Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells (original) (raw)
Summary
Checkpoint inhibitors have revolutionized cancer treatment. However, only a minority of patients respond to these immunotherapies. Here, we report that blocking the inhibitory NKG2A receptor enhances tumor immunity by promoting both natural killer (NK) and CD8+ T cell effector functions in mice and humans. Monalizumab, a humanized anti-NKG2A antibody, enhanced NK cell activity against various tumor cells and rescued CD8+ T cell function in combination with PD-x axis blockade. Monalizumab also stimulated NK cell activity against antibody-coated target cells. Interim results of a phase II trial of monalizumab plus cetuximab in previously treated squamous cell carcinoma of the head and neck showed a 31% objective response rate. Most common adverse events were fatigue (17%), pyrexia (13%), and headache (10%). NKG2A targeting with monalizumab is thus a novel checkpoint inhibitory mechanism promoting anti-tumor immunity by enhancing the activity of both T and NK cells, which may complement first-generation immunotherapies against cancer.
Keywords: immunce checkpoint inhibitor, cancer immunotherapy, inhibitory receptors, therapeutic monoclonal antibodies, lymphocytes, natural killer cells, CD8+ T cells
Graphical Abstract
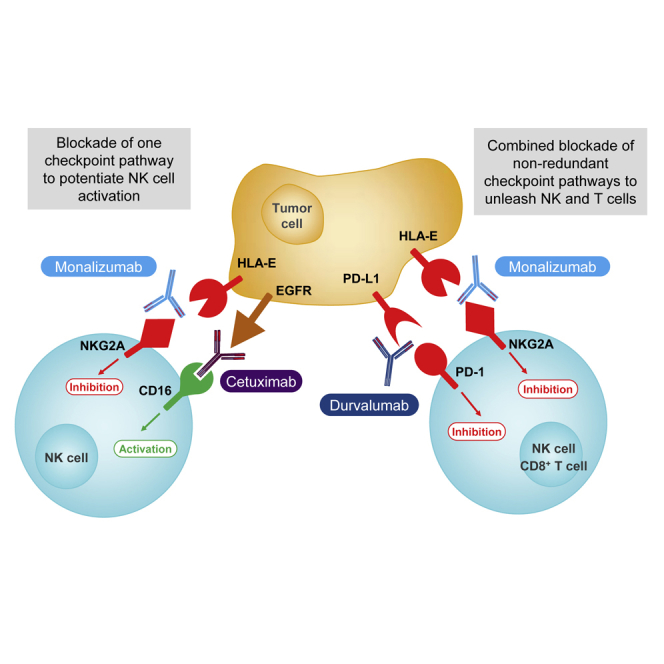
Highlights
- •
Blocking NKG2A unleashes both T and NK cell effector functions - •
Combined blocking of the NKG2A and the PD-1 axis promotes anti-tumor immunity - •
Blocking NKG2A and triggering CD16 illustrates the efficacy of dual checkpoint therapy
Blocking of the NKG2A inhibitory receptor unleashes both T and NK cells, and demonstrates anti-tumor efficacy in combination with anti-EGFR or with anti-PD-x antibodies.
Introduction
Immuno-oncology has revolutionized cancer treatment (Okazaki and Honjo, 2007, Okazaki et al., 2013, Baumeister et al., 2016, Schumacher and Schreiber, 2015, Sharma and Allison, 2015a, Sharma and Allison, 2015b). Unprecedented improvements in tumor control have been achieved using therapeutic monoclonal antibodies (mAbs) that block immune inhibitory “checkpoint” receptors. In particular, mAbs directed against the PD-1 (programmed-cell death protein 1)/PD-L1 (programmed -cell death ligand 1) axis (PDx) in monotherapy or combination therapy have been approved for the treatment of several indications, including metastatic melanoma, non-small-cell lung cancer, kidney cancer, bladder cancer, Hodgkin lymphoma, and solid tumors that are microsatellite instability-high or mismatched repair-deficient (Okazaki and Honjo, 2007, Okazaki et al., 2013, Baumeister et al., 2016, Schumacher and Schreiber, 2015, Sharma and Allison, 2015a, Sharma and Allison, 2015b). Such treatment often yields durable benefits, and, in most patients, toxicity can be controlled. However, only a subset of the patients treated with antibodies specific for PD-1 or PD-L1 display a strong response, and the cancers of a substantial fraction of patients are resistant to these immune checkpoint inhibitors (ICI). Therefore, one of the major challenges in immuno-oncology is understanding the mechanisms of resistance to ICI, to increase the proportion of patients benefiting from such treatment and to control treatment toxicity. One approach that could be used is to identify novel molecular targets, the modulation of which boosts anti-tumor immunity. Blocking other inhibitory pathways of effector lymphocytes, such as T cells and NK cells, is attracting considerable research interest in this context.
Cell surface receptors harboring intracytoplasmic tyrosine-based inhibitory motifs (ITIMs) are particularly relevant in this respect. These motifs are phosphorylated and recruit the phosphatases (SHP-1/2 or SHIP) responsible for transmitting the inhibition signal to immune effector cells (Daëron et al., 2008). Bioinformatics analyses of the human genome have predicted the presence of more than 300 type I and type II integral membrane proteins containing at least one ITIM domain (Daëron et al., 2008), but only a few of these receptors are currently targeted in therapeutic approaches.
NKG2A is an ITIM-bearing receptor expressed on both T and NK cells. Approximately half of peripheral blood NK cells express NKG2A (André et al., 1999, Mahapatra et al., 2017, Manser and Uhrberg, 2016) and its expression on NK cells can be upregulated upon stimulation with cytokines, such as interleukin-15 (IL-15) (Brady et al., 2004, Mori et al., 1998). In healthy individuals, around 5% of human peripheral blood CD8+ T cells express cell-surface NKG2A at steady state, but this expression can be upregulated by chronic antigenic stimulation (Bertone et al., 1999, McMahon et al., 2002, Mingari et al., 1998, Sheu et al., 2005). NKG2A is expressed at the cell surface as a heterodimer with CD94 in humans and mice and recognizes the non-classical class I major histocompatibility complex (MHC-I) molecules human leukocyte antigen (HLA)-E in humans and Qa-1b in mice. Binding of NKG2A/CD94 to its cognate ligand inhibits T and NK cell effector functions (Le Dréan et al., 1998, Rapaport et al., 2015). This inhibition is dependent on the recruitment of the SHP-1 tyrosine phosphatase to the tyrosine-phosphorylated form of the ITIM in NKG2A (Viant et al., 2014).
Here, we show that NKG2A blockade enhances the anti-tumor immunity mediated by NK and CD8+ T cells. We developed a humanized anti-NKG2A immunoglobulin G (IgG) 4-blocking mAb (monalizumab), and we describe its anti-tumor efficacy in vitro and in vivo when used as a single agent or in combination with other therapeutic antibodies, such as durvalumab, blocking PD-L1, or cetuximab, directed against the epidermal growth factor receptor (EGFR), which is expressed by tumor cells.
Results
NKG2A Blockade Promotes Anti-tumor Immunity
We assessed the impact of NKG2A on cytotoxic lymphocyte activity by using BALB/c B cell lymphoma A20 cells, which express the non-classical MHC-I Qa-1b molecule, the mouse homolog of HLA-E, and generating the corresponding Qa-1b-knockout cells (Figure S1A). The growth rates of parental and Qa-1b-deficient A20 cells were similar in vitro (data not shown). As expected, the frequency of activated NKG2A+ NK cells—assessed based on the expression of CD107a, a degranulation marker—was higher in cocultures with Qa-1b-deficient A20 cells than in cocultures with parental cells (data not shown). Following their subcutaneous injection into syngeneic BALB/c mice, wild-type A20 B cell lymphoma cells progressively grew in all mice (Figure 1A, left panel). By contrast, 70% of the mice into which genetically engineered Qa-1b-deficient A20 cells were injected did not display tumor growth (Figure 1A, right panel). Both NK cells and CD8+ T cells were required to control tumor growth, because the administration of anti-asialo-GM1 and anti-CD8α mAbs, respectively, into tumor-bearing mice abolished the control of parental and Qa-1b-deficient tumor growth and led to premature death (Figures 1B and 1C). These results validate Qa-1b as a potentially useful target.
Figure S1.

NKG2A Is an Inhibitory Receptor that Blocks the Anti-tumor Efficacy of NK and CD8+ T Cells, Related to Figure 1
(A) FACS histograms showing Qa-1b expression on A20 and A20 Qa-1b KO cells after stimulation with IFN-γ. White histograms: isotype control; gray histograms: anti-Qa-1b mAb. Numbers indicate the median fluorescence intensity.
(B) NK cells were co-cultured with Qa-1b-deficient YAC-1 or Qa-1b-expressing A20 cells or targets in the presence of an anti-NKG2A mAb (m20d5) or an isotype control (IC). CD107a degranulation was measured and is represented on box and whiskers plots, with crosses to represent the mean values. The data presented are the pooled results of three independent experiments (n = 7). Wilcoxon matched-pairs signed rank test, ∗p = 0.0156.
(C) NKG2A+PD-1+CD8+ TILs were stimulated in vitro with A20 tumor cells in the presence of the indicated mAbs. The frequencies of CD107a-producing cells are shown. The data presented are the pooled results of four independent experiments (n = 15). One-way ANOVA followed by Dunn’s test, ∗p = 0.043, ∗∗p = 0.0014, ∗∗∗p = 0.0005, ∗∗∗∗p < 0.0001.
Figure 1.

NKG2A Is an Inhibitory Receptor that Blocks the Anti-tumor Efficacy of NK and CD8+ T Cells
(A) Qa-1b-sufficient or -deficient A20 tumor cells were engrafted subcutaneously (s.c.) in BALB/c mice.
(B) BALB/c mice were treated with an anti-aGM1 pAbs or with control rabbit serum, an anti-CD8α mAb, or rat IgG2b isotype control and then subcutaneously engrafted with A20 tumor cells. Graphs show tumor growth in each individual mouse and combined survival curves. Complete regressions are indicated. log rank test, ∗∗p = 0.0020; ns, no significant.
(C) Experiment similar to that in (B), but with Qa-1b KO A20 tumor cells. Complete regressions are indicated. log rank test, ∗∗∗p = 0.0002 (NK cell depletion) and ∗∗∗p = 0.0006 (CD8+ T cell depletion).
See also Figure S1.
We then dissected the immune response to A20 in the tumor bed by analyzing tumor-infiltrating lymphocytes (TILs). A20 tumors were found to be infiltrated by NK and CD8+ T cells. ∼60% of tumor-infiltrating NK cells expressed the NKG2A receptor (Figure 2A). We also monitored PD-1 expression, because the immune control of A20 tumors has been reported to be partially dependent on PD-1 (Sagiv-Barfi et al., 2015). The expression of PD-1, either alone or together with NKG2A, was barely detectable on the surface of tumor-infiltrating NK cells. We did not observe NKG2A expression on the surface of CD8+ T cells from the spleen, and few cells expressed PD-1 (∼0.5%) (Figure 2A). However, PD-1+ CD8+ T cells accounted for ∼45% of TILs. Importantly, NKG2A was also expressed on the surface of around half the PD-1+ CD8+ TILs. In this model, we also observed that double-positive PD-1+ NKG2A+ CD8+ TILs displayed higher levels of PD-1 and NKG2A expression at their surface than cells positive only for PD-1 or for NKG2A (Figure 2A). Very few CD8+ TILs (∼2%) expressed NKG2A without PD-1.
Figure 2.

Combined Blockade of NKG2A and PD-1/PD-L1 Promotes Anti-tumor Immunity in A20 Tumor-Bearing BALB/c Mice
(A) Flow cytometry characterization of NK and CD8+ TILs 19 days after A20 tumor cells engraftment. The spleen was used as control. Upper panels: representative fluorescence-activated cell sorting (FACS) profiles of PD-1 and NKG2A expression on NK and CD8+ T cells in the spleen and the tumor bed. Lower panels: pie chart analysis (mean ± SD). The data presented are the pooled results of three independent experiments (n = 12).
(B) A20 tumor cells were engrafted in BALB/c mice. Tumor-bearing mice were then treated at 3- to 4-day intervals with an isotype control (IC), anti-NKG2A, anti-PD-L1, or a combination of these last two mAbs. Graphs show tumor growth in each individual mouse and combined survival curves. The data presented are the pooled results of three independent experiments. Complete regression are indicated. log rank test, ∗∗p = 0.0087; ∗∗∗p = 0.0001; ∗∗∗∗p < 0.0001.
(C) Experiment similar to that described in (B) but with treatment of the mice with an anti-asialo-GM1 pAbs or an anti-CD8α mAb 1 day before the initiation of immunotherapy with the combination of anti-NKG2A and anti-PD-L1 mAbs. Graphs show tumor growth in each individual and combined survival curves. Complete regression are indicated. log rank test, ∗p < 0.0016; ∗∗p < 0.01; ∗∗∗p = 0.0001.
See also Figure S2.
We then investigated whether NKG2A blockade could promote anti-tumor immunity. We generated a recombinant mouse version of the rat anti-mouse NKG2A antibody 20d5 (Vance et al., 1999). We confirmed that the blockade of NKG2A in vitro promoted the expression of CD107a by NK cells cocultured with Qa-1B+ A20 tumors, but not with Qa-1B− YAC-1 target cells (Figure S1B). When used as single agents in vitro, anti-NKG2A and anti-PD-L1 mAbs only modestly improved ex vivo tumor-infiltrating CD8+ T cell effector activities after restimulation with A20 cells (Figure S1C). By contrast, the use of anti-NKG2A and anti-PD-L1 mAbs in combination increased the frequency of CD107a-expressing NKG2A+ PD-1+ CD8+ TILs.
We further investigated the effects of immunotherapy with anti-NKG2A and anti-PD-L1 mAbs by treating A20 tumor-bearing mice with anti-NKG2A mAb, anti-PD-L1 mAb, or a combination of both blocking reagents (Figure 2B). In this experimental setting, anti-NKG2A mAb did not rescue mice from death when used as a single agent when compared to control group. By contrast, anti-PD-L1 mAb rescued ∼40% of tumor-bearing mice from death, as shown by comparison with untreated mice. Interestingly, a combination of anti-NKG2A and anti-PD-L1 mAbs had a synergistic effect, improving the control of tumor growth and rescuing ∼75% of the mice from death (Figure 2B). The results obtained for mice treated with anti-asialo-GM1 or anti-CD8α antibodies also demonstrated that the anti-tumor effect of the anti-NKG2A/PD-L1 mAbs combination therapy was dependent on both NK and CD8+ T cells (Figure 2C). Thus, the combination of a blocking anti-NKG2A mAb with a blocking anti-PD-L1 mAb had a therapeutic anti-tumor effect, because it unleashed NK cells and CD8+ T cells in the A20 model. Similar results were obtained with a combination of anti-NKG2A/PD-1 mAbs (Figure S2).
Figure S2.

The Combined Blockade of NKG2A and PD-1/PD-L1 Promotes Anti-tumor Immunity in A20 Tumor-Bearing BALB/c Mice, Related to Figure 2
(A) A20 tumor cells were engrafted in BALB/c mice. Tumor-bearing mice were then treated at three- to four-day intervals with isotype control (IC) antibody, anti-NKG2A antibody, anti-PD-1 antibody or a combination of these last two antibodies. Graphs show tumor growth in each individual and combined survival curves. The data presented are the pooled results of two independent experiments. Log-rank test, ∗∗p = 0.0087, ∗∗∗p = 0.0001, ∗∗∗∗p < 0.0001.
(B) Experiment similar to that described in (A) but with treatment of the mice with an anti-asialo-GM1 pAbs or an anti-CD8α mAb one day before the initiation of immunotherapy. Graphs show tumor growth in each individual and combined survival curves. Log Rank test, ∗p < 0.0016, ∗∗p < 0.01, ∗∗∗p = 0.0001.
Combined Blockade of NKG2A and PD-L1 Promotes the Generation of Protective Anti-tumor Memory
We investigated the anti-tumor therapeutic properties of the anti-NKG2A mAb further by using this antibody to treat C57BL/6 mice bearing another tumor, i.e., the subcutaneously injected RMA-Rae-1β T lymphoma. Like A20 cells, RMA-Rae-1β tumor cells express Qa-1b and PD-L1 (Figure S3). The frequency of NKG2A+ NK cells in the tumor was higher than that in the spleen, but tumor-infiltrating NK cells did not express PD-1, as observed in the A20 model (Figure 3A). We found that ∼20% of total CD8+ TILs expressed NKG2A, but not PD-1; ∼15% expressed both these molecules; and ∼20% expressed PD-1, but not NKG2A (Figure 3A). Neither anti-NKG2A mAb nor anti-PD-L1 monotherapy was effective in RMA-Rae-1β tumor-bearing mice (Figure 3B). However, treatment with a combination of mAbs against NKG2A and PD-L1 resulted in tumor growth control in 45% of the tumor-bearing mice, which were rescued from death. The combination therapy acted through the release of a CD8+ T cell, but not an NK cell inhibition, as the injection of a depleting anti-CD8α mAb, but not anti-NK1.1 mAbs, abolished tumor growth control and impaired mouse survival (Figure 3C).
Figure S3.

Qa-1b and PD-L1 Expression on RMA Rae-1β Tumor Cells, Related to Figure 3
FACS histograms showing Qa-1b and PD-L1 expression on RMA Rae-1β tumor cells. White histograms: isotype control; gray histograms: anti-Qa-1b or anti-PD-L1 mAbs. Numbers indicate the median fluorescence intensity.
Figure 3.

Combined Blockade of NKG2A and PD-1/PD-L1 Promotes Anti-Tumor Immunity in RMA Rae-1β Tumor-Bearing C57BL/6J Mice
(A) RMA Rae-1β tumor cells were injected subcutaneously into C57BL/6J mice. Flow cytometry characterization of NK and CD8+ TILs 12 days post-injection, with the spleen used as the standard. Upper panels: representative FACS profiles of PD-1 and NKG2A expression at the surface of NK and CD8+ T cells in the spleen and the tumor bed. Lower panels: pie chart analysis (mean ± SD). The data presented are the pooled results of two independent experiments (n = 8 mice).
(B) RMA Rae-1β tumor-bearing C57BL/6J mice were treated with IC antibodies, anti-NKG2A, anti-PD-L1, or a combination of these last two mAbs. Graphs show tumor growth in each individual mouse and combined survival curves. The data presented are the pooled results of four independent experiments. Complete regressions are indicated. log rank test, ∗∗∗∗p < 0.0001.
(C) Experiment similar to that in (B), except that the mice were treated with anti-NK1.1 mAb or anti-aCD8α mAb 1 day before the initiation of immunotherapy with the combination of anti-NKG2A and anti-PD-L1 mAbs. Graphs show tumor growth in each individual mouse and combined survival curves. Complete regressions are indicated. log rank test, ∗∗p = 0.0024.
(D) Upper left panels: FACS profiles of CD44 and CD62L expression on CD8+ T cells in the spleen of naive (no tumor) mice, mice receiving their first injection of RMA Rae-1β tumor cells (RMA Rae-1β), and mice previously injected with RMA Rae-1β tumors, cured by immunotherapy and rechallenged (RMA Rae-1β + mAbs rechallenged). Percentages of naive (CD44−CD62L+), central memory (TCM, CD44+CD62L+), effector memory (TEM, CD44+CD62L−) and effector CD8+ T cells (eff, CD44−CD62L−) are indicated. Upper right panel: absolute numbers of effector memory CD8+ T cells in the spleen are shown. Lines represent medians. Lower panels: RMA Rae-1β tumor-bearing C57BL/6J mice were treated with IC antibody or with a combination of anti-NKG2A and anti-PD-L1 mAbs. Mice cured by immunotherapy (n = 13) were rechallenged subcutaneously with RMA-Rae-1β tumor cells after 70 days. Untreated C57BL/6J mice (n = 15) also received injections of RMA-Rae-1β cells as a control. The graphs show tumor growth in each individual mouse. The data presented are the pooled results of two independent experiments.
See also Figure S3.
We observed the generation of CD62L− CD44+ effector memory CD8+ T cells in the spleens of mice in which RMA-Rae-1β tumors were implanted and then cured by immunotherapy, but not in the spleens of untreated mice (Figure 3D). Accordingly, RMA-Rae-1β tumor cells were completely rejected when injected into mice that had already been injected with the tumor and cured by treatment with anti-NKG2A and anti-PD-L1 mAbs, whereas the injection of these cells led to unchecked tumor growth in untreated mice (Figure 3D). Therefore, in addition to curing mice of their implanted tumors, blocking NKG2A in combination with another ICI can promote durable protective anti-tumor CD8+ T cell memory response in a preclinical mouse model.
HLA-E and NKG2A Expression in Human Tumors
We then monitored the expression of NKG2A and HLA-E at the surface of several human tumors, to identify the indications for which anti-NKG2A therapeutic blocking mAbs might promote anti-tumor immunity in cancer patients. HLA-E was found to be widely expressed on the surfaces of several human tumors. We observed HLA-E expression in lung, pancreas, stomach, colon, head and neck, and liver tumor tissues (Figure 4; Table S1). By contrast, PD-L1 expression was restricted to a fraction of lung, stomach, and colon tumors (Figure S4). HLA-E was strongly expressed by squamous cell carcinoma of the head and neck (SCCHN) and colorectal carcinoma (CRC) (Figures 4A–4C), in which we also detected NKG2A-positive cells. NKG2A-positive cells and HLA-E expression were also found in ovarian, endometrial, CRC, and cervical cancer (Figure 4C). NKp46+ NK and CD8+ TILs were also present in all these tumors. We investigated SCCHN more closely by flow cytometry and detected high frequencies of CD8+ TILs expressing PD-1 and co-expressing both PD-1 and NKG2A in the tumor (Figure 4D). NKG2A-expressing NK cells were also present at high frequency, and some of these cells had a PD-1+ NKG2A+ phenotype. Similar results were obtained for CRC and lung tumors (data not shown). Thus, several tumors expressed HLA-E and were infiltrated with NK and CD8+ TILs expressing NKG2A. Therefore, we reasoned that NKG2A blockade, either alone or together with the use of other checkpoint inhibitors, such as anti-PD-1/PD-L1 antibodies, might improve the anti-tumor efficacy of NK and CD8+ TILs in cancer patients.
Figure 4.

CD8+, NKp46+ and NKG2A+ Immune Cells Are Present in Several Types of HLA-E-Expressing Solid Cancers
(A) Representative example of HLA-E and NKG2A expression on frozen sections from SCCHN and CRC cancer samples. Bright-field images were inverted, and RGB channel splitting was performed. Pseudocolors were attributed to each marker (blue for hematoxylin and green for HLA-E or NKG2A). Scale bars represent 500 μm for low magnification or 50 μm for right inserts.
(B) Semiquantitative analysis of HLA-E expression on formalin-fixed paraffin-embedded (FFPE) SCCHN samples (n = 65). HLA-E expression was assessed on tumor cells (TC), lymphocytes (Ly), and endothelial cells (Endo). Score 1 = 1%–33%; score 2 = 34%–66%; score 3 ≥ 66% of positive cell.
(C) Semiquantitative analysis of NKG2A-, NKp46-, and CD8-positive cells and of HLA-E expression on colorectal cancer (n = 48), ovarian cancer (n = 40), endometrial cancer (n = 40) and cervical cancer (n = 17). CD8, NKp46 and NKG2A cells were quantified in the tumor nest (TN) and stroma (S). HLA-E expression was assessed separately on tumor cells (TC) and lymphocytes (Ly.). Score 1 = 1%–33%; Score 2 = 34%–66%; Score 3 ≥ 66% of positive cells.
(D) Percentages of NK cells (upper panels) and CD8+ T cells (lower panels) expressing NKG2A and PD-1 in SCCHN cancer samples. Cells from WB (whole blood, n = 23), LN (normal lymph node, n = 6), meta LN (metastatic lymph node, n = 12), adj (healthy tissue adjacent to the tumor, n = 8), and tumor (n = 13) were analyzed by flow cytometry. Box and whiskers plot, in which the means are indicated by crosses. Kruskal-Wallis analysis followed by Dunn’s multiple comparisons test. ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001; ∗∗∗∗p < 0.0001.
See also Figure S4 and Table S1.
Figure S4.

HLA-E and PD-L1 Expression in Solid Cancers, Related to Figure 4
Formalin-fixed paraffin-embedded (FFPE) tissue microarrays were stained by immunohistochemistry with anti-HLA-E (clone MEM-E/02) or anti-PD-L1 (clone E1L3N) antibodies. For each indication, scatterplots represent the percentage of the area stained for HLA-E or PD-L1 for each spot. Quantification was performed with Halo (Indicalabs). Colored bars indicate the mean (±SD). Lung (n = 45), pancreas (n = 79), stomach (n = 76), colon (n = 109), H&N (n = 70) and liver tumors (n = 106) were studied.
Generation and Characterization of a Chimeric Blocking mAb Directed against Human NKG2A
A murine anti-human NKG2A IgG1 mAb clone, Z270, was generated in a previous study (Sivori et al., 1996). We humanized this antibody by fusion with an IgG4 with a single point mutation in the Fc heavy chain to prevent the formation of half-antibodies and screened the selected humanized clones for binding to CD94-NKG2A with an affinity similar to that of the original murine mAb. The selected humanized clone was named monalizumab (IPH2201/NNC141-0100). Importantly, unlike other anti-NKG2A mAbs described to date, monalizumab is specific for human NKG2A, as it bound human NKG2A+ cells, but not Ba/F3-transfected cells expressing human NKG2C, the activating isoform of NKG2A (Figure S5A). The EC50 calculated by whole blood titration was 4.5 ng/mL for NKG2A+ NK cells and 11.4 ng/mL for NKG2A+ CD8+ T cells (Figure S5B). Finally, another critical feature of monalizumab resides in its capacity to inhibit the binding of HLA-E tetramers to human NK cells expressing NKG2A (Figure S5C).
Figure S5.

Characterization of Monalizumab, Related to Figures 5 and 6
(A) Titration of monalizumab on Ba/F3 cells expressing human NKG2A or NKG2C. Monalizumab was detected with a secondary antibody conjugated to PE.
(B) Titration of monalizumab on NKG2A+ NK and CD8+ T cells from the PBMCs of healthy donors.
(C) Monalizumab inhibits the binding of HLA-E tetramers to CD94/NKG2A receptors on human peripheral NK cells. Human PBMCs (n = 4 with NKG2C+ NK cells < 3.5%) were incubated with various concentrations of monalizumab (ranging from 0 to 10 μg/mL), washed and then incubated with PE-conjugated HLA-E tetramers. NK cells were defined as CD56+ CD3- lymphocytes. The percentage inhibition was calculated as follows: inhibition (%) = 100 – 100∗[(MFI PE (at X μg/mL of monalizumab) – MFI PE FMO)/(MFI PE (at 0 μg/mL of monalizumab) - MFI FMO)]. MFI: mean fluorescence intensity; FMO: fluorescence minus one.
Monalizumab Promotes the Anti-tumor Cell Activities of Human NK Cells and CD8+ T Cells
We then sought to assess the blocking activity of monalizumab on effector cells by monitoring the cell surface expression of CD107 by NKG2A+ NK cells cocultured with K562 tumor target cells expressing HLA-E (Figures 5A and 5B). The prototypic K562 cells, which lack HLA-E, activated NK cells, but forced HLA-E expression on K562 cells decreased the frequency of CD107+ NKG2A+ NK cells. The addition of monalizumab to the assay restored the production of CD107 by NKG2A+ NK cells to the levels observed with parental K562 targets (Figure 5A). In addition, monalizumab treatment led to increased frequencies of CD107 expression and IFN-γ production when IL-2-activated NK cells were co-cultured with K562-HLA-E+ targets (Figure 5B). We then assessed the anti-tumor efficacy of monalizumab in co-cultures of NK cells with tumor cell lines with different levels of HLA-E expression (Figure S6). Monalizumab increased the frequency of activated NKG2A+ NK cells, as assessed by measuring the cell-surface induction of CD107 and CD137 (4-1BB), an activation-induced costimulatory molecule, in co-cultures with three different SCCHN cell lines and three different ovarian tumor cell lines, although this stimulation was weaker for the CAL-27 and Caov-2 cell lines (Figure S6).
Figure 5.

Monalizumab and Durvalumab Unleash NK and CD8+ T Cell Function In Vitro
(A) NK cells were co-cultured with K562 or K562 cells expressing HLA-E in the presence or absence of monalizumab. The frequencies of CD107-positive NK cells are shown. Box and whiskers plot, with the means indicated by crosses. N = 8. The whiskers are drawn down to the 25th percentile minus 1.5 times IQR (interquartile range) and up to the 75th percentile plus 1.5 times IQR. Friedman analysis followed by Dunn’s multiple comparisons test. ∗∗p = 0.006; ∗∗∗p = 0.0001.
(B) Purified 7 days IL-2-activated NK cells were co-cultured or K562 cells expressing HLA-E in the presence or absence of monalizumab. The frequencies of CD107 and IFN-γ-positive NK cells are shown. Non-parametric Wilcoxon matched-pairs rank test. N = 6; ∗p = 0.0313.
(C) NK cells were stimulated in vitro with IL-15 for 9 days. The data shown are the frequencies of NK cells expressing NKG2A or PD-1 before (day 0) and after (day 9) culture.
(D) The NK cells generated in (C) were co-cultured with K562 cells expressing HLA-E or co-expressing HLA-E and PD-L1 without (control) or with monalizumab (mona), durvalumab (durva), or both these antibodies (combo). The data shown are the frequencies of CD107-expressing NKG2A+ PD-1+or PD-1− NK cells. Box and whiskers plot, with the means indicated by crosses like in Figure 5A. N = 13 donors. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
(E) CD8+ T cells were co-cultured in vitro with monocytes in the presence of IL-15 and Flu peptide for 9 days. Top panel: one representative dot plot showing the frequency of Flu tetramer positive (TMr+) CD8+ T cells after culture (n = 14). Bottom panel: frequencies of NKG2A+ and/or PD-1+ cells after gating on TMr+ CD8+ T cells (n = 14).
(F) The CD8+ T cells generated in (E) were co-cultured with Flu peptide-pulsed K562 cells expressing PD-L1, HLA-E and HLA-A2 without (control) or with monalizumab (mona), durvalumab (durva), or both antibodies (combo). The data shown are the frequencies of CD107-expressing (upper panels) and IFN-γ-secreting (lower panels) NKG2A+ or NKG2A− CD8+ T cells (n = 17). The whiskers are drawn like in Figure 5A. ∗p ≤ 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
(G) The CD8+ T cells generated in (E) were co-cultured with Flu peptide-pulsed K562 cells expressing PD-L1, HLA-E, and HLA-A2 loaded with Cr51 without (control) or with monalizumab (mona), durvalumab (durva), or both antibodies (combo). The data shown are the frequencies of K562 target cell lysis. N = 14 donors. The whiskers are drawn like in Figure 5A. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
See also Figures S5 and S6.
Figure S6.

Monalizumab Unleash NK Cell Function In Vitro, Related to Figure 5
(A) FACS profiles showing HLA-E expression at the cell surface in SCCHN cell lines. White histograms: isotype control; gray histograms: anti-HLA-E mAb. NK cells were co-cultured with the cell line indicated, in the presence or absence of monalizumab. The frequencies of CD107- and CD137-producing NKG2A+ NK cells are shown. Each donor is represented by a single dot. Wilcoxon matched-pairs signed-rank test, ∗ p < 0.05, ∗∗ p < 0.01.
(B) FACS profiles showing HLA-E expression at the cell surface in ovarian cancer cell lines. White histograms: isotype control; gray histograms: anti-HLA-E mAb. NK cells were co-cultured with the cell line indicated (after overnight stimulation of the cell line with IFN-γ) in the presence or absence of monalizumab. The frequencies of CD107- and CD137-producing NKG2A+ NK cells are shown. Each donor is represented by a single dot. Wilcoxon matched-pairs signed-rank test, ∗ p < 0.05, ∗∗ p < 0.01.
The anti-NKG2A mAb and the anti-PD-L1 mAb had synergistic effects in our preclinical mouse tumor models. We therefore assessed the effects of a combination of monalizumab and durvalumab on NK cell activity against K562 cells co-expressing HLA-E and PD-L1 in vitro. NKG2A+ PD-1+ NK cells were generated by chronically stimulating various donor PBMCs with IL-15 (Figure 5C). The anti-NKG2A monalizumab, used as a single agent, increased the frequencies of CD107+ NKG2A+ PD-1- NK cells in cocultures with K562-HLA-E or K562-HLA-E-PD-L1 cells (Figure 5D). Addition of durvalumab did not improve NK cell reactivity in this assay. When used as a single agent, monalizumab also improved CD107 expression by NKG2A+ PD-1+ NK cells cocultured with K562-HLA-E targets. The use of monalizumab or durvalumab as single agents only modestly increased the reactivity of NKG2A+ PD-1+ NK cells cocultured with K562-HLA-E-PD-L1 cells, whereas these two antibodies had additive effects when used in combination. Thus, monalizumab efficiently released the inhibition conferred by the engagement of the inhibitory receptor NKG2A. In combination with other ICI, monalizumab has additive effects, promoting NK-cell effector functions.
We assessed the boosting effect of monalizumab on CD8+ T cell functions in more detail because, in our preclinical model, many CD8+ TILs expressed NKG2A (Figures 2A and 3A). We aimed to generate antigen-specific NKG2A+ CD8+ T cells in vitro through chronic stimulation with IL-15, monocytes and antigenic peptides derived from human influenza virus (Flu) (Figure 5E). The Flu-specific CD8+ T cells obtained after nine days of culture harbored different phenotypes. In addition to PD-1+ NKG2A− Flu-specific CD8+ T cells, a substantial fraction of the Flu-specific CD8+ T cells co-expressed PD-1 and NKG2A (Figure 5E). Cells were then cocultured with Flu peptide-pulsed K562-HLA-A2 cells expressing or not expressing the inhibitory ligands HLA-E and PD-L1. The addition of monalizumab or durvalumab modestly increased the frequency of CD107+NKG2A+ Flu-specific-CD8+ T cells (Figure 5F). However, the combination of monalizumab with durvalumab improved CD8+ T cell activity as assessed by CD107 expression and IFN-γ production, and by the killing of target cells in a chromium release assay (Figures 5F and 5G). Thus, monalizumab can promote activation and effector functions of both NK cells and CD8+ T cells, and this effect is more marked when it is used in combination with durvalumab.
Monalizumab Promotes Human NK Cell Antibody-Dependent Cell-Mediated Cytotoxicity
We then evaluated the potential of monalizumab to promote NK-cell effector functions when combined with other commonly used anti-tumor reagents, such as those promoting antibody-dependent cell-mediated cytotoxicity (ADCC). The anti-EGFR mAb cetuximab is used to treat advanced and recurrent and/or metastatic SCCHN and metastatic CRC. Cetuximab mobilizes adaptive and innate immunity against tumor cells, partly by promoting ADCC (Ferris et al., 2018). HLA-E membrane expression in CRC could inhibit cetuximab-mediated cellular cytotoxicity (Levy et al., 2009). We used a combination of monalizumab and cetuximab to stimulate NK cells against an SCCHN cell line in vitro (Figure 6A, left panel), and monitored the induction of CD137 as a marker of NK cell activation including ADCC. This combination of mAbs amplified the activation of NK cells, as shown by the higher frequencies of CD137+ NK cells. Monalizumab also enhanced the NK cell-mediated ADCC by the anti-CD20 mAb obinutuzumab in cocultures with B cell lines expressing MHC class I (Figure 6A, right panel). Thus, the anti-NKG2A mAb monalizumab can amplify the beneficial effects of other IO treatments, such as those promoting ADCC.
Figure 6.

Monalizumab Enhances Human NK Cell-Mediated ADCC and Anti-tumor Activity of Monalizumab and Cetuximab
(A) Left panel: NK cells from healthy donors were co-cultured with the CAL-27 SCCHN cell line in the presence or absence of monalizumab (Mona) or cetuximab (Ctx). The data shown are the frequencies of CD137-expressing NKG2A+ NK cells after 24 hr. N = 13. Student t test comparing the Mona + Cetux combination with Ctx as single agent ∗∗∗∗p < 0.0001. Right panel: NK cells from healthy donors were co-cultured with 721.221 cells expressing HLA-Cw3 and HLA-Cw4 in the presence or absence of monalizumab (Mona) or obinutuzumab (Obz). The data shown are the frequencies of CD137-expressing NKG2A+ NK cells after 24 hr. N = 12. Student’s t test comparing the Mona + Obz combination with Obz as a single agent. ∗∗∗∗p < 0.0001.
(B) Waterfall plot of the largest change in target lesion relative to the baseline.
(C) Spider plot of the largest change in target lesion relative to the baseline.
The patient who died early, due to disease progression, before the first assessment is not represented in these graphs. In accordance with RECIST 1.1, a confirmation of response was required.
(D) Example of a partial response after treatment with the Mona + Cetux combination in a patient with recurrent oral cavity cancer (left masticator space) previously treated by surgery, chemotherapy (cisplatin), and radiation therapy.
See also Table 1.
Tumor Control by a Combination of Monalizumab and Cetuximab in Patients with SCCHN
We found that combinations of NKG2A-blocking mAbs with other IO treatments, such as anti-PD-1 mAbs, anti-PD-L1 mAbs or cetuximab, had additive effects on anti-tumor immunity in preclinical experimental settings in vitro and in vivo. These results provide a scientific rationale for evaluations of the efficacy and safety of monalizumab in cancer patients. SCCHN tumors were strongly positive for HLA-E and were infiltrated with CD8+ T cells and NK cells, which may express NKG2A (Figures 4A, 4B, and 4D). Cetuximab is used in the standard care regimen for SCCHN (Vermorken et al., 2007). We therefore assessed the safety and efficacy of the combination of monalizumab and cetuximab in patients with previously treated recurrent or metastatic (R/M) SCCHN in a phase II clinical trial (NCT02643550). We evaluated five doses of monalizumab (0.4, 1, 2, 4, 10 mg/kg every 2 weeks) in combination with the approved dose of cetuximab (400 mg/m2 loading dose and then 250 mg/m2 weekly). The maximum tolerated dose was not reached and the highest dose of monalizumab tested (10 mg/kg) was used for expansion of the phase II cohort. We used a one-stage Fleming design with futility analysis after the first 11 patients; the overall phase II study will include 40 patients. The characteristics of the patients are shown in Table 1. As of March 9, 2018, 31 patients with R/M SCCHN were treated and evaluable for safety, of which 26 patients were evaluable for efficacy while the remaining patients were studied too early for assessment. All 31 patients had been previously treated with platinum-based chemotherapy, and 24 patients received one or two systemic treatment regimens. Fourteen patients had already received immunotherapies, and three had been already treated with cetuximab for locally advanced disease and had been free from progressive disease for at least 4 months. Safety was the primary endpoint of part I and objective response rate (ORR) of part II. The combination was well tolerated. Most of the adverse events (AE) observed (93%) were of grades 1–2 severity, rapidly reversible and easily manageable. The most common monalizumab-related AEs were fatigue (17%), pyrexia (13%), and headache (10%). Other monalizumab-related AEs (interstitial lung disease, colitis and hypophosphatemia) were reported in 1 patient each. The most frequent AEs reported for cetuximab in previous studies (Vermorken et al., 2007) were skin disorders (rash, 49%; acne, 26%; nail disorders, 16%; dry skin, 14%), and these effects were not exacerbated by monalizumab. No infusion-related reactions were observed (patients received premedication for cetuximab as specified on the label). No treatment-related death was reported. No new or unusual signs suggestive of poor safety were observed with the combination of monalizumab and cetuximab. We thus concluded that the safety profile of the combination was similar to that for the two single agents.
Table 1.
Characteristics of Patients with Recurrent or Metastatic SCCHN, Enrolled in the NCT02643550 Phase II Clinical Trial
| Patient Characteristics, N = 31 | N (%) | |
|---|---|---|
| Age, median [range] | 64 [34–76] | |
| Sex | female | 10 (32) |
| male | 21 (68) | |
| ECOG | 0 | 12 (39) |
| 1 | 19 (61) | |
| HPV status | positive | 4 (13) |
| negative | 15 (48) | |
| to be determined | 12 (39) | |
| Tobacco | never | 6 (19) |
| former | 20 (65) | |
| current | 5 (16) | |
| Tumor site | oral cavity | 14 (45) |
| oropharynx | 10 (32) | |
| larynx | 4 (13) | |
| hypopharynx | 2 (6) | |
| nasopharynx | 1 (3) | |
| Type of recurrence | local | 18 (58) |
| distant | 13 (42) | |
| Prior lines of systemic therapy (overall) | Number of Previous Lines | |
| 1 | 16 (52) | |
| 2 | 10 (32) | |
| 3 | 5 (16) | |
| prior platinum | 31 (100) | |
| prior IO | 14 (45) | |
| prior cetuximab | 3 (10) |
Interim treatment efficacy results for the phase II trial showed that treatment with the monalizumab and cetuximab combination resulted in a confirmed RECIST partial response in 8 of 26 patients (31%), stable disease (SD) in 14 of 26 (54%) and progressive disease (PD) in 3 of 26 (11%) patients and one patient died from progressive disease at week 8 without post-baseline imaging (Figures 6B–6D). The lesion disappeared in one patient, as shown in Figure 6D. Assuming an ORR of 25%, using 10% as the cutoff for inactivity, α = 0.05, and a power of 0.76, the predefined number of eight responses required to declare a positive result for the trial already was reached. Two of the eight patients with confirmed responses had previously received immunotherapy. At the time of the analysis, the median response duration was not reached; six responding patients were still on treatment. Median follow-up time was 129 days: 17 patients (55%) were still on treatment, 14 patients (45%) had stopped treatment, because of progressive disease in 12 (38%), and adverse event in one and on the decision of the investigator in the final case. The activity of single agent cetuximab in recurrent and/or metastatic SCCHN is limited with a 13% ORR, a median duration of response (DoR) of 4 months, a median progression-free survival (PFS) of 2.3 months, and a median overall survival (OS) of 5.6 months (Vermorken et al., 2007, Vermorken et al., 2008). Even if no formal comparison can be made, the activity of monalizumab combined to cetuximab appears higher than historical data for cetuximab alone, although cross-trial comparisons of this kind should be interpreted with caution due to the small number of patients involved and possible differences in patient population and trial methodology. Overall, these data showed that the combination therapy of monalizumab with cetuximab has promise for the treatment of patients with SCCHN with expected toxicity profile of either agent alone.
Discussion
Immune checkpoint inhibitors have greatly improved the control of several types of cancer, but the efficacy of these treatments needs to be further improved, as does the ability to control their toxicity. One way of achieving this goal would be to identify critical immune checkpoints other than PD-1 and CTLA-4 for targeting by therapeutic antibodies to promote effective immune responses to cancers. Most immunomodulatory strategies to date have focused on enhancing T cell responses, but there has been a recent surge of interest in harnessing the relatively underexplored NK cell compartment for therapeutic interventions (Cerwenka and Lanier, 2018, Guillerey and Smyth, 2016, Rautela et al., 2018, Vivier et al., 2012, Chiossone et al., 2018). The manipulation of NK cells in cancer is designed to initiate a multilayered immune response culminating in protective and long-lasting immunity to tumors based on a number of different cell types, including T cells.
Here, we focus on the NKG2A receptor, a well-known ITIM-bearing inhibitory receptor expressed on both T and NK cells (López-Botet et al., 2000, Moretta et al., 2001), emitting inhibitory signals transduced via the protein tyrosine phosphatase SHP-1 (Viant et al., 2014). The abundance of NKG2A+CD8+ T cells is low in human blood, but NKG2A expression can be induced at the surface of CD8+ T cells upon activation (Braud et al., 2003). The targeting of NKG2A with a blocking antibody would therefore have the unique advantage of enhancing T and NK cell responses. Another advantage of targeting NKG2A is the safety of this approach, as no abnormalities have been reported in mouse strains lacking CD94 (Vance et al., 1999, Orr et al., 2010), which forms a heterodimer with NKG2A. These mice therefore lack cell-surface NKG2A expression.
One critical point for such an approach is the expression of NKG2A and HLA-E during cancer. We have shown that the NKG2A receptor is expressed on NK and T cells in the tumor bed in many human cancers and we have also shown that its ligand, HLA-E, is frequently overexpressed in tumors. By contrast, classical MHC-I expression is often weak on tumor cells, and this downregulation has been recognized as a major mechanism by which tumor cells escape T cell control (Garrido et al., 2017, Sharma et al., 2017). Unlike classical HLA class I molecules, HLA-E continues to be expressed on the surface of tumor cells, often even more strongly than on healthy cells, in patients with solid tumors or leukemia/lymphoma (Benson et al., 2012, Mamessier et al., 2011, Platonova et al., 2011, Talebian Yazdi et al., 2016). This conservation of expression likely results from the dependence of cell-surface HLA-E expression on many distinct peptides, including the leader peptides of HLA-A, -B, or -C. Downregulation of HLA-E expression therefore would require at least the elimination of three types of HLA molecules. Our data for NKG2A expression are consistent with earlier reports on tumor-infiltrating NK and T cells in melanoma and breast and cervical cancers (Mamessier et al., 2011, Sheu et al., 2005). A complementary study showed in several tumor mouse models that expression of NKG2A is associated with worse clinical outcome (van Montfoort et al., 2018).
One of the key findings of our studies is the demonstration that NKG2A is often co-expressed with PD-1 on CD8+ T cells. PD-1 expression is a hallmark of exhausted CD8+ T cells (Hashimoto et al., 2018). This result therefore suggested that NKG2A expression might constitute an additional brake to release for reversing CD8+ T cell exhaustion. The regulation of NKG2A expression on both NK and CD8+ T cells remains to be dissected in detail. Nevertheless, unlike PD-1 expression, which can be observed on the surface of CD8+ T cells from whole blood or lymph nodes from cancer patients, the number of NKG2A-expressing CD8+ T cells was selectively increased at the tumor bed or adjacent tissue. These results suggest that signals derived specifically from the tumor would be required to induce, or to sustain NKG2A expression.
We also found that HLA-E was more frequently expressed than PD-L1 in several types of cancer. This finding is consistent with previous suggestions that HLA-E expression may account for some of the lack of responsiveness to anti-PD-x observed in Merkel cell carcinoma (Paulson et al., 2018) and in an in vivo CRISPR screening program that identified Qa-1b (the mouse HLA-E ortholog) as a cancer immunotherapy target, because Qa-1b loss-of-function increased the efficacy of immunotherapy by PD-1 blockade (Manguso et al., 2017). These data support the use of a combination of mAbs blocking the PD-x and NKG2A/HLA-E inhibitory pathways. Our results in mice indicate that NKG2A pathway blockade does indeed improve tumor control when combined with a blockade of the PD-1/PD-L1 inhibitory pathway. We also demonstrated the generation of protective memory CD8+ T cells in mice into which RMA-Rae-1β tumors were implanted and then cured by combined PD-L1 and NKG2A blockade. Thus, our preclinical results provide a rationale for combining monalizumab and durvalumab into a novel immunotherapy for cancer patients. Importantly, such a clinical trial is ongoing (NCT02671435) and very recently preliminary safety and efficacy data were reported (Segal et al., 2018). Briefly, the dose escalation part of the study demonstrated the feasibility of combining the two agents with no new safety signals noted beyond the known safety profile for each individual agent. The initial clinical activity data from a cohort expansion in pretreated (median of three previous lines of systemic therapy) microsatellite stable colorectal cancer (MSS CRC; n = 39) demonstrated an ORR of 8% (median duration of response of 16.1 weeks) and a disease control rate (DCR) at 16 weeks of 31% (Segal et al., 2018). Although these results are very preliminary, they are an example of potential therapeutic opportunities for immunotherapy in MSS CRC, a setting in which immune checkpoint-based therapy has, so far, failed to demonstrate any consistent and meaningful clinical benefit.
Combining a blockade of inhibitory signals with the delivery of activating signals should improve the efficacy of immunotherapies. Many possible approaches of this type are being tested, including the triggering of innate immunity via the delivery of TLR ligands (Du et al., 2016), activation of the STING pathway at the tumor bed (Corrales et al., 2016), treatment with antibodies targeting activating cell surface receptors (Callahan et al., 2016, Muntasell et al., 2017), and the use of engineered forms of cytokines, such pegylated IL-2 (Charych et al., 2017, Charych et al., 2016) and IL-2 variants (Sockolosky et al., 2018). Antibodies directed against tumor cells also could be used to stimulate the immune response to tumor cells, thereby helping to eliminate cancer. The mode of action of these treatments differs between antibodies, but efficacy is partly dependent on ADCC, as for rituximab, an anti–CD20 mAb used to treat non-Hodgkin lymphoma and chronic lymphocytic leukemia (Cartron and Watier, 2017). Other antibodies are also used to stimulate the immune system via ADCC. One such antibody is cetuximab, which is used in metastatic CRC and SCCHN. We showed in vitro that NKG2A blockade with monalizumab boosts NK cell-mediated ADCC against cetuximab-coated SCCHN tumor cells. Consistent with these data, treatment with a combination of monalizumab and cetuximab was found to be effective in the NCT02643550 phase II clinical trial for SCCHN. Importantly, little, if any, toxicity was associated with blocking NKG2A and especially no signs of auto-immunity. The encouraging results obtained for SCCHN in NCT02643550 and for MSS CRC in NCT02671435 require consolidation in further clinical trials, but they constitute a key step toward the use of monalizumab in combination treatments against cancer. Once the samples from all the patients enrolled in the monalizumab clinical trials will be accessible, translational studies will be performed. In particular, it will be addressed whether the expression of HLA-E at the tumor bed is linked to the HLA types of the patients (Horowitz et al., 2016, Ramsuran et al., 2018) and to the clinical outcome.
In conclusion, we report here the full characterization of a first-in-class immune checkpoint inhibitor, monalizumab. This therapeutic antibody has several key features. First, it enhances the antitumor activities of both T and NK cells, by blocking the inhibitory function of NKG2A. Second, the ligand of NKG2A is the non-classical MHC class I molecule HLA-E, which is frequently overexpressed on human tumors, providing a mechanism of resistance to lymphocyte activation at the tumor bed. Third, monalizumab is well tolerated in humans and has yielded encouraging efficacy results in clinical trials assessing its use in combination with cetuximab in SCCHN, and in combination with durvalumab in MSS CRC, two clinical conditions with low ORRs, for which therapeutic options are very limited. Therefore, anti-NKG2A mAb is a promising checkpoint inhibitor that promotes antitumor immunity by enhancing the activities of both T and NK cells. Interestingly, NKG2A has been shown to contribute to the inhibition HIV-infected target cell clearance by NK cells (Ramsuran et al., 2018). Thus, beyond cancer, the therapeutic blockade of NKG2A/HLA-E interaction by monalizumab may be beneficial in patients with HIV disease, and in other conditions that remain to be explored.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Ultra-LEAF purified anti-Asialo-GM1 (Poly21460) | BioLegend | Cat#146002; RRID:AB_2562206 |
| Anti-mouse CD8α (YTS169.4) | Bioxcell | Cat#BE0117; RRID: AB_10950145 |
| Rat IgG2b, κ isotype control (LTF-2) | Bioxcell | Cat#BE0090; RRID:AB_1107780 |
| Anti-mouse CD279 (RMP1-14) | Bioxcell | Cat#BE0146; RRID: AB_10949053 |
| Rat IgG2a, κ isotype control (2A3) | Bioxcell | Cat#BE0089 RRID: AB_1107769 |
| Anti-mouse IFN-γ (XMG1.2) | Bioxcell | Cat#BE0055; RRID: AB_1107694 |
| Rat IgG1, κ isotype control (HRPN) | Bioxcell | Cat#BE0088 RRID: AB_1107775 |
| Anti-mouse NK1.1 (PK136) | Bioxcell | Cat#BE0036; RRID: AB_1107737 |
| Mouse IgG2a isotype control | Innate Pharma | N/A |
| Anti-mouse NKG2A (Fc silent mousified 20d5) | Innate Pharma | N/A |
| Mouse IgG1 isotype control (Fc silent) | Innate Pharma | N/A |
| Anti-mouse CD3 BV650 (17A2) | BioLegend | Cat#100229: RRID:AB_11204249 |
| Anti-mouse CD8α V500 (53-6.7) | BD Biosciences | Cat#560776; RRID:AB_1937317 |
| Anti-mouse CD16/CD32 (2.4G2) | BD Biosciences | Cat#553142; RRID:AB_394657 |
| Anti-mouse CD44 PE (IM7) | BD Biosciences | Cat#553134; RRID:AB_394649 |
| Anti-mouse CD45 FITC (30F11) | Miltenyi Biotec | Cat#130-102-491; RRID:AB_2659919 |
| Anti-mouse CD62L APC-Cy7 (MEL-14) | BD Biosciences | Cat#560514; RRID:AB_10611861 |
| Anti-mouse CD107a FITC (1D4B) | BD Biosciences | Cat#553793; RRID:AB_395057 |
| Anti-mouse CD274 (clone AB740080) | MedImmune | N/A |
| Anti-mouse CD274 PE (clone MIH-5) | Thermo Fisher | Cat#12-5982-81; RRID:AB_466088 |
| Anti-mouse CD279 PE/Dazzle 594 (RMP1-30) | BioLegend | Cat#109116; RRID:AB_2566548 |
| Anti-mouse CD335 BV605 (29A1.4) | BD Biosciences | Cat#564069 RRID:NA |
| Anti-mouse CD335 AlexaFluor 647(29A1.4) | Innate Pharma | N/A |
| Anti-mouse Qa-1b APC (6A8.6F10.1A6) | Miltenyi Biotec | Cat#130-104-252; RRID:AB_2653302 |
| Anti-mouse NKG2A/C/E VioBlue (20d5) | Miltenyi Biotec | Cat#130-105-566; RRID:AB_2653010 |
| Anti-human CD159a (Z270) | Innate Pharma | N/A |
| Anti-HLA-E (3D12) | BioLegend | Cat#342602; RRID:AB_1659247 |
| Anti-human CD335 (clone 9E2) | BD Biosciences | Cat#557911; RRID:AB_396933 |
| Anti-human CD8α (clone C8/144B) | Dako | Cat#M710301-2 RRID:AB_2075537 |
| Anti-human CD274 (clone E1L3N) | Cell Signaling Technology | Cat#13684S RRID:AB_2687655 |
| Rabbit IgG isotype control (EPR25A) | Abcam | Cat#ab172730; RRID:AB_2687931 |
| Mouse IgG1 isotype control (DAK-GO1) | Dako | Cat#X0931 RRID:AB_577451 |
| Goat anti mouse IgG (H+L) PE | Jackson ImmunoResearch Labs | Cat#115-116-146; RRID:AB_2338629 |
| Anti-human CD3 AmCyan (SK7) | BD Biosciences | Cat#339186; RRID:AB_647353 |
| Anti-human CD3 FITC (UCHT1) | Beckman Coulter | Cat#A07746 RRID:AB_131010 |
| Anti-human CD3 PercPCy5.5 (SP34-2) | BD Biosciences | Cat#552852; RRID:AB_394 |
| Anti-human CD8 APCCy7 (SK1) | BD Biosciences | Cat#348803 RRID:AB_400391 |
| Anti-human CD56 V450 (B159) | BD Biosciences | Cat#560360; RRID:AB_1645578 |
| Anti-human CD56 PEVio770 (REA196) | Miltenyi Biotec | Cat#130-100-676; RRID:AB_2658738 |
| Anti-human purified FcγRI/CD64 (10.1) | R and D Systems | Cat#MAB1257; RRID:AB_2293973 |
| Anti-human CD107a FITC (H4A3) | BD Biosciences | Cat#555800; RRID:AB_396134 |
| Anti-human CD107a APC (H4A3) | Miltenyi Biotec | Cat#130-095-510; RRID:AB_10828544 |
| Anti-human CD107b FITC (H4B4) | BD Biosciences | Cat#555804; RRID:AB_396138 |
| Anti-human CD107b APC (H4B4) | Miltenyi Biotec | Cat#130-103-960; RRID:AB_2654504 |
| Anti-human CD137 | Miltenyi Biotec | Cat#130-094-821 RRID:AB_10829768 |
| Anti-human CD159a PE (Z199) | Beckman Coulter | Cat#IM3291U; RRID:AB_10643228 |
| Anti-human CD159a APC (Z199) | Beckman Coulter | Cat#A60797 RRID:AB_10643105 |
| Anti-human CD279 BV421 (EH12.2H7) | BioLegend | Cat#329920; RRID:AB_10960742 |
| Anti-human IFN-γ PercPCy5.5 (4S.B3) | Thermo Fisher Scientific | Cat#45-7319-42; RRID:AB_10718246 |
| Anti-human IFN-γ APC (B27) | BD Biosciences | Cat#554702; RRID:AB_398580 |
| Anti-human IgG4 (Fc) PE (clone HP6025) | Southern Biotech | Cat#920009; RRID:AB_2629781 |
| Monalizumab | Innate Pharma | N/A |
| Durvalumab | AstraZeneca | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Normal Rabbit Serum | Abcam | Cat#ab7787 |
| Streptavidin PE | SouthernBiotech | Cat#7100-09 |
| Ammonium chloride solution | Stemcell | Cat#07800 |
| BD Cytofix Fixation Buffer | BD Biosciences | Cat#554655 |
| Recombinant human IL-2 (PROLEUKIN) | NOVARTIS | N/A |
| Recombinant human IL-15 | RD | Cat#247-IL/CF |
| Mouse IFN-γ | Peprotech | Cat#315-05-2500G |
| Biotinylated-HLA-E monomers | Novo Nordisk | N/A |
| Flu peptide (H-GILGFVFTL-OH) | JTP | Cat#MP-58-66 |
| Protein transport inhibitor GolgiStop | Becton Dickinson | Cat#554724 |
| Critical Commercial Assays | ||
| Tumor Dissociation Kit, mouse | Miltenyi Biotec | Cat#130-096-730 |
| CD19 MicroBeads, mouse | Miltenyi Biotec | Cat#130-052-201 |
| CD14 Microbeads, human | Miltenyi Biotec | Cat#130-050-201 |
| CD3 MicroBeads, human | Miltenyi Biotec | Cat#130-050-101 |
| CD56 MicroBeads, human | Miltenyi Biotec | Cat#130-050-401 |
| CD8+ T cell negative selection kit | Stemcell | Cat#17953 |
| LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit | Molecular Probes | Cat#L34976 |
| LIVE/DEAD Fixable Red Dead Cell Stain Kit | Molecular Probes | Cat#L34972 |
| LIVE/DEAD Fixable BlueDead Cell Stain Kit | Molecular Probes | Cat#L34962 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Molecular Probes | Cat#L34966 |
| MHC Multimer-APC | Immudex | Cat#WB2161 |
| Experimental Models: Cell Lines | ||
| CAL-27 | DSMZ, Germany | Cat#ACC-446; RRID:CVCL_1107 |
| H-N | DSMZ, Germany | Cat#ACC-417; RRID:CVCL_1283 |
| FaDu | ATCC, USA | Cat#HTB-43; RRID:CVCL_1218 |
| Caov-3 | ATCC, USA | Cat#HTB-75: RRID:CVCL_0201 |
| Colo-704 | DSMZ, Germany | Cat#ACC-198; RRID:CVCL_1994 |
| K562-HLA-E | Novo Nordisk | N/A |
| K562-HLA-E-PD-L1 | Innate Pharma | N/A |
| K562-HLA-E-HLA-A2-PD-L1 | Innate Pharma | N/A |
| A20 | ATCC | Cat#TIB-208; RRID:CVCL_1940 |
| A20 Qa-1b KO | Innate Pharma | N/A |
| RMA-Rae-1β | Pr. D. Raulet | N/A |
| YAC-1 | ATCC | Cat#TIB-160; RRID:CVCL_2244 |
| Experimental Models: Organisms/Strains | ||
| BALB/c mice | Janvier Laboratories | Cat#BALB/cRj SOPF RRID:IMSR_HAR:1255 |
| C57BL/6J mice | Janvier Laboratories | Cat#C57BL/6Rj SOPF RRID:IMSR_JAX:000664 |
| Software and Algorithms | ||
| GraphPad Prism version 7.00 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| BD FACSDiva Software 8.0 | BD Biosciences | |
| FlowJo version 10.2 | FlowJo LLC | https://www.flowjo.com/solutions/flowjo |
| Halo 2.0 | Indica Labs | http://www.indicalab.com/ |
| Fiji (ImageJ) | https://fiji.sc/ |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be addressed to the Lead Contact Eric Vivier (vivier@ciml.univ-mrs.fr).
Experimental Model and Subject Details
Mice
C57BL/6J and BALB/c mice were reared at Janvier Laboratories under specific pathogen-free conditions. Female mice were used at eight to 12 weeks of age and were allowed to acclimate to the housing facility for at least one week. All animal experiments were performed in accordance with the rules of the Innate Pharma ethics and animal welfare committees.
Cell lines
For A20 Qa-1b KO cell line, gene editing was used to introduce a targeted deletion in the H2-T23 gene in the A20 mouse cell line. To control the targeted deletion of Qa-1b by flow cytometry, the A20 cell line were grown with 125 UI/mL mouse IFN-γ during 24 hr to induce the Qa-1b cell surface expression. The pool of A20 edited cells were stained with the anti-mouse-Qa-1b antibody and the negative cells were sorted.
K562-HLA-E cell line was transduced with HLA-A2 lentivirus. Positive cells were sorted by flow cytometry and expanded in culture. The human PD-L1 gene (AAI13735.1) was cloned into an expression vector. The K562-HLA-E and the K562 HLA-E/HLA-A2 cell lines were transfected by nucleofection using the 4D-NucleofectorTM system from Lonza (Program FF-120, SF solution). The day after transfection, the hygromycin selection was added to the nucleofected cells at 400μg/ml.
Human primary cells
Peripheral blood samples from healthy donors were obtained from Etablissement Francais du Sang (EFS, Marseille) under a written consent obtained from each volunteer.
Peripheral blood, metastatic and non-metastatic lymph node, tumors and juxta tumor tissues from SCCHN patients were obtained at the time of surgical resection under Institut Curie approved protocol and written informed consent from each patient. Patients’ characteristics are summarized in Table S1.
Human trial information
Patients had to meet all of the following criteria for inclusion in the study: (i) age ≥ 18 years; (ii) histologically or cytologically-confirmed, HPV (+) or HPV (-) squamous cell carcinoma of the nasopharynx (WHO type 1), oropharynx, hypopharynx, larynx (supraglottis, glottis, subglottis) or oral cavity; (iii) recurrent or metastatic disease, documented by imaging (CT scan, MRI, X-ray) and/or physical examination; (iv) no prior treatment by cetuximab except if given for primary treatment (locally advanced disease) with no progressive disease for at least 4 months following the end of prior cetuximab treatment.
Method Details
Monoclonal anti-mouse NKG2A antibody
The mouse version of 20D5 antibody was generated by a classical CDR grafting method. The variable domain sequences of the heavy and light chains of the original rat antibody were first aligned with the mouse genome sequence. The CDR1, CDR2 and CDR3 regions of the variable heavy chain and of the variable light chain domains of the rat antibody were then inserted into the closest matching mouse sequences identified. The variable domain of the mousified light chain was inserted in frame with a mouse Ck domain into a eukaryotic expression vector to generate the light chain. The variable domain of the mousified heavy chain was inserted in frame with the constant domains of a mouse IgG1 harboring the N297Q mutation. CHO cells were then transfected with both the heavy- and light-chain constructs, to produce the antibody, which was then purified from the supernatant with Protein-A Sepharose beads.
In vivo murine tumor experiments
For A20 tumor experiments, 5x106 cells in endotoxin-free PBS were injected subcutaneously (s.c.) into the flank of BALB/c recipient mice. For RMA-Rae-1β experiments, 5x105 cells were injected s.c. into the flank of C57BL/6J recipient mice. Tumor size was monitored with a digital caliper (Mitutoyo) every three to four days and is expressed as a volume ((length x width2) /2). Mice were sacrificed when the tumor reached a size of 2000 mm3. For the purposes of Kaplan-Meier curves, mice were considered dead when tumor volume exceeded 1800 mm3.
For NK cell depletion, 100 μL of polyclonal anti-asialo-GM1 (Poly21460, Biolegend) antibody were injected i.p. into BALB/c mice once weekly. Normal rabbit serum was administrated as control. For C57BL/6J mice, 100 μg of anti-NK1.1 (PK136 clone) antibody were injected i.v. every 10 days. An isotype control was used as a control.
For the depletion of CD8+T cells in BALB/c and C57BL/6J mice, 400 μg of depleting anti-CD8α (YTS 169.4) mAb were administered i.v. for the initial injection and then 200 μg once weekly for two subsequent injections. A rat IgG2b, k (Bio X cell) was used as control. Immune cell depletions were initiated as indicated in the figure legends.
Treatments with checkpoint blockade antibodies were initiated when tumor volume was between 50 and 100 mm3. Anti-PD-L1 mAb (mouse IgG1) was administered by i.p. injection, at a dose of 50 μg for A20 tumors and 200 μg for RMA-Rae-1β tumors, twice weekly for three weeks. Mice received 200 μg of anti-NKG2A mAb by i.v. injection three times, at three- to four-day intervals. Anti-PD-1 mAb (rat IgG2a, RMP1-14 clone) was administered by three i.p. injections of 200 μg every 3-4 days.
Mice were killed for tumor-infiltrating lymphocyte (TIL) analysis 19 days after tumor cell injection for A20 and 12 days after tumor cell injection for RMA Rae-1β.
Flow cytometry analysis of splenic and tumor-infiltrating mouse lymphocytes (TILs)
Tumors were dissected into small pieces and digested with enzymes A and D from the Mouse Tumor Dissociation Kit (Miltenyi Biotec) and program 37C_m_TDK_1 of the GentleMACS Octo Dissociator with heaters (Miltenyi Biotec). Digested tumors were then filtered through a 70 μm-pore size MACS SmartStrainer (Miltenyi Biotec) and washed with RPMI 1640 medium. For A20 tumors, this step was followed by purification with mouse CD19 Microbeads (Miltenyi Biotec).
Spleens were mechanically dissociated with GentleMACS dissociator, in RPMI 1640 medium supplemented with 2% FBS. Dissociated spleens were passed through 70 μm-pore size pre-separation filters (Miltenyi Biotec) and washed with RPMI 1640 medium. Red blood cells were lysed after 5 min of incubation on ice with ammonium chloride solution (Stemcell) and then washed with RPMI 1640 medium.
We then incubated 1x106 or fewer cells per A20 tumor and 2-10 × 106 cells per RMA Rae-1β tumor or 10 × 106 splenocytes with 5 μg/mL blocking anti-mouse CD16/CD32 antibody (clone 2.4G2, BD PharMingen) at 4°C for 5 min before surface staining with an antibody cocktail at 4°C for 30 min in 100 μL. Cells were then washed twice with PBS, stained with LIVE/DEAD Fixable Dead Cell Stain Kit (Molecular Probes) at 4°C for 15 min, and washed twice with staining buffer (0.2% BSA 2 mM EDTA 0.02% NaN3 in PBS). Finally, cells were fixed by incubation in BD Cytofix Fixation Buffer (BD Biosciences) at 4°C for 30 min. Samples were then analyzed with a BD LSR Fortessa X-20 cell analyzer and BD FACSDiva Software version 8.0.
Functional assays with BALB/c splenocytes and A20 TILs
Splenocytes from BALB/c mice stimulated i.p. with 50 μg of poly I:C and A20 TILs were prepared as previously described. They were incubated with YAC-1 (Qa-1b-) or IFN-γ-simulated A20 (Qa-1b+) tumor cells, with an effector/target ratio of 10:1 (for splenic effector cells) or 1:1 (for A20 TILs), for 4 hr at 37°C, under an atmosphere containing 5% CO2, in the presence of monensin (Golgi Stop) and 5 μg/mL anti-NKG2A mAb or 5 μg/mL anti-PD-L1 mAb, or both. The frequencies of CD107a-producing NKG2A+ NK and NKG2A+PD-1+ CD8+ T cells were assessed by flow cytometry.
Immunohistochemistry
OCT-embedded frozen human tissue blocks were cut into 5 to 7 μm-thick sections on Superfrost Plus glass slides. After tissue rehydration with PBS, endogenous peroxidases were blocked by incubation with 0.3% hydrogen peroxide for 15-30 min at room temperature. Tissue sections were blocked with 5% goat serum and stained for 1 h at room temperature with the following unconjugated primary Abs: anti-NKG2A (clone Z270, Innate Pharma), anti-HLA-E (clone 3D12, Biolegend), anti-NKp46 (clone 9E2, Becton Dickinson) or anti-CD8 (clone C8/144B, Dako). Anti-NKG2A, anti-HLA-E and anti-NKp46 Abs were used at a concentration of 5 μg/mL and anti-CD8 mAb was used at a concentration of 1 μg/mL. Matching control mAb (mouse IgG1 isotype control; clone DAK-GO1, Dako) staining was performed in parallel. The signal was then amplified with the EnVision/HRP kit (K4001, Dako) for 30 min at room temperature. Enzymatic activity was detected with 3,3′-diaminobenzidine (SK-4100, Vector Laboratories) substrate. Tissue sections were then counterstained with hematoxylin II followed by bluing reagent (Roche, Ventana). Finally, tissues were washed with tap water, dehydrated in ethanol and cleared in xylene. Slides were mounted in Pertex and scanned on a Nanozoomer 2.0-HT slide scanner (Hamamatsu). Positive and negative controls (frozen cell pellets) were included in each run.
Formalin-fixed paraffin-embedded (FFPE) commercial human tissue microarrays (USBiomax) were stained on a Benchmark automaton (Ventana). After pretreatment with Cell Conditioning 1 (Ventana), sections were incubated for 1 hr at 37°C with anti-HLA-E (clone MEM-E/02, Exbio) or anti-PD-L1 (clone E1L3N, Cell Signaling Technology) mAbs at concentrations of 1 μg/mL and 10 μg/mL, respectively. An anti-mouse/rabbit IgG detection system (UltraView Universal DAB Detection Kit, Ventana) was used for amplification and primary Ab detection. Tissues were counterstained with hematoxylin, washed, dehydrated, cleared, mounted and coverslipped. Slides were scanned on a Nanozoomer 2.0-HT slide scanner (Hamamatsu). Positive and negative controls (FFPE cell pellets) were included in each run. When needed, and only for representation purpose, a color deconvolution algorithm was applied to bright-field images using ImageJ color deconvolution plugin (Ruifrok AC, Johnston DA. Quantification of histochemical staining by color deconvolution. Anal Quant Cytol Histol 23: 291-299, 2001) and resulting components images were inverted. Finally, pseudocolors were attributed to hematoxylin counterstain and HLA-E or NKG2A markers.
Five different cohorts (frozen tissues): colorectal (n = 48), ovarian (n = 40), endometrial (n = 41) and cervical cancers (n = 17) and one cohort of head and neck (n = 65) (FFPE tissues) were used. Staining was interpreted and scored by a trained pathologist. The following scoring method was used: score 0 = no positive cells; score 1 = between 1 and 33% of positive cells; score 2 = between 34 and 66% of positive cells and score 3 ≥ 67% of positive cells. HLA-E expression was assessed separately on tumor cells and lymphocytes and CD8+, NKp46+ and NKG2A+ cells quantified separately in the tumor nest and the stroma. For FFPE tissues, tissue microarrays of lung (n = 45), pancreas (n = 79), stomach (n = 76), colon (n = 109), head and neck (n = 70) and liver tumors (n = 106) were stained. The cytonuclear and area quantification modules (Halo, Indica Labs) were used to quantify NKp46+ cell density (number of positive cells/mm2) and the area stained for PD-L1 (area of positive tissue/total area of the tissue of interest, expressed as a percentage).
Monocyte-CD8 T cell cocultures
Human PBMCs were thawed or freshly isolated from the peripheral blood of healthy volunteers. Monocytes were purified by positive selection with the CD14 microbeads kit. The CD14-negative fraction was retained for the isolation of CD8+ T cells with a CD8+ T cell negative selection kit. Monocytes and CD8+ T cells were cocultured, in a 1:1 ratio, in IMDM supplemented with 1% penicillin-streptomycin and 10% human AB serum, in 24-well plates. rhIL-15 (10 ng/mL) and flu peptide (5 μg/mL) were added to cultures. Cells were cultured for 9 days at 37°C with replenishment with rhIL-15-supplemented medium every three days.
Long-term culture of PBMCs with IL-15 for the generation of PD-1+ NK cells
Human PBMCs were isolated from buffy coats by gradient density centrifugation in Pancoll Lymphocyte Separating Medium. PBMCs were used to seed 24-well culture plates at a density of 2x106 cell/mL per well, in complete culture medium (IMDM + 1% penicillin-streptomycin and 10% human AB serum), at a final volume of 1 mL per well containing IL-15 at a final concentration of 10 ng/mL. Cells were cultured for 9 days at 37°C with replenishment with rhIL-15-supplemented medium every three days.
Flu-specific CD8+ T cell staining
The percentage of flu-specific CD8+ T cells was determined by staining on day 0 before coculture and on day 9 of monocyte-CD8+ T cell coculture. Cells were transferred to a 5 mL tube, centrifuged and incubated with MHC multimer conjugated with APC for 10 min, in the dark, at room temperature. A mixture of antibodies was then added and incubated with the cells for 30 min at 4°C in the dark. The antibodies used were: anti-CD3-PerCP, anti-CD8-APC, anti-PD-1 BV421, and anti-NKG2A-PE antibodies. Dead cells were excluded with Live/Dead Fixable Aqua stain. After incubation, the cells were washed and analyzed by flow cytometry.
Functional assay with CD8+ T cells (expression of CD107/IFN-γ)
CD8+ T cells were recovered from co-cultures with monocytes after 9 days in the presence of rhIL-15 and flu peptide. Cells were transferred to 96-well plates and incubated with target cell lines (K562 transfected to express HLA-A2, HLA-E and PD-L1). Target cells were first pulsed with 1 μg/mL flu peptide for 1 h at 37°C and washed three times before incubation with CD8+ T cells and antibodies, such as monalizumab (20 μg/mL), durvalumab (20 μg/mL) and anti-human CD64 antibody (10 μg/mL). Golgi Stop was added. Plates were centrifuged and incubated for 4 hr at 37°C. The plates were then washed before re-staining with monalizumab at a concentration of 1 μg/mL. Cells were washed twice, incubated with anti-human IgG4 (Fc)-PE, washed twice and then stained with a mixture of antibodies (anti-CD3-FITC, anti-CD8-APC-Cy7, anti-PD1-BV421, anti-CD107a-APC, anti-CD107b-APC) and Live/Dead Fixable Aqua stain. Cells were washed twice and incubated overnight with BD Cytofix/Cytoperm. The next day, cells were washed twice and the intracellular staining was performed for 30 min at 4°C with anti-IFN-γ-PercPCy5.5 antibody. Plates were read on a flow cytometer to analyze the percentages of CD107+ and IFN-γ-producing CD8+ T cells.
Preparation of human TILs
Tissues were mechanically dissociated before enzymatic digestion using collagenase I (2 mg/mL, Sigma C0130), DNase I (25μg/mL, Roche 10104159001) and hyaluronidase (2mg/mL, SIGMA H3506). Red cells from dissociated tissues and whole blood were lysed (eBioscience 00-4300-54) and dead cells stained with Live/Dead Aqua. Cells were stained with the antibodies detailed in the Key Resources Table. Samples were analyzed by flow cytometry (BD LSR FORTESSA) using the BD FACS DIVA software 8.0. Data were analyzed on FlowJo V10.02.
Functional assay with CD8+ T cells (cytotoxic assay)
Flu-specific CD8+ T cell lytic activity was assessed against K562 HLA-E HLA-A2 PD-L1 cells using a standard chromium release assay. K562 cells co-expressing HLA-E HLA-A2 and PD-L1 were first pulsed with 1 μg/mL flu peptide for 1 h at 37°C and then washed three times before loading with 51Cr for 1 h. After washing, cells were plated with CD8+ T cells at 2:1 effector-to-target (E:T) cell ratio in presence of anti-human CD64 antibody (10 μg/mL) and in presence or absence of monalizumab (20 μg/mL), durvalumab (20 μg/mL) or isotype control antibodies. After 4 h at 37°C, 50 μL of supernatant was harvested and counted in a Topcount microplate scintillation gamma counter. Data were acquired in counts per minute. Specific lysis was calculated according to the formula [(test release − spontaneous release)/(maximum release − spontaneous release)] × 100. Spontaneous release represents 51Cr release from target cells in medium alone, and maximum release is the 51Cr release from target cells lysed in medium 2% Triton X-100, each measured in at least six replicate wells.
Phenotypic analysis of NK cells in day 9 cultures
The phenotype of NK cells in rhIL-15-supplemented cultures of PBMCs was analyzed on days 0 and 9 with the following panel of antibodies: anti-CD3-PercPCy5.5, anti-CD56-PE-Vio770, anti-CD8-APC-Cy7, anti-PD1-BV421, anti-NKG2A-APC and the Live/Dead Fixable Aqua stain kit.
Functional assays with peripheral human NK cells
The effector cells used were total PBMCs, purified human NK cells, nine-day culture of PBMCs with rhIL-15 or seven-day culture with rhIL-2 (PROLEUKIN) of enriched peripheral blood NK cells by CD3 depletion followed by CD56 positive selection. NK cells were incubated at a ratio of 1:1 or 5:2 with target cell lines (SCCHN, ovarian cancer cell lines or K562 cells transfected to express HLA-E and/or PDL-1). Cells were dispensed into 96-well plates with or without single antibodies or combinations of antibodies: monalizumab (10 μg/mL), durvalumab (10 μg/mL), cetuximab (0.1 μg/mL), obinutuzumab (1 μg/mL). Anti-human CD64 antibody (10 μg/mL) was added to saturate FcR. Plates were centrifuged and incubated for 4 hr (CD107) or 24 hr (CD137) at 37°C. The cells were incubated with monalizumab (1 μg/mL) for 30 min at 4°C followed with anti-human IgG4 (Fc)-PE to detect total surface NKG2A. Cells were then incubated with a mixture of antibodies (anti-CD3-FITC, anti-CD56-PE- Vio770 and anti-PD1-BV421 antibodies or anti-CD3-AmCyan and anti-CD56-V450) together with either anti-CD107a and anti-CD107b antibodies or anti-CD137 antibody and Live/Dead Fixable stain. For IFN-γ detection, cells were fixed and intracellular stained with anti-IFN-γ-APC antibody. Cells were washed twice and resuspended in BD CellFix for analysis on a FACSCanto II or a LSRII UV flow cytometer.
Inhibition of HLA-E tetramers binding
Solution of biotinylated-HLA-E monomers (5nM solution, kindly provided by Novo Nordisk) was slowly mixed with PE-conjugated streptavidin (Southern Biotechnology) at a ratio 1/4 and then kept at 4°C until use. Competition of monalizumab and PE-conjugated tetramers was performed on peripheral blood from healthy donors (EFS). 100μL of whole blood was incubated for 1 h at 37°C with 50μL PBS containing PE-HLA-E tetramers (0.1μL) and monalizumab (ranging from 30μg/mL to 0,001μg/mL, 1/3 dilution) and then washed with PBS. Peripheral blood cells were then stained with anti-CD3-PercpCy5.5 (BD Biosciences), anti-CD45-APC (BD Biosciences), anti-CD3-PC7 (BD Biosciences) and anti-human IgG4 Fc-FITC (HP6025, Southern Biotechnology). Red blood cells were lyzed with Optilyse C solution (Beckman Coulter). Acquisition was performed on a FACSCanto II flow cytometer.
Trial design
Phase II study of monalizumab, a first-in-class NKG2A monoclonal antibody, in combination with cetuximab in previously treated recurrent or metastatic squamous cell carcinoma of the head and neck (NCT02643550) is a multicenter, international (US and France), open label, single arm study to evaluate the antitumor activity of monalizumab in combination with cetuximab. Five dose levels of monalizumab (0.4, 1, 2, 4, 10 mg/kg every 2 weeks) in combination with the approved dosage of cetuximab (400 mg/m2 load then 250 mg/m2 weekly) were explored. The highest dose tested (10 mg/kg) was used for the phase II cohort expansion; the overall phase II study will include 40 patients. The trial consisted of 2 parts: a phase Ib dose escalation part of monalizumab combined with cetuximab followed by a cohort expansion phase II part of cetuximab + monalizumab. The decision to conduct the phase II part was made by the sponsor after Safety Committee review and was based on all the available data from phase I part (safety, efficacy, PK and PD) and from the monalizumab overall program.
Phase Ib - Dose escalation part
This part was open to pre-treated patients (regardless of the number of previous treatment lines) with recurrent and/or metastatic SCCHN. A 3+3 design was used and 5 escalating dose levels of monalizumab were explored with fixed doses of cetuximab. If the Maximum Tolerated Dose (MTD) was not reached, monalizumab would be tested in the phase II part at a dose of 10 mg/kg. Primary endpoint: DLT (Dose Limiting Toxicity) of monalizumab combined with cetuximab was assessed using National Cancer Institute’s Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03
Phase II - Cohort expansion
The cohort was expanded with cetuximab and monalizumab given at the dose recommended in phase Ib. This part was restricted to patients with recurrent and/or metastatic SCCHN who had received up to 2 prior systemic regimens for incurable SCCHN. A Gehan-like stopping rule for futility was used after 11 evaluable patients. A maximum of 40 patients were included. Primary endpoint: Objective Response Rate (ORR) was estimated according to RECIST 1.1.
Quantification and Statistical Analysis
Details on statistics used can be found in figure legends. All statistical analyses were performed using GraphPad Prism software version 7 (GraphPad). Kaplan Meier was used for survival representations. When sample size was large enough, normality of populations was tested using d’Agostino-Pearson omnibus normality test. When normality was not reached, statistical significance between two paired samples populations was determined using Wilcoxon matched-pair signed rank test, statistical analyses for three or more groups. N represents the number of samples used in the experiments. Samples are shown with means or medians with or without error bars showing the SD.
Significance was assumed with ∗p < 0.05; ∗∗p ≤ 0.01; ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Acknowledgments
We thank Mohammed Dar (Mediumme) for critically reviewing the manuscript; François Romagné (MI-mAbs), Fabien Chanuc, Nicolas Fuseri, and Flavien Caraguel (Innate Pharma) for help and advice during these studies. The E.V. lab was supported by funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (TILC, grant agreement no. 694502), the Agence Nationale de la Recherche, Equipe Labellisée “La Ligue,” Ligue Nationale contre le Cancer, MSDAvenir, Innate Pharma, and institutional grants to the CIML (INSERM, CNRS, and Aix-Marseille University) and Marseille Immunopole.
Author Contributions
P.A., R.Z., and E.V. conceived the project. P.A., C.D., C.S., M.B., C.B., L.G., and O.L. conceived the experiments. J.F., A.B.-C., P.D., and R.B.C. conceived the clinical study. C.H. and O.L. provided access to the human samples. C.B.-C., E.B., J.L., S.G., A.M., B.R., T.A., V.B., and A.I.L. performed the experiments. P.A., C.D., C.S., C.B.-C., G.H., R.R., A.M., H.G., M.J.-K., Y.M., R.H., A.B.-C., P.D., E.N.-M. and E.V. analyzed the data. P.A, E.N.-M. and E.V. wrote the manuscript.
Declaration of Interests
P.A., C.D., C.S., C.B.-C., J.L., T.A., M.B., C.B., L.G., A.M., B.R., R.R., V.B., E.B., A.B-C., R.Z., P.D., Y.M, and E.V. are employees of Innate Pharma. H.G. M.J.-K., and R.H. are employees of MedImmune. P.A., C.D., M.B. have a patent related to this work.
Published: November 29, 2018
Footnotes
Contributor Information
Pascale André, Email: pascale.andre@innate-pharma.fr.
Eric Vivier, Email: vivier@ciml.univ-mrs.fr.
Supplemental Information
Table S1. Characteristics of Patients with SCCHN Analyzed for NKG2A and PD-1 Expression on NK and CD8+ T Cells, Related to Figure 6
References
- André P., Brunet C., Guia S., Gallais H., Sampol J., Vivier E., Dignat-George F. Differential regulation of killer cell Ig-like receptors and CD94 lectin-like dimers on NK and T lymphocytes from HIV-1-infected individuals. Eur. J. Immunol. 1999;29:1076–1085. doi: 10.1002/(SICI)1521-4141(199904)29:04<1076::AID-IMMU1076>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Baumeister S.H., Freeman G.J., Dranoff G., Sharpe A.H. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 2016;34:539–573. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- Benson D.M., Jr., Hofmeister C.C., Padmanabhan S., Suvannasankha A., Jagannath S., Abonour R., Bakan C., Andre P., Efebera Y., Tiollier J. A phase 1 trial of the anti-KIR antibody IPH2101 in patients with relapsed/refractory multiple myeloma. Blood. 2012;120:4324–4333. doi: 10.1182/blood-2012-06-438028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertone S., Schiavetti F., Bellomo R., Vitale C., Ponte M., Moretta L., Mingari M.C. Transforming growth factor-beta-induced expression of CD94/NKG2A inhibitory receptors in human T lymphocytes. Eur. J. Immunol. 1999;29:23–29. doi: 10.1002/(SICI)1521-4141(199901)29:01<23::AID-IMMU23>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Brady J., Hayakawa Y., Smyth M.J., Nutt S.L. IL-21 induces the functional maturation of murine NK cells. J. Immunol. 2004;172:2048–2058. doi: 10.4049/jimmunol.172.4.2048. [DOI] [PubMed] [Google Scholar]
- Braud V.M., Aldemir H., Breart B., Ferlin W.G. Expression of CD94-NKG2A inhibitory receptor is restricted to a subset of CD8+ T cells. Trends Immunol. 2003;24:162–164. doi: 10.1016/s1471-4906(03)00064-4. [DOI] [PubMed] [Google Scholar]
- Callahan M.K., Postow M.A., Wolchok J.D. Targeting T cell co-receptors for cancer therapy. Immunity. 2016;44:1069–1078. doi: 10.1016/j.immuni.2016.04.023. [DOI] [PubMed] [Google Scholar]
- Cartron G., Watier H. Obinutuzumab: what is there to learn from clinical trials? Blood. 2017;130:581–589. doi: 10.1182/blood-2017-03-771832. [DOI] [PubMed] [Google Scholar]
- Cerwenka A., Lanier L.L. Natural killers join the fight against cancer. Science. 2018;359:1460–1461. doi: 10.1126/science.aat2184. [DOI] [PubMed] [Google Scholar]
- Charych D.H., Hoch U., Langowski J.L., Lee S.R., Addepalli M.K., Kirk P.B., Sheng D., Liu X., Sims P.W., VanderVeen L.A. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin. Cancer Res. 2016;22:680–690. doi: 10.1158/1078-0432.CCR-15-1631. [DOI] [PubMed] [Google Scholar]
- Charych D., Khalili S., Dixit V., Kirk P., Chang T., Langowski J., Rubas W., Doberstein S.K., Eldon M., Hoch U., Zalevsky J. Modeling the receptor pharmacology, pharmacokinetics, and pharmacodynamics of NKTR-214, a kinetically-controlled interleukin-2 (IL2) receptor agonist for cancer immunotherapy. PLoS ONE. 2017;12:e0179431. doi: 10.1371/journal.pone.0179431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiossone L., Dumas P.-Y., Vienne M., Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018;18:671–688. doi: 10.1038/s41577-018-0061-z. [DOI] [PubMed] [Google Scholar]
- Corrales L., McWhirter S.M., Dubensky T.W., Jr., Gajewski T.F. The host STING pathway at the interface of cancer and immunity. J. Clin. Invest. 2016;126:2404–2411. doi: 10.1172/JCI86892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daëron M., Jaeger S., Du Pasquier L., Vivier E. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol. Rev. 2008;224:11–43. doi: 10.1111/j.1600-065X.2008.00666.x. [DOI] [PubMed] [Google Scholar]
- Du B., Jiang Q.-L., Cleveland J., Liu B.-R., Zhang D. Targeting Toll-like receptors against cancer. J. Cancer Metastasis Treat. 2016;2:463–470. [Google Scholar]
- Ferris R.L., Lenz H.J., Trotta A.M., García-Foncillas J., Schulten J., Audhuy F., Merlano M., Milano G. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: Harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat. Rev. 2018;63:48–60. doi: 10.1016/j.ctrv.2017.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido F., Ruiz-Cabello F., Aptsiauri N. Rejection versus escape: the tumor MHC dilemma. Cancer Immunol. Immunother. 2017;66:259–271. doi: 10.1007/s00262-016-1947-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillerey C., Smyth M.J. NK cells and cancer immunoediting. Curr. Top. Microbiol. Immunol. 2016;395:115–145. doi: 10.1007/82_2015_446. [DOI] [PubMed] [Google Scholar]
- Hashimoto M., Kamphorst A.O., Im S.J., Kissick H.T., Pillai R.N., Ramalingam S.S., Araki K., Ahmed R. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu. Rev. Med. 2018;69:301–318. doi: 10.1146/annurev-med-012017-043208. [DOI] [PubMed] [Google Scholar]
- Horowitz A., Djaoud Z., Nemat-Gorgani N., Blokhuis J., Hilton H.G., Beziat V., Malmberg K.J., Norman P.J., Guethlein L.A., Parham P. Class I HLA haplotypes form two schools that educate NK cells in different ways. Sci. Immunol. 2016;3 doi: 10.1126/sciimmunol.aag1672. pii: eaag1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Dréan E., Vély F., Olcese L., Cambiaggi A., Guia S., Krystal G., Gervois N., Moretta A., Jotereau F., Vivier E. Inhibition of antigen-induced T cell response and antibody-induced NK cell cytotoxicity by NKG2A: association of NKG2A with SHP-1 and SHP-2 protein-tyrosine phosphatases. Eur. J. Immunol. 1998;28:264–276. doi: 10.1002/(SICI)1521-4141(199801)28:01<264::AID-IMMU264>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Levy E.M., Sycz G., Arriaga J.M., Barrio M.M., von Euw E.M., Morales S.B., González M., Mordoh J., Bianchini M. Cetuximab-mediated cellular cytotoxicity is inhibited by HLA-E membrane expression in colon cancer cells. Innate Immun. 2009;15:91–100. doi: 10.1177/1753425908101404. [DOI] [PubMed] [Google Scholar]
- López-Botet M., Llano M., Navarro F., Bellón T. NK cell recognition of non-classical HLA class I molecules. Semin. Immunol. 2000;12:109–119. doi: 10.1006/smim.2000.0213. [DOI] [PubMed] [Google Scholar]
- Mahapatra S., Mace E.M., Minard C.G., Forbes L.R., Vargas-Hernandez A., Duryea T.K., Makedonas G., Banerjee P.P., Shearer W.T., Orange J.S. High-resolution phenotyping identifies NK cell subsets that distinguish healthy children from adults. PloS ONE. 2017;12:e0181134. doi: 10.1371/journal.pone.0181134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamessier E., Sylvain A., Thibult M.L., Houvenaeghel G., Jacquemier J., Castellano R., Gonçalves A., André P., Romagné F., Thibault G. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Invest. 2011;121:3609–3622. doi: 10.1172/JCI45816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manguso R.T., Pope H.W., Zimmer M.D., Brown F.D., Yates K.B., Miller B.C., Collins N.B., Bi K., LaFleur M.W., Juneja V.R. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–418. doi: 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manser A.R., Uhrberg M. Age-related changes in natural killer cell repertoires: impact on NK cell function and immune surveillance. Cancer Immunol. Immunother. 2016;65:417–426. doi: 10.1007/s00262-015-1750-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon C.W., Zajac A.J., Jamieson A.M., Corral L., Hammer G.E., Ahmed R., Raulet D.H. Viral and bacterial infections induce expression of multiple NK cell receptors in responding CD8(+) T cells. J. Immunol. 2002;169:1444–1452. doi: 10.4049/jimmunol.169.3.1444. [DOI] [PubMed] [Google Scholar]
- Mingari M.C., Ponte M., Bertone S., Schiavetti F., Vitale C., Bellomo R., Moretta A., Moretta L. HLA class I-specific inhibitory receptors in human T lymphocytes: interleukin 15-induced expression of CD94/NKG2A in superantigen- or alloantigen-activated CD8+ T cells. Proc. Natl. Acad. Sci. USA. 1998;95:1172–1177. doi: 10.1073/pnas.95.3.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretta A., Bottino C., Vitale M., Pende D., Cantoni C., Mingari M.C., Biassoni R., Moretta L. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 2001;19:197–223. doi: 10.1146/annurev.immunol.19.1.197. [DOI] [PubMed] [Google Scholar]
- Mori S., Jewett A., Cavalcanti M., Murakami-Mori K., Nakamura S., Bonavida B. Differential regulation of human NK cell-associated gene expression following activation by IL-2, IFN-alpha and PMA/ionomycin. Int. J. Oncol. 1998;12:1165–1170. doi: 10.3892/ijo.12.5.1165. [DOI] [PubMed] [Google Scholar]
- Muntasell A., Ochoa M.C., Cordeiro L., Berraondo P., López-Díaz de Cerio A., Cabo M., López-Botet M., Melero I. Targeting NK-cell checkpoints for cancer immunotherapy. Curr. Opin. Immunol. 2017;45:73–81. doi: 10.1016/j.coi.2017.01.003. [DOI] [PubMed] [Google Scholar]
- Okazaki T., Chikuma S., Iwai Y., Fagarasan S., Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013;14:1212–1218. doi: 10.1038/ni.2762. [DOI] [PubMed] [Google Scholar]
- Okazaki T., Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- Orr M.T., Wu J., Fang M., Sigal L.J., Spee P., Egebjerg T., Dissen E., Fossum S., Phillips J.H., Lanier L.L. Development and function of CD94-deficient natural killer cells. PLoS One. 2010;5 doi: 10.1371/journal.pone.0015184. e15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson K.G., Park S.Y., Vandeven N.A., Lachance K., Thomas H., Chapuis A.G., Harms K.L., Thompson J.A., Bhatia S., Stang A. Merkel cell carcinoma: current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018;78:457–463.e2. doi: 10.1016/j.jaad.2017.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platonova S., Cherfils-Vicini J., Damotte D., Crozet L., Vieillard V., Validire P., André P., Dieu-Nosjean M.C., Alifano M., Régnard J.F. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011;71:5412–5422. doi: 10.1158/0008-5472.CAN-10-4179. [DOI] [PubMed] [Google Scholar]
- Ramsuran V., Naranbhai V., Horowitz A., Qi Y., Martin M.P., Yuki Y., Gao X., Walker-Sperling V., Del Prete G.Q., Schneider D.K. Elevated HLA-A expression impairs HIV control through inhibition of NKG2A-expressing cells. Science. 2018;359:86–90. doi: 10.1126/science.aam8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport A.S., Schriewer J., Gilfillan S., Hembrador E., Crump R., Plougastel B.F., Wang Y., Le Friec G., Gao J., Cella M. The inhibitory receptor NKG2A sustains virus-specific CD8+ T cells in response to a lethal poxvirus infection. Immunity. 2015;43:1112–1124. doi: 10.1016/j.immuni.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautela J., Souza-Fonseca-Guimaraes F., Hediyeh-Zadeh S., Delconte R.B., Davis M.J., Huntington N.D. Molecular insight into targeting the NK cell immune response to cancer. Immunol. Cell Biol. 2018;96:477–484. doi: 10.1111/imcb.12045. [DOI] [PubMed] [Google Scholar]
- Sagiv-Barfi I., Kohrt H.E., Czerwinski D.K., Ng P.P., Chang B.Y., Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA. 2015;112:E966–E972. doi: 10.1073/pnas.1500712112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher T.N., Schreiber R.D. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Segal N.H., Naidoo J., Curigliano G., Patel S., Sahebjam S., Papadopoulos K.P., Gordon M.S., Wang D., Rueda A.G., Song X. First-in-human dose escalation of monalizumab plus durvalumab, with expansion in patients with metastatic microsatellite-stable colorectal cancer. J. Clin. Oncol. 2018;36(Suppl 15) 3540–3540. [Google Scholar]
- Sharma P., Allison J.P. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Sharma P., Allison J.P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Hu-Lieskovan S., Wargo J.A., Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu B.C., Chiou S.H., Lin H.H., Chow S.N., Huang S.C., Ho H.N., Hsu S.M. Up-regulation of inhibitory natural killer receptors CD94/NKG2A with suppressed intracellular perforin expression of tumor-infiltrating CD8+ T lymphocytes in human cervical carcinoma. Cancer Res. 2005;65:2921–2929. doi: 10.1158/0008-5472.CAN-04-2108. [DOI] [PubMed] [Google Scholar]
- Sivori S., Vitale M., Bottino C., Marcenaro E., Sanseverino L., Parolini S., Moretta L., Moretta A. CD94 functions as a natural killer cell inhibitory receptor for different HLA class I alleles: identification of the inhibitory form of CD94 by the use of novel monoclonal antibodies. Eur. J. Immunol. 1996;26:2487–2492. doi: 10.1002/eji.1830261032. [DOI] [PubMed] [Google Scholar]
- Sockolosky J.T., Trotta E., Parisi G., Picton L., Su L.L., Le A.C., Chhabra A., Silveria S.L., George B.M., King I.C. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. 2018;359:1037–1042. doi: 10.1126/science.aar3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talebian Yazdi M., van Riet S., van Schadewijk A., Fiocco M., van Hall T., Taube C., Hiemstra P.S., van der Burg S.H. The positive prognostic effect of stromal CD8+ tumor-infiltrating T cells is restrained by the expression of HLA-E in non-small cell lung carcinoma. Oncotarget. 2016;7:3477–3488. doi: 10.18632/oncotarget.6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Montfoort N., Borst L., Korrer M.J., Sluijter M., Marijt K., Santegoets S., van Ham V., Ehrsan I., Pornpimol-Charoenteng K., Zervakis G. NKG2A blockade potentiates CD8 T-cell immunity induced by cancer vaccines. Cell. 2018;175 doi: 10.1016/j.cell.2018.10.028. Published online November 29, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance R.E., Jamieson A.M., Raulet D.H. Recognition of the class Ib molecule Qa-1(b) by putative activating receptors CD94/NKG2C and CD94/NKG2E on mouse natural killer cells. J. Exp. Med. 1999;190:1801–1812. doi: 10.1084/jem.190.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermorken J.B., Trigo J., Hitt R., Koralewski P., Diaz-Rubio E., Rolland F., Knecht R., Amellal N., Schueler A., Baselga J. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J. Clin. Oncol. 2007;25:2171–2177. doi: 10.1200/JCO.2006.06.7447. [DOI] [PubMed] [Google Scholar]
- Vermorken J.B., Herbst R.S., Leon X., Amellal N., Baselga J. Overview of the efficacy of cetuximab in recurrent and/or metastatic squamous cell carcinoma of the head and neck in patients who previously failed platinum-based therapies. Cancer. 2008;112:2710–2719. doi: 10.1002/cncr.23442. [DOI] [PubMed] [Google Scholar]
- Viant C., Fenis A., Chicanne G., Payrastre B., Ugolini S., Vivier E. SHP-1-mediated inhibitory signals promote responsiveness and anti-tumour functions of natural killer cells. Nat. Commun. 2014;5:5108. doi: 10.1038/ncomms6108. [DOI] [PubMed] [Google Scholar]
- Vivier E., Ugolini S., Blaise D., Chabannon C., Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012;12:239–252. doi: 10.1038/nri3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of Patients with SCCHN Analyzed for NKG2A and PD-1 Expression on NK and CD8+ T Cells, Related to Figure 6