Molecular Dissection of FUS Points at Synergistic Effect of Low-Complexity Domains in Toxicity (original) (raw)
Summary
RNA-binding protein aggregation is a pathological hallmark of several neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). To gain better insight into the molecular interactions underlying this process, we investigated FUS, which is mutated and aggregated in both ALS and FTLD. We generated a Drosophila model of FUS toxicity and identified a previously unrecognized synergistic effect between the N-terminal prion-like domain and the C-terminal arginine-rich domain to mediate toxicity. Although the prion-like domain is generally considered to mediate aggregation of FUS, we find that arginine residues in the C-terminal low-complexity domain are also required for maturation of FUS in cellular stress granules. These data highlight an important role for arginine-rich domains in the pathology of RNA-binding proteins.
Keywords: amyotrophic lateral sclerosis, frontotemporal lobar degeneration, phase transition, LLPS, protein aggregation, FUS, prion-like domain, low-complexity domain, intrinsically disordered protein
Graphical Abstract
Highlights
- •
Both QGSY and RGG2 domains are necessary for FUS-induced neurodegeneration in flies - •
Arginine-rich domains interact with QGSY hydrogels and liquid droplets - •
RGG2 arginines promote phase separation of FUS in vitro and in cells - •
FUS phase separation behavior in vitro correlates with neurodegeneration in vivo
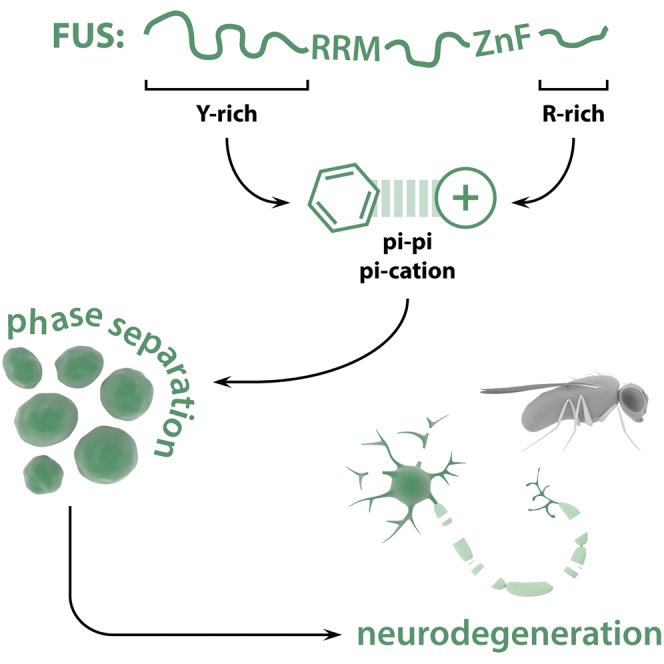
Protein aggregation is a hallmark of ALS. Bogaert et al. describe the molecular interactions between disordered regions of the FUS protein driving its liquid phase behavior, maturation, and neurotoxicity. These findings highlight the physicochemical interactions driving FUS phase separation and give us insights into its misregulation in disease.
Introduction
Amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) are two adult-onset neurodegenerative disorders affecting motor neurons and cortical neurons, respectively. Despite affecting different neuronal populations, leading to different symptoms, these two diseases are believed to constitute two extremes of a disease spectrum, with patients often presenting with symptoms of both (Swinnen and Robberecht, 2014). Apart from the clinical overlap, there are also genetic and pathological similarities. The RNA-binding protein TAR DNA-binding protein 43 (TDP-43) was identified as the major component of the ubiquitin-positive protein aggregates observed in most ALS patients (∼97%) and in patients of the FTLD-TDP subtype (∼45%) (Ling et al., 2013, Neumann et al., 2006). Moreover, mutations in the TAR DNA-binding protein gene (TARDBP), the gene encoding TDP-43, are a cause of both ALS and FTLD (Borroni et al., 2009, Kabashi et al., 2008, Sreedharan et al., 2008). These findings have directed the attention to problems in ribostasis, which is the misregulation of RNA-binding proteins (RBPs) and their responsive RNAs, as a major disease mechanism in ALS-FTLD (Ramaswami et al., 2013). Further support for this hypothesis has come from the discovery of other RBPs mutated and aggregated in these diseases. Besides TDP-43, FUS has a prime role in ALS-FTLD. Mutations in FUS are a cause of ALS and FTLD (Borroni et al., 2009, Kwiatkowski et al., 2009, Vance et al., 2009), and patients carrying FUS mutations present with cytoplasmic protein aggregates of FUS. Interestingly, wild-type FUS aggregates are also found in 10% of FTLD cases (Neumann et al., 2009). Deciphering how and why RBPs, such as FUS, start aggregating will be key to understanding the disease and could lead to therapeutic strategies.
Several RBPs involved in ALS-FTLD, including TDP-43 and FUS, are involved in stress granule metabolism (Boeynaems et al., 2016, Li et al., 2013, Ramaswami et al., 2013). Under normal conditions, these proteins reside in the nucleus where they regulate transcription and splicing (Ling et al., 2013). However, upon cellular stress, these proteins localize to the cytoplasm where they assemble in membrane-less organelles called stress granules (SGs) (Jain et al., 2016, Kedersha et al., 2013). As these assemblies are very similar in protein content compared to the ubiquitin-positive aggregates in ALS-FTLD, it has been suggested that SGs could serve as stepping stones toward pathological aggregation in the disease (Boeynaems et al., 2016, Li et al., 2013, Ramaswami et al., 2013). Interestingly, SGs are dynamic and reversible assemblies opposed to solid aggregates. Indeed, in recent years it has been found that SGs, and other membrane-less organelles, form by a process of phase separation (Boeynaems et al., 2017, Burke et al., 2015, Jain et al., 2016, Kato et al., 2012, Lee et al., 2016, Lin et al., 2015, Molliex et al., 2015, Murakami et al., 2015, Patel et al., 2015). Low-complexity domains (LCDs) in these proteins promote their demixing from the surrounding cytoplasm through weak multivalent interactions (Boeynaems et al., 2018, Brangwynne et al., 2015). Phase separation of FUS has been largely attributed to its N-terminal QGSY LCD through hydrophobic interactions between degenerate [G/S]Y[G/S] repeats (Burke et al., 2015, Kato et al., 2012, Lin et al., 2015, Murakami et al., 2015, Patel et al., 2015, Schwartz et al., 2013). This domain is also called a prion-like domain (PrLD), given the strong sequence similarity with yeast prions (King et al., 2012). Interestingly, such PrLDs seem to be pivotal in the aggregation of other ALS-FTLD-related RBPs as well (Couthouis et al., 2011, Couthouis et al., 2012, Kim et al., 2013, Lin et al., 2015, Molliex et al., 2015). Moreover, both FUS droplets and hydrogels formed in the test tube have been shown to undergo a switch to irreversible fibrillarization, potentially resembling the maturation of dynamic SGs into solid aggregates (Murakami et al., 2015, Patel et al., 2015).
Besides the QGSY PrLD, FUS has other domains that could play an important role in the disease. We generated a Drosophila model of FUS toxicity and performed a systematic domain deletion screen to identify FUS domains mediating toxicity of the full-length protein. As expected, deleting the QGSY domain rescued toxicity. However, the ΔQGSY construct showed some residual toxicity, suggesting that other domains are indeed important for the toxic function of FUS. We found that the RGG2 LCD is essential for toxicity, showing a synergistic effect of the N- and C-terminal LCDs in FUS toxicity. Both domains also strongly interacted in both FUS hydrogels and liquid droplets, and the strength of this interaction correlated with the toxicity of the deletion constructs in _Drosophil_a. In addition, we confirmed the importance of the RGG2 domain in phase separation and the maturation of FUS in mammalian SGs. More specifically, we identified the arginine residues in this domain as being mediators of both phase separation and toxicity.
Results
Wild-Type and Mutant FUS Induce Motor Neuron Degeneration and Motor Defects in Drosophila
To study the molecular determinants of FUS toxicity in an in vivo animal system, we generated a FUS Drosophila model. We generated fly lines expressing wild-type and NLS mutant FUS using site-directed integration. Ubiquitous expression of FUS was lethal and yielded no fly offspring (Table S1). Motor neuron expression (D42 Gal4) resulted in partial developmental lethality (Figure 1A). Flies raised at 25°C had problems with eclosion from the pupal case (Figure 1A). The few escapers displayed an immature phenotype, characterized by a soft cuticle, disoriented scutellar bristles, and unexpanded wings (Figure 1B). This phenotype was previously observed in TDP-43 fly models and is caused by degeneration of a specific neuronal population in the fly ventral nerve cord (Vanden Broeck et al., 2013). Lowering FUS expression levels specifically in the hatching phase, by placing pharate adult pupae at 18°C, attenuated toxicity, indicating that FUS toxicity is dose dependent (Table S2).
Figure 1.

FUS Expression Induces Neurodegeneration and Motor Defects in Both Developmental and Adult Stages
(A and B) Motor neuron expression using the D42 gal4 driver of WT and mutant FUS induces eclosion defects (A) and immature phenotypes (B) in Drosophila. Pictures give an example of a fly unable to eclose from the pupal case, and an escaper illustrating the immature phenotype.
(C) Ubiquitous, pan-neuronal and motor neuron-specific adult-onset expression of WT and mutant FUS reduces fly survival.
(D) Adult-onset FUS expression in the motor neurons induces neurodegeneration and affects flight ability. Extent of motor dysfunction is correlated to loss of neurons. Survival graphs: log-rank Mantel-Cox; all other graphs: Kruskal-Wallis, Dunn’s multiple comparison.
∗∗p < 0.01, ∗∗∗∗p < 0.0001.
To overcome developmental phenotypes, we used the inducible Gal80 system to restrict FUS expression to adult flies. Ubiquitous adult-onset expression of FUS resulted in a severely shortened life span (Figure 1C). Restricting expression of FUS to neurons or specifically motor neurons similarly shortened the life span, highlighting the neuronal component of FUS toxicity (Figure 1C). Furthermore, these flies showed an age-dependent, progressive motor performance defect measured by two different motor tests: flying ability (Figure 1D) and negative geotaxis (Figure S1A). Additionally, upon aging, these flies displayed an erect wing phenotype (Figure S1B), which has been previously observed in other fly models of neurodegeneration (Yang et al., 2006). Age-dependent motor defects correlated with a reduction of motor neurons in the ventral nerve cord (Figure 1D). These experiments show that the phenotypes in pharate adults are predictive of adult-onset phenotypes and can be used as an easy and reliable readout for FUS toxicity.
We did not observe a significant difference in toxicity between wild-type and mutant forms of FUS expressed at similar levels, although the P525L mutant had a trend toward lower protein levels (Figures S1C and S1E–S1G). We also ruled out any potential effect on the expression of the Drosophila FUS ortholog cabeza (caz) (Figures S1D and S1H). We next investigated the subcellular localization of the human FUS proteins in the fly tergotrochanter motor neuron. In young animals, all FUS variants were mainly nuclear, whereas upon aging the ratio of cytoplasmic versus nuclear FUS increased (Figures S1I–S1K). The fact that we found no differences between wild-type and mutant FUS is in contrast to previous findings (Lanson et al., 2011). However, it is important to note that we used site-directed integration instead of random integration, which rules out any genomic effects in our experiments. Another recently generated fly model also finds no difference between FUS wild-type (WT) and P525L mutant toxicity (Marrone et al., 2018).
The QSGY and the RGG2 Domains Are Necessary for FUS Toxicity
To evaluate the molecular determinants of FUS toxicity, we systematically deleted all domains from the full-length protein and expressed these deletion constructs in Drosophila (Figure 2A). Three domains strongly reduced FUS toxicity upon deletion: the N-terminal QSGY PrLD, the C-terminal RGG2 domain, and the nuclear localization signal (NLS) domain were crucial to induce toxicity in fly motor neurons (Figure 2B). We did not detect any effect of disrupting the RRM domain on toxicity, contrary to previous reports in yeast (Sun et al., 2011) and fly (Daigle et al., 2013). While we deleted the entire RRM domain, these two reports made use of F-L mutants in the RRM binding site, which could explain this discrepancy.
Figure 2.

Molecular Dissection of FUS Toxicity
(A) Scheme illustrating different domain deletions tested.
(B) Deletion RGG2 and NLS domains rescues FUS toxicity on both eclosion rates and the appearance of the immature phenotype, whereas deletion of QGSY only rescues eclosion rates. Kruskal-Wallis, Dunn’s multiple comparison.
∗∗∗∗p < 0.0001.
Of the pupae expressing ΔQGSY in their motor neurons, 62.7% were able to eclose in contrast to only 4% for full-length FUS-expressing animals. However, 83.1% of the flies that did eclose showed the immature phenotype seen in FUS-expressing flies. Upon adult onset expression, QGSY deletion rescued the shortened survival seen in FUS-expressing flies (Figure S2A). However, upon aging, these flies showed a clear reduction in motor performance (Figure S2B) and displayed the erect wing phenotype (data not shown). This indicates that the QSGY domain contributes to FUS toxicity, but its deletion does not fully abolish toxicity of the protein.
Flies expressing a protein lacking the RGG2 domain (ΔRGG2) were able to eclose similar to control flies (98.1% and 90.8%, respectively) and did not present with the immature phenotype (Figure 2B). In addition, no differences in life span or motor performance were observed upon adult-onset expression of ΔRGG2 (Figures S2C and S2D). These data indicate that the RGG2 domain is pivotal for FUS toxicity. Deletion of the NLS domain reduced toxicity of FUS as well, although not as strongly as deletion of the QGSY or RGG2 domain (Figure 2B). Deleting the NLS from the full-length protein reduced the pupal lethality to 49.7%, and only 15.2% of flies had an immature phenotype. In addition, we did not observe any reduction in life span or motor performance upon adult-onset expression (Figures S2E and S2F).
None of these phenotypes was due to lowered expression of the deletion construct compared to full-length FUS, or any effects on caz expression (Figures S3A–S3C). As expected, deletion mutants lacking the RGG2 and NLS distributed significantly more to the cytoplasm in young fly motor neurons compared to full-length FUS (Figure S3D). All other mutants distributed in a similar way in the cell as the full-length protein (Figure S3D). Of note, none of these deletion constructs was exclusively present in the nucleus or cytoplasm, suggesting that there was still shuttling across the nuclear membrane.
The QSGY Domain Differentiates between FUS and caz Toxicity
FUS and caz show a strong conservation at the level of domain structure and amino acid composition (Figure 3A). However, caz lacks the QGSY domain, which is most pronounced in vertebrates (Figure S4), and this domain constitutes the PrLD in FUS, which is important in FUS aggregation and toxicity (Kato et al., 2012, Patel et al., 2015, Shelkovnikova et al., 2014, Sun et al., 2011). Flies overexpressing caz were unable to eclose and died in the pharate adult stage (Figure 2B). However, mutating the NLS of caz (P398L, orthologous to P525L) completely abolished eclosion problems and maturation defects (Figure 3B). This observation was in contrast to what we previously observed for the human proteins, where both WT and NLS mutants were toxic. We reasoned that this difference could stem from the absence of the QGSY domain in caz. Indeed, addition of the QGSY domain to the mutant caz protein (hence, humanizing the Drosophila protein) created a toxic chimeric protein reminiscent of full-length FUS P525L toxicity (Figure 3B).
Figure 3.

N- and C-Terminal LCDs Are Sufficient to Replicate Full-Length Toxicity
(A) Comparison between caz and FUS. Both proteins share a similar domain architecture, protein disorder, and amino acid composition, but caz lacks the QGSY domain. Amino acid composition was calculated for each domain.
(B) Humanizing caz shows that addition of the FUS QGSY domain to the benign caz NLS mutant makes the chimeric protein toxic again.
(C) Drosophilizing FUS shows that deleting QGSY generates a protein with similar properties to the benign caz NLS mutant.
(D and E) Schematic representation of the constructs containing different regions of FUS that were used to create transgenic flies (D) and the effect on toxicity (E). Only when fused together do the N- and C-terminal LCDs cause toxicity.
For (B)–(E), Kruskal-Wallis, Dunn’s multiple comparison. ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
We next performed the reciprocal experiment and “Drosophilized” the human FUS protein (Figure 3C). As discussed above, deletion of the QGSY domain reduced toxicity of FUS expression. Interestingly, introducing an NLS mutation (R521G) in ΔQGSY FUS, hereby resembling even more the non-toxic caz NLS mutant, further reduced toxicity (Figure 3C). Furthermore, adult flies expressing the ΔQGSY-NLS mutant showed no reduction in life span, and the residual motor problems observed with the ΔQGSY mutant were absent in flies expressing the ΔQGSY-NLS mutant (Figures S5A–S5F).
The N-Terminal and C-Terminal LCDs Are Sufficient for FUS Toxicity
Our data showed that both the C- and N-terminal LCDs are required for FUS toxicity. To investigate whether these domains would also be sufficient for toxicity, we generated different fly lines, which allowed us to express combinations of the N-terminal and C-terminal domains (Figure 3D). Overexpression of the QSGY domain and the C terminus alone or in trans was not toxic to fly motor neurons (Figure 3E). Similarly, a fusion of the QGSY to the NLS domain was non-toxic (Figure 3E). However, toxicity was completely restored when fusing the QGSY to the C-terminal domain through a flexible protein linker (GGGGSx3) (Chen et al., 2013). When we used a rigid protein linker (EAAARx4), this toxicity was again slightly reduced (Figure 3E). These data clearly show that the QGSY and C-terminal domain are sufficient to recapitulate full-length FUS toxicity, but they must act in cis and require some conformational flexibility.
Strength of FUS N-C-Terminal Interaction In Vitro Correlates with Toxicity In Vivo
Our data indicate that FUS requires its two LCDs, both the prion-like QGSY and the arginine-rich RGG2, in cis for toxicity. These LCDs are known to act as binding modules and mediate phase separation of FUS (Boeynaems et al., 2017, Burke et al., 2015, Kato et al., 2012, Murakami et al., 2015, Patel et al., 2015, Schwartz et al., 2013, Shelkovnikova et al., 2014). Of interest, we and others have previously shown that arginine-rich domains can bind PrLDs, likely through pi-cation, pi-pi, or both types of interactions between tyrosine and arginine residues (Boeynaems et al., 2017, Lee et al., 2016, Lin et al., 2016).
To test this hypothesis, we evaluated the binding of different C-terminal deletion constructs (Figure 4A) to hydrogels (Kato et al., 2012) and droplets (Burke et al., 2015), which are formed by the prion-like QGSY domain. Both assays gave very similar results, and deleting RGG1 or RGG2 most drastically impaired binding (Figure 4B). Compellingly, when we plotted toxicity of a deletion construct in Drosophila against the strength of the in vitro interaction, this returned a clear linear correlation (Figure 4C). The RGG1 deletion was an outlier in this correlation. However, we must note that, in the in vitro experiment, we simply test the ability of the C-terminal domain to bind the PrLD. Hence, even though RGG1 and RGG2 share a similar binding propensity when tested out of the context of the full-length protein, their specific position in the sequence is of major importance to toxicity. As RGG1 is located in the full-length protein at a more central position, and shielded at both ends by two folded domains, that is, the FUS RRM and zinc finger, we hypothesize that the RGG1 domain has not as much conformational flexibility as the RGG2 located at the C-terminal end of the protein.
Figure 4.

RGG Domains Are Required for Efficient Binding of the FUS C-Terminal Domain to Phase-Separated QGSY Hydrogels and Liquid Droplets
(A) Scheme illustrating the C-terminal deletion constructs tested for QGSY droplet and hydrogel binding.
(B) The RGG domains are the main domains affecting binding of GFP fusion constructs to mCherry-QGSY hydrogels or QGSY droplets. Asterisk indicates higher exposure setting to show that there is still some residual binding of single ΔRGG mutants to the hydrogel.
(C) Partitioning of the C-terminal deletion constructs in the QGSY droplets and hydrogels in vitro correlates with the toxicity of the corresponding full-length deletion construct in fly. The red fit takes into account all values; the blue fit considers ΔRGG1 as an outlier.
Arginine Residues in RGG2 Are Required for FUS Toxicity
Given the observation that the RGG2 domain (Figure 5A) was important for toxicity and binding to PrLDs, we hypothesized that the arginines could be important for the function and toxicity of FUS. We wondered whether this function of the RGG2 to bind to hydrogels and droplets would also be mimicked in a more physiological setting. We first generated recombinant full-length FUS and a mutant replacing all arginines in RGG2 by glycines. Both WT and mutant FUS could phase separate in the test tube, as indicated by the spontaneous droplet formation (Figure 5B). However, the RGG2 mutant did so at dramatically reduced levels (Figure 5C), confirming the importance of the RGG2 domain to FUS phase separation. In a next step, we tested this in a cellular setting. When expressed in mammalian cells, FUS localized to SGs induced by arsenite treatment (Figures 5D and 5E). Of note, since modifying RGG2 interferes with nucleocytoplasmic targeting, we used a FUS NLS mutant (P525L) as a control. Upon deletion of the RGG2 domain, SG enrichment was strongly reduced, and mutating all arginines in RGG2 to glycines gave similar results (Figure 5D), indicating that the arginine residues are indeed the key mediators of the function of this domain. When we evaluated the dynamics of FUS in these induced SGs, we found that the RGG2 mutant was more dynamic and had a smaller insoluble fraction (=1-plateau) than the FUS NLS mutant (Figure 5F). Besides the arsenite-induced SGs, overexpression of FUS also resulted in spontaneous SGs assembly. These granules were noticeably larger (Figure 5E) and represent a more mature type of SG since they formed over the course of a 24-hr transfection period. The dynamics of FUS in these mature SGs was significantly reduced, and they had a larger insoluble fraction (Figure 5G), suggesting that FUS can become more insoluble upon SG aging, as previously observed for in vitro FUS droplets (Patel et al., 2015). Again, mutating RGG2 increased the dynamics and the fractional recovery of FUS again, and the dynamics of the FUS RGG2 mutant were unaltered compared to those of young SGs (half-life, unpaired t test, p = 0.15). These data show that, in a cellular context, the RGG2 domain is involved in SG maturation, a process suggested as a stepping stone toward FUS aggregation.
Figure 5.

Arginines in the RGG2 Domain Are Involved in the SG Targeting and Maturation of FUS
(A) Comparison between charge plots of QGSY and RGG2 domains. Mutated arginines of RGG2 are highlighted in green.
(B) RGG2 mutant full-length FUS shows reduced liquid droplet formation. Note that droplets over time fuse and wet the glass surface.
(C) Quantification of FUS phase separation; SEM.
(D) Deletion of RGG2 or mutating RGG2 arginines have a similar effect on the SG enrichment of FUS. Whiskers indicate 95% confidence interval; Kruskal-Wallis, Dunn’s multiple comparison.
(E) Examples of arsenite-induced and spontaneous SGs positive for FUS and the general SG marker G3BP1.
(F and G) Mutation of RGG2 arginines decreases the immobile fraction (1-plateau) and half-life of FUS in SGs induced by arsenite treatment (F), as well as SGs induced by FUS overexpression (G). FRAP graphs: two-way ANOVA; plateau and half-life comparisons: unpaired t test. Error bars, mean ± SEM.
(H) Mutating RGG2 arginines partially rescues FUS toxicity in fly. Kruskal-Wallis, Dunn’s multiple comparison.
∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001.
Going back to our Drosophila model, we found that mutating all RGG2 arginines into glycines strongly reduced toxicity compared to FUS NLS mutant (P525L), mimicking the complete RGG2 deletion (Figure 5H). This shows that also in our Drosophila model the arginine-mediated function of RGG2 is required for toxicity.
Discussion
Since the discovery of disease-causing mutations in FUS, much effort has gone into the investigation of why this protein starts aggregating in ALS-FTLD. Similar to TDP-43 and other heterogeneous nuclear ribonucleoproteins (hnRNPs), FUS has a PrLD (King et al., 2012). While the exact functions of such LCDs were unknown at the time, they were considered to be responsible for the high aggregation propensity of these proteins. Indeed, the PrLD of FUS is necessary and sufficient for in vitro fibrillarization (Schwartz et al., 2013, Sun et al., 2011). It was recently discovered that PrLDs, and other LCDs, can undergo phase separation (Boeynaems et al., 2017, Burke et al., 2015, Jain et al., 2016, Lee et al., 2016, Lin et al., 2015, Molliex et al., 2015, Murakami et al., 2015, Patel et al., 2015) or sol-gel transitions (Kato et al., 2012, Murakami et al., 2015). These findings suggest that these domains are involved in the biogenesis of membrane-less organelles, as formation of them also seems to be mediated by similar phenomena (Brangwynne et al., 2015). However, this functionality seems to come at a cost. Phase separation of such domains could make them more aggregation-prone given their high local concentrations inside membrane-less organelles. Indeed, it has been suggested before that SGs could act as seeds of aggregation (Boeynaems et al., 2016, Li et al., 2013, Ramaswami et al., 2013), and, interestingly, FUS droplets and hydrogels do mature to solid aggregates in vitro (Murakami et al., 2015, Patel et al., 2015). Hence, understanding the processes and interactions that mediate phase separation, and the liquid-to-solid switch of these proteins, can be expected to yield insights into the molecular underpinnings of RBP pathology in ALS-FTLD.
Besides the prion-like QGSY domain, FUS has other domains that may be important for its functioning and toxicity. To investigate this, we generated a Drosophila FUS toxicity model characterized by neurodegeneration and motor phenotypes as described before (Jäckel et al., 2015, Lanson et al., 2011). This facile readout allowed us to investigate which FUS domains were important for this phenotype. Deleting the QGSY PrLD only partially reduced toxicity, indicating the importance of additional domains to FUS toxicity. Indeed, RGG2 deletion completely abolished toxicity. This domain has been implicated before in FUS pathology, more specifically by modulating the potency of the FUS NLS (Dormann et al., 2010, Dormann et al., 2012). However, since mutating the NLS domain did not alter the toxicity of full-length FUS, and deletion of the full NLS domain only partially affected toxicity, we ruled out that the effect of RGG2 deletion is solely mediated by altered nucleocytoplasmic localization. Interestingly, two other reports have shown that the RGG domains could aid in the aggregation of FUS (Schwartz et al., 2013, Sun et al., 2011), and it has been suggested that their RNA binding capability may be involved in this process (Ozdilek et al., 2017, Schwartz et al., 2013).
As both the QGSY and RGG2 domain were identified as key players in the toxicity, we wondered whether both would be necessary and sufficient for toxicity. Analysis of the evolutionary conservation of FUS showed that the prion-like QGSY domain is absent in the fruit fly, and discriminated between FUS and caz toxicity. Therefore, we wondered whether the QGSY and the C-terminal LCD should be functionally linked to cause toxicity. Indeed, using fusion proteins, we showed that both LCDs are required in cis for toxicity and prefer a flexible conformation.
As we and others have recently shown that arginine-rich domains can interact with PrLDs (Boeynaems et al., 2017, Lee et al., 2016, Lin et al., 2016), we wondered whether this would also be the case for FUS. We investigated the interaction of the C-terminal LCD to hydrogels and droplets formed by the QGSY domain. Scanning the C-terminal domain with deletions allowed us to map the exact domains responsible for this interaction. We identified the RGG2 domain as a major player in the N-C-terminal interaction of FUS, and the strength of the interaction was correlated with toxicity in our Drosophila model. One exception was the RGG1 domain. Although RGG1 has similar binding capabilities as RGG2, deletion of the domain was redundant in our fly model. This finding strongly indicates that the position along the sequence dramatically affects the properties of these RGG domains.
Given the prime role for RGG2 in phase separation in vitro and toxicity in fly, we wondered whether this domain could affect phase separation and pathological hallmarks in a mammalian setting. We first found that simply mutating all arginines to glycines in RGG2 dramatically reduced full-length FUS phase separation in vitro. Subsequently, we showed that this mutant displays reduced SG targeting in mammalian cells. Additionally, the maturation of FUS upon SG aging was significantly reduced. This observation suggests that the RGG2 domain is a key mediator of the maturation of dynamic FUS assemblies, which is suggested to parallel its pathological aggregation.
In conclusion, we identified FUS RGG2 as a key domain in the toxicity and phase separation of FUS. The RGG2 domain has previously been implicated in FUS pathology, by modulating the potency of its NLS sequence or promoting aggregation. We now identify a role for this domain. The arginines in RGG2 were crucial for the maturation of FUS in cellular SGs, which suggests that the RGG2 domain plays a prime role in deleterious FUS aggregation, in addition to the PrLD. Interestingly, a series of recent studies have as well implicated the RGG2 domain in the phase behavior and aggregation of FUS. The RGG2-NLS region serves as the binding site for nuclear import factors, and while this had been already implicated in FUS nuclear translocation (Dormann et al., 2010, Dormann et al., 2012), it was now shown that transportin-1 binding to the RGG2-NLS region can also prevent and even reverse FUS phase separation and aggregation (Guo et al., 2018, Hofweber et al., 2018, Qamar et al., 2018, Yoshizawa et al., 2018). Additionally, methylation of RGG2 arginines also reduced FUS phase separation, likely via interfering with pi-cation or pi-pi interactions between N-terminal tyrosines and C-terminal arginines (Hofweber et al., 2018, Qamar et al., 2018). All of these data point at molecular interactions mediating liquid-to-solid switches of phase separated RBPs and shed light on the complex underpinnings of RBP pathology in neurodegenerative disease.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FUS | Bethyl Laboratories | A300-302A |
| Anti-Tubulin | Cell Signaling | #2125 |
| Anti-FLAG (mouse) | Sigma-Aldrich | F3165 |
| Anti-FLAG (rabbit) | Cell Signaling | #2368S |
| Anti-TIA1 | Santa Cruz | sc-1751 |
| Bacterial and Virus Strains | ||
| E. coli BL21-CodonPlus(DE3)-RIL cells | Agilent | #230240 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FUS LC | Dr. Nicolas Fawzi | Burke et al., 2015 |
| mCherry-FUS LC | Dr. Masato Kato | Kato et al., 2012 |
| FUS full-length | Dr. James Shorter | Guo et al., 2018 |
| FUS RGGmut full-length | Dr. James Shorter | This study |
| cOmplete protease inhibitor cocktail | Sigma-Aldrich | 11697498001 |
| Arsenite | Sigma-Aldrich | S7400 |
| Lipofectamine 3000 | Thermo Scientific | L3000015 |
| NucBlue | Thermo Scientific | R37605 |
| Prolong Gold Antifade | Thermo Scientific | P36934 |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC | ATCC CCL-2 |
| U2OS mCherry-G3BP1 | Dr. Nancy Kedersha | Kedersha et al., 2016 |
| Experimental Models: Organisms/Strains | ||
| Transgenic FUS fly lines | Genetivision | N/A |
| P{Act5C-GAL4}25FO1 | Bloomington Stock Center | Bl4414 |
| P{GD4553} nsyb-GAL4 | Vienna Drosophila Research Center | VDRC44011 |
| w[∗]; P{w[+mW.hs] = GawB}D42 | Bloomington Stock Center | Bl8816 |
| P{w[+mW.hs] = GawB}elav[C155] | Bloomington Stock Center | Bl458 |
| y[1] w[∗]; P{w[+mW.hs] = shakB(lethal)-GAL4.4.1}2GL15 | Bloomington Stock Center | Bl51633 |
| w[1118] | Bloomington Stock Center | Bl5905 |
| w[∗]; P{w[+mC] = tubP-GAL80[ts]}2/TM2 | Bloomington Stock Center | Bl7017 |
| Oligonucleotides | ||
| Primers | IDT | N/A |
| Recombinant DNA | ||
| CMV6 FUS-FLAG WT plasmid | Origene | TP301808 |
| CMV6 FUS-FLAG MT plasmids | Biomatic | This study |
| FUS-GFP plasmid | Dr. Simon Alberti | Patel et al., 2015 |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Ludo Van Den Bosch (ludo.vandenbosch@kuleuven.vib.be).
Experimental Model and Subject Details
Fly Culture Conditions, Stocks, and Transgenic Lines
Drosophila melanogaster strains were maintained on standard yeast, cornmeal, and agar-based medium (5% glucose, 5% yeast extract, 3.5% wheat flour, 0.8% agar) in a 12 hr light/dark rhythm. The w1118 (Canton-S10) line was used as control. Crosses for adult offspring frequencies and eclosion assay were performed at 25°C. Crosses for assessment of adult fly motor performance/activity, lifespan were set at 18°C to complete normal development. Progeny was collected daily and the flies were transferred to 25°C or 29°C. Flies were put on 29°C if the driver line was combined with a ubiquitously expressed temperature-sensitive Gal80 inhibitor (tub-Gal80ts), to induce expression.
GAL4 driver lines P{Act5C-GAL4}25FO1 (act5C-GAL4), P{GD4553} (nsyb-GAL4), P{GawB}elav[C155] (c155-GAL4), P{shakB(lethal)-GAL4.4.1}2GL152 (shakB-GAL4) and P{GawB}D42 (D42-GAL4) were obtained from the Bloomington Stock center. UAS-mCD8-GFP stock was kindly provided by Randall S. Hewes (University of Oklahoma).
All FUS constructs were sequence verified and injected into Drosophila embryos by Genetivision (Houston, TX, USA). Genomic landing site VK31 was used for site-specific transgene insertion on the third chromosome. Untagged transgenes were used to exclude possible alterations of FUS function or subcellular localization.
For all our experiments the w1118 was used as a control. Although this line does not overexpress a non-toxic protein, we have previously shown that this line behaves similarly to fly lines expressing non-toxic proteins (Boeynaems et al., 2016). We have also never observed differences between w1118 and VK31 control lines.
Cell Culture and Transfection
G3BP-mCherry U2OS (Dr. Paul Anderson and Dr. Nancy Kedersha, Harvard Medical School) and HeLa cells (ATCC CCL-2) were grown at 37°C in a humidified atmosphere with 5% CO2 for 24 hr. Cells were transiently transfected using Lipofectamine 3000 (Invitrogen) according to manufacturer’s instructions.
Methods Details
FUS Plasmids
All deletion and RGG mutant FUS constructs were obtained from Biomatik (Ontario, Canada). GFP tagged FUS constructs were a kind gift from Dr. Simon Alberti (Max Planck Institute, Germany). C-terminally Flag tagged FUS was purchased from Origene (Maryland, USA). NLS mutants were made by side directed mutagenesis kit from Stratagene (California, USA) according manufacture procedures.
Offspring Frequencies
To calculate adult offspring frequencies, the F1 generation eclosing from crosses was counted for 9 days, every 2-3 days, starting from the first day of eclosion. The percentage of balanced negative to balanced positive flies was calculated.
Eclosion Assay
Pharate adult flies were transferred to a Petri dish and monitored for 72 hr at 25°C or at 18°C. The percentage of eclosed flies was defined as ratio of total empty pupal cases to total pupal cases. Enclosed flies were scored for immature phenotype. If flies displayed signs of immaturity, they were scored affected. Bar plots were generated by giving each eclosing or affected fly a score of 1, and each uneclosed or unaffected fly a score of 0.
Motor Performance Assays
To assay adult motor performance, negative geotaxis and flight ability were analyzed.
To conduct the negative geotaxis assay, flies were placed one hr in advance in a transparent tube in the dark, under red light. The flies were then tapped to the bottom and scored according to their ability to climb. A fly was scored successful (score 1) when it reached the top of the 30 cm vial in 45 s. 50 −100 flies per genotype were tested.
Flight ability was measured using the ‘cylinder drop assay’. Flies were gently tapped down on top of a 34 cm cylinder using a funnel. Flies unable to fly immediately dropped to the bottom whereas flies able to hold on to the wall of the container were considered fliers. Flies were ranked according to their flying abilities into 6 categories (0 = unable to fly and 6 = found at the top of the container). 50 −100 flies per genotype were tested.
Lifespan Analysis
Ten newly hatched flies were placed on standard food and were transferred to a new vial every three days. After each transfer the surviving flies were counted. The density of flies per vial was controlled for, and female and male flies were separated. 50-100 flies per genotype were analyzed.
Immunohistochemistry of Ventral Nerve Cords
Immunostaining on ventral nerve cords was performed using standard techniques. The following primary antibodies were used: mouse monoclonal anti-GFP, 1:100 (Roche, Basel, Switzerland); rat anti-elav 7E8A10, 1:100 (Developmental Studies hybridoma bank); rabbit anti-FUS A300-292A, 1:100 (Bethyl Laboratories, Texas, USA); rabbit anti-FUS A300-293A, 1:100 (Bethyl Laboratories, Texas, USA) and rabbit anti-FUS A300-294A, 1:100 (Bethyl Laboratories, Texas, USA).
Secondary antibodies conjugated to Alexa488 and Alexa555 were used at 1:200 (Invitrogen, Carlsbad, California, USA). Ventral nerve cords were mounted in Vectashield mounting medium (Vector Laboratories, Burlingame, CA, USA) and analyzed with a Fluoview FV1000 confocal microscope (Olympus). Images of multiple fluorescent-labeled ventral nerve cords were obtained by sequential scanning of each channel at equal laser intensity unless noted otherwise.
Motor Neuron Degeneration
FUS constructs were expressed in motor neurons labeled with a membrane bound GFP. The ventral nerve cord was dissected at different time points and stained for GFP. A confocal image was taken every 5μm using with a Fluoview FV1000 confocal microscope (Olympus). The number of motor neurons was counted in the TH1-TH2 region of the ventral nerve cord.
In Vivo Subcellular Localization
FUS variants were expressed terotrochanter (TTM) motor neurons using ShakB-GAL4 to drive expression. We opted to measure the ratio in TTM neurons as they have a large cytoplasm and are motor neurons. To measure FUS cytoplasmic/nuclear ratios in terotrochanter motor neurons (TTM) neurons, Fiji Software was used to measure the fluorescence intensity (mean gray values) of confocal images. Images were made with equal laser intensity unless noted otherwise with a Fluoview FV1000 confocal microscope (Olympus). Ten TTM neurons of different VNCs were analyzed. Mean gray values of nucleus and cytoplasm were measured and the cytoplasmic versus nuclear intensity ratio was calculated. Statistical analysis was done using one-way ANOVA with post hoc Bonferroni’s multiple comparison test.
Transgene Expression Levels Using ddPCR
Fifty fly heads (Tubulin-GAL4 driven expression) or 30 larval central nervous systems (D42-GAL4 driven expression) were mixed with 1 mL TriPure reagent (Sigma Aldrich, St. Louis, MO, USA) and ground with a pestle. Total RNA was isolated by using standard procedures. cDNA was generated from 1 μg of RNA of each sample by using SuperScript III First-Strand Synthesis SuperMix (Thermo Fisher Scientific, Waltham, MA, UAS) according to manufacturer conditions. ddPCR was performed on an QX200 Droplet Digital PCR System with Probe mix (Biorad, Hercules, CA, USA) with primers designed by IDT PrimerQuest Tool (IDT, Coralville, Iowa). Expression levels of transcripts from the various samples were normalized to the housekeeping gene Rap2L.
| Transgene | Forward primer (5′-3′) | Probe (5′-3′) | Reversed primer (5′-3′) |
|---|---|---|---|
| FUS (C-terminal assay) | TGG ACA GCA GCA AAG CTA TAA | AGC AGA ACC AGT ACA ACA GCA GCA | CTT GGC CAT AGT TAC CTC CAC |
| FUS (QSGY assay) | GAC AGC AGA GTT ACA GTG GTT ATA G | CCA TAG CCT GAA GTG TCC GTG GAC | GGC TCT GGC CAT AAG AAG AAT AG |
| caz | CCG TGA TGG TGA CTG GAA AT | AAT AAC ACC AAC TTT GCC TGG CGC | CCC TTG GGA GTC TTA CAT CTA TTG |
| Rap2L | GAA CGA TGG TGG CGA ATA CT | ATC GAG GCA TCT GCA AAG GAT CGG | ATC CAC CGC TGA AGG TAA TG |
Transgene Expression Levels Using Western Blot
Thirty larval central nervous systems were homogenized on ice in radioimmune precipitation buffer (RIPA: 0.1% sodium dodecyl sulfate [SDS], 150 mM NaCl, 0.5% Na, 1% NP-40, 50 mM Tris-HCl [pH 8.0]) supplemented with protease inhibitor (Complete Protease inhibitor, Roche, Indianapolis, IN, USA). Homogenates were incubated on ice for 20 min, sonicated, and cleared at 14,000 rpm for 20 min at 4°C. Supernatants were used for immunoblotting. Protein concentrations were measured by BCA assay (Perbio Science N.V.). Twenty micrograms of protein was loaded on a 10% Nupage Bis-Tris gel (Invitrogen) and blotted on a polyvinylidene difluoride (PVDF) membrane. Membranes were blocked in 5% skimmed milk in PBS and probed with primary antibodies. Immunodetection was performed with specific secondary antibodies conjugated to horseradish peroxidase and the ECL-plus chemiluminescent detection system (Amersham Biosciences, Little Chalfont, UK) with bands quantified on a LAS-3000 Station (GE Healthcare, Chicago, IL, USA). The signal was normalized to the signal obtained from tubulin for quantification. Antibodies: rabbit anti-FUS (Bethyl laboratories, A300-302A) and rabbit anti-tubulin (Cell signaling, #2125).
Hydrogel Binding Assay
Purified FUS LC domain (residue 2-214) fused to GFP or mCherry was prepared as described previously (Kato et al., 2012). Hydrogel droplets of mCherry:FUS LC domain were prepared as described elsewhere (Kato et al., 2017). The GFP-FUS C-terminal deletion mutants (Figure 3A) were constructed in pHis-parallel-GFP vector by amplify and ligate the DNA fragments from the parental deletion constructs (Figure 1E), and overexpressed in E. coli BL21(DE3) and purified with Ni-NTA resin as described before (Kato et al., 2012). These proteins were diluted in 1 mL gelation buffer containing 20 mM Tris-HCl pH 7.5, 200 mM NaCl, 20 mM BME, 0.5 mM EDTA and 0.1 mM PMSF at the final concentration of 1 μM and poured into the mCherry:FUS LC hydrogel dishes. The hydrogel dishes were incubated at 4°C for 24 hr. GFP and mCherry signals in the hydrogel droplets were visualized by Leica SP5 confocal microscope. Signal intensities were analyzed with ImageJ.
FUS LC Domain Droplet Assay
FUS LC domain was expressed in E. coli and purified as described previously (Burke et al., 2015). FUS LC droplets were generated by diluting the stock solution to the 200 μM in 50 mM MES buffer 250 mM NaCl at pH 5. 1 μM of GFP-FUS constructs were added to the FUS LC droplets. Fluorescent droplets were incubated in plastic Cell Counter slides (Bio-Rad) at room temperature. Chambers were sealed using nail varnish to prevent evaporation. Pictures were taken on a Zeiss LSM 780 Meta NLO confocal microscope. Droplet enrichment was calculated as the ratio of the mean fluorescence intensity in the droplet over the mean fluorescence intensity of the background.
Full-Length FUS Droplet Assay
FUS WT and FUS RGG mutant proteins were expressed from MBP-fusion constructs using the pMAL-TEV vector, which is a pMAL (New England BioLabs, Ipswich, MA) modified to contain a TEV cleavage site kindly provided by Yuh Min Chook (UTSW). Proteins were expressed from E. coli BL21-CodonPlus(DE3)-RIL cells (Agilent) by induction with 1mM IPTG for 16 hr at 15°C. Bacteria were lysed by sonication in buffer containing 20mM HEPES pH 7.5, 2mM EDTA, 50mM NaCl, 2mM DTT, 10% glycerol, with protease inhibitors (cOmplete, EDTA-free, Roche Applied Science). MBP-FUS proteins were purified by affinity chromatography using amylose resin (New England BioLabs, Ipswich, MA), eluted with buffer containing 20mM HEPES pH 7.5, 2mM EDTA, 50mM NaCl, 2mM DTT, 10% glycerol, 10 mM maltose and further purified by heparin-affinity chromatography to disrupt association with nucleic acid.
For droplet forming experiments, MBP-FUS was buffer exchanged into buffer containing 50mM Tris pH 7.4, 1mM DTT, 50mM KCl, 0.5% glycerol. To induce droplet formation, TEV protease (Invitrogen) was added into 4μM MBP-FUS and liquid-liquid phase separation was monitored by absorbance at 395nm at room temperature using a TECAN plate reader. Parallel experiments were set up in a different plate and at indicated time point, 10μl samples were spotted onto a coverslip and imaged by Differential interference contrast (DIC) microscopy.
Immunohistochemistry and Microscopy of Stress Granules
Cells were fixed 24 hr after transfection in 4% formaldehyde in PBS post fixed with methanol followed by standard immunostaining. Following antibodies were used: anti-FLAG (F3165, Sigma), rabbit anti-FLAG (#2368S, Cell Signaling), and goat anti-TIA1 (sc-1751, Santa Cruz). AlexaFluor 488 and AlexaFluor 555 secondary antibodies (Life Technologies) were used. Slides were mounted using ProLong Gold antifade reagent (Life Technologies). Confocal images were obtained using a Zeiss LSM 780 Meta NLO confocal microscope. Images were analyzed, formatted, and quantified with Fiji software.
Stress granules were induced by incubating the cells for 1 hr with 0.5 mM NaAsO2 (Sigma).
Stress Granule Enrichment
Stress granule enrichment in HeLa cells was quantified as the ratio of fluorescence intensity in the stress granule over the intensity of the surrounding cytoplasm. All stress granules per cell were quantified, of at least 30 cells from 3 independent experiments.
Stress Granule FRAP Measurements
Stable mCherry-G3BP1 U2OS cells were cultured in glass bottom dishes (Ibidi) and transfected with GFP-FUS constructs as described above. After 24 hr spontaneous granules (i.e., matured granules), positive for both mCherry and GFP, were analyzed. Induced (“young”) granules represent stress granules formed only after treatment for 1 hr with 0.5mM NaAsO2 (Sigma). GFP granules were bleached and fluorescence recovery after bleaching was monitored using Zen software on a Zeiss LSM 780 Meta NLO confocal microscope. Raw data were background subtracted and normalized using Excel, and plotted using Prism software. FRAP curves were fitted with a one phase exponential curve.
Quantification and Statistical Analysis
Statistical parameters and distributions are reported in the Figures and corresponding Figure Legends. Statistical analysis was performed in Excel or GraphPad Prism.
Acknowledgments
We would like to thank Dr. Nicolas L. Fawzi (Brown University, USA) for his kind gift of recombinant FUS QGSY protein, Dr. Simon Alberti for GFP-FUS expression plasmids (Max Planck Institute, Germany), Dr. Yuh Min Chook for the MBP-FUS expression plasmid (UTSW, USA), Dr. Paul Anderson and Dr. Nancy Kedersha (Harvard Medical School, USA) for the G3BP-mCherry cell line, and Dr. Erik Storkebaum (Radboud University, the Netherlands) for his help with the Drosophila work. Research was funded by KU Leuven, VIB, and the European Research Council in the context of the European Seventh Framework Programme (FP7/2007-2013 Euro-MOTOR, Grant agreement 259867 and ERC Grant Agreement 340429), the Thierry Latran Foundation, the Fund for Scientific Research Flanders (FWO-Vlaanderen), the Interuniversity Attraction Poles Programme P7/16 initiated by the Belgian Science Policy Office, the Association Belge contre les Maladies Neuro-Musculaires (ABMM), the ALS Liga (Belgium), and the “Opening the Future” Fund. L.G. was supported by an EMF/AFAR fellowship, an AARF, and a Target ALS Springboard Fellowship. J. Shorter was supported by Target ALS, the Packard Center for ALS Research, ALSA, and NIH Grant R21NS090205. W.R. is supported through the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders and the “Hart voor ALS” Fund, KU Leuven. P.V.D. is supported by the Alzheimer Research Foundation (SAO-FRA), the Flemish Government-initiated Flanders Impulse Program on Networks for Dementia Research (VIND), and the European Union Joint Programme-Neurodegenerative Disease Research (JPND) Project RiMod-FTD. The Switch laboratory is additionally supported by grants from the European Research Council under the European Union’s Horizon 2020 Framework Programme ERC Grant Agreement 647458 (MANGO) to J. Schymkowitz, the KU Leuven (“Industrieel Onderzoeksfonds”), and the Flanders Agency for Innovation by Science and Technology (IWT) (SBO Grant 60839). S.B. received a PhD fellowship from the Agency for Innovation by Science and Technology (IWT) and an EMBO long-term fellowship. E.B. was supported by a post-doctoral fellowship from FWO-Vlaanderen and an ALSA grant (Grant ID 2169). P.V.D. holds a senior clinical investigatorship from FWO-Vlaanderen.
Author Contributions
E.B., S.B., M.K., L.G., and N.W. planned and performed the experiments. J. Steyaert, W.S., W.H., N.H., J. Schymkowitz, F.R., J. Shorter, P.C., W.R., and P.V.D. provided technical support, reagents, and feedback on the project. M.K., L.G., T.R.C., J. Schymkowitz, F.R., J. Shorter, and P.C. provided ideas for the project and participated in writing the paper. S.B., E.B., and L.V.D.B. wrote the paper.
Declaration of Interests
The authors declare no competing interests.
Published: July 17, 2018
Footnotes
Contributor Information
Steven Boeynaems, Email: steven.boeynaems@kuleuven.vib.be.
Ludo Van Den Bosch, Email: ludo.vandenbosch@kuleuven.vib.be.
Supplemental Information
Document S1. Figures S1–S5 and Tables S1 and S2
Document S2. Article plus Supplemental Information
References
- Boeynaems S., Bogaert E., Van Damme P., Van Den Bosch L. Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016;132:159–173. doi: 10.1007/s00401-016-1586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S., Bogaert E., Kovacs D., Konijnenberg A., Timmerman E., Volkov A., Guharoy M., De Decker M., Jaspers T., Ryan V.H. Phase separation of C9orf72 dipeptide repeats perturbs stress granule dynamics. Mol. Cell. 2017;65:1044–1055.e5. doi: 10.1016/j.molcel.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S., Alberti S., Fawzi N.L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L. Protein phase separation: a new phase in cell biology. Trends Cell Biol. 2018;28:420–435. doi: 10.1016/j.tcb.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borroni B., Bonvicini C., Alberici A., Buratti E., Agosti C., Archetti S., Papetti A., Stuani C., Di Luca M., Gennarelli M., Padovani A. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 2009;30:E974–E983. doi: 10.1002/humu.21100. [DOI] [PubMed] [Google Scholar]
- Brangwynne C.P., Tompa P., Pappu R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015;11:899–904. [Google Scholar]
- Burke K.A., Janke A.M., Rhine C.L., Fawzi N.L. Residue-by-residue view of in vitro FUS granules that bind the C-terminal domain of RNA polymerase II. Mol. Cell. 2015;60:231–241. doi: 10.1016/j.molcel.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Zaro J.L., Shen W.C. Fusion protein linkers: property, design and functionality. Adv. Drug Deliv. Rev. 2013;65:1357–1369. doi: 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J., Hart M.P., Shorter J., DeJesus-Hernandez M., Erion R., Oristano R., Liu A.X., Ramos D., Jethava N., Hosangadi D. A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl. Acad. Sci. USA. 2011;108:20881–20890. doi: 10.1073/pnas.1109434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J., Hart M.P., Erion R., King O.D., Diaz Z., Nakaya T., Ibrahim F., Kim H.J., Mojsilovic-Petrovic J., Panossian S. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012;21:2899–2911. doi: 10.1093/hmg/dds116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle J.G., Lanson N.A., Jr., Smith R.B., Casci I., Maltare A., Monaghan J., Nichols C.D., Kryndushkin D., Shewmaker F., Pandey U.B. RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations. Hum. Mol. Genet. 2013;22:1193–1205. doi: 10.1093/hmg/dds526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D., Rodde R., Edbauer D., Bentmann E., Fischer I., Hruscha A., Than M.E., Mackenzie I.R., Capell A., Schmid B. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D., Madl T., Valori C.F., Bentmann E., Tahirovic S., Abou-Ajram C., Kremmer E., Ansorge O., Mackenzie I.R., Neumann M., Haass C. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012;31:4258–4275. doi: 10.1038/emboj.2012.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L., Kim H.J., Wang H., Monaghan J., Freyermuth F., Sung J.C., O’Donovan K., Fare C.M., Diaz Z., Singh N. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell. 2018;173:677–692.e20. doi: 10.1016/j.cell.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofweber M., Hutten S., Bourgeois B., Spreitzer E., Niedner-Boblenz A., Schifferer M., Ruepp M.D., Simons M., Niessing D., Madl T. Phase separation of FUS is suppressed by its nuclear import receptor and arginine methylation. Cell. 2018;173:706–719.e13. doi: 10.1016/j.cell.2018.03.004. [DOI] [PubMed] [Google Scholar]
- Jäckel S., Summerer A.K., Thömmes C.M., Pan X., Voigt A., Schulz J.B., Rasse T.M., Dormann D., Haass C., Kahle P.J. Nuclear import factor transportin and arginine methyltransferase 1 modify FUS neurotoxicity in Drosophila. Neurobiol. Dis. 2015;74:76–88. doi: 10.1016/j.nbd.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Jain S., Wheeler J.R., Walters R.W., Agrawal A., Barsic A., Parker R. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell. 2016;164:487–498. doi: 10.1016/j.cell.2015.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E., Valdmanis P.N., Dion P., Spiegelman D., McConkey B.J., Vande Velde C., Bouchard J.P., Lacomblez L., Pochigaeva K., Salachas F. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kato M., Han T.W., Xie S., Shi K., Du X., Wu L.C., Mirzaei H., Goldsmith E.J., Longgood J., Pei J. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M., Lin Y., McKnight S.L. Cross-β polymerization and hydrogel formation by low-complexity sequence proteins. Methods. 2017;126:3–11. doi: 10.1016/j.ymeth.2017.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N., Ivanov P., Anderson P. Stress granules and cell signaling: more than just a passing phase? Trends Biochem. Sci. 2013;38:494–506. doi: 10.1016/j.tibs.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha, N., Panas, M.D., Achron, C.A., Lyons, S., Tisdale, S., Hickman, T., Thomas, M., Lieberman, J., McInerney, G.M., Ivanov, P., and Anderson, P. (2016). G3BP-Caprin1-USP10 complexes mediate stress granule condensation and associate with 40S subunits. J Cell Biol. 212, 845–860. [DOI] [PMC free article] [PubMed]
- Kim H.J., Kim N.C., Wang Y.D., Scarborough E.A., Moore J., Diaz Z., MacLea K.S., Freibaum B., Li S., Molliex A. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King O.D., Gitler A.D., Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski T.J., Jr., Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Lanson N.A., Jr., Maltare A., King H., Smith R., Kim J.H., Taylor J.P., Lloyd T.E., Pandey U.B. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet. 2011;20:2510–2523. doi: 10.1093/hmg/ddr150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.H., Zhang P., Kim H.J., Mitrea D.M., Sarkar M., Freibaum B.D., Cika J., Coughlin M., Messing J., Molliex A. C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane-less organelles. Cell. 2016;167:774–788.e17. doi: 10.1016/j.cell.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.R., King O.D., Shorter J., Gitler A.D. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 2013;201:361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Protter D.S., Rosen M.K., Parker R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell. 2015;60:208–219. doi: 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y., Mori E., Kato M., Xiang S., Wu L., Kwon I., McKnight S.L. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target lc domain polymers. Cell. 2016;167:789–802.e12. doi: 10.1016/j.cell.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S.C., Polymenidou M., Cleveland D.W. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrone L., Poser I., Casci I., Japtok J., Reinhardt P., Janosch A., Andree C., Lee H.O., Moebius C., Koerner E. Isogenic FUS-eGFP iPSC reporter lines enable quantification of FUS stress granule pathology that is rescued by drugs inducing autophagy. Stem Cell Reports. 2018;10:375–389. doi: 10.1016/j.stemcr.2017.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A., Temirov J., Lee J., Coughlin M., Kanagaraj A.P., Kim H.J., Mittag T., Taylor J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133. doi: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T., Qamar S., Lin J.Q., Schierle G.S., Rees E., Miyashita A., Costa A.R., Dodd R.B., Chan F.T., Michel C.H. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron. 2015;88:678–690. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Neumann M., Rademakers R., Roeber S., Baker M., Kretzschmar H.A., Mackenzie I.R.A. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdilek B.A., Thompson V.F., Ahmed N.S., White C.I., Batey R.T., Schwartz J.C. Intrinsically disordered RGG/RG domains mediate degenerate specificity in RNA binding. Nucleic Acids Res. 2017;45:7984–7996. doi: 10.1093/nar/gkx460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A., Lee H.O., Jawerth L., Maharana S., Jahnel M., Hein M.Y., Stoynov S., Mahamid J., Saha S., Franzmann T.M. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Qamar S., Wang G., Randle S.J., Ruggeri F.S., Varela J.A., Lin J.Q., Phillips E.C., Miyashita A., Williams D., Strohl F. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-pi interactions. Cell. 2018;173:720–734.e15. doi: 10.1016/j.cell.2018.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami M., Taylor J.P., Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz J.C., Wang X., Podell E.R., Cech T.R. RNA seeds higher-order assembly of FUS protein. Cell Rep. 2013;5:918–925. doi: 10.1016/j.celrep.2013.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova T.A., Robinson H.K., Southcombe J.A., Ninkina N., Buchman V.L. Multistep process of FUS aggregation in the cell cytoplasm involves RNA-dependent and RNA-independent mechanisms. Hum. Mol. Genet. 2014;23:5211–5226. doi: 10.1093/hmg/ddu243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J., Blair I.P., Tripathi V.B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J.C., Williams K.L., Buratti E. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z., Diaz Z., Fang X., Hart M.P., Chesi A., Shorter J., Gitler A.D. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9:e1000614. doi: 10.1371/journal.pbio.1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen B., Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014;10:661–670. doi: 10.1038/nrneurol.2014.184. [DOI] [PubMed] [Google Scholar]
- Vance C., Rogelj B., Hortobágyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Broeck L., Naval-Sánchez M., Adachi Y., Diaper D., Dourlen P., Chapuis J., Kleinberger G., Gistelinck M., Van Broeckhoven C., Lambert J.C. TDP-43 loss-of-function causes neuronal loss due to defective steroid receptor-mediated gene program switching in Drosophila. Cell Rep. 2013;3:160–172. doi: 10.1016/j.celrep.2012.12.014. [DOI] [PubMed] [Google Scholar]
- Yang Y., Gehrke S., Imai Y., Huang Z., Ouyang Y., Wang J.W., Yang L., Beal M.F., Vogel H., Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. USA. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizawa T., Ali R., Jiou J., Fung H.Y.J., Burke K.A., Kim S.J., Lin Y., Peeples W.B., Saltzberg D., Soniat M. Nuclear import receptor inhibits phase separation of FUS through binding to multiple sites. Cell. 2018;173:693–705.e22. doi: 10.1016/j.cell.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1–S5 and Tables S1 and S2
Document S2. Article plus Supplemental Information