Defining and Detecting Complex Peak Relationships in Mass Spectral Data: The Mz.unity Algorithm (original) (raw)
. Author manuscript; available in PMC: 2019 Mar 21.
Abstract
Analysis of a single analyte by mass spectrometry can result in the detection of more than 100 degenerate peaks. These degenerate peaks complicate spectral interpretation and are challenging to annotate. In mass spectrometry-based metabolomics, this degeneracy leads to inflated false discovery rates, data sets containing an order of magnitude more features than analytes, and an inefficient use of resources during data analysis. Although software has been introduced to annotate spectral degeneracy, current approaches are unable to represent several important classes of peak relationships. These include heterodimers and higher complex adducts, distal fragments, relationships between peaks in different polarities, and complex adducts between features and background peaks. Here we outline sources of peak degeneracy in mass spectra that are not annotated by current approaches and introduce a software package called mz.unity to detect these relationships in accurate mass data. Using mz.unity, we find that data sets contain many more complex relationships than we anticipated. Examples include the adduct of glutamate and nicotinamide adenine dinucleotide (NAD), fragments of NAD detected in the same or opposite polarities, and the adduct of glutamate and a background peak. Further, the complex relationships we identify show that several assumptions commonly made when interpreting mass spectral degeneracy do not hold in general. These contributions provide new tools and insight to aid in the annotation of complex spectral relationships and provide a foundation for improved data set identification. Mz.unity is an R package and is freely available at https://github.com/nathaniel-mahieu/mz.unity as well as our laboratory Web site http://pattilab.wustl.edu/software/.
Graphical Abstract
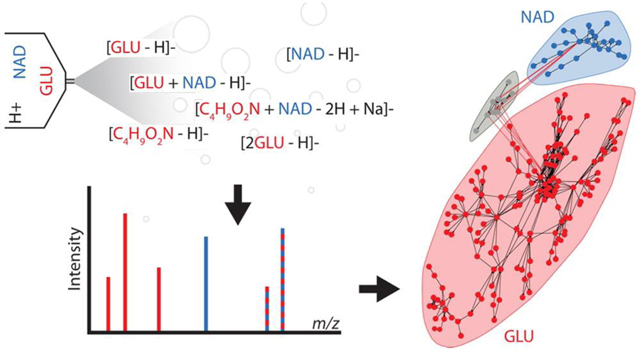
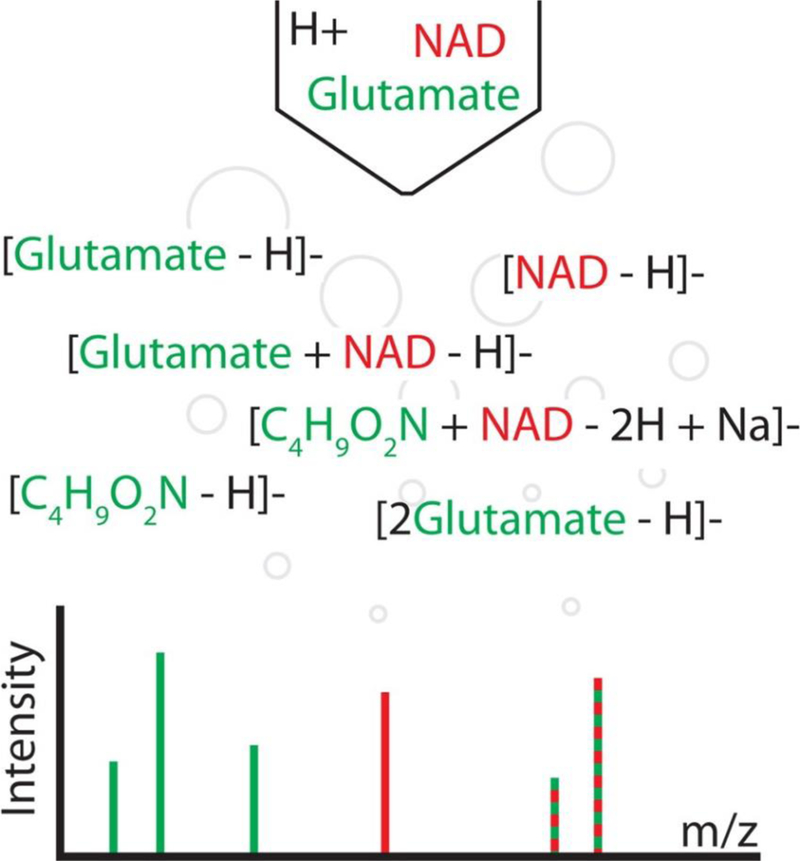
Adduction, fragmentation, and the natural abundance of heavy isotopes can cause a single analyte to generate more than 100 spectral peaks in mass spectrometry-based data sets. This is referred to as peak degeneracy, and it is a major source of the complexity that confounds data interpretation. Spectral peak degeneracy is challenging to annotate, and its complexity can exceed the ability of manual annotation in many cases. More recently, automated solutions have been developed to aid in the annotation of mass spectral data.(1–6) However, current annotation approaches fail to account for the full gamut of possible peak relationships. Further, several common assumptions made in these annotation approaches do not hold in general. Here we present mz-sum, a complete framework for describing complex peak relationships in mass spectrometry data, and mz.unity, an R package that enables the search and exploration of these relationships.
Sources of Degeneracy
The exact conditions under which a mass spectrum is collected have a strong influence on the peaks and types of peak relationships observed. The majority of spectral degeneracy is generated during ionization, which is the process by which analytes are converted from bulk-phase, neutral species to gas-phase ions. Electrospray ionization (ESI) is one commonly employed ionization technique. Here we focus on the peak relationships associated with ESI for clarity, but these approaches can be tailored to any ionization technique.
During ESI, analytes undergo various transformations before being detected as mass spectral peaks (Figure 1). The set of possible transformations provide the scope of the peak annotation problem. ESI involves the spray of analyte solution through a charged needle generating gas-phase droplets that evaporate until charged gas-phase compounds remain.(7) Two general types of analyte transformations are produced in this process, adduction and fragmentation.
Figure 1:
Illustaration of analyte transformations resulting in the detection of degenerate spectral peaks. Only two analytes are present, but they contribute to a total of six peaks.
Multiple chemical species that remain noncovalently bound after droplet evaporation are called an adduct. The adduct is a single gas-phase ion and will give rise to a single peak, but its formula is the combination of multiple distinct species. In the simplest case, the second chemical species is a proton, but other species such as sodium and solvent molecules can also form adducts. In general, any species present during ionization can adduct with any other species (this includes other analytes), see Figure 1 and the Glutamate NAD adduct.
In contrast, fragmentation is the breakage of bonds prior to MS detection. Often only one of the portions liberated during a single bond cleavage event is detected, but in some cases both are present in the resulting mass spectrum.(8) Bond cleavages can occur at various locations in a molecule, and therefore, a single structure can generate many fragment species.
An important contrast between the annotation of adducts and fragments is the constraint on possible relationships. For adduction, the space of possible relationships is limited by the species present at the time of ionization. Because a mass spectrum provides an exceptional record of present species, we can reasonably limit our search to those species. In contrast, fragment relationships are limited only to subformula of the parent and are therefore more challenging to annotate.(9, 10) In this work, we use mz.unity to putatively annotate two specific subsets of fragments discussed below.
Isotopes are a third source of degeneracy that are independent of the ionization process. Elements such as carbon are found in nature with varying numbers of neutrons (e.g., 12C and 13C). This natural abundance of heavy isotopes causes a single chemical formula to give rise to multiple masses, each corresponding to various numbers of heavy elements. Each of these heavy forms will be detected as a distinct mass peak.
Definitions:
Analyte, the chemical species which is of interest in the analysis, often a metabolite species but can include other molecules such as environmental exposures (e.g., pesticides); peak, a mass-to-charge ratio and intensity pair found in a mass spectrum; feature, a peak which has a Gaussian-like shape (a signal which rises and falls smoothly around a local maximum) in the chromatographic time domain; background peak, a peak which does not have a Gaussian-like shape in the chromatographic time domain; mer, an adduct between two analytes—includes homodimers, heterodimers, and higher _n_-mers; distal fragment, a fragment whose corresponding neutral loss also appears as a peak in the mass spectrum; granular-mz, mass and charge pairs supplied by the user to the mz.unity algorithm—these represent specific analyte transformations that combine to make peak relationships; complex relationships, mass spectral peak relationships between three or more detected peaks or relationships between peaks having multiple polarities (i.e., positive ions, negative ions, or neutral masses).
Motivation
Interpretation of a mass spectrum necessitates the annotation of relationships between degenerate peaks such as isotopes, adducts, and fragments. Critical to the field of metabolomics in particular is the annotation and removal of these degenerate peaks while preserving those that correspond to unique metabolites. Annotation has many benefits for metabolomics: (i) redundant features can be removed, reducing the size of the data set by more than an order of magnitude; (ii) the concomitant reduction in statistical tests performed allows for a less stringent multiple hypothesis testing correction; (iii) confidence in the validity of a detected peak is increased when degenerate peaks are also detected; (iv) annotated relationships can inform metabolite identification steps; (v) investigative efforts can be directed to unique analytes. Although we will focus on examples of peak degeneracy in metabolite mass spectra in this work, we point out that annotation is also important to other fields in addition to metabolomics. In proteomics, for example, annotation prior to selection of ions for MS/MS may reduce instrument cycles spent on degenerate peaks and therefore increase proteome coverage.(11–13) In trace impurity analysis, annotation can explain unknown peaks. In approaches that rely on feature counting, such as the evaluation of organic compound diversity on meteorites, annotation is critical to obtain realistic estimates of the total number of unique analytes detected.(14)
Current annotation tools utilize rule tables to describe possible peak relationships.(1, 15) A rule table is a list of transformations that neutral analytes may undergo prior to detection. Rules are applied to spectral peaks, and a relationship is asserted if two rule-peak pairs predict the same neutral mass. Unfortunately, this approach can only represent a subset of peak relationships. Limitations arise because many spectral peaks do not correspond to a single, underlying neutral mass. Thus, relationships between three or more peaks (as is the case for fragments and multiple-analyte-adducts) cannot be expressed or searched. Current rule tables are also not charge-aware and therefore can only annotate relationships of the same polarity. The limited scope of rule tables precludes the annotation of many putative peak relationships and, therefore, invites a more comprehensive approach to annotation.
To enable comprehensive spectral annotation, we detail two contributions here: mz-sum and mz.unity. Mz-sum is the simple concept that all peak relationships can be described as gain and/or loss of charged formulas. Mz.unity builds on this concept to enumerate all possible peak relationships in a charge-aware manner. Mz.unity is a software package implementing the peak relationship search and tools to plot and explore putative annotations. Together, mz-sum and mz.unity enable the detection of additional complex relationships that are not annotated by current approaches. The purpose of mz.unity is to find and return all putative peak relationships within a specified mass error. For example, the adduct of glutamate and nicotinamide adenine dinucleotide (NAD), fragments of NAD, and peaks detected in different polarities. While mz.unity is a functional tool for exploring spectra and programmatically evaluating relationships within them, we note that it is not an automated annotation solution and assessment of confidence in any specific peak relationship requires information beyond mass and charge. However, this contribution provides the groundwork necessary to enable automated annotation solutions to be developed in the future.
Experimental Methods
Notation and the Mz-sum Framework
Chemical species having mass “_m_” and charge “_z_” are denoted [_m_]z. For clarity, a mass can be referred to by a chemical formula or a compound name. When names are used the neutral, monoisotopic mass is implied. Thus, the following are equivalent: [146.0459]1–, [C5H8NO4]1–, and [glutamate – H]1–. Brackets are used to denote chemical species and can represent either detected mass spectral peaks or any additional formulas. Each set of brackets represents a distinct species. Conversions may be noted within brackets that describe the nature of the species. In the case of [Glu – H2O – H]1−, we are referring to a glutamate species after water loss and deprotonation.
Annotation seeks to find relationships between detected mass and charge species (peaks). Relationships are represented by equations of brackets that balance the mass and charge on each side. These equations anchor one or more [_m_]z peaks in the context of other detected peaks and gained and lost mass and charge. From the gained masses and charges, specific transformations can be inferred. For example, the description of a glutamate–acetate adduct can be written as the following equation: [C5H8NO4 – H]1– + [CH4CO2 – H]1– + [H]1+ = [C6H12NO6 – H]1–. Mz-sum is the basic assertion that any valid peak relationship will satisfy mass and charge balance and can be represented by such an equation. With this groundwork in place, it is now possible to define a search for all peak relationships.
Description of the Mz.unity Algorithm
Given a list of species with masses and charges [_m_]z, mz.unity searches for combinations of peaks that satisfy mass and charge balance (a description of the search problem can be found in Supporting Information Supplement S1). Additional parameters specify the combinatorial depth with which to search the supplied [_m_]z and the acceptable mass error. As follows from the discussion of mz-sum above, this search pattern is general enough to find any type of peak relationship. Below are examples of the general relationship types detected by mz.unity. Notably, each of these lies beyond the scope of previous annotation software. Though compound names are written for clarity, the actual search is performed by using accurate mass.
Complex adducts:
[glutamate−H]1−+[NAD−H]1−+[H]1+=[glutamate+NAD−H]1−
Distal fragments:
[fragment123.0453]1++[fragment540.0536]1−−[H]1+=[NAD−H]1−
Isotopes:
[glutamate−H]1−−[14N]0+[15N]0=[15N1−glutamate−H]1−
The mz.unity search can be tailored to a specific set of relationships by supplying “granular-mz” to the search. These user-supplied granular-mz represent undetected species which relate spectral peaks. In the case of adduction, many species present in solution will not be represented in the mass spectrum. This is because spectra have low and high mass cutoffs and only record ionizable species. Such granular-mz in the case of adduction would include small ions such as [H]1+, additives such as [acetate]0, and solvents such as [acetonitrile]0. In the adduction and fragmentation examples above, [H]1+ was a supplied granular-mz.
The most general relationship search would include granular-mz corresponding to the atoms C, H, N, O, P, and S, as well as an electron. This set of species would be sufficient to link every peak to every other peak, but in almost all cases these relationships would be arbitrary, linking unrelated analytes. By limiting the set of granular-mz, the mz.unity search can be limited to a specific condition or relationship type. In the case of ESI spectra, we seek to relate peaks that are degenerate. This leads to the use of granular-mz that represent transformations occurring during the analysis process.
Many fragments cannot be annotated by mz.unity because fragmentation is unique to each analyte and challenging to predict. There are two cases in which mz.unity can detect fragments. When a molecule has two distal charge sites and fragmentation occurs between them, both portions of the molecule will be detected. This is especially true when spectra from both polarities are included as demonstrated in the Fragment Annotations section. In this case, the relationship can be detected by mz.unity, even across polarities (see the distal fragment example above). The second set of detectable fragments are those which occur often under the experimental conditions employed (i.e., common fragments). Common fragments can be supplied as granular-mz and searched like any other relationship.
Output of the Mz.unity Algorithm
The output of an mz.unity search is a matrix (Table 1). Cells reference the supplied [_m_]z pairs involved in the relationship. Each row represents a relationship. Within each row, columns prefixed with “B.” and “M.” correspond to the peaks and granular-mz that sum to the peak referenced in column “A”. The mass error associated with each relationship is also reported. A convenient visualization of this output is a graph structure (Figure 2). In this representation, nodes are peaks and edges are the detected relationships.
Table 1.
Output of Mz. Unitya
| A | B.1 | .. | B.n | M.1 | ... | M.n | ppm |
|---|---|---|---|---|---|---|---|
| 12 | 1 | 1 | - | 11 | - | - | 0.52 |
| 1 | 29 | - | - | 11 | 17 | - | 0.59 |
| … | … | … | … | … | … | … | … |
Figure 2.
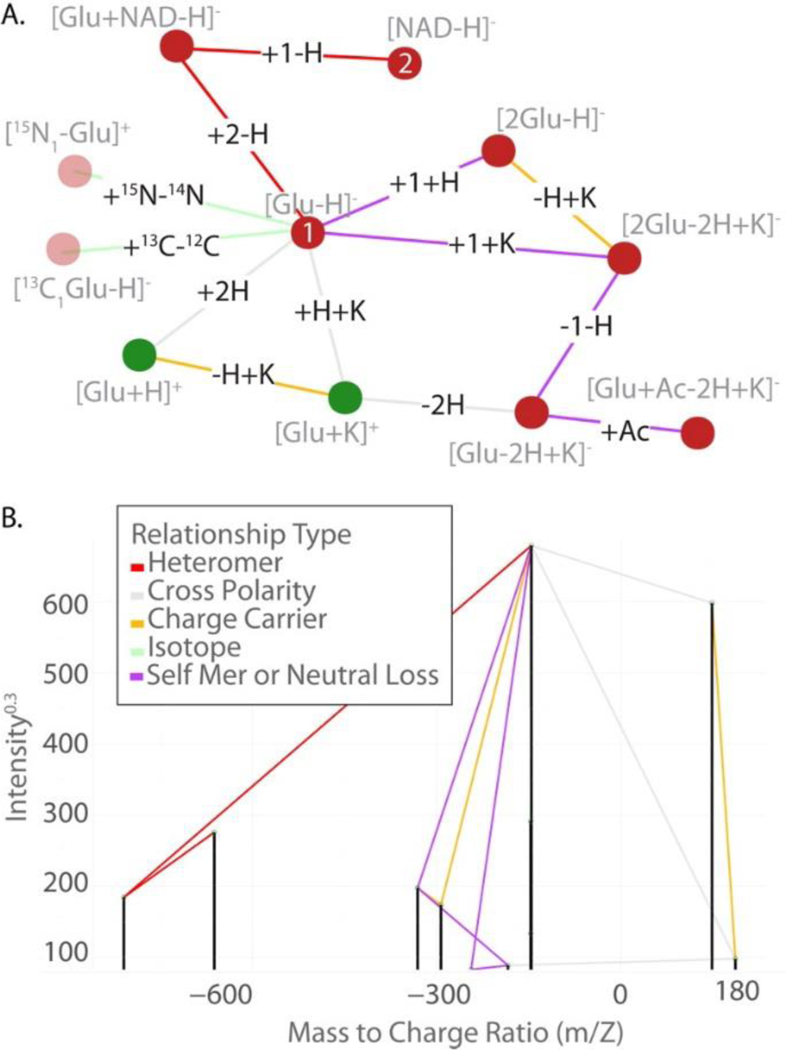
(A) Output of mz.unity represented as a graph structure. Edges represent peak relationships. (B) The modification relating the peaks is noted as text on each edge..Nodes represent detected m/z peaks. (C) The identity of each is noted with gray text by each node. Nodes are colored by polarity: positive (green) (D) and negative (red) Edges are colored by relationship type: charge carrier (yellow), cross-polarity (gray), (E) self-mer (purple), isotopic (green) and heterometer (red) (B) The graph structure in panel A superimposed on (F) the mass spectrum of the relevant peaks. Intensity in this graph is scaled as /0.3 so that the small peaks are visible.
Availability and Implementation
The mz.unity project is written in R and is available at http://github.com/nathaniel-mahieu/mz.unityas well as our laboratory Web site http://pattilab.wustl.edu/software/. Installation instructions, usage examples, data, and analyses presented in this paper can be found in the repository.
Limitations of Mz-sum and Mz.unity
Two limitations of mass- and charge-based annotation are mass measurement error and relationships that have multiple interpretations. Overcoming these limitations requires additional information beyond mass and charge.
Imperfect Mass Information
As described above, the search for appropriately summing masses and charges is a proxy for finding sets of peaks that represent equivalent formulas. Ideally, this search would be performed by using a peak’s underlying formula, but in practice this is not possible. All empirical mass measurements are made with imperfect accuracy, preventing a one-to-one mapping of mass to formula.(16) Thus, a single mass can represent many possible formulas, and this leads to relationships implied by formula mass that do not actually have equivalent formulas. As mass error increases, the number of false positive relationships will also increase. Similarly, the number of combinations of peaks increases rapidly as the number of peaks increases. The combinatorial explosion can quickly overwhelm the specificity offered by accurate masses. This limitation makes annotation of direct infusion data and spectra with over 5000 peaks challenging.(17, 18)
Relationship Ambiguity
Even with perfect formula information, some peak relationships have multiple interpretations that cannot be resolved without additional information. Common neutral losses such as a [H2O]0 loss could relate either a fragment analyte pair or two distinct analytes. Consider the following two interpretations of a relationship between peaks [133.0142]1– and [115.0037]1–. [malate−H]--[H2O]0=[malate-H2O]-(A)[malate-H]--[H2O]0=[fumarate-H]-(B)
In case A, the smaller peak is a fragment and the two peaks are degenerate, while in case B both peaks are distinct analytes. The two interpretations of this relationship are identical in terms of mass and charge, and additional information is required to determine which is true.
Similarly, fragment vs adduct is challenging to discriminate on the basis of mass and charge alone. In both cases, two formulas sum to a third. Consider tyrosine and coumaric acid, [163.0401]1– + [NH3]0 = [180.0666]1–. This could represent a fragment of tyrosine, in which case the [180.0666]1–peak would be the relevant ion. Alternatively, this could be an ammonium adduct of coumaric acid, in which case the [163.0401]1– peak would be the relevant ion. This ambiguity is true of all distal fragment and mer relationships. The two competing interpretations imply the relevance of different peaks: fragmentation events imply the heavier peak’s relevance, while mer relationships imply the relevance of the two lighter peaks.
Data Set Generation
For evaluation of mz.unity, we experimentally generated spectra in positive and negative polarity by using the Q-Exactive Plus mass spectrometer and the HESI-II ion source coupled to an Agilent 1260 capillary flow liquid chromatography system. Spectra were collected with the following settings: aux gas, 15; sheath gas, 30; counter gas, 0; capillary temperature, 310 °C; sheath gas temperature, 200 °C; spray voltage, 3.2 kV; needle diameter, 34 gauge; s-lens, 65 V; mass range, 85–1165 Da; resolution, 140 000; microscans, 1; max injection time; 200 ms; automatic gain control target, 3 × 106. Hydrophilic interaction liquid chromatography (HILIC) was performed as described previously with the Phenomenex Luna NH2 column (1.0 mm × 150 mm, 3 um) and a flow rate of 50 μL/min.(19) Spectra were collected in negative and positive ion mode during two different injections. Solvents were the following: A, 95% water + 20 mM ammonium hydroxide + 20 mM ammonium acetate; B, 100% acetonitrile. An injection volume of 1 μL was used with a linear gradient of (minutes, %A) 0, 5; 40, 100; 50, 100; 50.5, 40; 54.5, 15; 55, 5; 65, 5.
Spectra were taken from a data set of Escherichia coli (E. coli) strain K12, MG1655 metabolic extract. This design allowed us to inspect real-world data, including coelution and background ions. Metabolic extract was generated as described previously.(20) Briefly, cultures of E. coli were harvested by pelleting 10 mL of culture at OD600 = 1.0. Pellets were extracted by using 1 mL of 2:2:1 methanol/acetonitrile/water, and reconstituted in 100 μL of 1:1 acetonitrile/water.
Liquid chromatography/mass spectrometry (LC/MS)-based techniques generate a series of mass spectra. Peaks that appear in several sequential spectra with a Gaussian-like profile are termed features (peaks whose intensity rises and falls around a regional maximum over chromatographic time). Chromatographic feature detection was performed on the data set by using the centWave algorithm.(21) Features eluting from 21 to 22 min were used as a test set (FG). This included features from both positive and negative analyses. The set of background peaks (BG) was obtained by retaining all mass spectral peaks appearing in 80% of the scans within this range, regardless of peak shape. Peak lists used for annotation can be found in Supporting Information Supplements S2–S6, and a spectrum can be found in Supporting Information Supplement S7.
Standards of glutamate and NAD were then analyzed by direct infusion to validate the detected relationships. A solution of NAD and glutamate (both at 50 μg/mL in buffer A) was infused at 10 μL/min, and spectra were collected at a resolving power of 280 000 in both positive and negative mode.
Data Set Annotation
Mass spectra from an LC/MS analysis of E. coli metabolic extract were searched for relationships by using mz.unity. Several mz.unity searches were performed, each for different relationship types. In brief, the following relationships were searched by altering the supplied granular formulas and search depth: isotopes, charge carriers, neutral gains, cross-polarity, common fragments, distal fragments, and mers. Isotopes were detected and omitted from later searches. Charge states were assumed to be to 1 unless carbon isotope support for a higher charge state existed. Searches were performed with a ppm error limit of 2 ppm per observed mass. Exact parameters for each search, including supplied granular formulas and search depth, can be found in Supporting Information Supplements S7 and S8. Putative relationships detected by mz.unity were visualized as graphs and spectral graphs (Figure 3) by using built-in plotting functionality. The graph of relationships was parsed to reveal sets of peaks generated by a single analyte. From the relationship graph, fine isotopic patterns were extracted.
Figure 3:
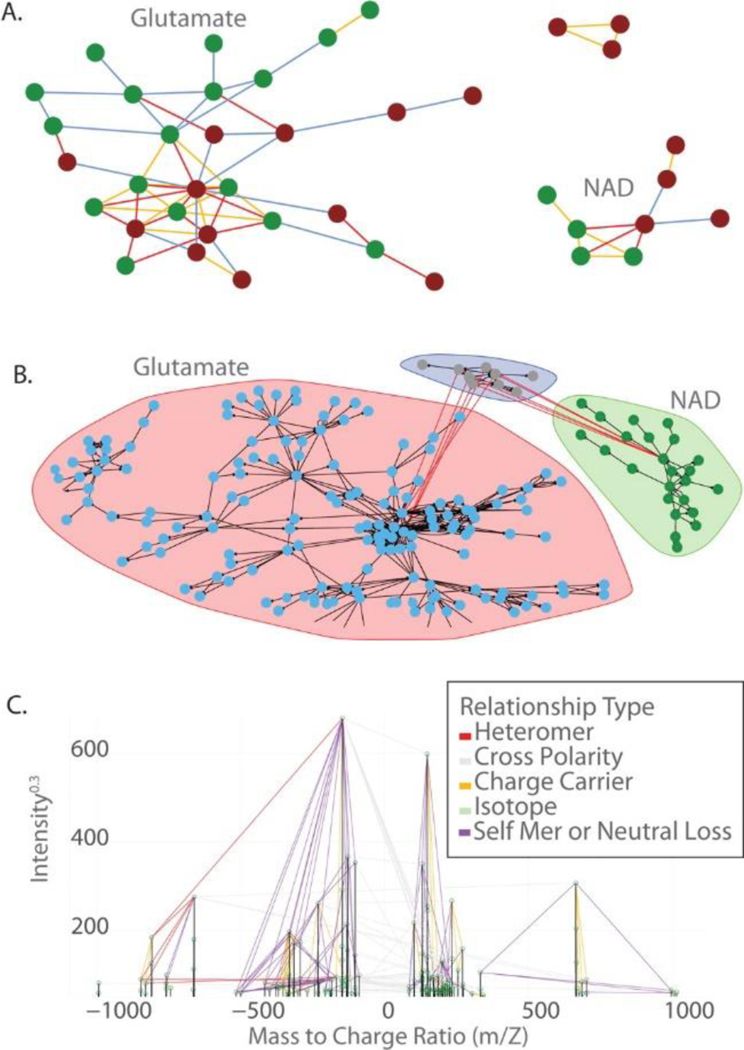
Visualizing peak relationships from the analytes, glutamate and NAD. (A) Annotation of simple relationships between NAD and glutamate. Each node is an m/z peak, and each edge is a detected relationship. (B) Visualization showing the result of the annotation of complex relationships. Peaks derived from the Glu-NAD heteromer are shown in the blue area. This plot includes isotopes, heteromers, homomers, charge carriers, and neutral losses but omits fragments and background mers. (C) The spectral graph of the results is shown in panel B
Results and Discussion
Annotation of a Spectrum Containing Glutamate and NAD
We demonstrate mz.unity, our charge-aware framework for detecting and exploring peak relationships, with a set of peaks observed from the LC/ESI/MS analysis of an E. coli extract. The extract was a complex mixture of small molecule analytes that gave rise to approximately 46 000 total features when analyzed in both positive and negative polarities. The spectrum used to evaluate mz.unity was a composite taken from the time range of 21–22 min consisting of 454 features (peaks with a Gaussian-like shape in the chromatographic domain) and 2212 background peaks. This spectrum was annotated with incremental relationship searches covering various relationship types.
Two groups of peaks were considered, features and background peaks. In LC/MS techniques, all detected analytes of interest appear as features and therefore annotation typically seeks to remove redundancy from the set of peaks that are features. Still, to fully annotate the features, background peaks must be considered as participants in adduct formation. The chromatographic domain was used only to classify mass peaks as features or background peaks, and mz.unity analysis relied only on the mass and charge of the classified peaks.
We consider three general types of relationships in this discussion of results. Simple annotations relate two detected peaks through supplied, granular-mz. Distal fragment and mer relationships relate three or more detected peaks and some number of granular-mz. Finally, background relationships are mers formed between features and the background peaks. All relationships were searched, combining both positive and negative polarities.
Simple Annotations
Isotope searches detected 64 monoisotopic features having isotopic support. This isotopic support consisted of 101 isotopic features identified in 141 relationships. The remaining 289 features lacked isotopes, indicating low abundance or various types of detector noise. Fine isotopic structure of analytes could be annotated below ~300 Da where resolution permitted.
Charge-aware search, as implemented in mz.unity, allowed for relationships between positive and negative mode ions to be detected simply. These included relationships like [Glu – 2H + K]1– + [2H]2+ = [Glu + K]1+. The charge-aware search also enabled the inclusion of a neutral mass, [glutamate]0, in the search and easy retrieval of all transformations of this specific mass. In targeted mining approaches, the annotation search can be seeded with relevant analyte neutral masses for simple compound spectra generation. Charge carrier searches between the 64 monoisotopic features with isotopic support detected 104 relationships, 52 of which were cross-polarity relationships (Figure 3A).
Ambiguous relationships have two interpretations that are indistinguishable by mass and charge alone. These relationships can be drawn between two distinct analytes as well as analyte–fragment or analyte–adduct pairs. We detected 91 ambiguous neutral losses corresponding to loss of [NH3]0 and [H2O]0. Manual review of these ambiguous relationships suggested that each of these were true neutral losses and not distinct analytes. Review consisted of evaluating chromatographic peak shape and the elution time of the possible derivative analytes as well as fragmentation spectra of the putative parent. An example confirmation was the relationship [Glu – H]1– – [H2O]0 = [128.0351]1–, which was confirmed by using the fragmentation spectrum of a glutamate standard as seen in Supporting Information Supplement S10. The automated resolution of ambiguous relationships is one of the challenges that remains to be addressed by an automated annotation solution.
Unambiguous simple relationships included additional neutral losses and several adducts common to this chromatography such as [CH3CN]0 and [SiO3H2]0. These relationships are unambiguous as the fragments are rare and the related formulas are unlikely to coelute. Within the 454 features, 193 additional neutral relationships were detected. A breakdown of these neutral relationships can be found in Table 2. This annotation of simple relationships reduced the 64 isotopically supported features to 34 feature groups.
Table 2.
Distribution of Common Neutral Losses Detected
| formula | count |
|---|---|
| - H2O | 50 |
| - CO2 | 19 |
| - NH3 | 41 |
| + HCOOH | 20 |
| + CH3COOH | 15 |
| + CH3CN | 47 |
| + CH3OH | 32 |
| - CO | 43 |
| + H3PO4 | 3 |
| + SiO3H2 | 6 |
| + SiO4H4 | 4 |
| + SiC2H6O | 4 |
Annotation thus far is similar to annotations provided by traditional rule tables. The only extension we have provided at this point is the inclusion of charge awareness that enabled the linkage of analytes from positive and negative mode as well as neutrals. We extend annotation beyond the traditional annotation scope in the next section.
Mer and Distal Fragment Annotations
A novel set of annotated relationships included mers and distal fragments. Both of these relationship types follow the pattern of relating three or more detected features (i.e., represent complex relationships). This contrasts with approaches based on rule tables that are limited to two detected features. The distinction between mer and distal fragment is in the interpretation; distal fragments imply that the heavier feature is the original analyte while mers imply that the lighter features are the original analyte. In the absence of tools to classify relationships as mers or fragments, we have presented summaries of these searches.
Searching for analyte–analyte complex relationships asserted 420 relationships between 263 analyte peaks (analyte peaks include peaks from features and background). When these were examined, examples of both distal fragmentation and analyte–analyte adduction were seen. For example, a distal fragment pair of NAD was found: [123.0553]1+ + [540.0536]1– – [H]1+ = [NAD – H]1– and confirmed by MS/MS as per the section on Fragment Annotations. The analyte–analyte adduct [glutamate – H]1– + [NAD – H]1– + [H]1+ = [glutamate + NAD – H]1– was also detected (Figure 3B). The reduction of complex relationships into analyte groups relies on classification of the relationship as mer or distal fragment. Accordingly, we cannot present known analyte groups.
As described above, mass measurement error contributes to false positive peak relationships. Combinatorial searching for peak relationships can rapidly exceed the specificity offered by the mass accuracy of the technique. Ultimately, a solution to probabilistically evaluate each putative relationship is needed for automated annotation. In the absence of this solution, we have manually evaluated a portion of putative relationships to control for the possibility of false positives. Known constituents of the spectrum were checked for incorrect relationships. If the search produced a significant number of false positive relationships, we expected to find these peaks implicated in incorrect relationships. The peaks corresponding to glutamate and NAD had no false positive relationships, indicating that in general these results are valid.
Similarly, mers between analytes and background peaks were searched. Ideally, this search should exclude the possibility of fragment relationships because fragments would appear as features. In practice, some fragment features are detected but not recorded as features and thus enter the background pool. For this reason, we again omit the generation of analyte groups. A search of relationships with background peaks resulted in 474 relationships between 373 peaks. Of those 373 peaks, 129 were background peaks and 244 were features. We show an example of a background mer relationship later in the section on the Application to 2-Hydroxyglutarate Metabolism. A summary of the detected relationships is shown in Table 3.
Table 3.
Distribution of the Types of Relationships Detected
| relationship type | count |
|---|---|
| background mer | 474 |
| cross-polarity mer/fragment | 137 |
| single-polarity mer/fragment | 283 |
| neutral loss | 284 |
| cross-polarity | 52 |
| charge carrier | 52 |
Fragment Annotations
To examine the ability of mz.unity to detect fragments, we collected the targeted fragmentation spectrum of a neat NAD standard (Supporting Information Supplement S9). This obviates the possibility of mer formation because only the NAD precursor m/z was experimentally selected by the quadrupole for fragmentation. Fragment annotation is enabled by mz.unity’s charge-aware complex relationship searches. Spectra from a variety of collision energies and both positive and negative polarity were deisotoped and combined into a composite spectrum consisting of 283 peaks (two of which were the protonated and deprotonated parent peaks). Fragment relationships were detected within this composite spectrum.
Mz.unity detected 404 pairs of fragmentation relationships (Figure 4, parts A and B). These are pairs of detected fragments that correspond to the two liberated portions of the parent ion (Figure 4A). Interestingly, mz.unity’s charge-aware annotation is a major advantage for this type of search. In 250 of the detected fragment relationships, one fragment portion was detected in positive mode while the second fragment portion was detected in negative mode (Figure 4C). We also evaluated how intensity impacted the probability of finding both fragment halves. As expected, more intense fragments were more likely to result in a detected pair (Figure 4D). This implies that the number of annotated fragments will be dependent on the sensitivity of the instrument.
Figure 4:
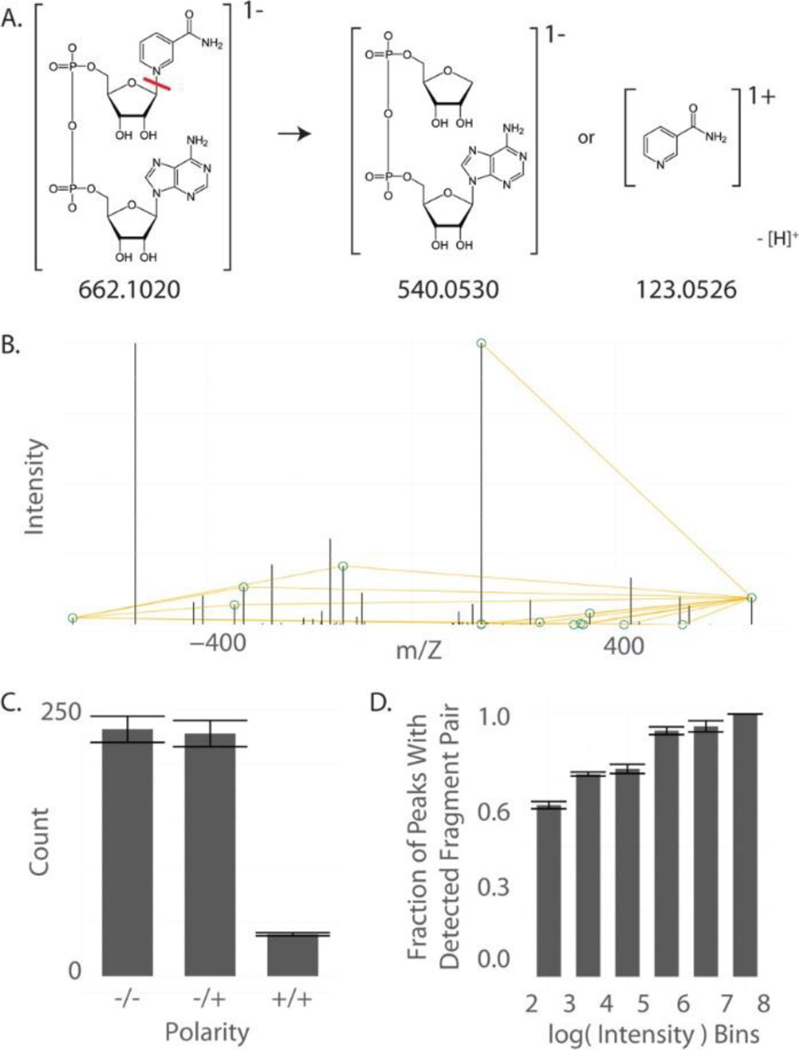
Distal fragment searches. (A) Schematic of NAD fragmentation resulting in two distal fragments. (B) The fragmentation spectrum of NAD and the pairs of distal fragments that sum to the positive and negative molecular ions. (C) The number of fragment pairs detected in each polarity. Most fragments were detected by combining positive and negative polarities. (D) The portion of peaks with detected distal fragments at varying intensity.
We supplemented the distal fragment search with several common fragments that were unable to be detected on our mass spectrometer due to their low mass. In their neutral form, these were [H2O]0, [NH3]0, [CO2]0, and [CO]0. The possibility of ambiguous relationships was excluded because this was a targeted MS/MS experiment omitting other analyte species. These common neutral losses resulted in the annotation of 86 additional fragmentation relationships.
Of the original 283 peaks in the fragmentation spectrum of NAD, a combination of common neutral loss and distal fragment annotation included 171 peaks (60% of all detected fragments). The remaining fragments were both not in our list of common fragments and lacked a detectable distal second half. Annotation of this type of fragment remains an open challenge to future annotation techniques.
Annotation Summary
This work represents the most thorough annotation of a complex LC/ESI/MS spectrum to date and has important implications for the analysis of metabolomic data. We show that commonly occurring complex spectral relationships lie beyond the scope of previous annotation approaches. Consequently, the amount of spectral degeneracy in mass spectrometry-based data sets has been underestimated. The two analytes in this spectrum provide a somewhat contrasting picture of this degeneracy. Both glutamate and NAD were of relatively high abundance with intensities of 1 × 109and 3 × 108, respectively. Although they were present at similar intensities, glutamate produced 98 peaks and NAD only produced 23. The results presented here underscore the need for thorough analysis of metabolomic data sets to ensure that the myriad of redundant peaks and noise sources do not obscure relevant analytes.
Application to 2-Hydroxyglutarate Metabolism
Although additional work is required to implement mz.unity as an automated annotation solution on a comprehensive scale, even in its current form mz.unity provides a powerful resource for interpreting LC/MS-based untargeted metabolomic data. In this section, we provide one brief example application to highlight the utility of our mz.unity software package in processing untargeted metabolomic results.
The metabolite 2-hydroxyglutarate (2HG) is known to accumulate in several types of cancer due to gain-of-function mutations in isocitrate dehydrogenase 1 and 2.(22–24) However, the biochemical effects of 2HG accumulation are incompletely understood. We were interested in testing the hypothesis that cancer pathogenesis might be at least partially mediated by the downstream metabolism of 2HG.
We first needed to determine if 2HG is transformed into downstream products in cells. This was accomplished by comprehensively tracking the transformation of uniformly labeled 13C 2HG (U-13C 2HG) into downstream metabolites.(25, 26) From the thousands of features we screened by untargeted metabolomics, we found 10 features that were greater than 5-fold enriched with 13C carbon compared to natural-abundance samples.
To investigate the identity of these 10 enriched features, we first analyzed the data with the rule-table based annotation package CAMERA.(1) CAMERA indicated that 6 of the 10 features were adducts of 2HG, leaving 4 of the 10 features to represent biochemical transformations of 2HG. Importantly, this result seemed to support the metabolism of 2HG into downstream products. Therefore, we applied the conventional untargeted metabolomic workflow to identify these features as unique metabolites. When the accurate mass and MS2 data did not match those in databases, we began to explore the exciting possibility that these features might represent novel “unknown” metabolites. Fortunately, before committing to this path, we further analyzed the data with mz.unity to search for complex relationships and fragments. With mz.unity, we discovered that the remaining four features were indeed complex adducts and fragments of 2HG (Supporting Information Figure S11). The mz.unity result fundamentally altered the conclusion of our experiment, showing that 2HG is not readily metabolized in the cells we tested. This brief example illustrates how the mz.unity software package can be used in untargeted metabolomic workflows to analyze and refine lists of potentially interesting features.
Observed Failures of Current Annotation Assumptions
In-depth analysis of the aforementioned data sets revealed several assumptions made by current annotation approaches that do not hold in practice. The application of these assumptions therefore prevents the annotation of several relationships in our data sets.
EIC Correlation
Analytes detected by LC/MS techniques elute over sequential spectra with a Gaussian-like profile. A common assumption made by current annotation approaches is that related features will have similar peak shapes. This similarity is commonly measured as the Pearson product moment correlation (Pearson’s r) between the extracted ion chromatograms (EICs) of the two peaks.(10)Two risks exist: high correlation and assertion of a relationship between unrelated peaks, and low correlation and segregation of related peaks. We find both of these cases to be common in our data sets. We present two cases in which related peaks exhibit low correlation.
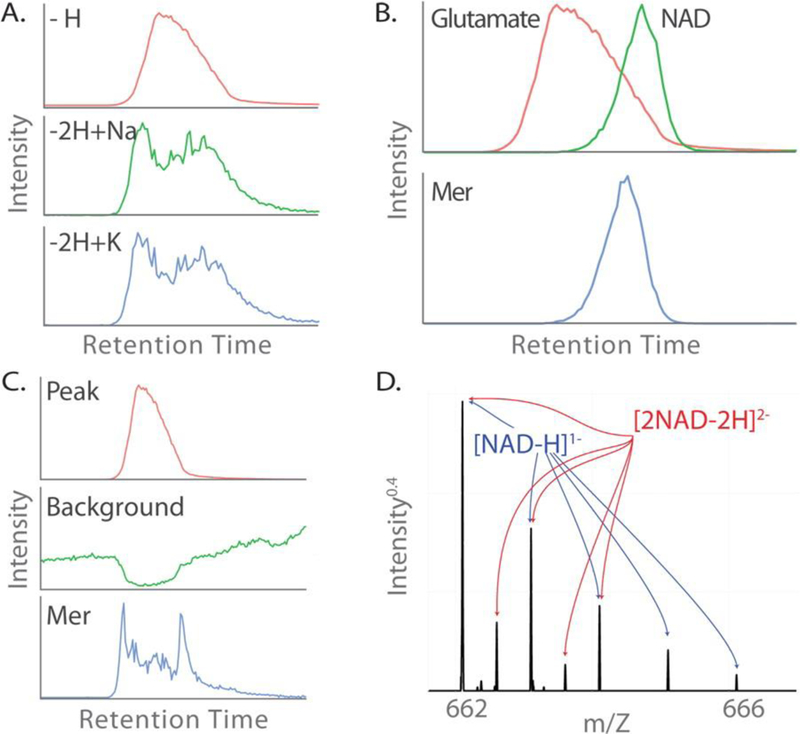
Figure 5A shows three salt adducts of glutamate (Glu) that were annotated by mz.unity in our data set: [Glu – H]1–, [Glu – 2H + Na]1–, and [Glu – 2H + K]1– corresponding to m/z 146.0455, 168.0276, and 184.0015, respectively. The EIC of the deprotonated form exhibits a smooth peak shape typical of our chromatography, but the EICs of both salt adducts exhibit a strikingly different profile. Each initially rise in tandem with the elution of the deprotonated form but quickly plateau. It is clear that each of these salt adducts is related to the [Glu – H]1– peak, yet their correlation is far below useful cutoffs (r of 0.59 and 0.53, respectively).(27)
Figure 5:
Surprising annotation examples. (A) The sodium (middle) and potassium (bottom) adducts of glutamate exhibit different peak shapes than the deprotonated form (top) (Pearson’s r of 0.59 and 0.53). (B) Overlapping peaks glutamate and NAD (top) adduct to form a glutamate-NAD mer (bottom). (C) An artifactual peak (bottom) is formed from the adduction of glutamate (top) and a background peak that lacks a chromatographic peak shape (middle). (D) A single m/z peak with two charge states and two formulas. The base peak at 662 is composed of [NAD - H]- and [2NAD - 2H]2-, as evidenced by the annotated isotopic packet. Masses, intensities, and retention times can be found in Supporting Information, Figure S12.
A second example of poor EIC correlation between related peaks occurs when two adducting species elute at different times. This is the case in the adduction of glutamate and NAD to form the GluNAD adduct. As can be seen in Figure 5B, glutamate and NAD have a very low correlation (_r_of 0.09), yet these two ions are related through the glutamate–NAD mer (GluNAD). The heterodimer GluNAD also does not correlate well with either of its parent species (r of 0.34 and 0.78, respectively). Interestingly, the convolution of the glutamate and NAD EIC traces exhibits strong correlation with that of the mer (r of 0.97), suggesting a possible improvement to this test. Importantly, when EIC correlation is used to group detected features prior to relationship detection, the identification of relationships such as these is precluded.
Background Ions
Peaks lacking a chromatographic peak shape (i.e., background peaks) represent chemical species that can be involved in the ionization process. Current annotation approaches consider only ions displaying a chromatographic peak shape, and in doing so they fail to annotate relationships that involve background ions. Background ions have various sources including column bleed, previously eluted compounds washing off the column, solvent impurities, and other contaminants. It is important to emphasize that background ions contribute to detected features with chromatographic peak shapes. As shown in Figure 5C, the adduction of a bona fide feature with a background ion results in a feature with a peak shape. With current annotation approaches, this background-derived artifact would be confused as an additional analyte during later processing. Annotation of this feature is only possible when background peaks are considered during the annotation process.
The adducts in Figure 5A and the background ion in Figure 5C demonstrate characteristics of ion suppression. This general term refers to the reduction in the intensity of a signal due to the presence of other species. It is interesting to note that reduction in the signal of the background ion may not necessarily be due to the mechanisms conventionally thought to underlie ion suppression. Rather than competition for charge or alteration of droplet dynamics, an additional source of “suppression” could be the scavenging of the monomer signal by other adduct signals. The result being that the same number of species are ionized and detected, but the distribution of signal among masses is altered. This is visible in the background trace in which the signal of the dimer necessarily takes signal from the background peak; notably this phenomena may also contribute to non-linearity as peaks reach high intensities. The complexity of this type of ion suppression is further indicated by the adducts in Figure 5A. Adduct formation during droplet shrinkage is a dynamic chemical process involving multiple species. As concentrations change over the course of analyte elution, rates and equilibria will also be altered. In the case of the salt adducts above, it is possible that glutamate sequestered all available salt or alternatively dimer formation became more favorable than the monomer production. The link between adduct formation and ion suppression warrants further study.
Charge States Assignment
A mass spectral peak is generally taken to represent a single species. Figure 5D demonstrates that this is not true in general, but rather, it is possible to detect a single m/z peak which corresponds to two distinct formulas. This is common in the case of multiply charged dimers. In the spectrum of NAD found in Figure 5D, two distinct isotopic envelopes can be seen. The major pattern is the result of [NAD – H]1–. The second pattern has spacing of (13C – 12C)/2, representing a compound of charge state 2–. This pattern is produced by the ion [2NAD – 2H]2–. The m/z of these two ions is identical, 662.1020, but both species have a different charge state, different formulas, and therefore different mass. The assignment of a single charge state can only explain one of the isotopic envelopes. Full annotation requires the consideration of multiple charge states.
Future Directions
Increases in the mass accuracy and resolving power of mass spectrometers have enabled more thorough analyses of metabolomic data sets. The tools described here, mz-sum and mz.unity, leverage these advances to provide a comprehensive list of possible spectral relationships. Still, several relationship classes require information beyond mass and charge to make definitive annotation assignments. Both ambiguous relationships and fragment/mer relationships have multiple interpretations that cannot be distinguished based on mass and charge alone.
We see four distinct challenges remaining for an automated annotation solution: (i) discrimination between distal fragments and adducts; (ii) discrimination between fragments and distinct analytes; (iii) annotation of rare, nondistal fragments; (iv) evaluation of confidence in each asserted relationship. Metabolomic data sets offer many rich sources of information to tackle these challenges. Peak intensity, chromatographic profile, mass decomposition, isotope pattern, convolution of adduct-constitutent’s isotopic patterns, and the web of putative relationships are all expected to offer predictive power in the context of these problems. Network-based optimization problems and probabilistic assessments have addressed similar problems like fragmentation tree calculation and analyte identification with much success.(2, 9, 28)
A challenge distinct from annotation is the prediction of underlying neutral masses that give rise to the spectrum. The web of annotated relationships and additional information sources can be combined to assert the masses and identities of the untransformed analytes. These untransformed masses are of interest for metabolite identification and data interpretation in the context of biochemistry. Ultimately, an automated annotation solution will allow faster and more robust metabolomic data analysis while also enabling reliable analyte identification.
Conclusions
Current approaches fail to annotate a significant fraction of relationships in mass spectrometry-based data sets. We have shown that metabolites such as glutamate produce 100 or more spectral peaks, yet current approaches annotate only a fraction of these. This resulting peak degeneracy is a major challenge to the further analysis of MS data, requiring time-intensive manual curation and increasing the number of false positive and misleading hits. Here we have presented mz-sum and mz.unity, which provide a novel framework for assessing these complex mass spectral relationships and enable identification of degenerate peaks that would not be found with current annotation approaches.
Referring to relationships as mz-sums accurately represents any possible analyte transformation, including complex and cross-polarity relationships. Consideration of all possible analyte transformations is critical to building thorough and robust data set annotation tools for several fields, including metabolomics.(14) Here we have expanded upon the relationship approaches based on rule tables by developing the mz.unity R package. While current annotation approaches are based on common and universal transformations, the true set of possible relationships searched for by mz.unity is much broader, encompassing both complex adducts and distal fragments. Mz.unity is both a convenient tool for manual annotation and interpretation of mass spectra as well as a step toward automated annotation of omic scale data sets.
Supplementary Material
S1-S12
Acknowledgment
This work was supported by funding from NIH Grants R01 ES022181 (G.J.P.) and R21 CA191097 (G.J.P.) as well as grants from the Alfred P. Sloan Foundation (G.J.P.), the Camille & Henry Dreyfus Foundation (G.J.P.), and the Pew Scholars Program in the Biomedical Sciences (G.J.P.). We thank R. Yost for insightful discussion at the preliminary stages of this project.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b01702.
References
- 1.Kuhl C; Tautenhahn R; Böttcher C; Larson TR; Neumann S Anal. Chem. 2012, 84 (1) 283– 289 DOI: 10.1021/ac202450g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daly R; Rogers S; Wandy J; Jankevics A; Burgess KEV; Breitling R Bioinformatics 2014, 30 (19)2764–2771 DOI: 10.1093/bioinformatics/btu370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandez-Albert F; Llorach R; Andres-Lacueva C; Perera A Bioinformatics 2014, 30 (13) 1937– 1939 DOI: 10.1093/bioinformatics/btu136 [DOI] [PubMed] [Google Scholar]
- 4.Zhang W; Chang J; Lei Z; Huhman D; Sumner LW; Zhao PX Anal. Chem. 2014, 86, 6245 DOI: 10.1021/ac501162k [DOI] [PubMed] [Google Scholar]
- 5.Rabiner LR . Acoust. Soc. Am. 1978, 63 (S1) S79 DOI: 10.1121/1.2016831 [DOI] [Google Scholar]
- 6.Wang M; Yu G; Mechref Y; Ressom HW In Proceedings of the 2013 IEEE International Conference on Bioinformatics and Biomedicine, Shanghai, China, Dec 18–21, 2013; Li G-Z; Hu X; Kim S; Ressom H;Hughes M; Liu B; McLachlan G; Liebman M; Sun H, Eds.; IEEE: Piscataway, NJ, 2013; pp 16–22. [Google Scholar]
- 7.Fenn J; Mann M; Meng C; Wong S; Whitehouse C Science (Washington, DC, U. S.) 1989, 246 (4926)64–71 DOI: 10.1126/science.2675315 [DOI] [PubMed] [Google Scholar]
- 8.Wolf S; Schmidt S; Müller-Hannemann M; Neumann S BMC Bioinf. 2010, 11 (1) 148 DOI: 10.1186/1471-2105-11-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Böcker S; Dührkop KJ Cheminf. 2016, 8 (1) 5 DOI: 10.1186/s13321-016-0116-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ipsen A; Want EJ; Lindon JC; Ebbels TMD Anal. Chem. 2010, 82 (5) 1766– 1778 DOI: 10.1021/ac902361f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zerck A; Nordhoff E; Resemann A; Mirgorodskaya E; Suckau D; Reinert K; Lehrach H; Gobom JJ Proteome Res. 2009, 8 (7) 3239–3251 DOI: 10.1021/pr800835x [DOI] [PubMed] [Google Scholar]
- 12.Liu H; Sadygov RG; Yates JR Anal. Chem. 2004, 76, 4193 DOI: 10.1021/ac0498563 [DOI] [PubMed] [Google Scholar]
- 13.Elias JE; Gygi SP Nat. Methods 2007, 4 (3) 207–214 DOI: 10.1038/nmeth1019 [DOI] [PubMed] [Google Scholar]
- 14.Schmitt-Kopplin P; Gabelica Z; Gougeon RD; Fekete A; Kanawati B; Harir M; Gebefuegi I;Eckel G; Hertkorn N Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (7) 2763–2768 DOI: 10.1073/pnas.0912157107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Draper J; Enot DP; Parker D; Beckmann M; Snowdon S; Lin W; Zubair H BMC Bioinf. 2009, 10 (1)227 DOI: 10.1186/1471-2105-10-227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers S; Scheltema RA; Girolami M; Breitling R Bioinformatics 2009, 25 (4) 512–518 DOI: 10.1093/bioinformatics/btn642 [DOI] [PubMed] [Google Scholar]
- 17.Junot C; Madalinski G; Tabet J-C; Ezan E Analyst 2010, 135 (9) 2203–2219 DOI: 10.1039/c0an00021c [DOI] [PubMed] [Google Scholar]
- 18.Gross RW; Han X Am. J. Physiol. - Endocrinol. Metab. 2009, 297 (2) E297–E303 DOI: 10.1152/ajpendo.90970.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivanisevic J; Zhu Z-J; Plate L; Tautenhahn R; Chen S; O’Brien PJ; Johnson CH; Marletta MA;Patti GJ; Siuzdak G Anal. Chem. 2013, 85 (14) 6876–6884 DOI: 10.1021/ac401140h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahieu NG; Huang X; Chen Y-J; Patti GJ Anal. Chem. 2014, 86 (19) 9583–9589 DOI: 10.1021/ac503092d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tautenhahn R; Böttcher C; Neumann S BMC Bioinf. 2008, 9 (1) 504 DOI: 10.1186/1471-2105-9-504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang L; White DW; Gross S; Bennett BD; Bittinger MA; Driggers EM; Fantin VR; Jang HG;Jin S; Keenan MC; Marks KM; Prins RM; Ward PS; Yen KE; Liau LM; Rabinowitz JD;Cantley LC; Thompson CB; Vander Heiden MG; Su SM Nature 2010, 465 (7300) 966–966 DOI: 10.1038/nature09132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward PS; Patel J; Wise DR; Abdel-Wahab O; Bennett BD; Coller HA; Cross JR; Fantin VR;Hedvat CV; Perl AE; Rabinowitz JD; Carroll M; Su SM; Sharp KA; Levine RL;Thompson CB Cancer Cell 2010, 17 (3) 225–234 DOI: 10.1016/j.ccr.2010.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu W; Yang H; Liu Y; Yang Y; Wang P; Kim S-H; Ito S; Yang C; Wang P; Xiao M-T; Liu L;Jiang W; Liu J; Zhang J; Wang B; Frye S; Zhang Y; Xu Y; Lei Q; Guan K-L; Zhao S; Xiong Y Cancer Cell 2011, 19 (1) 17–30 DOI: 10.1016/j.ccr.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelman SJ; Mahieu NG; Cho K; Llufrio EM; Wencewicz TA; Patti GJ Cancer Metab. 2015, 3,13 DOI: 10.1186/s40170-015-0139-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang X; Chen Y-J; Cho K; Nikolskiy I; Crawford PA; Patti GJ Anal. Chem. 2014, 86, 1632 DOI: 10.1021/ac403384n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pape J; Vikse KL; Janusson E; Taylor N; McIndoe JS Int. J. Mass Spectrom. 2014, 373, 66–71 DOI: 10.1016/j.ijms.2014.09.009 [DOI] [Google Scholar]
- 28.Shen H; Dührkop K; Böcker S; Rousu J Bioinformatics 2014, 30 (12) i157–i164 DOI: 10.1093/bioinformatics/btu275 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1-S12