell-mediated chronic viral control. Cell Rep. 2015;13:1118–1124. doi: 10.1016/j.celrep.2015.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolkers MC, et al. Nab2 regulates secondary CD8+ T-cell responses through control of TRAIL expression. Blood. 2012;119:798–804. doi: 10.1182/blood-2011-08-373910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vijayanand P, et al. Invariant natural killer T cells in asthma and chronic obstructive pulmonary disease. N Engl J Med. 2007;356:1410–1422. doi: 10.1056/NEJMoa064691. [DOI] [PubMed] [Google Scholar]
- 52.Engel I, et al. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nat Immunol. 2016;17:728–739. doi: 10.1038/ni.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marsh D, et al. Stromal features are predictive of disease mortality in oral cancer patients. J Pathol. 2011;223:470–481. doi: 10.1002/path.2830. [DOI] [PubMed] [Google Scholar]
- 54.Ward MJ, et al. Tumour-infiltrating lymphocytes predict for outcome in HPV-positive oropharyngeal cancer. Br J Cancer. 2014;110:489–500. doi: 10.1038/bjc.2013.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seumois G, et al. Transcriptional profiling of Th2 cells identifies pathogenic features associated with asthma. J Immunol. 2016;197:655–664. doi: 10.4049/jimmunol.1600397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Picelli S, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- 57.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bolotin DA, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015;12:380–381. doi: 10.1038/nmeth.3364. [DOI] [PubMed] [Google Scholar]
- 60.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Safford M, et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol. 2005;6:472–480. doi: 10.1038/ni1193. [DOI] [PubMed] [Google Scholar]
- 62.Fridman AL, Tainsky MA. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene. 2008;27:5975–5987. doi: 10.1038/onc.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp Table1
Supp Table2
Supp Table6
Supp Table7
Supp figures 1-7
Supp table10
Supp table3
Supp table4
Supp table5
Supp table8
Supp table9
Data Availability Statement
Sequencing data are deposited into the Gene Expression Omnibus (accession code GSE90730). Source data files for Figures 5 and 6 and Supplementary Figures 5 and 7 are available online.