Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting (original) (raw)
. Author manuscript; available in PMC: 2020 Mar 18.
Published in final edited form as: Cancer Cell. 2019 Mar 18;35(3):489–503.e8. doi: 10.1016/j.ccell.2019.02.003
Summary
Many potential targets for CAR-T cells in solid tumors are expressed in some normal tissues, raising concern for off-tumor toxicity. Following lymphodepletion, CAR-T cells targeting the tumor-associated antigen ROR1 lysed tumors in mice but induced lethal bone marrow failure due to recognition of ROR1+ stromal cells. To improve selectivity, we engineered T cells with synthetic Notch (synNotch) receptors specific for EpCAM or B7-H3, which are expressed on ROR1+ tumor cells but not ROR1+ stromal cells. SynNotch receptors induced ROR1 CAR expression selectively within the tumor, resulting in tumor regression without toxicity when tumor cells were segregated from, but not when co-localized with, normal ROR1+ cells. This strategy, thus, permits safe targeting of tumors that are sufficiently separated from normal cells.
Keywords: chimeric antigen receptor, T cells, immunotherapy, toxicity, ROR1, synthetic Notch receptors, combinatorial antigen recognition, logic-gating
Graphical Abstract
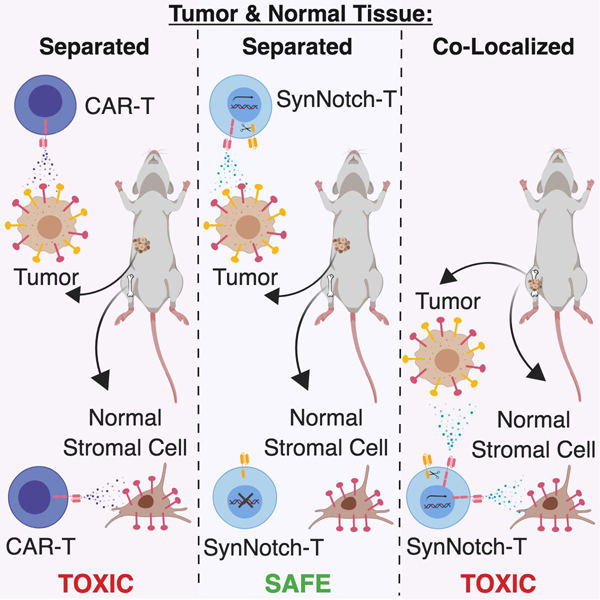
Srivastava et al. show that ROR1-targeted CAR-T cells expressing synthetic Notch receptors for EpCAM or B7-H3, which are expressed on tumor cells but not stromal cells, induce tumor regression without toxicity when ROR1+ tumor cells and ROR1+ normal cells are segregated, but not when they are co-localized.
Introduction
Adoptive cell therapy (ACT) is a promising cancer treatment that involves transferring T cells that are engineered with a synthetic chimeric antigen receptor (CAR) or exogenous T cell receptor (TCR) to redirect T cell activity toward a tumor antigen (Rosenberg and Restifo, 2015). CAR-T cells have shown efficacy in B cell malignancies, where lineage markers like CD19 can be targeted without serious complications from depletion of normal CD19+ B cells (Kochenderfer and Rosenberg, 2013). An obstacle to extending CAR-T cells to solid tumors is identifying cell surface antigens that are expressed on tumor cells but not on critical normal tissues (Srivastava and Riddell, 2018). The mutant EGFRvIII molecule in glioblastoma is tumor-specific but its heterogeneous expression has hindered therapeutic efficacy of EGFRvIII CAR-T cells (O’Rourke et al., 2017). The vast majority of other candidate CAR targets in solid tumors such as HER2, GD2, and mesothelin are co-expressed on various normal tissues, making toxicity a risk of immunotherapy against such molecules (Brudno and Kochenderfer, 2016).
Receptor tyrosine kinase-like orphan receptor 1 (ROR1) is expressed in embryonic development and exhibits high and homogenous cell surface expression in many epithelial tumors and some B cell malignancies (Balakrishnan et al., 2017; Ho et al., 2012). In many tumors, ROR1 expression is associated with poor prognosis (Chien et al., 2016; Zheng et al., 2016) and efforts to target ROR1 with immunotherapy are in progress (Choi et al., 2018; 2015). T cells that express a ROR1 CAR improved survival in xenograft models of ROR1+ human tumors (Hudecek et al., 2013). However, our lab demonstrated that ROR1 was expressed in some normal tissues, raising concerns that targeting ROR1 in patients may cause toxicity (Balakrishnan et al., 2017; Hudecek et al., 2010).
Molecules like ROR1 that are expressed on both tumor and normal tissues might be targeted safely using combinatorial antigen recognition, where engagement of two different antigens is required to elicit full T cell activity (Kloss et al., 2013; Wilkie et al., 2012). One implementation of such an “AND” gate was described by Roybal et al., where a synthetic Notch (synNotch) receptor specific for antigen A induced expression of a CAR specific for antigen B, such that CAR-T cell activity was only elicited against tumors where both antigens A and B were present (Roybal et al., 2016). This study demonstrated that in principle, logic-gated CARs could discriminate single- and dual-positive tumor xenografts using model antigens like GFP and CD19 but did not address whether such a design could provide sufficiently stringent regulation of CAR expression to prevent toxicity to normal endogenous tissue(s) in a clinically relevant model of off-tumor toxicity.
We sought to develop a model of CAR-T cell toxicity to evaluate how T cells might be instructed to discriminate tumor cells and normal tissues. In particular, we sought to understand whether regulated expression of the CAR using synNotch receptors could mitigate toxicity to organs easily accessible to CAR-T cells and in close proximity to tumors without impairing efficacy against a solid tumor.
Results
ROR1 CAR-T cells induce lethal toxicity in pre-conditioned mice
The R12 CAR targeting human ROR1 used in previous xenograft and non-human primate studies was not cross-reactive with murine ROR1 (mROR1) (Berger et al., 2015; Hudecek et al., 2013). Thus, to evaluate whether targeting ROR1 with CAR-T cells in mice caused toxicity, we transduced murine T cells with a 4–1BB/CD3ζ CAR comprised of an scFv specific for an epitope in the Kringle domain of ROR1 that is conserved in mouse and human (Figure 1A) (Hudecek et al., 2013; 2015). A truncated cell-surface murine CD19 (tCD19) was co-expressed with the CAR using a 2A ribosomal skip sequence to enable detection of tCD19+ CAR-T cells by flow cytometry. Adoptive transfer of ROR1 CAR-T cells or control T cells transduced with tCD19 alone into C57BL/6 or BALB/c mice did not induce toxicity, but engraftment of donor T cells was low (<0.02% of live cells) (Figure 1B). However, when mice were pre-conditioned with sub-lethal radiation (500 R) to deplete endogenous lymphocytes and increase engraftment of transferred T cells, all mice receiving ROR1 CAR-T cells but not control T cells progressively lost weight and died within two weeks (Figure 1C). Relative to control T cells, ROR1 CAR-T cells expressed high levels of PD-1 and were present in greater number in spleen, BM, peripheral lymph nodes, and liver, but not in lungs, kidney or pancreas (Figure 1D, Figure S1A). Consistent with this pattern of T cell distribution, serum chemistry of control and ROR1 CAR-T cell-treated mice indicated normal pancreatic, kidney, and parathyroid function (Figure S1B). ROR1 CAR-T cell-treated mice, however, exhibited a rapid decline in red blood cell (RBC) and platelet (PLT) counts (Figure 1E) and elevated serum levels of ALT and AST by 10 days post-transfer (Figure S1B).
Figure 1. ROR1 CAR-T cells induce lethal toxicity in mice pre-conditioned with radiation.
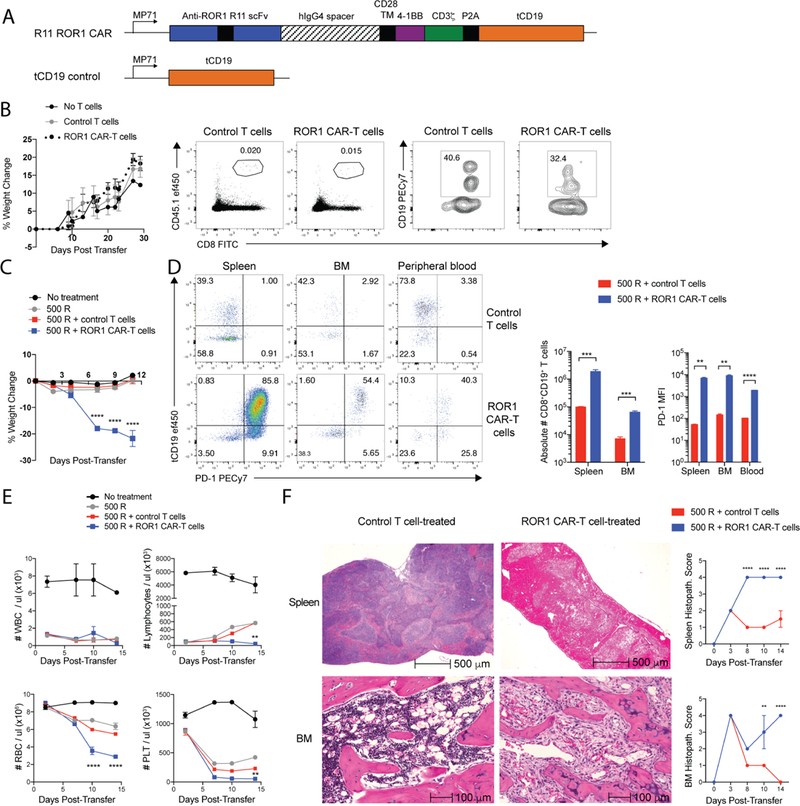
(A) Map of retroviral constructs used to generate ROR1 CAR and control T cells. TM = transmembrane. hIgG4 = human immunoglobulin G4. scFv = single chain variable fragment. (B) Percent change in body weight in BALB/c mice (left) and representative flow cytometric plots showing frequency of CD8+CD45.1+ donor T cells of live cells and frequency of CD8+tCD19+ cells of CD45.1+ donor T cells 7 days post-transfer (right). (C) Percent change in body weight in BALB/c mice. n=4 mice per group. Two-way ANOVA with Tukey’s post-test (ROR1 CAR-T vs control T at Day 7, 9, 11: p<0.00001). (D) Representative flow cytometric plots showing expression of tCD19 transduction marker and PD-1 on CD8+CD45.1+ donor T cells 4 days post-transfer (left) and summary of absolute number and PD-1 median fluorescence intensity (MFI) on CD45.1+CD8+tCD19+ control or ROR1 CAR-T cells 4 days post-transfer (right). n=4 mice per group. Unpaired Student’s two-way t-test (Abs #: spleen, p=0.0004; BM, p=0.0002; PD-1: spleen, p=0.0007; BM, p=0.0007; blood, p=0.000008). (E) White blood cell (WBC), lymphocyte, red blood cell (RBC), and platelet (PLT) counts in peripheral blood. n=4 mice per group. Two-way ANOVA with Tukey’s post-test (ROR1 CAR-T vs control T: Lymphocytes Day 14, p<0.005; RBC Day 10,14, p<0.00001; PLT Day 14, p<0.005). (F) Representative H&E stains of spleen and femur (BM) 14 days post-transfer (left) and histopathology scoring of control or ROR1 CAR-T cell-treated tissues (right). n=4 mice per group. Two-way ANOVA with Sidak post-test (Spleen Day 8,10,14, p<0.00001; BM Day 10, p=0.004; BM Day 14, p<0.00001). Data are representative of 5 independent experiments. All data are presented as the mean values ± SEM. See also Table S1 and Figures S1-S2.
Analysis of organ histology from treated mice identified significant pathology in the spleen and BM. Whereas spleen and BM from control T cell-treated mice showed hyperplasia and recovery of all hematopoietic lineages following radiation, spleens from mice treated with ROR1 CAR-T cells showed diffuse necrosis with a paucity of nucleated cells by 14 days after T cell transfer (Figure 1F; Table S1). Femurs from ROR1 CAR-T cell-treated mice exhibited myelofibrosis at the epiphyseal ends of the bone and markedly reduced hematopoiesis, with an absence of erythroid cells and megakaryocytes consistent with the marked decline in RBC and PLT counts (Figure 1F). By contrast, histologic changes in the lungs, pancreas, kidney, and small and large intestines were mild, and resolved to that observed with control T cells by 14 days (Figure S1C, S1D). Livers from control T cell-treated mice showed signs of extramedullary hematopoiesis (EMH), a normal mechanism of recovery from irradiation or chemotherapy, but livers from ROR1 CAR-T cell-treated mice showed a lack of EMH and focal coagulative necrosis (Figure S1C, S1E), consistent with ischemic and/or cytotoxic injury due to severe anemia or high serum IFNγ and IL-6 levels (Figure 1E, Figure S1F) (Greaves, 2012). We observed the same toxicities in mice pre-conditioned with 200 mg/kg cyclophosphamide (Cy) prior to ROR1 CAR-T cell infusion (Figure S2A-C). Thus, adoptive transfer of ROR1 CAR-T cells following lymphodepleting radiation or chemotherapy induced a consistent pattern of splenic and BM toxicity.
Toxicity is dependent on the intensity of lymphodepletion and T cell dose
Lymphodepletion and cell dose are important factors determining engraftment and toxicity of adoptively transferred T cells in humans (Hay et al., 2017). To test how CAR-T cell toxicity was affected by lymphodepletion, mice were given 100 R or 500 R of radiation or 100 mg/kg or 200 mg/kg of Cy prior to T cell transfer. All mice given lower doses of lymphodepletion exhibited reduced weight loss, all survived, and had less robust CAR-T cell expansion in the peripheral blood, suggesting that more intense lymphodepletion promoted greater CAR-T cell accumulation and toxicity (Figure 2A, 2B). Mice irradiated 500 R and given a lower dose of ROR1 CAR-T cells also exhibited reduced weight loss and all recovered (Figure 2C), with low-dose CAR-T cells in the peripheral blood contracting faster and upregulating PD-1 and TIM3 earlier than high-dose CAR-T cells (Figure 2E). These data demonstrate that toxicity from targeting ROR1 is dependent on achieving a threshold level of functional CAR-T cells in vivo, which is influenced by the intensity of lymphodepletion and cell dose.
Figure 2. ROR1 CAR-T cell-mediated toxicity is dependent on degree of lymphodepletion and dose of ROR1 CAR-T cells.
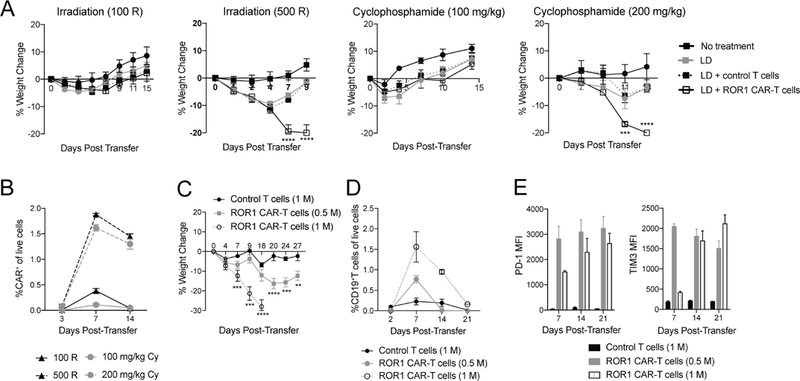
**(**A) Percent change in body weight in BALB/c mice lymphodepleted (LD) as indicated. n=4 mice per group. Two-way ANOVA with Tukey’s post-test (ROR1 CAR-T vs control T: 500 R Day 7, 9, p<0.00001; 200 mg/kg cyclophosphamide (Cy) Day 7, p=0.0004; 200 mg/kg Cy Day 9, p<0.00001). (B) Frequency of CD8+CD45.1+tCD19+ ROR1 CAR-T cells in peripheral blood of BALB/c mice receiving 100 R, 500 R, 100 mg/kg Cy, or 200 mg/kg Cy for LD. n=4 mice per group. (C-E) Percent change in body weight (C), frequency (D) and PD-1 and TIM3 expression (E) on CD8+CD45.1+tCD19+ donor T cells in peripheral blood of BALB/c mice irradiated 500 R and treated as indicated. n=4 mice per group. Two-way ANOVA with Tukey’s post-test (1 M CAR-T vs control T: Day 7, p=0.0003; Day 9,18, p<0.00001; 0.5 M CAR-T vs control T: Day 20, p<0.00001; Day 24, p=0.0005; Day 27, p=0.0044). Data are representative of 2 independent experiments. All data are presented as the mean values ± SEM. See also Figure S3.
Chemotherapy and radiation could have effects beyond inducing lymphodepletion that contribute to toxicity of ROR1 CAR-T cells. Thus, we transferred control or ROR1 CAR-T cells into Rag2 −/− mice without pre-conditioning to test whether lymphopenia alone was sufficient to induce toxicity. Interestingly, _Rag2_−/− mice did not develop weight loss, anemia, thrombocytopenia or mortality after CAR-T cell infusion (Figure S3A, S3B). ROR1 CAR-T cells accumulated in spleen and BM to levels comparable to those in mice that received chemotherapy or radiation, were PD-1+ and Ki67+ and eventually upregulated TIM3, suggesting that some ROR1+ target cells were being recognized in those tissues in the absence of preconditioning (Figure S3C, S3D). Although weight loss and hematopoietic toxicity did not develop, the splenic necrosis observed in WT irradiated mice receiving ROR1 CAR-T cells occurred in _Rag2_−/− mice (Figure S3E, S3F). Femurs from ROR1 CAR-T cell-treated mice showed only minor fibrosis at the epiphyseal ends but normal recovery of hematopoietic lineages throughout the shaft by 30 days after transfer, consistent with the normal RBC and PLT counts in both control- and CAR-treated _Rag2_−/− mice (Figure S3E). Thus, lymphopenia was sufficient to induce ROR1 CAR-mediated splenic necrosis but not BM failure or mortality, indicating that cytotoxic pre-conditioning created a context in which ROR1+ cells in the BM were critical for hematopoietic recovery. Indeed, Rag2 −/− mice pre-conditioned with 500 R radiation prior to transfer of ROR1 CAR-T cells developed rapid weight loss, anemia, thrombocytopenia, splenic necrosis and died (Figure S3G, S3H, S3I). These data demonstrate that the safety of CAR-T cells specific for an antigen expressed on normal tissues can be highly context dependent, and identify both the dose of T cells and the intensity of cytotoxic lymphodepletion as critical variables for revealing toxicity.
ROR1 CAR-T cell toxicity is dependent on ROR1 expression in non-hematopoietic cells
We next sought to identify the cell(s) targeted by ROR1 CAR-T cells. The CAR we used does not recognize ROR2 (Yang et al., 2011); however, Kringle domains are present in many proteins, and toxicity could be due to off-target recognition of cells expressing a homologous protein. To address this possibility, we generated Ror1 “knockout” (“ROR1-KO”) mice by crossing Ror1 fl/fl mice to EIIα-Cre mice, which express Cre in the early mouse embryo, resulting in deletion of Ror1 in all tissues (Ho et al., 2012). WT or ROR1-KO mice were irradiated (500 R) and received either control or ROR1 CAR-T cells. ROR1-KO mice that received ROR1 CAR-T cells did not exhibit any of the toxicities observed in WT mice, including weight loss, anemia, thrombocytopenia, and splenic necrosis, indicating that toxicity was due to recognition of ROR1 (Figure 3A).
Figure 3. ROR1 CAR-T cell-mediated toxicity is dependent on ROR1 expression in non-hematopoietic cells.
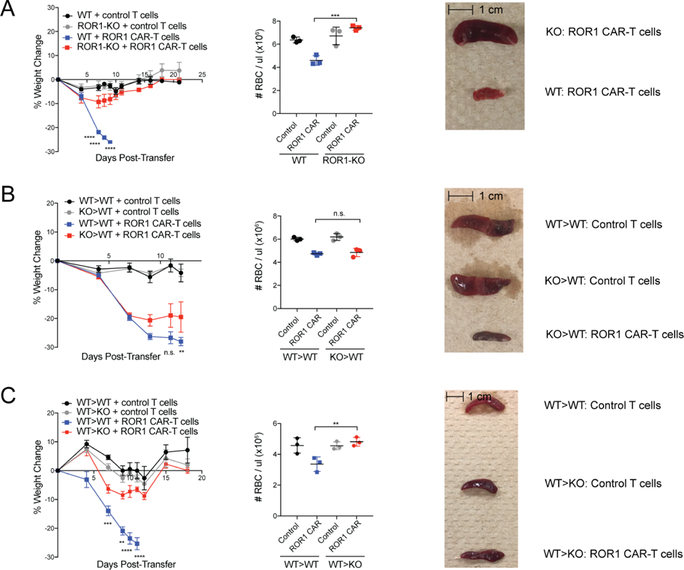
**(**A) Percent change in body weight (left), RBC count in peripheral blood (middle), and representative pictures of spleens 9 days post-transfer (right) from B6 Ror1 fl/fl (WT) or EIIα-Cre+Ror1 fl/fl (KO) mice treated with 500 R and control or ROR1 CAR-T cells. n=3 mice per group. Left: two-way ANOVA with Tukey post-test (WT+CAR-T vs. KO+CAR-T: Day 7,8,9, p<0.00001). Middle: one-way ANOVA with Tukey post-test (p=0.0003). (B) Percent change in body weight (left), RBC count in peripheral blood (middle), and representative pictures of spleens 15 days post-transfer (right) from WT>WT or ROR1-KO>WT BM chimeric mice treated with 500 R and control or ROR1 CAR-T cells. n=3 mice per group. Left: two-way ANOVA with Tukey post-test (WT>WT CAR-T vs. KO>WT CAR-T: n.s. = not significant). Middle: one-way ANOVA with Tukey post-test. (C) Percent change in body weight (left), RBC count in peripheral blood (middle), and representative pictures of spleens 40 days post-transfer (right) from WT>WT or WT>ROR1-KO BM chimeric mice treated with 500 R and control or ROR1 CAR-T cells. n=3 mice per group. Left: two-way ANOVA with Tukey post-test (WT>WT CAR-T vs. WT>KO CAR-T: Day 7, p=0.00013; Day 9, p=0.0045; Day 10,11, p<0.00001). Middle: one-way ANOVA with Tukey post-test (p=0.0044). Data are representative of 2 independent experiments. All data are presented as the mean values ± SEM.
To determine whether hematopoietic and/or nonhematopoietic cell types were targets of ROR1 CAR-T cells, we constructed reciprocal BM chimeras by lethally irradiating WT mice and reconstituting them with WT or ROR1-KO BM. After allowing 8 weeks for full hematopoietic reconstitution, mice were irradiated (500 R) and either control or ROR1 CAR-T cells were adoptively transferred. A comparable decline in RBC and PLT counts, weight loss, splenic necrosis, and myelofibrosis was observed in WT>WT and KO>WT chimeric mice treated with ROR1 CAR-T cells, indicating that hematopoietic ROR1 expression was not required for toxicity to spleen or BM. CAR-treated KO>WT mice showed slightly less severe weight loss than CAR-treated WT>WT mice (Figure 3B), suggesting that ROR1 expression in the hematopoietic compartment may contribute to disease severity, potentially due to expression of ROR1 on pre-B cells (Hudecek et al., 2010), which may provide an antigen source to drive CAR-T cell expansion.
The complementary experiment in which WT or ROR1-KO mice were reconstituted with WT BM showed that mice lacking ROR1 in non-hematopoietic cells were completely rescued from toxicity. WT>KO mice showed no significant weight loss, anemia, splenic necrosis or myelofibrosis, and all survived after receiving ROR1 CAR-T cells (Figure 3C). Thus, ROR1 expression in non-hematopoietic cells is necessary and sufficient for the lethal toxicity mediated by ROR1 CAR-T cells in mice.
Bone marrow and splenic stromal cells express ROR1 and are targeted by ROR1 CAR-T cells
We next examined what subset(s) of non-hematopoietic cells might be reognized by ROR1 CAR-T cells. Data from the Gene Expression Commons showed murine Ror1 expression in BM stromal cells (Seita et al., 2012), many of which provide survival signals to HSCs and promote hematopoiesis (Anthony and Link, 2014). We sorted different BM stromal subsets from WT mice and found high levels of Ror1 transcript in osteoblasts (OBL) and mesenchymal stem cells (MSC) but not in endothelial cells (EC) or hematopoietic progenitors. Transcript levels were further increased in MSC, but not OBL or EC, 48 hours after irradiation (500 R) (Figure 4A). ROR1 CAR-T cells secreted high levels of IFNγ upon co-culture with MSC derived from WT mice but not with MSC from ROR1-KO mice, indicating that WT MSC express sufficient ROR1 protein to activate CAR-T cells (Figure 4B). MSC were drastically reduced in femurs from CAR-T cell-treated mice compared to control mice, demonstrating these cells were targeted in vivo (Figure 4C). Pre-B cell progenitors, which are known to express ROR1 (Hudecek et al., 2010), were also absent from ROR1 CAR-T cell-treated mice (Figure 4C). By contrast, no differences in erythrocyte, megakaryocyte, or granulocyte/macrophage progenitors were observed control and ROR1 CAR-T cell-treated recipients, consistent with the BM chimera data (Figure 3B).
Figure 4. Bone marrow stromal cells express ROR1 and are targeted by ROR1 CAR-T cells.
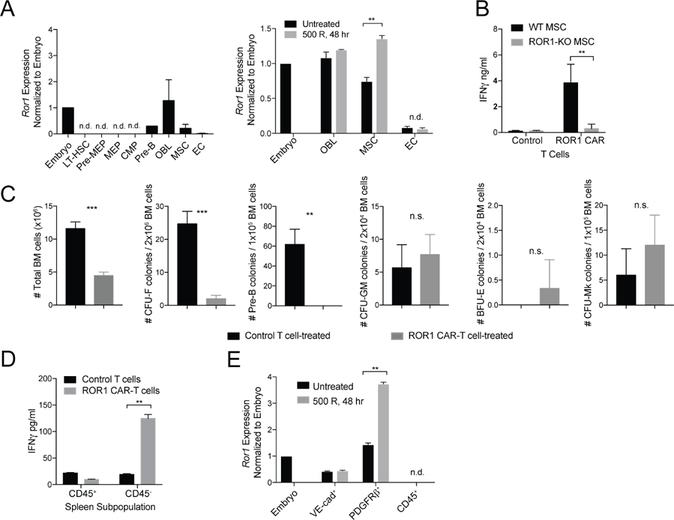
**(**A) qPCR analysis of Ror1 expression in sorted BM progenitors (left) and BM stromal cells (right) from BALB/c mice left untreated or irradiated 500 R 48 hr prior to euthanasia. Ror1 expression was normalized to Actb expression and expressed relative to Ror1 expression in E10.5 embryos. OBL = osteoblasts. MSC = mesenchymal stem cells. EC = endothelial cells. n=3 mice per group. (B) ELISA analysis of IFNγ production by CD8+ control or ROR1 CAR-T cells co-cultured for 48 hr with primary MSC derived from WT or ROR1-KO femurs and expanded in vitro for 14 days. Data are summarized from 3 independent experiments. Unpaired two-way Student’s t-test (p=0.0041). (C) Absolute number of cells in BM flushed from femurs and tibia of control or ROR1 CAR-T cell-treated mice 9 days post-transfer (left) and number of MSC, pre-B cell, granulocyte/macrophage, erythrocyte, and megakaryocyte colonies formed after culture of BM cells from femurs of control or ROR1 CAR-T cell-treated BALB/c mice collected 9 days post-transfer (right). n=3 mice per group. Unpaired two-way Student’s t-test (# BM cells, p=0.0004; MSC, p=0.0004; Pre-B, p=0.0023). (D) ELISA analysis of IFNγ production by CD8+ control or ROR1 CAR-T cells co-cultured for 48 hr with CD45+ or CD45−splenic cells. Data are summarized from 3 independent experiments. Unpaired two-way Student’s t-test (p=0.0049). (E) qPCR analysis of Ror1 expression in splenic subpopulations sorted from BALB/c mice left untreated or irradiated 500 R 48 hr prior to euthanasia. n.d. = not detected. Data are summarized from 3 independent experiments. All data are presented as the mean values ± SEM.
In the spleen, CD45−Ter119−VE-cadherin+ endothelial cells and CD45−Ter119−Tcf21+ perivascular stromal cells produce Scf and CXCL12 that support EMH in situations of stress to the BM (Inra et al., 2015). Microarray data indicated that Ror1 expression was enriched in Scf-producing spleen stromal cells relative to unenriched spleen cells, suggesting that elimination of these cells by ROR1 CAR-T cells may be responsible for the splenic necrosis we observed (Inra et al., 2015). ROR1 CAR-T cells secreted IFNγ upon co-culture with CD45−stromal but not CD45+ hematopoietic splenic cells, indicating recognition of a stromal population in the spleen (Figure 4D). We then sorted different stromal populations and analyzed Ror1 expression by RT-PCR. Most Tcf21+ stromal cells were uniformly PDGFRβ+; therefore we sorted for CD45−VE-cadherin+ splenic endothelial cells and CD45−PDGFRβ+ stromal cells from WT mice prior to and 48 hours after radiation and found Ror1 transcripts in both splenic stromal subsets but not in CD45+ hematopoietic cells from the spleen, with Ror1 transcript levels selectively increased in CD45−PDGFRβ+ stromal cells 48 hours after irradiation (Figure 4E). ROR1 CAR-T cells, thus, target splenic stromal cells and BM MSC, resulting in splenic necrosis, failure of EMH, and the inability to recover hematopoiesis after cytotoxic therapy.
SynNotch EpCAM-inducible ROR1 CAR-T cells selectively target EpCAM+ROR1+ tumors but not EpCAM−ROR1+ normal tissues
Identifying strategies that mitigate off-tumor toxicity to normal tissues without compromising on-tumor immune activity is an unresolved obstacle for CAR T cell therapy. An elegant combinatorial antigen-sensing approach was described in which recognition of one antigen by a synthetic Notch (synNotch) receptor releases an orthogonal transcription factor that drives expression of a CAR that is specific for a distinct antigen (Roybal et al., 2016). Such logic-gated expression confined CAR recognition to dual-positive tumors when single and dual-positive tumors expressing the model antigens CD19 and GFP were spatially separated by implantation on opposite flanks of a mouse. However, the half-life of CAR expression after disengagement of the synNotch receptor is ~8 hours, and T cells expressing sufficient levels of the CAR might migrate to neighboring tissues and mediate toxicity against single-positive normal cells in this time frame.
We searched for combinations of antigens to determine whether the synNotch approach could prevent toxicity mediated by ROR1 CAR-T cell recognition of cells in BM and spleen, where T cells frequently traffic. We found that EpCAM is highly expressed on murine 4T1 mammary carcinoma cells that we transduced to express murine ROR1, but is absent from ROR1+ BM and splenic stromal cells (Figure 5A). We therefore constructed a lentiviral vector encoding a myc-tagged EpCAM-specific synNotch receptor bearing a Gal4-VP64 intracellular transcription activation domain, and a second lentiviral vector in which the ROR1 4–1BB/CD3ζ CAR transgene was placed under control of the UAS promoter that is activated by Gal4-VP64 released after engagement of the synNotch receptor (Figure S4A). Truncated CD19 was included in the inducible CAR cassette as a surrogate marker of CAR expression. A constitutively expressed blue fluorescent protein (BFP) was placed downstream of the inducible CAR transgene to identify transduced T cells.
Figure 5. SynNotch EpCAM-inducible ROR1 CAR-T cells selectively target EpCAM+ROR1+ cells but not EpCAM+ROR1− or EpCAM−ROR1+ cells in vitro.
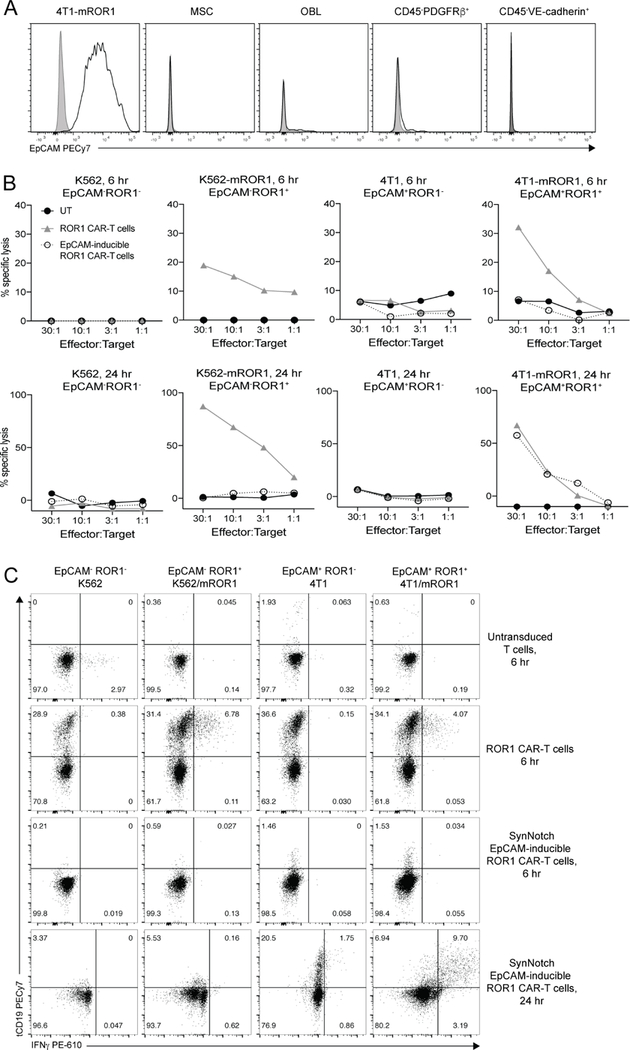
**(**A) Representative flow cytometric analysis of EpCAM expression on 4T1 tumor cells, MSC, osteoblasts (OBL), CD45−Ter119−PDGFRβ+ spleen cells, and CD45−Ter119−VE-cadherin+ spleen cells from BALB/c mice. Percent lysis of the indicated 51Cr-labeled tumor cells after 6 hr (top) or 24 hr (bottom) of co-culture with CD8+ untransduced (black), ROR1 CAR-T cells (gray), or EpCAM-inducible ROR1 CAR-T cells (open circles). (C) Intracellular cytokine analysis of tCD19 CAR marker and IFNγ expression in the indicated T cells co-cultured with tumor cells in the presence of Brefeldin A for the last 6 hr of culture. Data are representative of two independent experiments. See also Figures S4 and S5.
Murine CD8+ T cells were co-transduced with both lentiviral vectors and co-transduction verified by myc and BFP expression (Figure S4B). SynNotch T cells were enriched by FACS sorting BFP+ T cells such that ~30–60% of T cells were myc+BFP+ and carried the full EpCAM-synNotch/UAS-ROR1-CAR circuit while 40–70% of cells were myc−BFP+ and incapable of binding EpCAM or inducing ROR1 CAR expression (Figure S4B). We did not observe any tCD19 expression in the absence of synNotch engagement, indicating lack of leakiness of CAR expression (Figure S4B). For all experiments, transduction of ROR1 CAR-T cells was adjusted to achieve a frequency of tCD19+ ROR1 CAR-T cells that was similar to the frequency of myc+BFP+ synNotch T cells (Figure S4C).
We cultured BFP-sorted synNotch T cells, ROR1 CAR-T, and untransduced T cells with K562, K562-mROR1, 4T1, or 4T1-mROR1 cells to test recognition of ROR1 and EpCAM single- and dual-positive tumor cells. ROR1 CAR-T cells killed both EpCAM+ROR1+ and EpCAM−ROR1+ tumors in 6 hour and 24 hour co-cultures. By contrast, synNotch T cells only recognized tumor cells expressing both EpCAM and ROR1, and comparable lytic activity to ROR1 CAR-T cells required 24 hours of co-culture, consistent with previous reports showing 12–24 hours is required to fully upregulate CAR expression after engagement of the synNotch receptor (Figure 5B) (Roybal et al., 2016). Additionally, whereas the presence of EpCAM was sufficient to induce expression of the CAR marker tCD19 on synNotch T cells, tCD19+ T cells only produced IFNγ when ROR1 antigen was also present (Figure 5C).
Low target antigen expression is a tumor escape mechanism that may impair the ability of synNotch T cells to upregulate CAR expression. To test whether the expression level of the synNotch target affected the kinetics or level of CAR expression, we sorted EpCAMhi and EpCAMlow 4T1 tumor cells and co-cultured them with synNotch EpCAM-inducible ROR1 CAR-T cells. Although EpCAMhi 4T1 cells had >4-fold higher expression than EpCAMlow tumor cells, both populations induced CAR expression on synNotch T cells with similar kinetics and to similar levels, and synNotch T cells killed both 4T1-EpCAMhi-mROR1 and 4T1-EpCAMlow-mROR1 tumors equivalently well in vitro (Figure S5A-C). Low antigen expression, thus, may not necessarily impair the efficacy of synNotch T cells, though the threshold antigen level required to induce CAR expression may vary based on the affinity of the synNotch receptor.
We next tested whether T cells co-transduced with synNotch EpCAM-inducible ROR1 CAR constructs could mediate in vivo antitumor effects against ROR1+ breast cancer without toxicity to normal tissues. We inoculated mice with 4T1-mROR1 tumors in the mammary fat pad and after tumors were palpable, treated mice with 200 mg/kg Cy and either untransduced, ROR1 CAR-T cells, or BFP-sorted synNotch T cells. As previously observed, ROR1 CAR-T cells induced rapid weight loss within one week of transfer and induced severe anemia, thrombocytopenia, and splenic necrosis (Figure 6A–6C). By contrast, mice receiving synNotch T cells recovered weight, RBC and PLT counts with the same kinetics and showed similar splenic morphology as mice that received untransduced T cells. Although synNotch T cells did not induce toxicity to normal ROR1+ tissues, they mediated tumor control comparable to that observed with ROR1 CAR-T cells and had improved survival compared to mice receiving untransduced or ROR1 CAR-T cells, though mice receiving synNotch T cells died later due to tumor outgrowth (Figure 6D, 6E). ROR1 CAR-T cells accumulated in spleen, BM, and tumors, and expressed high levels of PD-1, consistent with local activation (Figure 6F). Although transferred synNotch T cells were detected in the spleen and BM, at least 10-fold fewer of these cells expressed the ROR1 CAR (Figure 6F, 6G). By contrast, synNotch T cells expressing the ROR1 CAR were present in the same high numbers as ROR1 CAR-T cells in tumors, and expressed similar levels of the CAR and markers of activation and proliferation, including PD-1 and Ki67 (Figure 6F, 6G). Thus, synNotch T cells exhibited tumor-selective accumulation, CAR expression, and anti-tumor activity without causing toxicity to critical ROR1+ cells in BM and spleen.
Figure 6. SynNotch EpCAM-inducible ROR1 CAR-T cells rescue toxicity to ROR1+ normal tissues while maintaining activity against ROR1+ tumors.
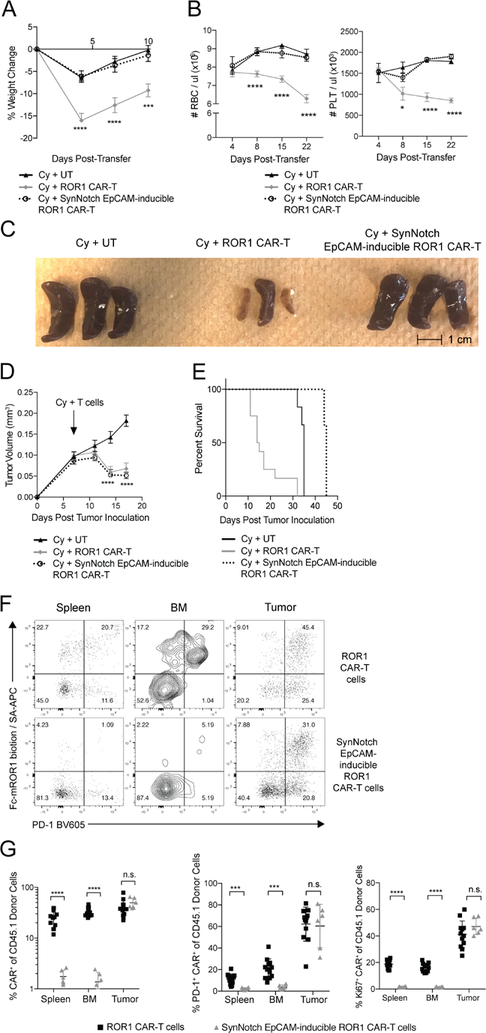
**(**A) Percent change in body weight in BALB/c mice inoculated with 4T1-mROR1 tumors and treated as indicated. n=6–12 mice per group. Two-way ANOVA with Tukey post-test (ROR1-CAR vs. EpCAM-inducible ROR1 CAR: Day 4, p<0.0001; Day 7, p<0.0001; Day 10, p=0.0002). (B) RBC and PLT counts from peripheral blood of tumor-bearing mice treated as indicated. n=6–12 mice per group. Two-way ANOVA with Tukey post-test (ROR1-CAR vs. EpCAM-inducible ROR1 CAR: RBC Day 4,8,15,22:, p<0.0001; PLT Day 8 p=0.0114; PLT Day 15, 22, p<0.0001). (C) Representative pictures of spleens 10 days post-transfer from tumor-bearing BALB/c mice treated as indicated. (D) Tumor volume in BALB/c mice treated as indicated. n=6–12 mice per group. Two-way ANOVA with Tukey post-test (ROR1-CAR vs. UT: Day 14, p<0.0001; Day 17, p<0.0001; EpCAM-inducible ROR1 CAR vs. UT: Day 14, p<0.0001; Day 17, p<0.0001; ROR1-CAR vs EpCAM-inducible ROR1 CAR: n.s). (E) Survival of mice treated as indicated. n=6–12. Log-rank Mantel-Cox test (UT vs. ROR1 CAR, p=0.0003; UT vs. EpCAM-inducible ROR1 CAR, p=0.0016; ROR1 CAR vs. EpCAM-inducible ROR1 CAR, p=0.0001). (F) Representative flow cytometric analysis of ROR1 CAR and PD-1 expression on CD8+CD45.1+ donor T cells from mice treated with Cy and the indicated T cells 10 days post-transfer. (G) Summary of frequency of ROR1 CAR+, CAR+Ki67+, and CAR+PD-1+ T cells of CD8+CD45.1+ donor T cells from mice treated with Cy and the indicated T cells 10 days post-transfer. n=6–12 mice per group. Unpaired two-way Student’s t-test (Frequency: spleen, p<0.0001; BM, p<0.0001; PD-1: spleen, p=0.0004; BM, p=0.0003; Ki67: spleen, p<0.0001; BM, p<0.0001). Data are summarized from 2 independent experiments. All data are presented as the mean values ± SEM.
SynNotch T cells fail to prevent toxicity in mice with a high burden of circulating tumor
SynNotch T cells were safe and effective in the 4T1 breast tumor model, but a potential limitation is the absence of spatial proximity of ROR1+ tumors to ROR1+ normal tissue. Thus, it is unclear if this type strategy would be equally effective when tumor cells are circulating or have metastasized to organs where normal ROR1+ cells reside. To test the role of spatial proximity, we inoculated immunodeficient NOD/SCID/γc −/− (NSG) mice with Raji lymphoma cells transfected to express human ROR1 (hROR1), which results in rapidly progressive metastatic disease in blood and BM. We transduced human T cells with the ROR1 CAR targeting mouse and hROR1, but with human 4–1BB/CD3ζ signaling domains and truncated human EGFR (tEGFR) as a transduction marker. Similar to B6 and BALB/c mice, treatment of irradiated non-tumor bearing NSG mice with human ROR1 CAR-T cells resulted in severe weight loss and anemia, and activated PD-1+ CAR-T cells accumulated in the spleen and BM (Figure S6A-D). We then transduced human T cells with a synNotch receptor specific for human CD19, such that synNotch T cells would upregulate the ROR1 CAR upon recognition of CD19 on the Raji tumor and would be capable of killing both human ROR1+ Raji and murine ROR1+ normal stromal cells (Figure 7A). When NSG mice bearing disseminated Raji/ROR1 tumors were treated with ROR1 CAR or synNotch T cells, synNotch T cells upregulated the ROR1 CAR systemically in the blood by day 7 after transfer, showing only a brief kinetic delay relative to constitutive ROR1 CAR-T cells, and mediated equivalent anti-tumor activity (Figure 7B,C). However, synNotch T cells induced similar levels of toxicity, as mice treated with either constitutive ROR1 CAR or synNotch T cells showed similarly decreased survival due to toxicity compared to control mice (Figure 7D). These data indicate that synNotch T cells are unable to distinguish tumor and normal tissue when tumor is co-localized with normal tissue expressing the therapeutic target, illustrating the importance of spatial segregation of tumor and normal tissue for success of this strategy.
Figure 7. SynNotch CD19-inducible ROR1 CAR-T cells are unable to rescue toxicity to ROR1+ normal tissues in the presence of circulating and bone marrow-resident CD19+ROR1+ Raji tumors.
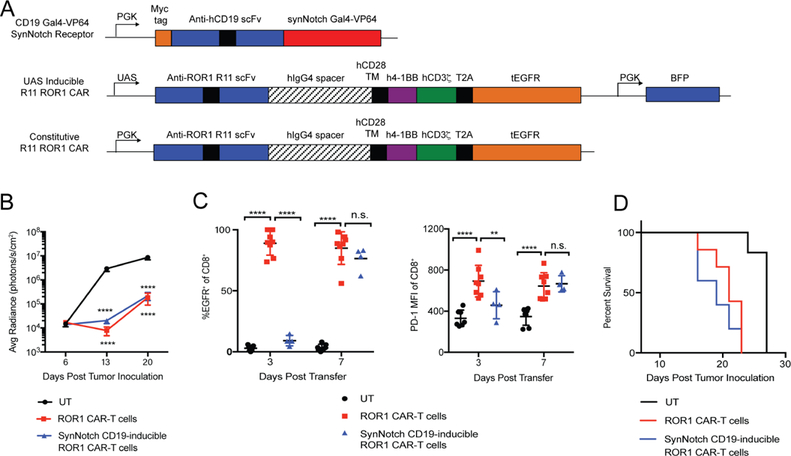
**(**A) Map of lentiviral constructs encoding CD19-Gal4VP64 synNotch Receptor, UAS inducible R11 ROR1 CAR, and constitutive R11 ROR1 CAR. (B) Raji tumor bioluminescence over time in NSG mice irradiated 250 R and treated with the indicated primary human T cells. n=6–7 per group. Two-way ANOVA with Tukey post-test (UT vs. ROR1 CAR-T: Day 13, 20, p<0.0001; UT vs. CD19-inducible ROR1 CAR-T: Day 13, 20, p<0.0001). (C) Summary of tEGFR CAR marker and PD-1 expression on the indicated donor CD8+CD45+ T cells in peripheral blood. n=4–7 per group. Two-way ANOVA with Tukey post-test (CAR: UT vs. ROR1 CAR, Day 3, 7, p<0.0001; ROR1 CAR vs. CD19-inducible ROR1 CAR, Day 3, p<0.0001; Day 7, n.s. PD-1: UT vs. ROR1 CAR, Day 3, 7, p<0.0001; ROR1 CAR vs. CD19-inducible ROR1 CAR, Day 3, p<0.0001; Day 7, n.s.). (D) Survival of tumor-bearing mice irradiated 250 R and treated with the indicated T cell groups. n=6–7 per group. Log-rank Mantel-Cox test (UT vs. ROR1 CAR, p=0.0007; UT vs. CD19-inducible ROR1 CAR, p=0.0007; ROR1 CAR vs. CD19-inducible ROR1 CAR, n.s.). Data are representative of 2 independent experiments. All data are presented as the mean values ± SEM. See also Figure S6.
B7-H3 is a clinically relevant synNotch target that restricts ROR1 CAR expression to tumors and rescues toxicity to normal ROR1+ tissues
We used an EpCAM-specific synNotch receptor in the 4T1 model because EpCAM is not co-expressed on ROR1+ BM or splenic stromal cells in mice. EpCAM is, however, co-expressed with ROR1 on some normal human epithelial tissues, suggesting EpCAM may not be optimal for clinical applications of synNotch. We examined other antigens that could be targeted by the synNotch receptor in humans and identified B7-H3 as a promising candidate, as it is often co-expressed with ROR1 in human breast, lung, and ovarian cancers (Figure S7A-S7D), but expressed at low levels in normal human tissues that express ROR1 (Uhlén et al., 2015). We designed a synNotch receptor specific for human B7-H3 that regulated expression of the ROR1 CAR in human T cells and induced selective IFNγ production and lysis of B7-H3+ROR1+ but not B7-H3−ROR1+ human tumor cells after 24 hours of co-culture (Figure S8A-S8C). By contrast, T cells expressing the ROR1 CAR constitutively produced IFNγ and killed both B7-H3+ROR1+ and B7-H3−ROR1+ tumor cells.
To test whether synNotch T cells could discriminate B7-H3+ROR1+ tumors from normal B7-H3−ROR1+ cells in vivo, we transplanted NSG mice subcutaneously with MDA-MB-231 human breast tumors that endogenously express ROR1 and B7-H3. Mice treated with human T cells expressing the constitutive ROR1 CAR had severe weight loss, anemia, and thrombocytopenia, whereas mice treated with synNotch T cells showed no toxicity but equivalent anti-tumor activity against MDA-MB-231 as observed with constitutive ROR1 CAR-T cells and improved survival (Figure 8A-8D). SynNotch T cells only upregulated ROR1 CAR expression at the tumor site and were PD-1+ and Ki67+, consistent with local activation by tumor (Fig 8E, 8F). Few ROR1 CAR+ synNotch T cells were detected in the spleen or BM and were PD1low (Figure 8E, 8F). By contrast, constitutive ROR1 CAR-T cells were present in tumor, spleen, and BM, and were PD-1+ and Ki67+ at all sites. Combinatorial targeting of B7-H3 and ROR1, thus, may be an effective therapeutic strategy in lung and breast cancer if toxicity is revealed in clinical trials with ROR1 CAR-T cells.
Figure 8. Primary human synNotch B7-H3-inducible ROR1 CAR-T cells rescue toxicity to ROR1+ normal tissues while maintaining activity against human ROR1+ tumors.
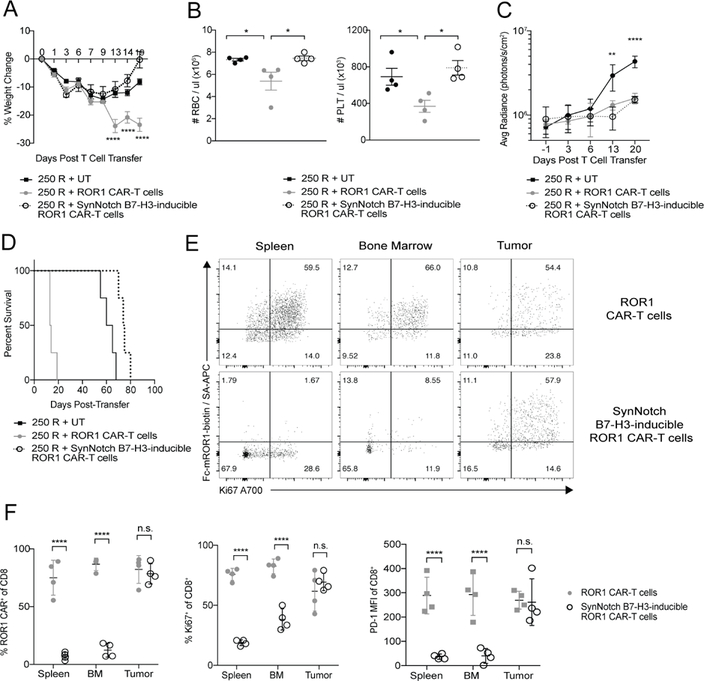
**(**A) Percent change in body weight in NSG mice implanted with MDA-MB-231 tumors, irradiated 250 R and treated with the indicated primary human T cells. n=4 mice per group. Two-way ANOVA with Tukey post-test (ROR1-CAR vs. EpCAM-inducible ROR1 CAR: Day 13, 14, 19, p<0.0001). (B) RBC and PLT counts from peripheral blood of tumor-bearing mice treated as indicated 20 days post-T cell transfer. n=4 mice per group. Two-way ANOVA with Tukey post-test (RBC: ROR1 CAR vs. EpCAM-inducible ROR1 CAR, p=0.0426; UT vs. ROR1 CAR, p=0.0418. PLT: ROR1 CAR vs. EpCAM-inducible ROR1 CAR, p=0.0117; UT vs. ROR1 CAR, p=0.0442). (C and D) Tumor bioluminescence (C) and survival (D) of NSG mice treated as indicated. n=4 mice per group. Two-way ANOVA with Tukey post-test (ROR1 CAR vs. UT: Day 13, p=0.0167; Day 20, p<0.0001. EpCAM-inducible ROR1 CAR vs. UT: Day 13, p=0.0029; Day 20, p<0.0001. ROR1 CAR vs EpCAM-inducible ROR1 CAR: n.s). (E) Representative flow cytometric analysis of ROR1 CAR and Ki67 expression on CD8+CD45+ donor T cells from NSG mice irradiated 250 R and given indicated T cells 22 days post-transfer. (F) Summary of frequency of ROR1 CAR+, Ki67+, and PD-1+ T cells of CD8+CD45+ donor T cells from mice irradiated 250 R and given indicated T cells 22 days post-transfer. n=4 mice per group. Unpaired two-way Student’s t-test (Frequency: spleen, p<0.00001; BM, p<0.00001; tumor, n.s. Ki67: spleen, p<0.0001; BM, p<0.0001; tumor, n.s. PD-1: spleen, p<0.0001; BM, p<0.0001; tumor, n.s.). Data are representative of 2 independent experiments. All data are presented as the mean values ± SEM. See also Figures S7 and S8.
Discussion
The identification of truly tumor-specific target antigens remains a barrier for treating common epithelial cancers with engineered T cells. Many groups are targeting molecules expressed by both tumor and normal cells, and this approach can cause life-threatening toxicity as observed in clinical trials with CAR-T cells targeting CAIX or HER2 due to recognition of cognate antigen on bile duct and lung epithelia, respectively (Lamers et al., 2013; 2006; Morgan et al., 2010). ROR1 is an attractive target for CAR-T cell therapy due to its expression in common solid tumors; however, expression on some normal tissues makes safety a concern (Balakrishnan et al., 2017). We examined CAR-mediated toxicity to endogenous ROR1+ normal tissue using a mouse model to define parameters where normal cells are susceptible to injury and to test strategies to improve safety. In this model, toxicity was dependent on the intensity of lymphodepletion prior to CAR-T cell transfer and T cell dose and was manifest by the elimination of ROR1+ BM and splenic stromal cells that are critical for hematopoietic recovery from cytotoxic stress induced by radiation or chemotherapy. Targeting these stromal cells resulted in severe anemia, thrombocytopenia, splenic necrosis, and mortality. Despite the rapid onset, severity, and location of toxicity, we show in both immunocompetent and immunodeficient mouse models that logic-gated ROR1 CAR expression in mouse and human T cells using synNotch receptors completely rescued toxicity to spleen and BM without diminishing anti-tumor activity against ROR1+ breast tumors. This rigorous model of T cell-mediated toxicity illustrates that combinatorial antigen sensing can be a powerful tool for improving safety of CAR-T cells.
The mechanism of toxicity from ROR1 CAR-T cells is due to recognition of ROR1+ BM and splenic stromal cells that support hematopoietic recovery from cytotoxic stress. MSC have been shown to play an important role in tissue repair and in supporting HSC survival and function following irradiation (Dimarino et al., 2013). Our data show that BM MSC upregulate ROR1 expression upon irradiation in vivo, are targets for ROR1 CAR-T cells, and decline selectively in CAR-treated mice. ROR1 has not previously been implicated in MSC function, but its homologue ROR2 is expressed on mouse and human MSC, and the ROR1/ROR2 ligand Wnt5a promotes MSC differentiation into osteoblasts (Liu et al., 2009). Analogous to MSC in the BM, PDGFRβ+ stromal cells in the spleen support EMH under conditions of stress to the BM and splenic regeneration (Inra et al., 2015; Tan and Watanabe, 2017). Destruction of these splenic cells may prevent tissue regeneration and EMH after cytotoxic therapy, eventually resulting in necrosis of the entire tissue. Some studies suggest that PDGFRβ+ stromal cells are mesenchymal in origin (Castagnaro et al., 2013), suggesting that MSC and PDGFRβ+ stromal cells are developmentally linked and may play parallel roles supporting hematopoiesis and tissue regeneration in their respective organs. Their elimination by ROR1 CAR-T cells destroys critical niches for HSC both inside and outside the BM, thereby preventing hematopoietic recovery from cytotoxic stress that is necessary for survival.
In humans and non-human primates (NHP), ROR1 is expressed in pancreatic islets, parathyroid, esophagus, and gastric mucosa, and there is evidence of ROR1 expression on human MSC (Balakrishnan et al., 2017; Potratz et al., 2016). No toxicity was observed using ROR1 CAR-T cells in NHP or with a monoclonal antibody targeting ROR1 administered to rats, cynomolgus monkeys, and humans (Berger et al., 2015; Choi et al., 2018; 2015), and both therapies have advanced to clinical trials. The lack of toxicity may be due to insufficient levels of ROR1 on normal cells for T cell or antibody recognition, poor T cell trafficking to ROR1+ tissues, or the lack of lymphodepleting cytotoxic therapy. Toxicity in mice with CAR-T cells targeting mROR1 was dependent on the intensity of lymphodepleting therapy, suggesting that toxicity may become apparent in clinical trials as T cell dose or intensity of preconditioning are increased. Our data caution that the absence of toxicity in clinical trials of ACT targeting antigens expressed on normal tissues should be interpreted only within the context of the regimen and T cell dose administered, and not taken to infer that the target is safe. Moreover, our findings illustrate that T cell recognition of rare normal cells such as MSC that are not easily evaluated for target expression by qPCR analysis of whole tissues or IHC of small subsections of tissue can be the cause of lethal toxicity.
Many strategies have been proposed to minimize off-tumor toxicity to normal tissues (Li and Zhao, 2017; Srivastava and Riddell, 2015), but these have not been tested in rigorous preclinical models of CAR or TCR therapy or in patients. Combinatorial antigen sensing or “AND” logic gates exploit a principle of natural T cell biology by integrating multiple signals to regulate the state of T cell activation and/or effector function. “Split signaling” receptors are one incarnation of this strategy that spatially separate CAR CD3ζ and costimulatory domains and depend on the two receptors being non-functional on their own, but functional in combination when both receptors simultaneously bind antigen (Grada et al., 2013; Wilkie et al., 2012). We found a ROR1 CAR carrying only the CD3ζ signaling domain still induced toxicity in irradiated mice (unpublished), indicating that a “split signaling” approach would not rescue toxicity without further modifications, such as lowering the affinity of the scFv to decrease the threshold for activation (Kloss et al., 2013). Like “split signaling” receptors, logic-gated synNotch receptors can mediate activity against dual-positive but not single-positive tumors (Roybal et al., 2016). Whereas “split signaling” receptors require both antigens be engaged nearly simultaneously to co-deliver signals 1 and 2 of T cell activation, there is a significant temporal delay between engagement of the synNotch receptor, induction of CAR expression, and degradation of the CAR following disengagement of the synNotch receptor. Consequently, there is a less stringent requirement for both target antigens to be co-expressed on the same target cell. In fact, mixtures of target cells expressing only antigen A or antigen B still elicit activity from synNotch CAR-T cells despite the absence of any dual-positive cells, indicating that spatial proximity is an important determinant of how well synNotch receptors discriminate single- and dual-positive cells (unpublished). The synNotch strategy, thus, functions more as an “IF/THEN” logic gate than a strict “AND” logic gate. This may be advantageous in the context of antigen loss, where heterogeneous expression of antigen A and B in a tumor might still elicit effective synNotch T CAR-T cell activity. Here, we show that logic-gated synNotch CARs requiring dual recognition of either EpCAM or B7-H3 with ROR1 can safely target murine and human ROR1+ breast tumors without toxicity to normal ROR1+ cells. Whereas targeting ROR1 directly with traditional CAR-T cells resulted in marked accumulation of CAR-T cells in BM and spleen, the vast majority of synNotch T cells found in spleen and BM did not express the ROR1 CAR, indicating limited recognition of EpCAM+ or B7-H3+ cells in those tissues and/or limited migration of ROR1 CAR+ synNotch T cells out of the tumor. Spleen and BM are easily accessible to T cells, and averting toxicity at these sites with synNotch logic-gated CARs provides a rigorous test of the efficacy of this approach.
There are potential situations where synNotch receptors may not be able to distinguish tumor and normal tissue. For example, extensive tumor metastases in BM could activate ROR1 CAR expression and lead to elimination of normal ROR1+ BM stromal cells. The breast cancer models used in our studies do not result in widespread marrow metastasis. However, using a disseminated ROR1+ lymphoma model where marrow involvement with tumor is uniform, we show that synNotch-regulated CAR expression was unable to rescue toxicity. The presence of metastatic disease in multiple distant organs may complicate how best to screen for candidate antigen pairs for logic-gating. Further studies will be required to understand whether tighter regulation of CAR degradation or additional logic gated control can improve tumor and normal cell discrimination. An additional limitation of the current synNotch system is the reliance on a non-human orthogonal transcription factor that could be immunogenic, and alternative transcription factors or structure-guided deimmunization may be necessary for clinical application.
Our results demonstrate that “AND” logic gates are a promising strategy to increase the portfolio of cell surface molecules on solid tumors that can be targeted safely with CAR-T cells. ROR1 is being evaluated as a CAR target in a clinical trial (NCT02706392) in lung and breast cancer. Logic-gating using synNotch receptors may be an effective strategy if toxicity emerges as a barrier with high ROR1 CAR T cell doses and/or more intense lymphodepletion. EpCAM may not be the ideal target for the synNotch component of the logic gate for clinical applications; however, our data show that B7-H3 is a suitable alternative synNotch target since it is frequently co-expressed with ROR1 on human breast, lung, and ovarian cancers but not on the same normal tissues as ROR1 (Uhlén et al., 2015). One can envision many additional pairs and more complex logic gates that impart functions in the local tumor environment that would broaden both the applicability and efficacy of CAR-T cells against solid tumors.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shivani Srivastava (ssrivas2@fhcrc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL/6J (B6), B6 CD45.1, BALB/c, BALB/c CD45.1, BALB/c Rag2 −/−, B6 EIIα-Cre, Ror1 l/flf, and NOD/SCID/γ_c_ −/− (NSG) mice were purchased from Jackson Laboratory. Ror1 fl/fl mice were backcrossed for three generations to B6 mice and subsequently crossed to EIIα-Cre mice. For xenograft studies, 8–10 week old age-matched and sex-matched male or female NSG mice were used. For all other experiments, 6–8 week old age-matched and sex-matched mice were used. Mice of the same sex were randomly assigned to experimental groups. All mice were housed and bred at the Fred Hutchinson Cancer Research Center (FHCRC) (Seattle, WA). All experiments were approved by the Institutional Animal Care and Use Committee of the FHCRC and were performed in accordance with the relevant institutional and national guidelines and regulations.
Human Subjects
Human breast, lung, and ovarian tissues were purchased from Cureline or as tissue microarrays (TMAs) from US-Biomax (BR1141 breast cancer, core J8; BCS04017a lung adenocarcinoma, core H5). The use of commercially obrtained human tissues was considered exempt by the FHCRC Institutional Review Board.
Cell Lines
K562, 4T1, Raji, and MDA-MB-231 cell lines were obtained from the American Type Culture Collection. SK-N-DZ cells were generously provided by Michael Jensen (Seattle Children’s Hospital). Lenti-X cells for lentiviral packaging were purchased from Clontech. Plat-E cells for retroviral packaging were purchased from Cell Biolabs. K562, Raji, and MDA-MB-231 cell lines were maintained in LCL media (RPMI 1640 with 5% FBS, 100 U/ml penicillin/streptomycin and 2 mM L-glutamine). 4T1 cell lines were maintained in complete RPMI (RPMI 1640 with 10% FBS, 1 mM sodium pyruvate, 1 mM HEPES, 100 U/ml penicillin/streptomycin, and 50 µM β-mercaptoethanol). SK-N-DZ, Lenti-X, and Plat-E cells were maintained in complete DMEM (DMEM with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin/streptomycin, 25 mM HEPES). 4T1-mROR1, K562-mROR1, and Raji-hROR1 cell lines were generated by retroviral transduction with full-length murine Ror1 cDNA (UniProt: Q9Z139) or human ROR1 cDNA (UniProt: Q01973) and subsequent FACS sorting of ROR1+ cells to >95% purity. MDA-MB-231 GFP-ffluc and Raji GFP-ffluc cell lines were generated by retroviral transduction with cDNA encoding GFP fused to firefly luciferase (ffluc) and subsequent FACS sorting of GFP+ cells to >95% purity. 4T1-EpCAMhi, 4T1-EpCAMlow, 4T1-mROR1-EpCAMhi, and 4T1-mROR1-EpCAMlow cell lines were generated by FACS sorting cells to >95% purity. All cells were tested bi-monthly for the absence of mycoplasma.
Primary Cell Cultures
Peripheral Blood T cells from Healthy Donors
Peripheral blood mononuclear cells (PBMC) were obtained from healthy adults (>18 years old) after written informed consent on research protocols approved by the FHCRC IRB. 400 ml of peripheral blood was collected by venipuncture and PBMC were isolated by density gradient using Lymphocyte Separation Media (Corning). Bulk CD8+ and/or CD4+ T cells were enriched from PBMC of normal donors using EasySep Human CD8 and CD4 T Cell Isolation kits (Stem Cell Technologies), respectively, and cultured in CTL media (RPMI 1640 with 10% human serum, 2 mM L-glutamine, 25 mM HEPES, 100 U/ml penicillin/streptomycin, 50 µM β-mercaptoethanol) supplemented with 50 U/ml recombinant human IL-2 (Prometheus).
Murine T Cells
Cell suspensions were prepared from spleen and peripheral lymph nodes by tissue disruption with glass slides and filtered through a 40 µm filter. Murine CD8+ and/or CD4+ T cells were enriched from spleens and peripheral lymph nodes of congenic CD45.1 B6 or BALB/c mice using EasySep mouse CD8+ and CD4+ T cell isolation kits (Stem Cell) and cultured in complete RPMI (RPMI 1640 with 10% FBS, 1 mM sodium pyruvate, 1 mM HEPES, 100 U/ml penicillin/streptomycin, and 50 µM β-mercaptoethanol).
Murine Mesenchymal Stem Cells (MSCs)
Femurs and tibia were crushed with a mortar and pestle and digested with 1 mg/ml collagenase type I (Worthington) and 200 U/ml DNase I (Stem Cell) for 1 hr at 37°C. Cells were filtered through a 40 µm filter, ACK lysed, and depleted of CD45+ cells using anti-CD45 microbeads (Miltenyi). MSCs were expanded in vitro using the MesenCult Expansion Kit (StemCell) by culturing in a hypoxic incubator (5% O2, 5% CO2, 37 °C) for 14 days.
METHOD DETAILS
Cloning of Murine and Human CAR Constructs
The mp71 retroviral vector was modified to encode either a murine ROR1-specific CAR or truncated murine CD19 (mp71-tCD19, UniProt: P25918, amino acids [aa] 1–321) for transduction of control murine T cells. The CAR possessed a murine CD8α signal peptide (UniProt: P01731, aa1–27), R11 scFv, modified human IgG4 long spacer with 4/2NQ mutations (Hudecek et al., 2015), murine CD28 transmembrane (UniProt: P31041, aa151–177), murine 4–1BB (UniProt: P20334, aa211–256), murine CD3ζ (UniProt: P24161, aa52–164), and was linked by a P2A ribosomal skip element to murine truncated tCD19 (mp71-R11–41BB-CD3ζ-P2A-tCD19). The HIV7 lentiviral vector was modified to encode a human ROR1-specific CAR possessing a human GM-CSFR signal peptide (UniProt: P15509, aa1–22), R11 scFv, modified human IgG4 long spacer with 4/2NQ mutations, human CD28 transmembrane (UniProt: P10747, aa153–179), human 4–1BB (UniProt: Q07011, aa214–255), and human CD3ζ (UniProt: P20963–3, aa52–163). The CAR sequence was linked by a T2A ribosomal skip element to human truncated EGFR (tEGFR) (Wang et al., 2011).
synNotch receptors and response elements were obtained from Addgene (Addgene plasmids #79123 and 79125). To generate an anti-EpCAM synNotch receptor, we synthesized an EpCAM-specific monoclonal antibody, G8.8, in VH-VL format with N-terminal murine CD8α signal peptide and myc tag (EQKLISEEDL) (Bourquin et al., 2015). To generate an anti-B7-H3 synNotch receptor, we synthesized a B7-H3-specific monoclonal antibody, BRCA69D, in VH-VL format with N-terminal human CD8α signal peptide (UniProt: P01732, aa1–21) and myc tag. Completed fragments were fused onto the synNotch-Gal4VP64 receptor backbone (Addgene plasmid #79125) in place of the CD19-specific scFv. To generate ROR1 CAR response elements, the mCherry gene segment in Addgene plasmid #79123 was replaced with the previously described murine or human ROR1 CAR transgenes. In this system, constitutive myc tag or BFP expression defined T cells transduced with the synNotch receptor and response element, respectively. tCD19 or tEGFR expression identified cells actively expressing the R11 ROR1 CAR. To generate constitutive CAR controls for synNotch experiments, the previously described murine or human ROR1 CARs were cloned into the pHR backbone under control of the PGK promoter. All cloning was performed using fusion PCR, Gibson assembly, and/or restriction enzyme digest. Plasmids were verified by restriction digest and capillary sequencing prior to use. synNotch receptor, response element, and CAR amino acid sequences are provided in Table S2.
Generation of Murine CAR-T Cells
Retrovirus was produced by transient transfection (Clontech) of Plat-E cells with the indicated MP71 vectors. 48 hr after transfection, viral supernatant was harvested and filtered through a 0.45-µm syringe filter (Millipore). 24-well non-tissue culture plates were coated with 12.5 µg/ml RetroNectin (TaKaRa) according to the manufacturer’s protocol, and plates were loaded with 1 ml filtered virus per well and centrifuged for 2 hr at 3000 g at 32 °C. Murine CD8 + and/or CD4+ T cells were enriched from spleens and peripheral lymph nodes of congenic CD45.1 B6 or BALB/c mice using untouched negative isolation kits (Stem Cell) and stimulated with 1 µg/ml each of plate-bound anti-CD3 and anti-CD28 (clone 145–2C11 and 37.51, respectively) for 24 hr in a 37 °C, 5% CO 2 incubator in complete RPMI (RPM1 1640, 10% heat inactivated FBS, 1 mM sodium pyruvate, 1 mM HEPES, 100 U/ml penicillin/streptomycin, 50 µM β-mercaptoethanol) supplemented with 50U/ml recombinant murine IL-2 (Peprotech). Murine T cells were harvested from anti-CD3/28-coated plates and resuspended to 1×106 cells/ml in complete RPMI supplemented with 50 U/ml IL-2 and anti-CD3/28 mouse T-activator Dynabeads (ThermoFisher) at a bead to cell ratio of 1:1. Viral supernatant was aspirated from RetroNectin-coated plates, plates were rinsed with PBS, and 1ml (1×106) T cells were added to each virus-coated well. Plates were then centrifuged at 800 g for 30 min at 32°C and returned to 37°C, 5% CO 2 incubators. A second transduction was performed as described the next day by harvesting another batch of viral supernatant from Plat-E cells 72 hr after transfection. T cells were subsequently harvested, counted, and resuspended in complete RPMI with 50 ng/ml IL-15 every 1–2 days after. 4–5 days after transduction, magnetic beads were removed and T cell transduction was measured by flow cytometry staining for tCD19 and/or ROR1 CAR. Transduction rates between control and ROR1 CAR-T cells were normalized by diluting cultures with untransduced T cells cultured in parallel such that the proportion of transduced cells was the same between control and CAR-T cell populations, and control and CAR-treated mice received the same number of transduced and untransduced T cells.
For synNotch experiments, lentivirus was produced by transient calcium phosphate transfection of the packaging cell line LentiX with the indicated pHR lentiviral vectors, psPAX2 (Addgene #12260), and pHIT123 ecotropic envelope (Soneoka et al., 1995). Viral supernatant was harvested 48 and 72 hr after transfection and filtered through a 0.45-µm pore filter. Lentivirus was concentrated 40X by mixing filtered lentivirus with 40% polyethylene glycol (PEG, Sigma) at a PEG to virus ratio of 1:3 for 1–2 hr at 4°C. The virus/PEG mixture was then centrifuged at 1500g for 45 min at 4°C, supernatant was aspirated, and the virus pellet was resuspended in 40X smaller volume of serum-free DMEM and snap-frozen in liquid nitrogen for long-term storage. Frozen virus was used to transduce murine T cells using the RetroNectin-based transduction protocol described above. When co-transduction of two constructs was desired, frozen virus from both vectors was mixed 1:1 and added to the same RetroNectin-coated well. BFP+ synNotch T cells were FACS sorted to enrich for T cells capable of inducing expression of the ROR1 CAR before adoptive transfer into mice.
Generation of Human CAR-T Cells
LentiX cells were transiently transfected with the indicated CAR vectors, psPAX2 (Addgene #12260), and pMD2.G (Addgene #12259) packaging plasmids. One day later (day 1), primary CD8+ and CD4+ T cells were activated with Dynabeads Human T-Activator CD3/28 (ThermoFisher) in fresh CTL medium (RPMI 1640 with 10% human serum, 2 mM L-glutamine, 25mM HEPES, penicillin/streptomycin (100 U/ml), 50 µM β-mercaptoethanol (Sigma)) with 50U/ml recombinant human IL-2 (Prometheus). On day 2, T cells were transduced by centrifugation at 800 g for 90 min at 32ºC with filtered lentiviral supernatant supplemented with 4.4 µg/mL polybrene (Millipore). Viral supernatant was replaced 8 hr later with fresh CTL medium with 50U/ml human IL-2, and T cells were further expanded with half media changes every 48 hr. Dynabeads were removed on day 6. On day 9–10, T cells were FACS sorted to >95% purity with the following markers: untransduced T cells: tEGFR−; ROR1 CAR-T cells: tEGFR+; synNotch T cells: myc+BFP+. Sorted T cells were subsequently expanded for 4–5 days in CTL medium with 50 U/ml human IL-2 prior to in vitro assays or adoptive transfer. CD8+ and CD4+ T cells were cultured and transduced separately.
In Vivo CAR-T Cell Toxicity Model
B6 or BALB/c mice were pre-conditioned with sublethal radiation (100 R or 500 R) or with intraperitoneal injection with cyclophosphamide (100 mg/kg or 200 mg/kg) and 5–6 hr later were injected intravenously by retro-orbital injection with 1×106 CD8+tCD19+ control or ROR1 CAR-T cells. For experiments with primary human T cells, NSG mice were pre-conditioned with sublethal radiation (250 R) and 5–6 hr later were injected intravenously by tail vein injection with 2×106 CD8+ untransduced or CD8+tEGFR+ ROR1 CAR-T cells. Mice were weighed just prior to pre-conditioning and 1–2 times per week thereafter. Percent weight change was calculated as: (weight at time X – weight at time 0) / (weight at time 0). All mice in each experiment were sacrificed when any individual mice showed clinical signs of severe disease or 20 percent weight loss.
Cell Isolation, Sorting, and Transfer
Cell suspensions were prepared from spleen and peripheral lymph nodes by tissue disruption with glass slides and filtered through a 40 µm filter. For bone marrow isolation, femurs and tibia were flushed with complete RPMI using a 27 gauge needle and cells were filtered with 40 µm filter. Liver, kidney, pancreas, lungs, and tumors were digested with 10mg/ml collagenase type IV (Worthington) for 1hr at 37°C with gentle agitat ion and then filtered through a 40 µm filter. Cells were lysed with ACK lysing buffer (Gibco) and resuspended as single cell suspensions for downstream analysis.
For sorting of hematopoietic bone marrow progenitors, bone marrow was isolated from femurs and tibia as described above, filtered through a 40 µm filter, and ACK lysed. Cells were subsequently stained and FACS sorted for subpopulations using the following markers: LT-HSC (Lin−Sca1+ckit+CD150+CD48−Flt3−), pre-MEP (Lin−Sca1−ckit+CD150+CD34+FcγRlo), MEP (Lin− Sca1−ckit+CD34−FcγR−CD150−), CMP (Lin−Sca1−ckit+CD150−CD34+FcγRlo), pre-B cells (CD45+Ter119−B220+), where lineage-negative (Lin−) cells are defined as CD3−CD4−CD8−B220− CD11b−CD11c−Gr-1−Ter119−. For sorting of bone marrow stromal cells, femurs and tibia were crushed with a mortar and pestle and digested with 1mg/ml collagenase type I (Worthington) and 200 U/ml DNase I (Stem Cell) for 1 hr at 37°C. Cells were filtered through a 40 µm filter, ACK lysed, and depleted of CD45+ cells using anti-CD45 microbeads (Miltenyi). Cells were subsequently stained and FACS sorted for subpopulations using the following markers: mesenchymal stem cells (CD45−Ter119−PDGFRα+CD51+CD31−), endothelial cells (CD45− Ter119−PDGFRα−CD51−CD31+), and osteoblasts (CD45−Ter119−PDGFRα−CD51+CD31−). For sorting of splenic stromal cells, spleens were cut into ~1 mm3 fragments using scissors and digested with 1 mg/ml collagenase type I (Worthington) and 200U/ml DNase I (Stem Cell) for 1 hr at 37°C. Cells were filtered through a 40 µm filter, ACK lysed, and depleted of CD45+ cells using anti-CD45 microbeads (Miltenyi). Cells were subsequently stained and FACS sorted for subpopulations using the following markers: PDGFRβ+ spleen stromal cells (CD45−Ter119− PDGFRβ+) and VE-cadherin+ spleen stromal cells (CD45−Ter119−VE-cadherin+). For analysis of endothelial cells, mice were injected intravenously into the retro-orbital venous sinus with 10 µg Alexa Fluor 660 conjugated anti-VE-cadherin antibody (BV13) 10 min before sacrifice. Samples were analyzed using a FACSAria II flow cytometer (BD Biosciences).
Co-Culture of Primary MSC and Splenic Stromal Cells with CAR-T Cells
Bone marrow stromal cells were prepared from femurs and tibia of WT or ROR1-KO mice as described above, and MSCs were expanded in vitro using the MesenCult Expansion Kit (StemCell) by culturing in a hypoxic incubator (5% O2, 5% CO2, 37°C) for 14 days. For isolation of splenic stromal cells, spleens were digested as described above and separated into CD45+ and CD45− fractions using anti-CD45 microbeads (Miltenyi). For co-culture experiments, 50,000 _in vitro_-expanded MSC or splenic cells were co-cultured with 50,000 CD8+ control or ROR1 CAR-T cells in 0.2 ml of complete RPMI in triplicate in 96-well U-bottomed plates (Costar) at 37°C, 5% CO 2. After 48 hr, supernatant was harvested and frozen at −20°C for long-term storage and analyzed for IFNγ expression using the Ready-Set-Go Mouse IFNγ ELISA Kit (eBioscience).
CBC, Serum Cytokine, and Serum Chemistry Analysis
Peripheral blood was collected by retro-orbital bleeds into serum separator tubes or EDTA FACS tubes. Serum separator tubes were incubated at room temperature for 30 min to allow blood to clot and then centrifuged at 15800xg for 5 min. Serum was either frozen at −80°C for multiple cytokine immunoassay by the FHCRC Immune Monitoring Core (Luminex) or submitted to Phoenix Central Labs for serum chemistry analysis. Blood in EDTA FACS tubes was submitted to Phoenix Central Labs for complete blood count analysis with differential.
Flow Cytometry
For live/dead staining, cells were stained using the Live/Dead Fixable Aqua Dead Cell stain kit (Invitrogen) according to the manufacturer’s protocol. For surface staining, cells were incubated at 4°C for 30 min in staining buffer (PBS, 2% FBS) with the following directly conjugated antibodies for murine proteins (from Biolegend unless otherwise specified): anti-CD4 (RM4–5), - CD8 (53–6.7), -CD45.1 (A20), -CD3 (145–2C11), -CD19 (eBio1D3, Thermo Fisher), -CD45 (30- F11), -PD-1 (29F.1A12), streptavidin (Thermo Fisher), -Ter119 (TER119), -B220 (RA3–6B2), - PDGFRα (APA5), PDGFRβ (APB5), -CD51 (RMV7), -CD31 (390), -CD11b (M1/70), -CD11c (N418), -Gr-1 (RB6–8C5), -Sca1 (D7), -cKit (2B8), -CD150 (mShad150, Thermo Fisher), -CD48 (HM48–1), -Flt3 (A2F10), -CD34 (RAM34, Thermo Fisher), -FcγR (93), -EpCAM (G8.8, Thermo Fisher), -myc (9B11, Cell Signaling), -TIM3 (RMT3–23), -CD44 (IM7, Thermo Fisher); or with the following directly conjugated antibodies for human proteins (from Biolegend unless otherwise specified: anti-CD45 (2D1), -CD8 (RPA-T8, BD), -CD4 (RPA-T4, Thermo Fisher), -PD-1 (EH12.2H7), -EGFR (AY13), -TIM3 (F38–2E2), -ROR1 (2A2, Miltenyi Biotec). Biotinylated IgG1 R11 and biotinylated recombinant Fc-mROR1 were used to measure mROR1 and ROR1 CAR expression, respectively (Yang et al., 2011). For intracellular staining, cells were surface stained as described, washed and permeabilized for 20 min with eBioscience Fix/Perm buffer at 4°C. Cells were stained for 30 min at 4°C with anti-mous e IFN-γ (XMG1; Thermo Fisher), -Ki67 (B56, BD Biosciences), and/or -human IFNγ (B27) in 1X Perm/Wash staining medium (eBioscience).
For intracellular cytokine staining following restimulation, 50,000 T cells were stimulated with 50,000 tumor cells or with 50ng/ml PMA and 1 µg/ml ionomycin in 0.2ml complete RPMI in triplicate in 96-well U-bottomed plates (Costar) at 37°C, 5% CO 2 for 6 hr or 24 hr. GolgiPlug (BD) was added to all wells according to the manufacturer’s protocol for the last 6 hr of culture before staining for flow cytometry as described above. Data were acquired on LSRII, Canto 2 or Symphony flow cytometers (BD Biosciences) and analyzed using FlowJo software (Treestar).
Immunohistochemistry and Disease Scoring
Tissues were immersion fixed in 10% neutral buffered formalin, paraffin embedded, cut into 5 µm sections, and stained with hematoxylin and eosin by the FHCRC Experimental Histopathology Core. Sections were scored semi-quantitatively from 0 to 4 for inflammation and toxicity in a blinded fashion. Changes typically associated with each grade for various tissues are described in Table S1. For ROR1 and B7-H3 staining, the following tissue microarrays (TMAs) were purchased from BioMax: BR1141 breast cancer, core J8; BCS04017a lung adenocarcinoma, core H5. Ovarian carcinoma TMA was generously provided by FHCRC Experimental Histopathology Core. Formalin-fixed paraffin-embedded tissues were sectioned onto charged slides, baked for 1 hr at 60°C, depara ffinized in xylene, and rehydrated in graded dilutions of ethanol to water. Antigen retrieval was achieved in a Decloaking Chamber (Biocare Medical, Pacheco, CA) with a Tris-EDTA solution (10mM Tris, 1 mM EDTA, 0.05% tween, pH 9.4) for 25 min at 110°C before staining the slides on a Biocare intelliPATH system. Endogenous peroxidase was blocked with 3% H2O2 for 5 min followed by protein blocking with Background Punisher (Biocare Medical) for 5 min. ROR1 primary antibody (2 μg/ml, mouse clone 6D4, Fred Hutchinson Cancer Research Center) was applied for 30 min then rinsed with TBS Automation Wash buffer (Biocare). OPAL Polymer HRP Mouse plus Rabbit secondary (PerkinElmer, Hopkington, MA) was applied for 10 min, washed, and followed by an Opal 570 tertiary TSA-amplification reagent (PerkinElmer) for 10 min. The primary and secondary antibodies were stripped away with an additional antigen retrieval step using DIVA Decloaker (Biocare) for 15 min before repeating the staining process using B7H3 (1 μg/ml, clone RBT-B7H3, Bio SB, Santa Barbara, CA) for the primary antibody and Opal 650 for the tertiary reagent. Slides were removed from the intelliPATH and counterstained with 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI, Sigma Aldrich, St. Louis, MO) and coverslipped with Prolong Gold Antifade reagent (Invitrogen/Life Technologies, Grand Island, NY). Slides were cured for 24 hr at room temperature before scanning on an Aperio FL whole slide scanner (Leica Biosystems, Buffalo Grove, IL). The DAPI, ROR1, and B7H3 were visualized using the DAPI, Cy3, and Cy5 filters, respectively. Cellular analysis of the images was performed with HALO image analysis software (Indica Labs, Corrales, NM).
Bone Marrow Chimeras
Femurs and tibia were isolated from B6 or EIIα-Cre/Ror1 fl/fl mice and flushed using a 27 gauge needle to isolate bone marrow cells. Bone marrow cells were filtered using 40 µm filters, ACK lysed, and depleted of CD4+ and CD8+ cells using anti-CD4 and anti-CD8 microbeads (Miltenyi Biotec). 2–4×106 T cell-depleted bone marrow cells were injected intravenously into lethally irradiated (1000 Rad) B6 or EIIα-Cre/Ror1 fl/fl mice. Mice were bled retro-orbitally 8 weeks after transplant and analyzed by flow cytometry to confirm reconstitution of hematopoietic lineages before use in adoptive transfer experiments.
CFU Assays
Femurs and tibia from control and ROR1 CAR-T cell-treated mice were harvested 9 days post-T cell transfer and submitted to ReachBio Inc. for CFU-GM, BFU-E, CFU-Mk, Pre-B CFC, and CFU-F assays.
qPCR
Cell populations were sorted directly into buffer RLT and RNA was extracted using the RNeasy Micro kit (QIAGEN). cDNA was generated using the iScript cDNA Synthesis kit (BioRad) according to the manufacturer’s instructions. Expression of Ror1 was analyzed in triplicate and normalized to mActb. Amplifications were performed for 50 cycles on an ABI Prism 7900 (Applied Biosystems) in a 20 μl reaction consisting of Power SYBR Green PCR Master Mix (Applied Biosystems), 5 ng of cDNA, and 500 nM gene-specific forward and reverse primers: Ror1, 5’-CAAAACCCGTCAGAGGACAGA-3’ and 5’ATGAAACGCACAGCGGAAAG-3’; Actb, 5’-CTGTCCCTGTATGCCTCTG-3’ and 5’-ATGTCACGCACGATTTCC-3’. The cycle threshold (Ct) was determined using SDS software (Applied Biosystems) and the level of gene expression calculated using the comparative Ct method (2(^Ct)).
Chromium Release Assay
Tumor cells were labeled with 51Cr (PerkinElmer) for 1 hr at 37°C and washed with c omplete RPMI. 1×103 51Cr-labeled target cells were plated per well in triplicate and co-cultured with T-cells at various effector to target (E:T) ratios for 24 hr in a 37°C, 5% CO 2 incubator. Supernatants were harvested for γ-counting after 6 hr and 24 hr of incubation and specific lysis calculated by comparing counts to standardized wells where target cells were lysed with NP40-based soap solution.
Mouse Tumor Models
For the 4T1 model, 1×105 4T1-mROR1 tumor cells were injected subcutaneously into the 4th right mammary fat pad of 6–8 week old BALB/c female mice. After 7 days, mice were injected intraperitoneally with 200mg/kg cyclophosphamide and 5–6 hr later injected with 5×105 CD8+ and CD4+ untransduced, ROR1 CAR, or synNotch EpCAM-inducible ROR1 CAR-T cells intravenously. SynNotch T cells were enriched by untouched sorting for BFP+ T cells, as described above, such that ~30% of sorted T cells were myc+BFP+ and carried the full EpCAM-synNotch/UAS-ROR1-CAR circuit. Transduction rate of constitutive ROR1 CAR-T cells was equalized to ~30% by diluting cultures with untransduced T cells cultured in parallel such ROR1 CAR- and synNotch-treated mice each received the same number of T cells capable of expressing the ROR1 CAR and the same number of total T cells. Tumor size was monitored using calipers, and tumor volume was calculated as (length in mm)*(width in mm)^2.
For the Raji model, 5×105 Raji-hROR1-GFP-ffluc tumor cells were injected intravenously via tail vein into 8–12 week old NSG mice. After 7 days, mice were irradiated (250 R) and 5–6 hr later injected intravenously with 2×106 primary human CD8+ untransduced, CD8+tEGFR+ ROR1 CAR-T, or CD8+myc+BFP+ synNotch CD19-inducible ROR1 CAR-T cells. For the MDA-MB-231 model, 5×105 MDA-MB-231 GFP-ffluc cells were injected subcutaneously in the right flank of 8–12 week old NSG mice. After 7 days, mice were irradiated 250 R and 5–6 hr later injected intravenously with 3×106 CD8+ and 3×106 CD4+ primary human untransduced, tEGFR+ ROR1 CAR-T, or myc+BFP+ synNotch B7-H3-inducible ROR1 CAR-T cells. For all NSG experiments, T cells were sorted to >95% purity and expanded in IL-2 for 4–5 days prior to injection. Tumor burden was monitored by IVIS bioluminescence imaging.
IVIS Imaging
For bioluminescence imaging of tumor growth, mice received intraperitoneal injections of luciferin substrate (Caliper Life Sciences) resuspended in PBS (15 µg/g body weight). Mice were anesthetized with isoflurane and imaged using an Xenogen IVIS Imaging System (Caliper) 10, 12 and 14 min after luciferin injection in small binning mode at an acquisition time of 1 s to 1 min to obtain unsaturated images. Luciferase activity was analyzed using Living Image Software (Caliper) and the photon flux analyzed within regions of interest that encompassed the entire body of each individual mouse.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as the mean values ± SEM. Statistical significance was determined by one-way ANOVA with Tukey’s post-test, two-way ANOVA with Tukey’s post-test, two-way ANOVA with Sidak post-test, log-rank Mantel-Cox test, or unpaired Student’s two-way t-test as indicated in figure legends using Prism software (Graphpad). Statistical significance was established at the levels of *, p<0.05; **, p<0.005; ***, p<0.0005; ****, p<0.0001.
Supplementary Material
2
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse B220 FITC | BioLegend | Cat# 103206, RRID:AB_312991 |
| Anti-mouse CD11b FITC | BioLegend | Cat# 101205, RRID:AB_312788 |
| Anti-mouse CD11c FITC | BioLegend | Cat# 117306, RRID:AB_313775 |
| Anti-mouse PDGFRα APC | BioLegend | Cat# 135908, RRID:AB_2043970 |
| Anti-mouse PDGFRβ PE | BioLegend | Cat# 136006, RRID:AB_1953271 |
| Anti-mouse CD150 PerCPeFluor710 | Thermo Fisher Scientific | Cat# 46–1502-82, RRID:AB_2016699 |
| Anti-mouse FcγR BV510 | BioLegend | Cat# 101333, RRID:AB_2563692 |
| Anti-mouse CD19 APC | Thermo Fisher Scientific | Cat# 17–0193-82, RRID:AB_1659676 |
| Anti-mouse CD19 eFluor450 | Thermo Fisher Scientific | Cat# 48–0193-80, RRID:AB_2637304 |
| Anti-mouse CD19 PECy7 | Thermo Fisher Scientific | Cat# 25–0193-82, RRID:AB_657663 |
| Anti-mouse CD3 APC-eFluor780 | Thermo Fisher Scientific | Cat# 47–0031-82, RRID:AB_11149861) |
| Anti-mouse CD3 FITC | BioLegend | Cat# 100306, RRID:AB_312671 |
| Anti-mouse CD31 PECy7 | BioLegend | Cat# 102417, RRID:AB_830756 |
| Anti-mouse CD34 AlexaFluor700 | Thermo Fisher Scientific | Cat# 56–0341-82, RRID:AB_493998 |
| Anti-mouse CD4 BUV496 | BD Biosciences | Cat# 564667, RRID:AB_2722549 |
| Anti-mouse CD4 FITC | BioLegend | Cat# 100510, RRID:AB_312713) |
| Anti-mouse CD45 FITC | BioLegend | Cat# 103108, RRID:AB_312973 |
| Anti-mouse CD45 PECy7 | BioLegend | Cat# 103114, RRID:AB_312979 |
| Anti-mouse CD45.1 BV711 | BioLegend | Cat# 110739, RRID:AB_2562605 |
| Anti-mouse CD45.1 PerCP-Cy5.5 | BioLegend | Cat# 110728, RRID:AB_893346 |
| Anti-mouse CD45.1 Pacific Blue | BioLegend | Cat# 110722, RRID:AB_492866 |
| Anti-mouse CD48 BV421 | BioLegend | Cat# 103427, RRID:AB_10895922 |
| Anti-mouse CD51 PE | BioLegend | Cat# 104105, RRID:AB_313074 |
| Anti-mouse CD8 FITC | BioLegend | Cat# 100705, RRID:AB_312744 |
| Anti-mouse CD8 APC-eFluor780 | Thermo Fisher Scientific | Cat# 9047–0087-120, RRID:AB_11181660 |
| Anti-mouse CD8 BUV395 | BD Biosciences | Cat# 563786, RRID:AB_2732919 |
| Anti-mouse cKit APC | BioLegend | Cat# 105811, RRID:AB_313220 |
| Anti-mouse EpCAM PECy7 | Thermo Fisher Scientific | Cat# 25–5791-80, RRID:AB_1724047) |
| Anti-mouse Flt3 PE | BioLegend | Cat# 135305, RRID:AB_1877218 |
| Anti-mouse Gr-1 FITC | BioLegend | Cat# 108405, RRID:AB_313370 |
| Anti-mouse IFN gamma PE-eFluor610 | Thermo Fisher Scientific | Cat# 61–7311-82, RRID:AB_2574662 |
| Anti-human Ki-67 AlexaFluor647 | BD Biosciences | Cat# 558615, RRID:AB_647130) |
| Anti-human Ki-67 AlexaFluor700 | BD Biosciences | Cat# 561277, RRID:AB_10611571 |
| Anti-myc AlexaFluor 647 | Cell Signaling Technology | Cat# 2233, RRID:AB_823474 |
| Anti-mouse PD-1 PECy7 | BioLegend | Cat# 135215, RRID:AB_10696422 |
| Anti-mouse PD-1 BV605 | BioLegend | Cat# 135219, RRID:AB_11125371 |
| Rat IgG2a kappa Isotype Control PECy7 | Thermo Fisher Scientific | Cat# 25–4321-82, RRID:AB_470200 |
| Anti-mouse Sca-1 PECy7 | BioLegend | Cat# 108113, RRID:AB_493597 |
| Streptavidin APC | Thermo Fisher Scientific | Cat# 17–4317-82 |
| Anti-mouse Ter119 BV421 | BioLegend | Cat# 116234, RRID:AB_2562917 |
| Anti-mouse Ter119 BV605 | BioLegend | Cat# 116235, RRID:AB_11204244 |
| Anti-mouse Ter119 FITC | BioLegend | Cat# 116206, RRID:AB_313707 |
| Anti-mouse Tim-3 PE | BioLegend | Cat# 119704, RRID:AB_345378 |
| Anti-mouse CD44 eFluor 660 | Thermo Fisher Scientific | Cat# 50–1441-80, RRID:AB_11219470 |
| Anti-human CD45 APC | BioLegend | Cat# 304011, RRID:AB_314399 |
| Anti-human CD8 BUV395 | BD Biosciences | Cat# 563795, RRID:AB_2722501 |
| Anti-human EGFR PECy7 | BioLegend | Cat# 352910, RRID:AB_2562159 |
| Anti-human PD-1 BV785 | BioLegend | Cat# 367431, RRID:AB_2721561 |
| Anti-human CD45 APC/Cy7 | BioLegend | Cat# 368515, RRID:AB_2566375 |
| Anti-human CD4 eFluor 450 | Thermo Fisher Scientific | Cat# 48–0049-42, RRID:AB_1272057 |
| Anti-human Tim-3 PE | BioLegend | Cat# 345005, RRID:AB_1877236 |
| Anti-human CD8 eFluor 450 | Thermo Fisher Scientific | Cat# 48–0088-42, RRID:AB_1272062 |
| Anti-human IFN gamma APC | BioLegend | Cat# 506510, RRID:AB_315443 |
| Anti-human ROR1 APC | Miltenyi Biotec | Cat# 130–117-942, RRID:AB_2733449 |
| Anti-ROR1 6D4 | Fred Hutchinson Cancer Research Center (Balakrishnan et al., 2017) | N/A |
| Anti-human B7-H3 | Bio SB | Cat# BSB2813 |
| Fc-mROR1 biotin | Christoph Rader, Scripps Research Institute (Yang et al., 2011) | N/A |
| R11 biotin | Christoph Rader, Scripps Research Institute (Yang et al., 2011) | N/A |
| Biological Samples | ||
| Breast cancer TMA, core J8 | BioMax | Cat #BR1141 |
| Lung adenocarcinoma TMA, core H5 | BioMax | Cat #BCS04017a |
| Ovarian cancer TMA | FHCRC Experimental Histopathology Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mouse T-Activator CD3/28 Dynabeads | Thermo Fisher Scientific | Cat #11452D |
| Human T-Activator CD3/28 Dynabeads | Thermo Fisher Scientific | Cat #11132D |
| Retronectin | Takara | Cat #T202 |
| Critical Commercial Assays | ||
| EasySep Human CD8+ T Cell Isolation Kit | Stem Cell Technologies | Cat #17953 |
| EasySep Human CD4+ T Cell Isolation Kit | Stem Cell Technologies | Cat #17952 |
| EasySep Mouse CD8+ T Cell Isolation Kit | Stem Cell Technologies | Cat #19853 |
| EasySep Mouse CD4+ T Cell Isolation Kit | Stem Cell Technologies | Cat #19852 |
| MesenCult Expansion Kit (Mouse) | Stem Cell Technologies | Cat #05513 |
| RNeasy Micro kit | QIAGEN | Cat #74004 |
| iScript cDNA Synthesis Kit | Bio-Rad | Cat #1708890 |
| Experimental Models: Cell Lines | ||
| K562 | ATCC | Cat# CCL-243, RRID:CVCL_0004 |
| 4T1 | ATCC | Cat# CRL-2539, RRID:CVCL_0125) |
| Raji | ATCC | Cat# CRL-7936, RRID:CVCL_0511 |
| MDA-MB-231 | ATCC | Cat# CRM-HTB-26, RRID:CVCL_0062 |
| SK-N-DZ | Michael Jensen, Seattle Children’s; ATCC | Cat# CRL-2149, RRID:CVCL_1701 |
| Lenti-X 293T | Clontech | Cat #632180 |
| Plat-E | Cell Biolabs | RRID:CVCL_B488 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 (B6): C57BL/6J | The Jackson Laboratory | Cat# JAX:000664, RRID:IMSR_JAX:000664 |
| Mouse: B6 CD45.1: B6.SJL-Ptprc a Pepc b/BoyJ | The Jackson Laboratory | Cat# JAX:002014, RRID:IMSR_JAX:002014 |
| Mouse: BALB/c: BALB/cByJ | The Jackson Laboratory | Cat# JAX:001026, RRID:IMSR_JAX:001026 |
| Mouse: BALB/c CD45.1: CByJ.SJL(B6)-Ptprc a/J | The Jackson Laboratory | Cat# JAX:006584, RRID:IMSR_JAX:006584 |
| Mouse: BALB/c Rag2 −/−: C.B6(Cg)-Rag2 tm1.1Cgn/J | The Jackson Laboratory | Cat# JAX:008448, RRID:IMSR_JAX:008448 |
| Mouse: EIIα-Cre: B6.FVB-Tg(EIIa-cre)C5379Lmgd/J | The Jackson Laboratory | Cat# JAX:003724, RRID:IMSR_JAX:003724) |
| Mouse: ROR1f/f: B6;129S4-Ror1 tm1.1Meg/J | The Jackson Laboratory | Cat# JAX:018353, RRID:IMSR_JAX:018353 |
| Mouse: NOD/SCID/ γc−/− (NSG): NOD.Cg-Prkdc scid Il2rg tm1Wjl/SzJ | The Jackson Laboratory | Cat# JAX:005557, RRID:IMSR_JAX:005557) |
| Oligonucleotides | ||
| Ror1 F primer: 5’-CAAAACCCGTCAGAGGACAGA-3’ | This paper | N/A |
| Ror1 R primer: 5’-ATGAAACGCACAGCGGAAAG-3’ | This paper | N/A |
| Actb F primer: 5’-CTGTCCCTGTATGCCTCTG-3’ | This paper | N/A |
| Actb R primer: 5’-ATGTCACGCACGATTTCC-3’ | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pMP71_R11_long42NQ_mCD28TM_m41BB_mCD3z_P2A_tCD19 | This paper | N/A |
| Plasmid: pMP71_tCD19 | This paper | N/A |
| Plasmid: epHIV7_R11_long42NQ_hCD28TM_h41BB_hCD3z_T2A_tEGFR | This paper | N/A |
| Plasmid: pHR_Gal4UAS_IRES_mC_pGK_tBFP | Roybal et al., 2016 | Addgene Plasmid #79123 |
| Plasmid: pHR_PGK_antiCD19_synNotch_Gal4VP64 | Roybal et al., 2016 | Addgene Plasmid #79125 |
| Plasmid: pHR_PGK_antiEpCAM_synNotch_Gal4VP64 | This paper | N/A |
| Plasmid: pHR_PGK_antiB7H3_synNotch_Gal4VP64 | This paper | N/A |
| Plasmid: pHR_Gal4UAS_IRES_R11-h41BB-hCD3z-T2A-tEGFR_pGK_tBFP | This paper | N/A |
| Plasmid: pHR_Gal4UAS_IRES_R11-m41BB-mCD3z-T2A-tCD19_pGK_tBFP | This paper | N/A |
| Plasmid: psPax2 | Addgene | Addgene Plasmid #12260 |
| Plasmid: pHIT123 | Soneoka et al., 1995 | N/A |
| Plasmid: pMD2.G | Addgene | Addgene Plasmid #12259 |
| Software and Algorithms | ||
| FlowJo v10 | TreeStar | https://www.flowjo.com/solutions/flowjo/downloads |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| HALO Image Analysis Software | Indica Labs | http://www.indicalab.com/halo/ |
| Living Image Software | Perkin Elmer | http://www.perkinelmer.com/product/li-software-for-spectrum-1-seat-add-on-128113 |
| Other | ||
| Power SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat #4367659 |
Signficance.
The identification of tumor-specific antigens remains an obstacle for T cell therapy for common epithelial cancers, as most candidate target molecules are also expressed in some normal tissues, making off-tumor toxicity a significant risk of therapy. Our study shows that combinatorial antigen sensing using synNotch receptors can avert lethal CAR-T cell-mediated toxicity to normal tissues if tumor and normal tissue are spatially segregated, but not if they are highly co-localized at the same site. Our work illustrates that synNotch receptors can be a powerful tool for improving the safety of CAR-T cells specific for clinically relevant targets and may dramatically increase the number of cell surface molecules that can be targeted safely in cancer immunotherapy.
Highlights.
- ROR1-specific CAR-T cells induce lethal toxicity after cytotoxic pre-conditioning
- Toxicity is due to attack of ROR1+ stroma needed for recovery from cytotoxic stress
- SynNotch receptors restrict CAR activity to ROR1+ tumors expressing synNotch ligands
- SynNotch receptors only rescue toxicity if tumor and normal cells are separated
Acknowledgements
We wish to thank Wendell Lim, Kole Roybal, Wolfgang Uckert, and Ashwini Balakrishnan for helpful discussions and reagents. We also thank Don Parrilla and Mercedez Jess for assistance with tumor xenograft experiments. Graphical abstract was created with BioRender. This work was supported by grants from the National Institutes of Health (R01CA114536 to S.R.R), Cancer Center Support Grant (P30 CA015704), and Juno Therapeutics. S.S. is a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute. A.I.S. is supported by the FHCRC Interdisciplinary Training Grant in Cancer Research.
Footnotes
Declaration of Interests
S.R.R. is a cofounder and has served as an advisor for Juno Therapeutics, a Celgene company, and holds equity in Celgene. S.R.R. has served on advisory boards for Adaptive Biotechnologies, Nohla, and Cell Medica. C.R. is named inventor on U.S. Patent 9,758,586 claiming anti-ROR1 monoclonal antibodies R11 and R12 and is on the advisory board of BNE-Therapeutics. No potential conflicts of interest were disclosed by the other authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anthony BA, Link DC, 2014. Regulation of hematopoietic stem cells by bone marrow stromal cells. Trends Immunol 35, 32–37. 10.1016/j.it.2013.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan A, Goodpaster T, Randolph-Habecker J, Hoffstrom BG, Jalikis FG, Koch LK, Berger C, Kosasih PL, Rajan A, Sommermeyer D, Porter PL, Riddell SR, 2017. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin. Cancer Res 23, 3061–3071. 10.1158/1078-0432.CCR-16-2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger C, Sommermeyer D, Hudecek M, Berger M, Balakrishnan A, Paszkiewicz PJ, Kosasih PL, Rader C, Riddell SR, 2015. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol Res 3, 206–216. 10.1158/2326-6066.CIR-14-0163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourquin C, Castoldi R, Endres S, Klein C, Kobold S, Niederfellner G, Sustmann C, Inc H-LR, 2015. Bispecific antibody molecules with antigen-transfected t-cells and their use in medicine
- Brudno JN, Kochenderfer JN, 2016. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127, 3321–3330. 10.1182/blood-2016-04-703751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagnaro L, Lenti E, Maruzzelli S, Spinardi L, Migliori E, Farinello D, Sitia G, Harrelson Z, Evans SM, Guidotti LG, Harvey RP, Brendolan A, 2013. Nkx2–5(+)islet1(+) mesenchymal precursors generate distinct spleen stromal cell subsets and participate in restoring stromal network integrity. Immunity 38, 782–791. 10.1016/j.immuni.2012.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien H-P, Ueng S-H, Chen S-C, Chang Y-S, Lin Y-C, Lo Y-F, Chang H-K, Chuang W-Y, Huang Y-T, Cheung Y-C, Shen S-C, Hsueh C, 2016. Expression of ROR1 has prognostic significance in triple negative breast cancer. Virchows Arch 468, 589–595. 10.1007/s00428-016-1911-3 [DOI] [PubMed] [Google Scholar]
- Choi MY, Widhopf GF, Ghia EM, Kidwell RL, Hasan MK, Yu J, Rassenti LZ, Chen L, Chen Y, Pittman E, Pu M, Messer K, Prussak CE, Castro JE, Jamieson C, Kipps TJ, 2018. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell 22, 951–959. 10.1016/j.stem.2018.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi MY, Widhopf GF, Wu CCN, Cui B, Lao F, Sadarangani A, Cavagnaro J, Prussak C, Carson DA, Jamieson C, Kipps TJ, 2015. Pre-clinical Specificity and Safety of UC-961, a First-In-Class Monoclonal Antibody Targeting ROR1. Clin Lymphoma Myeloma Leuk 15 Suppl, S167–9. 10.1016/j.clml.2015.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimarino AM, Caplan AI, Bonfield TL, 2013. Mesenchymal stem cells in tissue repair. Front Immunol 4, 201 10.3389/fimmu.2013.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schönfeld K, Koch J, Dotti G, Heslop HE, Gottschalk S, Wels WS, Baker ML, Ahmed N, 2013. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids 2, e105 10.1038/mtna.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves P, 2012. Histopathology of Preclinical Toxicity Studies Academic Press. [Google Scholar]
- Hay KA, Hanafi L-A, Li D, Gust J, Liles WC, Wurfel MM, López JA, Chen J, Chung D, Harju-Baker S, Cherian S, Chen X, Riddell SR, Maloney DG, Turtle CJ, 2017. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 130, 2295–2306. 10.1182/blood-2017-06-793141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho H-YH, Susman MW, Bikoff JB, Ryu YK, Jonas AM, Hu L, Kuruvilla R, Greenberg ME, 2012. Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc. Natl. Acad. Sci. U.S.A 109, 4044–4051. 10.1073/pnas.1200421109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudecek M, Lupo-Stanghellini M-T, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR, 2013. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res 19, 3153–3164. 10.1158/1078-0432.CCR-13-0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini M-T, Nishida T, Yamamoto TN, Bleakley M, Turtle CJ, Chang W-C, Greisman HA, Wood B, Maloney DG, Jensen MC, Rader C, Riddell SR, 2010. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood 116, 4532–4541. 10.1182/blood-2010-05-283309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR, 2015. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res 3, 125–135. 10.1158/2326-6066.CIR-14-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inra CN, Zhou BO, Acar M, Murphy MM, Richardson J, Zhao Z, Morrison SJ, 2015. A perisinusoidal niche for extramedullary haematopoiesis in the spleen. Nature 527, 466–471. 10.1038/nature15530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M, 2013. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol 31, 71–75. 10.1038/nbt.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Rosenberg SA, 2013. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol 10, 267–276. 10.1038/nrclinonc.2013.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, Bakker den M, Oosterwijk E, Debets R, Gratama JW, 2013. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol. Ther 21, 904–912. 10.1038/mt.2013.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E, 2006. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol 24, e20–2. 10.1200/JCO.2006.05.9964 [DOI] [PubMed] [Google Scholar]
- Li H, Zhao Y, 2017. Increasing the safety and efficacy of chimeric antigen receptor T cell therapy. Protein Cell 8, 573–589. 10.1007/s13238-017-0411-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Vijayakumar S, Grumolato L, Arroyave R, Qiao H, Akiri G, Aaronson SA, 2009. Canonical Wnts function as potent regulators of osteogenesis by human mesenchymal stem cells. J. Cell Biol 185, 67–75. 10.1083/jcb.200810137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA, 2010. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther 18, 843–851. 10.1038/mt.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, Isaacs R, Mohan S, Plesa G, Lacey SF, Navenot J-M, Zheng Z, Levine BL, Okada H, June CH, Brogdon JL, Maus MV, 2017. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9, eaaa0984 10.1126/scitranslmed.aaa0984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potratz J, Tillmanns A, Berning P, Korsching E, Schaefer C, Lechtape B, Schleithoff C, Unland R, Schäfer K-L, Müller-Tidow C, Jürgens H, Dirksen U, 2016. Receptor tyrosine kinase gene expression profiles of Ewing sarcomas reveal ROR1 as a potential therapeutic target in metastatic disease. Mol Oncol 10, 677–692. 10.1016/j.molonc.2015.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Restifo NP, 2015. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68. 10.1126/science.aaa4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA, 2016. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779. 10.1016/j.cell.2016.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seita J, Sahoo D, Rossi DJ, Bhattacharya D, Serwold T, Inlay MA, Ehrlich LIR, Fathman JW, Dill DL, Weissman IL, 2012. Gene Expression Commons: an open platform for absolute gene expression profiling. PLoS ONE 7, e40321 10.1371/journal.pone.0040321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneoka Y, Cannon PM, Ramsdale EE, Griffiths JC, Romano G, Kingsman SM, Kingsman AJ, 1995. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res 23, 628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Riddell SR, 2018. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J. Immunol 200, 459–468. 10.4049/jimmunol.1701155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Riddell SR, 2015. Engineering CAR-T cells: Design concepts. Trends Immunol 36, 494–502. 10.1016/j.it.2015.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JKH, Watanabe T, 2017. Stromal Cell Subsets Directing Neonatal Spleen Regeneration. Sci Rep 7, 40401 10.1038/srep40401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA-K, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist P-H, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, Feilitzen von K, Forsberg M, Persson L, Johansson F, Zwahlen M, Heijne von G, Nielsen J, Pontén F, 2015. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419–1260419. 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- Wang X, Chang W-C, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC, 2011. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 118, 1255–1263. 10.1182/blood-2011-02-337360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie S, van Schalkwyk MCI, Hobbs S, Davies DM, van der Stegen SJC, Pereira ACP, Burbridge SE, Box C, Eccles SA, Maher J, 2012. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol 32, 1059–1070. 10.1007/s10875-012-9689-9 [DOI] [PubMed] [Google Scholar]
- Yang J, Baskar S, Kwong KY, Kennedy MG, Wiestner A, Rader C, 2011. Therapeutic potential and challenges of targeting receptor tyrosine kinase ROR1 with monoclonal antibodies in B-cell malignancies. PLoS ONE 6, e21018 10.1371/journal.pone.0021018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y-Z, Ma R, Zhou J-K, Guo C-L, Wang Y-S, Li Z-G, Liu L-X, Peng Y, 2016. ROR1 is a novel prognostic biomarker in patients with lung adenocarcinoma. Sci Rep 6, 36447 10.1038/srep36447 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
2