CRISPR-Cas guides the future of genetic engineering (original) (raw)
. Author manuscript; available in PMC: 2019 Apr 9.
Published in final edited form as: Science. 2018 Aug 31;361(6405):866–869. doi: 10.1126/science.aat5011
Abstract
The diversity, modularity, and efficacy of CRISPR-Cas systems are driving a biotechnological revolution. RNA-guided Cas enzymes have been adopted as tools to manipulate the genomes of cultured cells, animals, and plants, accelerating the pace of fundamental research and enabling clinical and agricultural breakthroughs. We describe the basic mechanisms that set the CRISPR-Cas toolkit apart from other programmable gene-editing technologies, highlighting the diverse and naturally evolved systems now functionalized as biotechnologies. We discuss the rapidly evolving landscape of CRISPR-Cas applications, from gene editing to transcriptional regulation, imaging, and diagnostics. Continuing functional dissection and an expanding landscape of applications position CRISPR-Cas tools at the cutting edge of nucleic acid manipulation re-writing biology.
Introduction
Researchers have long pursued a means of efficiently manipulating DNA and RNA to tailor genes and their regulation. Genetic perturbation enables scientists to probe gene function or correct mutations but is often intractable due to a technical challenge: site-specific nucleic acid targeting. Targeted gene-editing has been achieved by induced double-stranded DNA (dsDNA) breaks in eukaryotic chromosomes (1), but with challenging technologies based on engineering direct protein-DNA recognition. The history recounting the discovery, development, and application of such engineered nucleic acid binding proteins including zinc fingers, TALENS, and meganucleases is rich in remarkable scientific feats (2). Over the past six years, however, transformative discoveries shaped the clustered regularly interspaced short palindromic repeat (CRISPR) CRISPR-associated (Cas) toolbox for genetic manipulation based on simpler RNA-guided DNA recognition. This toolbox now provides incredible scientific opportunities for curing genetic diseases, engineering desirable genetic traits, and new approaches to live-cell imaging, high-throughput functional genomic screens in addition to point-of-care diagnostics. In this review we summarize the basic mechanisms of RNA-guided single-component CRISPR-Cas systems and their general applications. The basis for the CRISPR revolution goes beyond inherent programmability, lending itself to the naturally-evolved diversity of systems that extend CRISPR-based technology beyond precision gene editing. To capture the broadened landscape of Cas applications and their impact as a force for revolution in molecular biology, where appropriate, we refer readers to recent reviews for a more detailed discussion.
Diverse RNA-programmable CRISPR-Cas enzymes
CRISPR-Cas systems provide microbes with RNA-guided adaptive immunity to foreign genetic elements by directing nucleases to bind and cut specific nucleic acid sequences (3–5) (Fig. 1). Through a process termed adaptation, microbes capture snippets of foreign genetic elements and incorporate them into their genomic CRISPR-array. Transcription of CRISPR arrays creates CRISPR-RNAs (crRNA) that bind to Cas nucleases and provide specificity by base-pairing with target nucleic acids (4, 5). Among the diverse naturally evolved CRISPRCas systems, those designated Class 2 constitute a single large RNA-guided Cas nuclease that mediates target interference or cleavage (reviewed in (6)).
Figure 1 -. Adaptive immunity by CRISPR-Cas systems.

a) Foreign genetic elements are acquired by Cas1/Cas2 and integrated into the CRISPR-array in a process broadly termed adaptation. b) The CRISPR array and associated Cas proteins are expressed. The CRISPR array is processed and Cas effector nucleases associate with a crRNA to form an active surveillance complex. c) The Cas effector nucleases target foreign genetic elements complementary to their crRNA leading to target interference and immunity.
The Class 2 Type II DNA-targeting endonuclease Cas9 (the first Cas effector to be harnessed for genome engineering) has several properties that ensure precise and efficient editing (Box 1, a). Cas9 assembles with only the intended guide RNA through specific recognition of the crRNA and its interaction with a trans-activating CRISPR-RNA (tracrRNA). In addition, the dual crRNA-tracrRNA can be fused into a chimeric single-guide RNA (sgRNA) creating a two-component system, Cas9 and its sgRNA (7). Finally, stable binding to target DNA adjacent to a specific motif (protospacer adjacent motif, PAM (8, 9)) with the correct nucleotide sequence acts as a switch, triggering Cas9 to introduce a dsDNA break (7). Scientists worldwide have deployed Cas9 due to this switchable nuclease activity and the ease of redirecting the enzyme by altering the sgRNA targeting region (or spacer sequence) (10–12).
Box 1 -. Schematic of Class 2 CRISPR-Cas systems.
a) Class 2 Type II CRISPR-Cas9 shown schematically with an sgRNA (blue) encoding a spacer (red) bound to a target dsDNA (black) proximal to a PAM (teal). Correct base-pairing activates the HNH and RuvC nuclease domains cleaving both strands (orange). b) Class 2 Type V CRISPR-Cas12a shown schematically with a crRNA (blue) encoding a spacer (red) bound to a complementary dsDNA target (black) proximal to a PAM (teal). Correct base-pairing activates the RuvC nuclease cleaving both strands and continuing with multiple-turnover general ssDNase activity (arrow). c) Class 2 Type VI CRISPR-Cas13a shown schematically with a crRNA (blue) encoding a spacer (red) bound to a complementary RNA target (black). Correct base-pairing activates HEPN-nuclease general ssRNase activity (arrow).
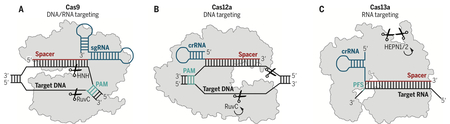
Although S. pyogenes (SpCas9) remains the most commonly used Cas effector, it is not alone in the evolutionary arms race against mobile genetic elements. Bacteria and archaea have evolved numerous functionally distinct CRISPR-Cas systems that maintain the programmable characteristics key to the success of SpCas9. Scientists have tapped the evolutionary diversity of Type II systems, incorporating divergent homologs and engineered variants of SpCas9 into an arsenal of genome editors. At the tail end of 2015, Class 2 systems expanded to include a number of candidate systems, systems later designated Type V CRISPR-Cas12a (formerly Cpf1) and Type VI CRISPR-Cas13a (formerly C2c2) (Box 1, b–c). Today, SpCas9 shares the spotlight with a diversity of Cas9 homologs, DNA-targeting Cas12, and RNA-targeting Cas13, all of which are programmable RNA-guided nucleases (reviewed in (13)). It is this inherent programmability present in a diversity of naturally evolved systems that extends CRISPR-Cas applicability beyond precision genome editing.
Applications of Cas-mediated genome editing
While the scope of Cas application has broadened, precision genome engineering remains at the forefront of the CRISPR revolution. Cas9 and Cas12a are RNA-guided nucleases that can induce genome editing by triggering dsDNA break repair at a specific site (Fig. 2a). Editing occurs after cellular DNA repair pathways resolve the break by nonhomologous end joining (NHEJ), which can introduce small insertions or deletions, or by homology-directed repair (HDR) with a donor sequence at the site of the dsDNA break (reviewed in (17)).
Figure 2 -. CRISPR-Cas systems allow genetic manipulation across the central dogma.

a) Cas9 and Cas12a are used for inducing dsDNA breaks for genome-editing. b) nCas9 can be fused to base-editors to modify nucleotides in dsDNA for genome-editing without introducing a dsDNA break. c) dCas9 can be fused to transcriptional activators, repressors, or epigenetic modifiers to regulate transcription. d) Cas9 and Cas13a can be used for targeted RNA interference. e) Cas13a fused to base-editors can be used to modify nucleotides in ssRNA. f-g) dCas9 or dCas13a can be fused to GFP to image DNA/RNA and RNA, respectively.
As tools for precision genome engineering, Cas9 and Cas12a work in a wide range of cell types and organisms. Cas-mediated gene editing has prompted genome-wide screens to probe basic biological function, in addition to identification and validation of potential drug targets in complex heritable diseases (reviewed in (18)). Agricultural applications of Casnucleases (reviewed in (19)) have produced modified crops that now have a streamlined path to the market (20). In the clinic, Cas-nucleases allow diseases with a known genetic basis to be treated, and in an era of high-throughput DNA sequencing, personalized to a patient’s disease etiology. Examples include gene editing to correct mutations or induce skipping of defective exons in Duchenne muscular dystrophy (DMD), strategies that are already showing efficacy in animal models (21, 22). Cas9 has also been used to inactivate defective genes that underlie neurological diseases including amyotrophic lateral sclerosis (23) and Huntington’s disease (24). Scientists have used Cas9 to eliminate an entire chromosome in aneuploid human pluripotent stem cells (25), to inactivate an endogenous retrovirus in pigs (26), and to engineer T cells as a prelude to developing advanced immunotherapies to target cancers (27). Furthermore, Cas9 has allowed targeting of the genetic basis for sickle cell disease (28) such that there are now established protocols for the correction of genetic defects in this cell type (29). Beyond such somatic cell editing, the potential to correct genetic mutations in human embryos is on the horizon, raising societal and ethical questions about creating heritable changes in the human germline (30).
However, it is important to note that precision editing remains challenging, particularly given competing repair outcomes (such as NHEJ) restraining the efficiency of more desirable HDR repair outcomes (31). An alternative approach utilizes Cas effectors fused to base-editors, limiting unintended edits and eliminating the requirement for repair templates. Distinct from DNA cleavage and repair, nickase Cas9 (nCas9) mediated base-editing carries a single base editor to a target locus facilitating base conversion without dsDNA cleavage (Fig. 2b) (reviewed in (32)). Recently, the toolbox of base-editors expanded to include a laboratory evolved deaminase enabling nCas9-mediated single-base editing to catalyze A-T to G-C transitions (33). The existing Cas9-mediated base-editors now enable researchers to create any of the four possible transition mutations at a specific genomic locus (33–36). While single base editors provide the potential to correct disease-causing mutations without inducing a dsDNA break, the technology requires further development to limit off-target editing. Looking forward, the next generation of Cas-mediated genome editors will likely include base-editors, ideally with base-editing activity conformationally coupled to Cas9 target DNA binding.
Transcriptional regulation with dCas9
Cas9 has proven to be a modular platform with functionally distinct DNA binding and nuclease activities. Decoupling DNA binding from the enzymatic activity of Cas9 by mutating the nuclease domains creates catalytically deficient Cas9 (dCas9), a functional scaffold for recruiting protein or RNA components to a specific locus to perturb transcription without permanently altering DNA (reviewed in (37, 38)) (Fig. 2c). The use of dCas9 has revolutionized functional genetic screening by enabling specific, rapid, and multiplexed genetic knockdowns in a range of cell types including immune cells and neurons (39, 40). These advances with dCas9 highlight the practicality of genomic perturbation without the risk of DNA damage, an attribute that has motivated studies in model systems to drive therapeutic development. For example, dCas9 fused to TET1, a demethylase, targeted to the dysregulated FMR1 locus reversed the phenotype of fragile X syndrome in neurons and mouse models (41). Gain-of-function studies have successfully implemented a modified dCas9 target gene activation system to treat type 1 diabetes, acute kidney injury, and murine muscular dystrophy (42). The ability to conduct suppressor screens and synthetic lethal screens in virtually any cell type offers the potential to discover gene functions, effector interactions, and pathways at a pace never before possible. However, challenges remain: dCas9-effector fusions have complex off-target effects due to the fused catalytic domains targeting neighboring or even unrelated loci. Additionally, unpredictable locus-specific effects on chromatin, and in turn transcription, can confound analysis and obscure causality (43). Future work should appropriately control for unpredictable locus-specific effects with systematic validation and should look to further improve specificity.
Post-transcriptional engineering with RNA-targeting Cas
As an alternative to permanent genetic alteration, Cas-effectors can be applied to transiently perturb the transcriptome through direct RNA-targeting (Fig 2d). Engineering SpCas9 to create a programmable RNA-targeting system with the use of a PAMmer (44) ushered in applications for RNA-targeting with Cas9 (RCas9). Targeting RCas9 to RNA can eliminate pathogenic RNA foci, rescue mRNA splicing defects, or attenuate polyQ-containing protein production from RNAs with trinucleotide CAG repeats (45). To date, the arsenal of RNA-targeting Cas9s has expanded to include related homologs with programmable RNA-targeting activity that is PAMmer independent (46–48). Given its success, Cas9 lends itself to further development for post-transcriptional engineering, such as fusions to single-base RNA modifiers to achieve site-specific RNA modifications.
Cas13 has also emerged as a highly versatile tool for RNA-targeting. Reconstituting Cas13a in E. coli (15) and in vitro (15, 16) established Type VI systems as an RNA-guided general RNase. Cas13a has been employed in vivo as a tool for specific knockdown in mammalian (49) and plant cells (50). Evolutionarily and functionally related to Cas13a, Cas13b enzymes have programmable RNase activity that has been functionalized for RNA interference and RNA editing in mammalian cells (51, 52) (Fig. 2e). More recently, CRISPRCas13d was identified (53, 54) and reconstituted for modulating splicing in vivo (53). RNA-targeting systems such as Cas9 and Cas13 support targeted RNA-guided research in addition to clinical applications akin to antisense oligonucleotide therapies for the treatment of acute non-mendelian pathologies avoiding the risks associated with permanent genetic modification. However, future studies are needed to determine how RNA-targeting Cas-effectors interface with a structured or protein-occluded RNA landscape and how trans-RNA cleavage by Cas13 is attenuated in vivo.
Programmable nucleic acid imaging
Correct spatiotemporal localization is critical to the function of specific genomic loci, mRNAs, and non-coding RNAs, with dysregulated molecular localization strongly implicated in disease. Current technologies for live cell imaging of genomic loci or nascent RNA are limited by the need for protein engineering or the introduction of targetable sequences into a transcript of interest. Leveraging dCas9, researchers have imaged repetitive genomic loci in live cells using dCas9 fused to fluorescent reporters (reviewed in (55)) (Fig. 2f). Exploiting the stringency of dCas9 PAM recognition, a method was developed that allows high-resolution single nucleotide polymorphism CRISPR live-cell imaging of DNA loci (56). However, widespread use of dCas9 to study localization of specific genomic loci has been limited by low signal-to-noise at non-repetitive genomic sequences. One strategy to overcome inadequate signal-to-noise involves appending multiple bacteriophage MS2 operator RNA hairpins (MS2 motifs) to the sgRNA (57). Tandem MS2 motifs act as high-affinity binding sites recruiting multiple MS2 motif binding proteins fused to a fluorescent reporter, effectively amplifying the signal to allow detection of a single dCas9-sgRNA bound to DNA in vivo (57). Leveraging RNA-targeting RCas9 has allowed researcher to track RNA in live cells (58) visualizing clinically relevant repeat expansion-containing transcripts (45) (Fig. 2g). With the growth of the RNA-guided RNA-targeting toolbox, RNA imaging tools now also include catalytically deficient Cas13a (dCas13a) (49). While both RCas9 and dCas13a show promise when targeted to repetitive elements, further development is required to realize either platform as a reliable tool for low abundance transcripts lacking in repetitive sequences. Furthermore, it is unclear if localizing large exogenous RNPs to transcripts might perturb cellular processes.
Nucleic acid detection and diagnostics
The RNA-guided nuclease activities of Cas13a and Cas12a have driven development of innovative tools for nucleic acid detection. Functionally distinct from Cas9, for both Cas13 and Cas12a a target nucleic acid (or activator RNA/DNA) activates general multiple turn over nuclease activity through correct base-pairing to the guide RNA (Box 1, b–c). Leveraging this switchable nuclease activity, Cas13a was first functionalized as a tool for detecting target RNA transcripts of interest in a pool of RNA by detecting its RNase activity (16). Expanding on this work, SHERLOCK was developed as a platform incorporating pre-amplification of the input material to create a tractable paper-based assay with improved sensitivity (59). Biochemical dissection identified that divergent Cas13a homologs have discrete crRNA and substrate preferences enabling orthogonal use to simultaneously detect two different transcripts (60). Similar dissection of Cas13b homologs revealed substrate preferences that supported expansion of the SHERLOCK platform, now SHERLOCKv2, to simultaneously detect Dengue and Zika virus ssRNA (61) in a readily deployable format (62).
Analogous to Cas13, Cas12a has evolved a functionally convergent switchable general nuclease that targets ssDNA (63). Exploiting this activity, DETECTR was developed as a CRISPR-based DNA detection and diagnostic platform (63). Coupled with isothermal preamplification, DETECTR was shown to rapidly and accurately detect clinically relevant types of human papillomavirus (64). SHERLOCKv2 integrated Cas12a-based DNA-targeting to detect either P. aeruginosa or S. aureus DNA targets in parallel to detection of RNA targets by Cas13a/b (61). Akin to Cas13, tapping the functional diversity of Cas12 systems may yield functional variants that enable further development of DNA-based diagnostics. Looking ahead the detection of a specific transcript using CRISPR-Cas is rapid and readily adaptable in the clinic, setting the stage for inexpensive point-of-care diagnostics.
Specificity and delivery of CRISPR-Cas
Unintended binding, modification, and cleavage of nucleic acids poses a significant challenge to all genome-editing technologies. Compared to the side-effects caused by offtarget interactions of small molecule drugs or antibody therapeutics, off-target Cas nuclease activity is especially deleterious due to the permanence of genome editing. Indeed, this further reinforces the necessity for nuclease specificity and targeted delivery. Researchers have made significant advances in evolving and engineering Cas enzymes (65, 66) or sgRNAs (67) to improve nuclease specificity. In addition, robust methods for predicting targeting outcomes (68) and achieving spatiotemporal gene regulation (69) provide comprehensive strategies to reduce off-targets. Beyond engineering the Cas nuclease, researchers are also developing a deeper understanding of cellular DNA repair to improve the likelihood of achieving a desired editing outcome (70).
Optimizing vehicles for efficient and specific delivery of the Cas payload remains a significant obstacle, particularly in light of immune responses to sgRNA and Cas9 in humans (71, 72). Within the lab, researchers have a number of options (electroporation, transfection, direct injection, viral-vectors) for delivering either the DNA encoding the Cas payload, the sgRNA and mRNA encoding the Cas proteins, or pre-formed RNPs to cells_ex vivo_ (73) or within immune privileged environments (24). Unfortunately, many of these options cannot be broadly translated in clinical settings where the specific requirements for efficient in vivo delivery vary with disease etiology. Furthermore, the large size of Cas nucleases bound to their guide-RNA presents a challenge for packaging within viral-based vectors. One strategy to solve this problem is leveraging smaller related Cas homologs, or minimized systems that support packaging into viral vectors (74, 75). Alternatively, functionalized nanomaterials enable specific delivery to a cell-type of interest. Indeed, recent studies have shown that directly injecting nanoparticles containing Cas9:sgRNA efficiently corrects the causative DMD mutation, leading to improved clinical phenotypes in mice (76). In all likelihood, the success of CRISPR-based therapeutics will depend on further development of suitable vehicles for delivering the Cas payload.
Conclusions and future directions
CRISPR-Cas based technologies provide an accessible and adaptable means to alter, regulate and visualize genomes, enabling biological research and biotechnological applications in a wide range of fields. CRISPR-Cas tools have vastly accelerated the pace of research, from understanding the genetics of previously unstudied organisms to discovering genes that contribute directly to disease. The field of Cas-based biotechnology is developing at a rapid pace with multiple Cas9-based clinical trials in progress or beginning soon with the results likely guiding future use for somatic cell editing both ex vivo and in patients. Outside of the clinic, agricultural applications of CRISPR-Cas9 are already creating products for various markets, leading to recent rulings by the USDA about their regulation (20). This ever-expanding repertoire of applications firmly positions the CRISPR-Cas toolkit at the cutting edge of genome editing and more broadly, genetic engineering.
Acknowledgments
We apologize to those whose work was not cited due to space limitations. We thank members of the Doudna lab for discussion. Special thanks to Steven C. Strutt, Joshua C. Cofsky, and Christof Fellmann for editing. J.A.D. receives funding from the HHMI, the NIH, the NSF, the Paul G. Allen Foundation, the Keck Foundation, and DARPA. J.A.D is an Investigator of the HHMI and executive director of the Innovative Genomics Institute at the University of California, Berkeley and the UCSF. J.A.D is a co-founder of Editas Medicine, Intellia Therapeutics, and Caribou Biosciences and a scientific adviser to Caribou, Intellia, eFFECTOR Therapeutics, and Driver. The Regents of the University of California have patents pending for CRISPR technologies on which the authors are inventors.
References
- 1.Jasin M, Rothstein R, Cold Spring Harb. Perspect. Biol 5, a012740–a012740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Urnov FD, Cris. J 1, 34–46 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Barrangou R et al. , Science (80-.) 315, 1709–1712 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Marraffini LA, Sontheimer EJ, Science (80-.) 322, 1843–1845 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brouns SJJ et al. , Science (80-.) 321, 960–964 (2008). [Google Scholar]
- 6.Koonin EV, Makarova KS, Zhang F, Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol 37 (2017), pp. 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jinek M et al. , Science (80-.) 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marraffini LA, Sontheimer EJ, Nature. 463, 568 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Almendros C, Microbiology. 155, 733–740 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Mali P et al. , Science (80-.) 339, 823–826 (2013). [Google Scholar]
- 11.Cong L et al. , Science (80-.) 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jinek M et al. , Elite. 2 (2013), doi: 10.7554/eLife.00471. [DOI] [Google Scholar]
- 13.Murugan K, Babu K, Sundaresan R, Rajan R, Sashital DG, Mol. Cell 68, 15–25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zetsche B et al. , Cell. 163, 759–771 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abudayyeh OO et al. , Science (80-.) 353, 5573–5579 (2016). [Google Scholar]
- 16.East-Seletsky A et al. , Nature. 538, 270–273 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pawelczak KS, Gavande NS, VanderVere-Carozza PS, Turchi JJ, ACS Chem. Biol, in press, doi: 10.1021/acschembio.7b00777. [DOI] [PubMed] [Google Scholar]
- 18.Fellmann C, Gowen BG, Lin P-C, Doudna JA, Corn JE, Nat. Rev. Drug Discov 16, 89–100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin K, Gao C, Qiu J-L, Nat. Plants 3, 17107 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Waltz E, Nat. Biotechnol 36, 6–7 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y et al. , Sci. Adv 3, e1602814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long C et al. , Sci. Adv 4, eaap9004 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaj T et al. , Sci. Adv 3, eaar3952 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Staahl BT et al. , Nat. Biotechnol 35, 431–434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zuo E et al. , Genome Biol. 18 (2017), doi: 10.1186/s13059-017-1354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niu D et al. , Science (80-.), eaan4187 (2017). [Google Scholar]
- 27.Rupp LJ et al. , Sci. Rep 7, 737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eyquem J et al. , Nature. 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bak RO, Dever DP, Porteus MH, Nat. Protoc 13, 358–376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma H et al. , Nature (2017), doi: 10.1038/nature23305. [DOI] [Google Scholar]
- 31.Canny MD et al. , Nat. Biotechnol 36, 95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hess GT, Tycko J, Yao D, Bassik MC, Mol. Cell 68, 26–43 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaudelli NM et al. , Nature (2017), doi: 10.1038/nature24644. [DOI] [Google Scholar]
- 34.Nishida K et al. , Science (80-.) 353, aaf8729–aaf8729 (2016). [Google Scholar]
- 35.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR, Nature. 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komor AC et al. , Sci. Adv 3, eaao4774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kampmann M, ACS Chem. Biol, in press, doi: 10.1021/acschembio.7b00657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pulecio J, Verma N, Mejía-Ramírez E, Huangfu D, Raya A, CRISPR/Cas9-Based Engineering of the Epigenome. Cell Stem Cell. 21 (2017), pp. 431–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bak RO et al. , Elite. 6 (2017), doi: 10.7554/eLife.27873. [DOI] [Google Scholar]
- 40.Zheng Y et al. , Nat. Neurosci (2018), doi: 10.1038/s41593-018-0077-5. [DOI] [Google Scholar]
- 41.Liu XS et al. , Cell. 172, 979–992.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao H-K et al. , Cell. 171, 1495–1507.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Howe FS et al. , Elite. 6 (2017), doi: 10.7554/eLife.29878. [DOI] [Google Scholar]
- 44.O’Connell MR et al. , Nature. 516, 263–266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Batra R et al. , Cell. 170, 899–912 e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strutt SC, Torrez RM, Kaya E, Negrete OA, Doudna JA, Elite. 7 (2018), doi: 10.7554/eLife.32724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rousseau BA, Hou Z, Gramelspacher MJ, Zhang Y, Mol. Cell (2018), doi: 10.1016/j.molcel.2018.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dugar G et al. , Mol. Cell 69, 893–905.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abudayyeh OO et al. , Nature. 550, 280–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aman R et al. , Genome Biol. 19, 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smargon AA et al. , Mol Cell (2017), doi: 10.1016/j.molcel.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cox DBT et al. , Science (80-.) 358, 1019–1027 (2017). [Google Scholar]
- 53.Konermann S et al. , Cell. 173, 665–676.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yan WX et al. , Mol. Cell 70, 327–339.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knight S, Tjian R, Doudna J, Angew. Chemie Int. Ed (2017), doi: 10.1002/anie.201709201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maass PG et al. , Nat. Struct. Mol. Biol 25, 176–184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin P et al. , Nat. Commun 8 (2017), doi: 10.1038/ncomms14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nelles DA et al. , Cell. 165, 488–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gootenberg JS et al. , Science (80-.) 356, 438–442 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.East-Seletsky A, O’Connell MR, Burstein D, Knott GJ, Doudna JA, Mol Cell. 66, 373–383 e3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gootenberg JS et al. , Science (80-.), eaaq0179 (2018). [Google Scholar]
- 62.Myhrvold C, Freije CA, Gootenberg JS, Omar O, Science (80-.) 360, 1–49 (2018). [Google Scholar]
- 63.Chen JS, Ma E, Harrington LB, Tian X, Doudna JA, bioRxiv, 226993 (2017). [Google Scholar]
- 64.Chen JS et al. , Science (80-.), eaar6245 (2018). [Google Scholar]
- 65.Hu JH et al. , Nature (2018), doi: 10.1038/nature26155. [DOI] [Google Scholar]
- 66.Chen JS et al. , Nature. 550, 407–410 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yin H et al. , Nat. Chem. Biol 14, 311–316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim HK et al. , Nat. Biotechnol (2018), doi: 10.1038/nbt.4061. [DOI] [Google Scholar]
- 69.Richter F et al. , Curr. Opin. Biotechnol 48, 119–126 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Kan Y, Ruis B, Takasugi T, Hendrickson EA, Genome Res. 27, 1099–1111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Charlesworth CT et al. , bioRxiv, 243345 (2018). [Google Scholar]
- 72.Kim S et al. , Genome Res. (2018), doi: 10.1101/gr.231936.117. [DOI] [Google Scholar]
- 73.Glass Z, Lee M, Li Y, Xu Q, Trends Biotechnol. 36, 173–185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ran FA et al. , Nature. 520, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim E et al. , Nat. Commun 8, 14500 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee K et al. , Nat. Biomed. Eng 1, 889–901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]