Antigen receptor repertoire profiling from RNA-seq data (original) (raw)
. Author manuscript; available in PMC: 2018 Oct 3.
Published in final edited form as: Nat Biotechnol. 2017 Oct 11;35(10):908–911. doi: 10.1038/nbt.3979
Somatic recombination and accumulation of mutations in V-D-J segments results in remarkable heterogeneity of T cell receptor (TCR) and immunoglobulin (Ig) repertoires1, 2. High-throughput profiling of immune receptors has become an important tool for studies of adaptive immunity and for the development of diagnostics, vaccines, and immunotherapies3–7. There are efficient molecular and software tools for the targeted sequencing of TCR and Ig repertoires6, 8 but the material and data for such analysis are not always available.
At the same time, TCR and Ig transcripts are also present in bulk RNA-seq data. Since transcriptome sequencing has become routine in both basic and clinical studies, it could serve as a source of functionally relevant information on immune receptor hypervariable regions (CDR3s) repertoires. Thus the massive repositories of RNA-seq data available from The Cancer Genome Atlas (TCGA, >10,000 tumor samples, https://gdc-portal.nci.nih.gov/) and other databases could be employed for immune repertoire profiling. Such analysis is of particular interest in cancer immunotherapy studies. Available tumor tissue is often limited, which precludes splitting the samples for separate transcriptome, TCR, and Ig profiling. Separate immune repertoire profiling also adds complexity and increases the costs for massive clinical studies.
Furthermore, transcriptomic analysis is often employed in comparative studies of functional T- and B-cell subsets9–13, and could additionally yield the immune receptor repertoires at no cost.
Several groups have reported tools for TCR or Ig repertoire extraction from bulk14–17 or single-cell18 RNA-seq data. However, a broadly applicable software that enables the accurate and efficient extraction of immune repertoires from RNA-seq was not available.
Here we developed a tool on the basis of MiXCR19, implementing a set of algorithms (see Online Methods, Supplementary Note 1, Supplementary Figures 1,2, Supplementary Tables 1,2) to extract as many true CDR3 sequences as possible, with nearly zero amount of CDR3-like false-positives. The key algorithms include: 1) a sensitive and highly selective aligner that employs fast algorithm but switches to a more sensitive modified Smith-Waterman/Needleman-Wunsch algorithm in ambiguous cases; 2) a partial alignments assembler that builds contigs from several initial alignments in a manner protected from artificial diversity generation (doi.org/10.5281/zenodo.804326); and 3) CDR3 extension (for TCRs but not Igs, due to possible presence of hypermutations) that fills in the edges of the CDR3 based on known information on the relevant germline gene segments. The resulting RNA-seq analysis pipeline employs the same MiXCR modules, the same error-correction algorithms, and has the same output format as for targeted TCR or Ig profiling. This allows unified processing and comparison of immune repertoires obtained from different types of raw sequencing data.
Software testing with _in silico_-generated data demonstrated the high extraction efficiency of MiXCR, with zero false-positive clones observed. TRUST software14, 17 efficiency was an order of magnitude lower, and the software generated a substantial number of false clonotypes (Online Methods, Supplementary Fig. 3).
To further verify the efficiency and specificity of extracting TCR CDR3 repertoires from RNA-seq data, we performed deep targeted profiling of TCR alpha (TRA) and beta (TRB) chain repertoires (TCR-seq) as described20 and 100+100 bp paired-end RNA-seq for the same RNA samples, obtained from surgically resected melanoma specimens from two patients, SPX6730 (ileocecal lymph node metastasis) and SPX8151 (small intestine resection).
We next assessed the dependence of the number of TCR-seq-confirmed RNA-seq-extracted clonotypes on their abundance estimated from TCR-seq data. MiXCR was able to extract all relatively large TRB-CDR3 clonotypes (frequency in repertoire > 0.15%) from the lymph node metastasis RNA-seq, even with the short paired-end reads (trimmed in silico to 50+50 bp). In contrast, TRUST failed to extract a considerable proportion of high-frequency clonotypes (Fig. 1a and Supplementary Fig. 2). Most MiXCR-reported clonotypes were confirmed by the control TCR-seq data, while only a minor fraction of CDR3s reported by TRUST could be confirmed (Fig. 1b). The clonotype frequencies extracted by MiXCR from the RNA-seq correlated with TCR-seq (Fig. 1c), demonstrating that RNA-seq-based profiling can be quantitative for large clonotypes in samples that contain a substantial number of T-cells.
Figure 1. Sensitivity and specificity of TCR and Ig repertoires extraction from tumor RNA-seq data.
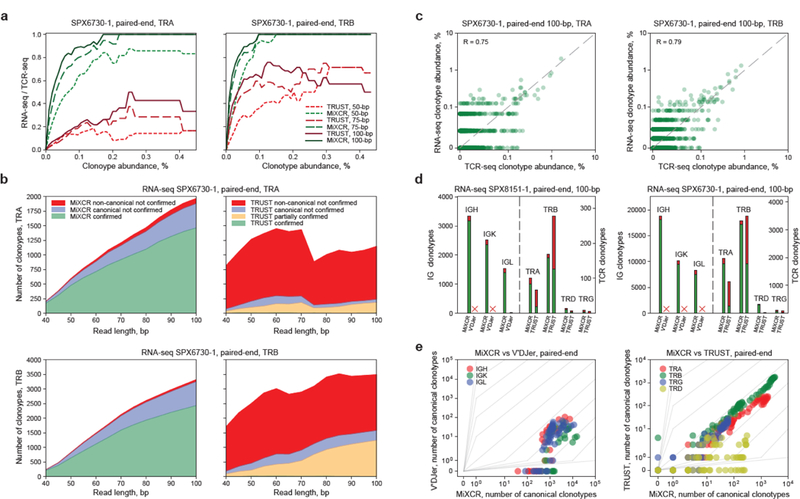
a. Dependence of the share of TCR-seq-confirmed clonotypes extracted from RNA-seq on clonotype abundance, tumor sample SPX6730. The x-axis corresponds to clonotype frequency A, as estimated from TCR-seq. The y-axis corresponds to the fraction of identified clonotypes in the total number of control clonotypes with abundance greater than A. b. Dependence of the number of TCR-seq-confirmed clonotypes (green), canonical unconfirmed clonotypes (blue), and non-canonical unconfirmed clonotypes (red) on the paired-end sequencing read length for SPX6730 sample. For TRUST data, orange denotes partially confirmed (allowing for truncation of 6 nucleotides) clonotypes. c. Correlation of TCR clonotype frequencies in MiXCR-extracted repertoires from TCR-seq and RNA-seq data from SPX6730 sample. d. Extraction of Ig and TCR CDR3 repertoires from SPX8151 and SPX6730 RNA-seq by MiXCR, TRUST and V’DJer. Cross indicates that analysis took too long (> 4 days running in 8 computing threads on Intel Xeon CPU E5–2683 v3 @ 2.00GHz with 50 GB of RAM). Canonical CDR3 clonotypes are shown in green, non-canonical clonotypes are shown in red. e. Extraction of Ig CDR3 repertoires with V’DJer and MiXCR (left) and TCR CDR3 repertoires with TRUST and MiXCR (right) from representative RNA-seq datasets. Each circle indicates the number of clonotypes extracted. Gray graduations indicate the fold-difference in the number of extracted clonotypes.
We also compared MiXCR with V’DJer software16 for the extraction of Ig repertoires. For some samples (including large SPX6730 and SPX8151 RNA-seq datasets) V’DJer failed to extract Ig heavy chain (IGH) and light chains (IGL, IGK) repertoires within four days using 8 threads on a Xeon E5–2683 CPU with 50 GB of RAM. MiXCR successfully extracted repertoires for IGH, IGK, IGL, TRA, and TRB chains from both melanoma samples, and outperformed both V’DJer and TRUST, yielding many-fold more canonical and minimal numbers of non-canonical clonotypes (Fig. 1d). We additionally used several samples analyzed previously by others14, 17. In all comparisons, MiXCR demonstrated superior sensitivity, extracting 10–1,000-fold more clonotypes compared to V’DJer (Fig. 1e). Both V’DJer and TRUST require substantially more hands-on time and implementation of third-party alignment tools with particular versions of the human genome and particular analysis settings, which are not clearly defined in the documentation and required optimization. The output from both tools irretrievably loses useful biological information (Supplementary Table 1).
In single T-cell transcriptome analysis, MiXCR outperformed TraCeR18 in efficiency of TRA and TRB chains detection (Supplementary Table 3).
Next, we extracted repertoires from TCGA 48+48-bp paired-end RNA-seq for 458 patients with cutaneous melanoma (SKCM) (see doi:10.6084/m9.figshare.4620739 for clonesets, Supplementary Note 2 for details). Notably, the obtained Ig repertoires were an order of magnitude larger than the TCRs, indicating the presence of intratumoral Ig-producing plasma cells.
High intratumoral IGH expression levels were associated with longer survival (Fig. 2a), in agreement with recent work showing a positive correlation of activated B-cell gene signatures with survival in SKCM21. High levels of IGH clonality (analyzed as in ref. 22) were also associated with longer survival (Fig. 2b, Supplementary Fig. 4a), and the two parameters had strong cumulative value. Patients with both high IGH expression and clonality had the best prognosis, while high IGH expression with low clonality was associated with poor prognosis (Fig. 2c).
Figure 2. Immune repertoire analysis in human melanoma and sorted mice T cells.
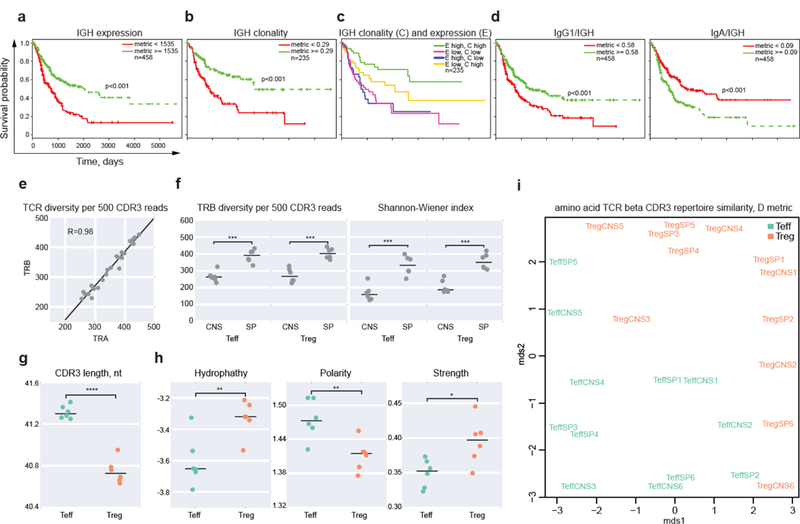
a-d. Kaplan–Meier plots depicting the survival probability over time relative to repertoire-related metrics for TCGA SKCM patient samples. Samples were divided into high (>=cutoff) and low (<cutoff) value cohorts based on the median for the relevant metric. a. IGH expression (reads per million). b. IGH clonality. Samples with at least 500 IGH CDR3 sequencing reads were included to minimize the influence of repertoire depth on clonality index. c. IGH expression and clonality. d. Kaplan-Meier plots showing survival probability relative to IgG1/IGH and IgA/IGH proportions. p values reflect log-rank test for survival difference between low and high metrics groups. n, number of patients. e-i. Comparative analysis of CD4 effector and regulatory T-cell repertoires. e. Correlation of observed TRA versus TRB CDR3 diversity per 500 unique CDR3-covering reads. f. TRB diversity observed per 500 randomly-sampled CDR3-containing reads (left), and estimated using the Shannon-Wiener index (right) for the spleen (SP) and central nervous system (CNS) samples from Foxp3 yfpcre mice with induced experimental autoimmune encephalomyelitis (EAE). g. Average TRB CDR3 lengths for Teff and Treg subsets in spleen, weighted for clonotype size. h. Functional characteristics of the middle portion of CDR3 amino acid sequence for Teff and Treg TRB repertoires in the spleen. Plots show hydropathy, polarity, and strength metrics as derived from VDJtools software, weighted for clonotype size. i. Multi dimensional space analysis of TRB CDR3 amino acid repertoire overlaps, as derived with VDJtools, metric D. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (paired, two-tailed t test).
In many patients, a single dominant intratumoral Ig clonotype occupied 30–80% of all Ig CDR3 sequences in both heavy and light chains repertoires. Hypermutating IGH CDR3 variants could be observed even in primary tumor samples (Supplementary Table 4, Supplementary Fig. 4b), which could reflect the presence of intratumoral germinal centers. Observation of extra-large Ig expansions and hypermutation processes in tumors raises the question of antigenic specificity of dominant Ig variants, for which functional heavy and light chain pairs can be identified based on frequency23. If such dominant intratumorally-produced Igs are tumor-specific24, exploration of their usefulness in precision immunotherapy—for example, in the context of chimeric antigen receptors (CARs25)—would be of considerable interest.
We found that high proportion of IgG1 of all IGH transcripts was associated with longer survival (Fig. 2d, Supplementary Fig. 4), suggesting that intratumorally-produced clonal IgG1 antibodies could exert opsonizing anti-tumor effects. High IgA/IGH proportion was associated with a negative prognosis, consistent with recent work showing that IgA-producing plasma cells function as potent immunosuppressors through the secretion of IL-10 and PD-L126. Collectively, these results indicate that intratumorally produced Igs may represent an important component of the anti-tumor response, and extraction of Ig clonal repertoires from RNA-seq data could be a potential source of clinically useful biomarkers for cancer immunotherapy.
To test the feasibility of extracting TCR repertoire sequences from sorted T cells, we performed 50+50-bp paired-end RNA-seq for the effector (Teff) and regulatory (Treg) CD4 T-cells from spleen and central nervous system (CNS) of Foxp3 yfpcre mice27 with induced experimental autoimmune encephalomyelitis (see Supplementary Note 3 for details). The near-100% abundance of T-cells in these samples allowed MiXCR to extract high-quality TCR repertoires comprising at average 1330/3295 TRA and 1489/3933 TRB unique functional CDR3 clonotypes/CDR3 sequencing reads per sample (Supplementary Table 5, doi:10.6084/m9.figshare.4620739).
Extracted repertoires were suitable for routine post-analysis using the VDJtools software28. To compare TCR diversity between these T cell subsets, we normalized samples by extracting 500 random CDR3-containing reads, representing fragments of unique template RNA molecules from each sample. The diversity correlated well between TRA and TRB repertoires (R>0.95, Fig. 2e), was similar between Teff and Treg cells, and was significantly lower in CNS compared to spleen (P<0.001, paired two-tailed t-test, Fig. 2f, reflecting the narrowed TCR repertoire in the CNS.
Treg cells were characterized by shorter TRB CDR3 lengths (Fig. 2g). The functional characteristics of the amino acids comprising the middle portion of CDR3 differed between Treg and Teff cells TRB repertoires (Fig. 2h). The higher interaction “strength” (a relative number of strongly interacting amino acids29) of Treg CDR3s is in keeping with the previously observed higher TCR affinity of Tregs for self-peptide:MHC complexes, which may enable Treg precursors to compete more efficiently for the limited amount of antigen presented on thymic antigen-presenting cells30–32. Analysis of amino acid TRB CDR3 repertoire overlaps revealed separate clustering of Teff and Treg cells. Co-clustering of the same T-cell subsets of multiple animals indicates their functional similarity across mice (Fig. 2i). We conclude that detailed and highly informative insights into the structure of TCR repertoires can be obtained from RNA-seq data of sorted T-cell subsets.
RNA-seq analysis is rapidly becoming routine, both in clinical practice and basic research, with implementation gaining steam as the cost of high-throughput sequencing decreases and read-length increases. The MiXCR pipeline now enables mining these datasets straightforwardly for immune repertoires profiling. We anticipate that in the near future RNA-seq-based profiling may replace targeted profiling of TCR and Ig repertoires in many applications.
The software, comprehensive user documentation and source code can be found at https://github.com/milaboratory/mixcr. Our software is continuously upgraded, and the source code and stable binary releases are freely available for non-commercial use. Human, mouse and rat references are built in, and IMGT (http://www.imgt.org/) references can also be used for analysis.
Online Methods
Back-to-back RNA-seq versus TCR-seq comparison
Tumor samples SPX6730 and SPX8151 were collected from two patients with advanced melanoma, following signed informed consent and after approval of the medical ethical committee at NKI-AVL. Material was a resection taken from the ileocecum. Morphological analysis showed that SPX6730 was an ileocecal lymph node metastasis, while SPX8151 was a small intestine resection.
Total RNA was extracted using TRIzol reagent (15596–018, Ambion life technologies) according to the manufacturer’s protocol. Each tissue sample in TRIzol was split on 3 portions. Extracted total RNA was purified using the MinElute Cleanup Kit (74204, Qiagen) according to the manufacturer’s instructions. Quality and quantity of the total RNA was assessed by the 2100 Bioanalyzer using a Nano chip (Agilent, Santa Clara, CA). Total RNA samples having RIN>8 were subjected to library generation. For the RNA-seq analysis, strand-specific libraries were generated from total RNA using the TruSeq Stranded mRNA sample preparation kit (Illumina Inc., San Diego, RS-122–2101/2) according to the manufacturer’s instructions (Illumina, Part # 15031047 Rev. E), in two replicas. The resulting libraries were sequenced with 100 bp paired-end reads on a HiSeq2500 using V4 chemistry (Illumina Inc., San Diego). The second portion of total RNA of each sample was used for the targeted TRA and TRB profiling as described20, using Illumina MiSeq paired end 150+150 bp sequencing.
The TCR-seq control data were analysed using standard MiXCR analysis pipeline for targeted TCR profiling data19. The resulting assembled clonal CDR3 sequences for TRA and TRB chains were used as control data.
Performance of extraction of repertoire data from RNA-seq heavily depends on read length and reads topology (paired-end or single-end). Each read from initial RNA-seq samples was in silico cut to several lengths from 40 bp to 100 bp with 5 bp increment to simulate RNA-seq samples with shorter reads. This way based on each initial 100+100 bp RNA-seq sample we generated 26 samples simulating different sequencing setups (40 bp, 45 bp, … 95 bp, 100 bp, single- / paired-end).
We compared MiXCR with TRUST software17. The prototype of TRUST was used in one of the widest14 studies on repertoire extraction from RNA-seq known to date. In the case of MiXCR we have directly tested the equality of CDR3 nucleotide sequences between RNA-seq and TCR-seq. TRUST does not assemble reported contigs into full CDR3 clonal sequences, so unique CDR3s reported by TRUST were used for comparison. The majority of CDR3s reported by TRUST are truncated in a nondeterministic way (dramatically complicating any further analysis), so strict CDR3 equality-based matching with control data gives almost no results. Thus, we used the following workaround to describe the performance of underlying TCR extraction algorithms: we allowed a partial match between CDR3s from control data and TRUST reported CDR3s. Namely we allowed N maximal possibly truncated nucleotides. TRUST CDR3 is considered to match some control CDR3 if it’s nucleotide sequence is contained in the control nt sequence (of course it can be equal) and the control sequence is not more than N nucleotides longer than the TRUST sequence. For example, for N = 6 the following TRUST sequences are considered as confirmed by the control:
Control CDR3: TGTGCCAGTAGTATAGAACATAGCAATCAGCCCCAGCATTTT TRUST matched CDR3: TGTGCCAGTAGTATAGAACATAGCAATCAGCCCCAGCATTTT TRUST matched CDR3: …GCCAGTAGTATAGAACATAGCAATCAGCCCCAGCAT… TRUST matched CDR3: ….CCAGTAGTATAGAACATAGCAATCAGCCCCAGCATTT. TRUST matched CDR3: TGTGCCAGTAGTATAGAACATAGCAATCAGCCCCAGCATT..
The value N = 6 was used for all calculations in this paper, this value was chosen because it gave biggest intersection with control data, while greater values showed substantial number of false-matches (matches of exclusively germline-derived regions).
The primary characteristic of repertoire extraction software for RNA-seq data is it’s ability to extract as many true CDR3 sequences as possible while preserving nearly zero false-positive CDR3 calls. For further verification of the efficiency and specificity of TCR CDR3 repertoires extraction on the RNA-seq samples with TCR-seq control we introduced the following metrics:
- Total number of reported RNA-seq CDR3 clonotypes
- Number of reported RNA-seq CDR3s that are also present in TCR-seq control Such verified CDR3’s are considered as definitely true-positive ones.
- Number of reported non-canonical RNA-seq CDR3s that are absent in TCR-seq control. CDR3s that found in RNA-seq sample but not found in TCR-seq control can be either true-positive or false-positive. It is natural to differentiate such clones in two groups: with canonical and with non-canonical amino acid sequences. The former are more likely true-positive, while the latter are more likely false-positive ones. This assumption is also supported by the fact that over 85% of clones in control TCR-seq samples have canonical CDR3 a.a. sequences:
Control sample TRA clonotypes TRB clonotypes % non-canonical TRA % non-canonical TRB SPX6730 168,572 269,648 13.2% 3.3% SPX8151 16,427 30,451 14.7% 6.7%
Canonical CDR3 amino acid sequence was defined as matching regex ^C[^_]*(?:[FW]|[FW]G.G)$, where [FW]G.G in the end of the sequence are allowed to match CDR3s from TRUST output with displaced right CDR3 boundary actually being canonical. No CDR3s with displaced right boundary were detected in MiXCR output.
TCR/Ig repertoires extraction from samples without control
Apart from the data generated specifically for this paper, we used several representative samples from TCGA (CDC file IDs: 9986414f-26b7–4c3b-90cd-feb59dc033a2 (old id: b6c7b112–81cb-4ae6-b604–1564eabc7aec), 0d674012–2baa-4b6e-afe5–735f19990a52 (old id: fed55b2d-2c04–4a5b-b714–38962c061f72), 92e1bb3e-7f6d-4128-bb6f-8cbbab1444a6 (old id 3d44d6c3–5dcf-42ee-8ebe-dcdaad30493c)) and SRA (SRR1813898, SRR1813883 и SRR2314045), which underwent in silico cut procedure from 40 bp to 50 bp with 5 bp increment. Additionally, each sample was analyzed in two ways: (i) taking both mates from paired-end reads (R1 and R2 fastq files; paired-end analysis) and (ii) taking only first mate (R1 fastq file; single-end analysis). This procedure gave us final set of 140 samples for analysis (including SPX6730 and SPX8141):
- SPX6730 and SPX8141 both have two replicas (SPX6730–1, SPX6730–2, SPX8141–1, SPX8141–2), in silico truncated from 40bp to 100bp with 5bp increment in paired-end and single-end modes --- 104 samples
- 3 TCGA samples, in silico truncated from 40bp to 50bp in paired-end and single-end modes --- 18 samples
- 3 SRA samples, in silico truncated from 40bp to 50bp in paired-end and single-end modes --- 18 samples
To test the performance of the software on samples without control data we calculated two metrics:
- Total number of reported CDR3 clonotypes
- Number of reported CDR3s with non-canonical sequence
In the case of MiXCR and TRUST which both can report CDR3 amino acid translation we considered canonical amino acid sequence as matching ^C[^_]*(?:[FW]|[FW]G.G)$, where [FW]G.G in the end of the sequence are allowed to match CDR3s from TRUST output with displaced right CDR3 boundary actually being canonical (no CDR3s with displaced right boundary detected in MiXCR output).
In the case of V’DJer which reports only CDR3 nucleotide sequences we used the following criteria of canonicity. Canonical CDR3 nucleotide sequence defined as:- a)
matching regex ^TG[TC].*(?:TT[TC]|TGG)$ pattern - b)
not matching (?:…)*(?:TAA|TAG|TGA)(?:…)* pattern - c)
sequence length is multiple of 3
- a)
For IGH, IGK, and IGL MiXCR extracted at average 2 orders of magnitude more CDR3 clonotypes than V’DJer, preserving small fraction of non-canonical clonotypes (https://doi.org/10.6084/m9.figshare.4620739)
MiXCR aligner optimization for RNA-seq data
Since there is no prior knowledge on true TCR/Ig sequences in real data and because some sources of false-positive sequences are not random (see section Reproducible false positives below), implementation of fully automated procedure for optimization of alignment algorithm is substantially complicated. To find the best aligner parameters for analysis of RNA-seq data we performed the following steps:
- Manual analysis of results obtained on real RNA-seq samples showed that in order to find scoring values that better discriminate between false-aligned non-TCR/Ig and true-aligned TCR/Ig sequences, V-gene alignment must be forcefully extended to the 5’ end of the sequence and J-gene alignment must be forcefully extended to the 3’ end of the sequence (even if it lowers total score of the alignment; in other words alignment must be global on the one side in respect to read sequence while being local on another side). Built-in MiXCR aligner (KAligner) can be setup to work in this regime by setting floatingLeftBound or floatingRightBound parameters to false. As a result, two modifications to the default parameters were made: - OvParameters.floatingLeftBound=false, -OjParameters.floatingRightBound=false. Similar parameter is also present in software like STAR33 (--alignEndsType parameter), but there it is not possible to specify different values for different genes.
- Multi-target optimization using NSGAII34 algorithm implemented in MOEA Framework (http://moeaframework.org/ ) was performed on all numeric parameters of V and J aligners as well as minimal scoring thresholds to find Pareto optimal set of parameters in terms of number of false-alignments calculated on RNA-seq samples known to have zero TCR/Ig-content (while actually containing trace amounts of target molecules and non-canonical transcripts from non-rearranged TCR/Ig loci) and true-positive TCR/Ig sequences generated using procedure and software reported in ref. 19. From this set, solution having best performance on true TCR/Ig data while having no real-false-positive alignments on control samples was manually selected.
- Further analysis of results obtained on real RNA-seq samples revealed rare false-positive alignments for several non-canonical transcripts from TCR/Ig loci. Scoring thresholds for V and J alignments as well as threshold on the total V,D,J and C alignments were adjusted to reliably exclude these false-positives. Adjustment did not substantially change overall sensitivity of the aligner.
Resulting parameter values can be found in “RNA-seq” section of parameter preset file (https://github.com/milaboratory/mixcr/blob/develop/src/main/resources/parameters/vdjcalignerparameters.json).
Results of false-alignment rate evaluation during our initial parameters optimization (not shown here) and results provided in section “**Performance of MiXCR and TRUST on in silico data**” below, had not reveal any false-alignments on a wide range of input dataset types, suggesting zero observed false-positive rate for MiXCR.
In silico generated data
We used in silico modeled RNA-seq data to estimate several characteristics of MiXCR and other software packages under consideration in maximally controlled setup. VDJ recombination model from Ref. 1 was used to generate TRB VDJ recombinations. Generator is available as easy to use software util – repseqio (https://github.com/repseqio/repseqio, doi:10.5281/zenodo.804326). We used well-known ART software package35 to model Illumina reads from generated TRB sequences. To introduce negative control sequences into our data we modeled reads from the collection of Gencode Human transcripts (ftp://ftp.sanger.ac.uk/pub/gencode/Gencode_human/release_26/gencode.v26.transcripts.fa.gz, Ref. 36). The following procedures were used in each case:
To simulate RNA-seq reads from rearranged TRB genes, fasta file containing first three exons of mature TRB gene transcript sequences were generated using repseqio tool according to model from ref. 1. Transcripts were repeated several times to simulate power-law distribution of clone abundances (Pareto distribution with parameters Xm = 0.9, alpha = 1.3). art_illumina tool was used to simulate RNA-seq Illumina reads from transcripts generated on the previous step.
Negative control (non-TCR) RNA-seq reads were simulated from human transcript sequence collection downloaded from Gencode using art_illumina tool.
Results from this chapter can be easily reproduced using publically available Docker (https://www.docker.com/community-edition) image (see section Data, software and source code below).
Performance of MiXCR and TRUST on in silico data
Using the procedure described above, we generated RNA-seq reads for 10^3 in silico TRB clones and randomly picked 10^3 reads (results are invariant to the number of clones in initial dataset due to the scale invariance of Pareto distribution used to model clonotype abundances).
For negative control we generated 10^8 non-TCR RNA-seq reads. Finally, we have prepared two datasets for analysis by MiXCR and TRUST:
- 10^8 non-TCR reads (negative control sample)
- Dataset obtained by mixing 10^8 of non-TCR reads with 10^3 TRB reads (approximately reproducing average RNA-seq TCR content)
The analysis was repeated for 50-bp, 75-bp and 100-bp read lengths. Each analysis was performed for both paired-end and single-end (using only R1) modes.
STAR aligner33 was used to produce BAM files for TRUST. For the script which runs the whole analysis (from in silico data generation to runnig of MiXCR and TRUST) as well as a Docker container see section Data, software and source code below).
CDR3s reported by MiXCR and TRUST were compared against the originally in silico generated clones allowing up to 3 mismatches or indels (mutations are allowed to account for possible mutations in CDR3s introduced by art_illumina).
Supplementary Fig. 3 shows the results of MiXCR and TRUST performance comparison for datasets described above. MiXCR shows consistently higher extraction efficiency. On a paired-end data MiXCR showed order of magnitude higher efficiency. Most CDR3 confirmed sequences for TRUST have three mismatches or indels differences from true CDR3 sequence, while nearly all MiXCR clonotypes are supported by the exact match. The major part of CDR3s reported by TRUST were not found in original set of synthetic clones, even with three mutations allowed. Moreover, TRUST have reported CDR3 records even for negative control data, supporting observation that it is prone to extract false CDR3 sequences from non-TCR targets (see section Reproducible false positives below).
Estimation of rates for false-overlaps and false-extensions on in silico generated data
In silico generated data was used to estimate potential false-diversity that may be introduced during partial alignments assembly (assemblePartial) and incomplete alignments extension for TCR reads (extendAlignments) steps on different types of input data. Individual overlaps and extensions performed by the software were analysed to calculate the values.
Datasets consisting entirely of TRB reads was generated using procedure described above. We tested all possible combinations of the following dataset parameters:
- clonotype count: 10^2, 10^3, 10^4
- number of reads: 10^3, 10^4, 10^5
- read length: 50, 75, 100
- art_illumina sequencing errors profile: HS20, HS25, NS50
Overlap events where emerged CDR3 was different from any of true known CDR3s of reads participated in overlap were defined as false-overlaps. Because further analysis performs quality-aware error-correction, and does not form new clones from CDR3 sequences having quality score less than 20 in some position, we also calculated number of false-overlaps having enough quality scores in CDR3 region to form new clonotypes during further pipeline steps.
For datasets with number of modelled clonotypes 10^2 and 10^3, which represent typical RNA-seq datasets, false-overlap rate was less than 0.04% and potentially introduced artificial diversity was no more than 0.006% across all combinations of other parameters, while in most cases we observed zero values for both characteristics. Worst observed value for false-overlap rate across all parameters including untypically diverse samples with 10^4 clones was 0.25% and maximal potentially introduced artificial diversity was less than 0.09%.
The worst overall artificial diversity introduced by false-overlaps was nearly two orders of magnitude less than typical artificial diversity introduced by sequencing and PCR errors, making it’s effect negligible for further analysis.
The same datasets were used for estimation of false-extension rate. Analysis of each individual extension event was performed. Extension was defined as false if subsequence introduced by the procedure was different from corresponding subsequence from originally generated CDR3 sequence for current alignment. Total 195461 extensions were performed by the software, and only two of them were inconsistent with the sequences expected from original CDR3. This gives approximately one in 10^5 false extension rate.
Another potential source of false-extensions is allelic variants of certain V/J segments, which are not present in the reference library. Unfortunately extent to which this phenomenon can affect performance of the procedure is very hard to estimate. Still, there is a possible solution for the issue, consisting of learning of allelic variants right from the target sample. This functionality is planned for implementation in the described software.
Calculation of potential repertoire information content for RNA-seq datasets
Because MiXCR RNA-seq mode showed best CDR3 extraction efficiency from RNA-seq data among software packages for this task known to date, we used it to estimate current technical limit for this procedure.
Analysis was performed on (i) melanoma samples SPX6730 and SPX8151 truncated in silico to 100, 75 and 50 bp, (ii) all samples for the functional characterization of TRA and TRB repertoires based on sorted T-cells RNA-seq, (iii) all the samples from ref. 18, and (iv) three samples from TCGA used in our comparison with TRUST. All samples were analysed by MiXCR in both paired- (PE) and single-end (SE) variants (by utilizing only R1 fastq file). Following characteristics were calculated:
- - sum of TRBC1 and TRBC2 expression levels in terms of reads count (calculated using Kallisto software package37). This characteristic was chosen instead of transcripts per million (TMP) because it is proportional to the total number of reads in the sample, which is also the case for the number of CDR3 calls, removing the need for any additional normalization.
- - number of MiXCR TRB CDR3 calls that left after all processing steps and participated in clonotype formation.
PE and SE samples with the same read length showed good correlation between two described characteristics. Supplementary Table 2 shows relation between TRBC coverage and CDR3 sequence yield for different sequencing setups.
Reproducible false positives
One of the main sources of false-positive alignment calls are fragments of mRNA molecules homologous to antibody or TCR hypervariable regions. Such false-positives are especially dangerous, because they could be reproduced in different samples, thereby providing misleading evidence about connection between TCR/Ig repertoires. In our experience, substantial part of such sequences came from unprocessed or wrongly spliced TCR/Ig or non-TCR/Ig mRNA molecules. Due to this fact, given that specificity of extraction software is low, absence or existence of certain false sequences in the output may e.g. be correlated with efficiency of nonsense-mediated mRNA decay, which in turn may lead to false conclusion on correlation between certain physiological conditions and TCR/IG repertoire composition. Many other similar sources of reproducible false-positives may also exist. In view of this we believe that high specificity of RNA-seq repertoire extraction software is crucial for obtaining reliable biological results.
In the case of TRUST a substantial part of reported contigs can be directly mapped to raw genome (i.e. clearly are false-positives) which shows low software specificity.
We have found TRUST assembled contigs for most of analysed datasets annotated, with corresponding false-positive CDR3, which entirely maps onto certain genomic regions in TRB and TRA loci. Such sequences presumably came from immature mRNA molecules, not fully processed by nonsense mediated decay and splicing. This observation suggest low software specificity and potential risk of observing false intersections between samples due to other than immunological physiological reasons.
Example of TRUST false-positive fasta record that was reproduced in two different patients (samples RNASeq_SPX6730–2 and RNASeq_SPX8151–1, 50 bp, paired-end) and fully maps to genomic intron region near TRBC1 gene, which shows that low specificity of the software may lead to false matches between repertoires:
RNASeq_SPX8151- 1_cut50_PAIREDEND.bam+est_clonal_freq=0.0243902439024+seq_length=50+est_lib_size=11717+TRB V5-404_TRBV4-301_TRBV1401_TRBV5-502_TRBV4-303_TRBV203_TRBV1302_TRBV7- 801_TRBV1902_TRBV7-204_TRBV7-201_TRBV5-801_TRBV10-201_TRBV6-601_TRBV7- 902_TRBV1903_TRBV4-201_TRBV5-503_TRBV5-802_TRBV6-401_TRBV7-905_TRBV6- 602_TRBV7-202_TRBV10-102_TRBV11-302_TRBV6-501_TRBV6-603_TRBV7-904_TRBV11- 201_TRBV11-203_TRBV7-803_TRBV11-301_TRBV6-101_TRBV7-401_TRBV7- 702_TRBV1603_TRBV4-102_TRBV1601_TRBV3-101_TRBV901_TRBV2701_TRBV7- 907_TRBV25-101_TRBV1402_TRBV5-401_TRBV903_TRBV12-501_TRBV7-301_TRBV5- 403_TRBV5-402_TRBV10-101_TRBV4-302_TRBV6-901_TRBV12-402_TRBV12-401_TRBV4- 202_TRBV7-701_TRBV5-601_TRBV6-801_TRBV7-802_TRBV6-301_TRBV12-301_TRBV7- 601_TRBV202_TRBV4-304_TRBV7-602_TRBV7-305_TRBV902_TRBV6-605_TRBV11- 303_TRBV11-101_TRBV5-501_TRBV5-101_TRBV6-604_TRBV3-102_TRBV201_TRBV4- 101_TRBV7-906_TRBV6-402_TRBV7-903_TRBV5-102_TRBV11-202_TRBV2801_TRBV6- 201_TRBV7-901_TRBV7-304_TRBV1301_TRBV1901_TRBV18*01++TRBC2|chr7:142498725- 142499111+CASLRPIPRALSH+minus_log_Eval=1.90138771133+TGTGCTTCATTACGGCCCATTCCCAGGGCT CTCTCTCACAC GGACATAGTTGTGCTTCATTACGGCCCATTCCCAGGGCTCTCTCTCACAC
Another example of TRUST false-positive fasta record that was reproduced in two different samples and fully maps to genomic intron region, which proofs that situation described above takes place for the TRUST software:
Sample#1: SRR2314045, paired-end, 45bp Sample#2: fed55b2d-2c04-4a5b-b714-38962c061f72, paired-end, 45bp Record#1:
SRR2314045_cut45_PAIREDEND.bam+est_clonal_freq=0.0238095238095+seq_length=45+est_lib_size =6832+TRAV2501_TRAV3501_TRAV2701++TRBC2|chr7:142498725- 142499111+CAGQRPCVDGHGQ+minus_log_Eval=3.0+TGTGCTGGTCAGCGCCCTTGTGTTGATGGCCATGGTC AAGAG CTCTTGACCATGGCCATCAACACAAGGGCGCTGACCAGCACAGCA Record#2: fed55b2d-2c04-4a5b-b714- 38962c061f72_cut45_PAIREDEND.bam+est_clonal_freq=0.0238095238095+seq_length=45+est_lib_size =53346+TRAV2501_TRAV3501_TRAV2701++TRBC2|chr7:142498725- 142499111+CAGQRPCVDGHGQ+minus_log_Eval=3.0+TGTGCTGGTCAGCGCCCTTGTGTTGATGGCCATGGTC AAGAG CTCTTGACCATGGCCATCAACACAAGGGCGCTGACCAGCACAGCA
Several other examples of such false-intersection between samples were found in TRUST results.
Analysis of Ig isotype proportions
We run Kallisto37 with default parameters and calculated estimated counts for all the transcripts of human transcriptome GRCh37.75 (http://www.ensembl.org/info/website/tutorials/grch37.html). Gene expressions were calculated as sum of estimated counts over its transcripts. Coverages of IgA isotype were taken from IGHA1 and IGHA2 genes. We calculated relative coverages dividing estimated count of each isotype by sum of estimated counts over all the isoforms. Pseudogenes IGHEP1, IGHEP2, IGHGP and IGHMBP2 demonstrated relatively low expression and were not taken into account.
RNA-seq from the sorted murine T-cell functional subsets
Six male Foxp3 yfpcr e mice27 (B6.129(Cg)-Foxp3 tm4(YFP/icre)Ayr/J) mice that were 10 weeks of age were used for EAE induction. Treatment of mice was performed according to protocol 08–10-023 approved by the Memorial Sloan Kettering Institute Institutional Animal Care and Use Committee. All mice were maintained in the Memorial Sloan Kettering Institute animal facility under specific pathogen-free conditions in accordance with institutional guidelines. Mice were immunized s.c. with 100 ug MOG35–55 peptide (GenScript) emulsified in CFA (Sigma) and injected i.p. with 200 ng pertussis toxin (EMD chemicals) on day 0 and day 2 post immunization. Spleen and pooled brain and spinal chord (CNS) were harvested at day 16 (peak of disease). CNS tissue was minced and digested for 45 minutes in the presence of Collagenase A and DNaseI (Sigma) and lymphocytes were enriched on a Percoll gradient (30%/37%/70%)(GE healthcare). CD4+ T-cells were isolated with the Dynabeads FlowComp Mouse CD4 kit (Life Technologies). Splenic and CNS derived effector T-cells (Teff: TCRb+CD4+Foxp3-(YFP-)CD62L-D44+) or effector regulatory T-cells (Treg: TCRb+CD4+Foxp3+(YFP+)CD62L-CD44+) were sorted into Trizol LS (Life Technologies) using a BD FACS-AriaII cell sorter. For splenic Treg and Teff subsets, we sorted 1×105 cells, for the CNS we sorted 4,000–24,000 Teff cells and 500–2,400 Treg cells per sample (Supplementary Table 5). RNA was extracted, amplified using SMARTer technology and libraries were prepared using Illumina TruSeq paired-end library preparation. Samples were sequenced on the Illumina HiSeq 2500 platform to an average depth of 25 × 106 50 bp paired-end reads per sample. PCR duplicates were removed from fastq files using clumpify utility from BBMap (https://sourceforge.net/projects/bbmap/), in order to count unique RNA fragments. Comparative post-analysis was performed using VDJtools software28.
Extraction of TCR and Ig repertoires from TCGA SKCM data
PCR duplicates were removed using FastUniq software38. Extraction was performed by MiXCR as described in the next section.
Running software
On each sample a standard analysis pipeline for MiXCR, TRUST and V’DJer was performed (since V’DJer works only with paired-end data, it was executed only on paired-end samples). The results of each run were analyzed for all available immunological chains (all TCR and Ig for MiXCR, TCR only for TRUST and Ig only for V’DJer). The following sections describe exact commands executed for each software:
MiXCR
Standard alignment procedure for RNA-seq data onto all IG/TCR loci preserving reads that not fully cover CDR3 (-OallowPartialAlignments=true).
Single-end analysis:
mixcr align -p RNA-seq -OallowPartialAlignments=true sample_R1.fastq.gz alignments.vdjca
Paired-end analysis:
mixcr align -p RNA-seq -OallowPartialAlignments=true sample_R1.fastq.gz sample_R2.fastq.gz alignments.vdjca
The following commands were the same for single- and paired- end analysis.
Performing two rounds of contig assembly from alignments not covering full CDR3 sequence
mixcr assemblePartial -p alignments.vdjca alignments_rescued_1.vdjca mixcr assemblePartial -p alignments_rescued_1.vdjca alignments_rescued_2.vdjca
Extend V/J junctions of TCRs with not fully covered sequence of CDR3 and unambiguously assigned with V and J genes, up to complete CDR3 sequence:
mixcr extendAlignments alignments_rescued_2.vdjca alignments_rescued_2_extended.vdjca
Assemble clonotypes for the final vdjca files:
mixcr assemble -ObadQualityThreshold=0 alignments_rescued_2_extended.vdjca alignments_rescued_2_extended.clns
Exporting clonotypes
mixcr exportClones -c ${CHAIN}
alignments_rescued_2_extended.clns
alignments_rescued_2_extended.clns.${CHAIN}.txt
Where ${CHAIN} should be replaced by a particular immunological chain (TRA/TRB/TRG/TRD for specific TCR chain, TCR for all TCR chains, IGH/IGK/IGL for specific IG chain, IG for all IG chains, ALL for all TCR and IG chains), or can be skipped in order to export clones for all possible immunological chains (both TCR and IG).
STAR
Running STAR on input samples:
./STAR --readFilesCommand zcat --runThreadN 8 --genomeDir /path/to/star/hg38 -- outSAMtype BAM SortedByCoordinate --outSAMunmapped Within --outStd BAM_SortedByCoordinate --readFilesIn sample_R1.fastq.gz [sample_R2.fastq.gz] > sample.sort.bam
Preparing BAM for analysis:
samtools index sample.sort.bam
TopHat
Running TopHat on input samples
tophat -p 8 /path/to/hg19 sample_R1.fastq.gz [sample_R2.fastq.gz]
Adding all unmapped reads to final bam file
samtools merge sample.unsorted.bam accepted_hits.bam unmapped.bam
Prepare bam file for analysis
samtools sort --threads 8 -o sample.sort.bam sample.unsorted.bam samtools index sample.sort.bam
TRUST
TopHat aligner was used to produce bam files in all tests except tests with in silico data (described in section “Performance of MiXCR and TRUST on in silico data”) where STAR aligner was used.
Running TRUST
python TRUST.py -f sample.sort.bam -a -H
V’DJer
STAR aligner was applied as described above to produce bam files for V’DJer.
Running V’DJer:
./vdjer --in sample.sort.bam --t 8 --ins 175 --chain ${CHAIN} --ref-dir vdjer_human_references/$(echo ${CHAIN} | tr ‘[:upper:]’ ‘[:lower:]’)” > sample.sam 2> sample.log
In the last command ${CHAIN} should be replaced with one of IGH, IGK, IGL (capital letters). V’DJer requires three separate runs to analyze all IG chains. Since the execution time of V’DJer on some samples is extremely large (for some samples V’DJer was running for a week in 8 threads occupying 50Gb of RAM and still did not succeed) we constrained V’DJer execution time by 11 hours (running in 8 computing threads). V’DJer analysis which took more than 11 hours we considered as failed due to timeout.
In order to build STAR reference for V’DJer we used recent hg38 from http://www.gencodegenes.org and the following command:
./STAR --runMode genomeGenerate --runThreadN 52 --genomeDir /path/to/star/hg38/ -- genomeFastaFiles GRCh38.p7.genome.fa --sjdbGTFfile gencode.v25.chr_patch_hapl_scaff.annotation.gtf
The software versions were as follows:
MiXCR: 2.1.3 (doi:10.5281/zenodo.804046) Samtools: 1.3.1 TopHat:2.1.1 TRUST17 STAR: 2.4.2a V’DJer: 0.12
All execution commands and software versions were approved by the authors of corresponding software (we have contacted to both TRUST and V’DJer developers).
Statistical tests used
On the Kaplan-Meier plots showing survival probability relative to IgG1/IGH and IgA/IGH proportions (Fig. 2a-d), p values reflect log-rank test for survival difference between low and high metrics groups, divided based on the median for the relevant metric. For the comparison of TCR CDR3 repertoires characteristics (Fig. 2f-h), paired two-tailed t-test was used.
Data availability and accession code availability
Raw sequencing data generated for the paper is deposited in SRA under BioProject id PRJNA371303. Analysis results using MiXCR, TRUST and V’DJer software are available at https://doi.org/10.6084/m9.figshare.4620739.
Software and source code described in this paper:
- MiXCR: 2.1.3 (doi:10.5281/zenodo.804046; https://github.com/milaboratory/mixcr/releases/tag/v2.1.3)
- RepSeq.IO util, version 1.2.8, including artificial clones generator (doi: 10.5281/zenodo.804326; https://github.com/repseqio/repseqio/releases/tag/v1.2.8)
- Docker image and scripts to reproduce in-silico experiments (published in DockerHub uder id: milaboratory/RNA-seq-paper:v1.0; source code https://github.com/milaboratory/mixcr-RNA-seq-paper/releases/tag/v1.0). See README.md from source code package for usage instruction.
Other software used in the paper:
- Samtools version 1.3.1 (https://github.com/samtools/samtools/releases/tag/1.3.1)
- TopHat version 2.1.1 (https://ccb.jhu.edu/software/tophat/downloads/tophat-2.1.1.Linux_x86_64.tar.gz )
- TRUST, version published with17
- STAR version 2.5.3a (https://github.com/alexdobin/STAR/releases/tag/2.5.3a)
- V’DJer version 0.12 (https://github.com/mozack/vdjer/releases/tag/v0.12)
- Kalisto version 0.43.1 (https://github.com/pachterlab/kallisto/releases/tag/v0.43.1)
Supplementary Material
Supplementary
Acknowledgments
Research supported by grant of the Ministry of Education and Science of the Russian Federation Number 14.W03.31.0005. Human melanoma SPX6730 and SPX8151 sample preparation supported by European Union’s Horizon 2020 Research and Innovation Programme Number 633592 (to T.N. Schumacher). Foxp3yfpcre mice sample preparation supported by NIH/NCI Cancer Center Support Grant P30 CA008748 (to A.Y. Rudensky).
Footnotes
Competing interests
MiLaboratory LLC develops MiXCR software and has exclusive rights for its commercial distribution.
References
- 1.Murugan A, Mora T, Walczak AM & Callan CG Jr. Proc Natl Acad Sci U S A 109, 16161–16166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elhanati Y et al. Philos Trans R Soc Lond B Biol Sci 370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greiff V, Miho E, Menzel U & Reddy ST Trends Immunol 36, 738–749 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Attaf M, Huseby E & Sewell AK Cell Mol Immunol 12, 391–399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyd SD & Crowe JE Jr. Curr Opin Immunol 40, 103–109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heather JM, Ismail M, Oakes T & Chain B Briefings in bioinformatics (2017). [DOI] [PMC free article] [PubMed]
- 7.Hackl H, Charoentong P, Finotello F & Trajanoski Z Nature reviews. Genetics 17, 441–458 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Georgiou G et al. Nat Biotechnol 32, 158–168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casero D et al. Nat Immunol 16, 1282–1291 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanduri K et al. Genome medicine 7, 122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinstein JS et al. Blood 124, 3719–3729 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pulko V et al. Nat Immunol 17, 966–975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bottcher JP et al. Nature communications 6, 8306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li B et al. Nature genetics 48, 725–732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown SD, Raeburn LA & Holt RA Genome medicine 7, 125 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mose LE et al. Bioinformatics 32, 3729–3734 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B et al. Nature genetics 49, 482–483 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stubbington MJ et al. Nat Methods 13, 329–332 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolotin DA et al. Nat Methods 12, 380–381 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Britanova OV et al. J Immunol 196, 5005–5013 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Charoentong P et al. Cell reports 18, 248–262 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Tumeh PC et al. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy ST et al. Nat Biotechnol 28, 965–969 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Kotlan B et al. J Immunol 175, 2278–2285 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Khalil DN, Smith EL, Brentjens RJ & Wolchok JD Nature reviews. Clinical oncology 13, 273–290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shalapour S et al. Nature 521, 94–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubtsov YP et al. Immunity 28, 546–558 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Shugay M et al. PLoS computational biology 11, e1004503 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kosmrlj A, Jha AK, Huseby ES, Kardar M & Chakraborty AK Proc Natl Acad Sci U S A 105, 16671–16676 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jordan MS et al. Nature immunology 2, 301–306 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Feng Y et al. Nature 528, 132–136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsieh CS, Zheng Y, Liang Y, Fontenot JD & Rudensky AY Nature immunology 7, 401–410 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Dobin A et al. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deb K, Anand A & Joshi D Evolutionary computation 10, 371–395 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Huang W, Li L, Myers JR & Marth GT Bioinformatics 28, 593–594 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harrow J et al. Genome Res 22, 1760–1774 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bray NL, Pimentel H, Melsted P & Pachter L Nat Biotechnol 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Xu H et al. PLoS One 7, e52249 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary
Data Availability Statement
Raw sequencing data generated for the paper is deposited in SRA under BioProject id PRJNA371303. Analysis results using MiXCR, TRUST and V’DJer software are available at https://doi.org/10.6084/m9.figshare.4620739.
Software and source code described in this paper:
- MiXCR: 2.1.3 (doi:10.5281/zenodo.804046; https://github.com/milaboratory/mixcr/releases/tag/v2.1.3)
- RepSeq.IO util, version 1.2.8, including artificial clones generator (doi: 10.5281/zenodo.804326; https://github.com/repseqio/repseqio/releases/tag/v1.2.8)
- Docker image and scripts to reproduce in-silico experiments (published in DockerHub uder id: milaboratory/RNA-seq-paper:v1.0; source code https://github.com/milaboratory/mixcr-RNA-seq-paper/releases/tag/v1.0). See README.md from source code package for usage instruction.
Other software used in the paper:
- Samtools version 1.3.1 (https://github.com/samtools/samtools/releases/tag/1.3.1)
- TopHat version 2.1.1 (https://ccb.jhu.edu/software/tophat/downloads/tophat-2.1.1.Linux_x86_64.tar.gz )
- TRUST, version published with17
- STAR version 2.5.3a (https://github.com/alexdobin/STAR/releases/tag/2.5.3a)
- V’DJer version 0.12 (https://github.com/mozack/vdjer/releases/tag/v0.12)
- Kalisto version 0.43.1 (https://github.com/pachterlab/kallisto/releases/tag/v0.43.1)