Neuropathology of Degenerative Cell Death in Caenorhabditis elegans (original) (raw)
Abstract
In Caenorhabditis elegans necrosis-like neuronal death is induced by gain-of-function (gf) mutations in two genes, mec-4 and deg-1, that encode proteins similar to subunits of the vertebrate amiloride-sensitive epithelial Na+ channel. We have determined the progress of cellular pathology in dying neurons via light and electron microscopy. The first detectable abnormality is an infolding of the plasma membrane and the production of small electron-dense whorls. Later, cytoplasmic vacuoles and larger membranous whorls form, and the cell swells. More slowly, chromatin aggregates and the nucleus invaginates. Mitochondria and Golgi are not dramatically affected until the final stages of cell death when organelles, and sometimes the cells themselves, lyse. Certain cells, including some muscle cells in deg-1 animals, express the abnormal gene products and display a few membrane abnormalities but do not die. These cells either express the mutant genes at lower levels, lack other proteins needed to form inappropriately functioning channels, or are better able to compensate for the toxic effects of the channels. Overall, the ultrastructural changes in these deaths suggest that enhanced membrane cycling precedes vacuolation and cell swelling. The pathology of mec-4(gf) and _deg-1(gf)_cells shares features with that of genetic disorders with alterations in channel subunits, such as hypokalemic periodic paralysis in humans and the weaver mouse, and with degenerative conditions, e.g., acute excitotoxic death. The initial pathology in all of these conditions may reflect attempts by affected cells to compensate for abnormal membrane proteins or functions.
Keywords: neurodegeneration, Caenorhabditis elegans, degenerin, mec-4, deg-1 neuropathology, necrosis, membrane cycling
Wyllie et al. (1980) suggested that cells die in two morphologically distinct patterns, apoptosis and necrosis, that are ubiquitous in Metazoans. In apoptosis, which occurs in normal development and homeostasis, the nucleus and cytoplasm condense, while cytoplasmic organelles maintain their integrity and DNA degrades into nucleosome-sized fragments. In necrosis, which commonly results from cell injury, cell membranes and organelles are disrupted, and the dying cells swell and lyse.
Programmed cell death in the nematode Caenorhabditis elegans_shares morphological and molecular similarities with apoptosis in higher organisms (Robertson and Thomson, 1982; Vaux et al., 1992; Miura et al., 1993; Yuan et al., 1993; Hengartner and Horvitz, 1994). Necrosis-like deaths in C. elegans can be induced by unusual gain-of-function (gf) mutations in two genes,mec-4 and deg-1, which cause swelling and degeneration of specific groups of neurons (Chalfie and Sulston, 1981;Chalfie and Wolinsky, 1990; García-Añoveros et al., 1995). These deaths are induced by a mechanism genetically distinct from programmed cell death (Hedgecock et al., 1983; Ellis and Horvitz, 1986; Chalfie and Wolinsky, 1990; Yuan and Horvitz, 1990). The_mec-4 and deg-1 genes encode proteins, called degenerins, that are similar to the subunits of the amiloride-sensitive epithelial Na+ channel (ENaC; Canessa et al., 1993, 1994;Chalfie et al., 1993; Waldmann et al., 1995). By analogy, the C. elegans degenerin proteins are presumed to form ion channels. The death-inducing substitutions in these proteins are thought to hyperactivate the channels, resulting in increased or altered ion flow and/or osmotic imbalance and consequent death (Chalfie and Wolinsky, 1990; Driscoll and Chalfie, 1991; Hong and Driscoll, 1994;García-Añoveros et al., 1995). A recent study of a similar mammalian protein, MDEG, supports this model (Waldmann et al., 1996).
To understand how degenerin defects lead to cell death and to compare this degeneration with other types of cell death, we have followed the progression of neuronal pathology in mec-4(gf) and_deg-1(gf)_ animals via light and electron microscopy. Degeneration seems to be initiated with enhanced endocytosis at the plasma membrane, the inferred location of the ion channels. The onset of the death depends on the dosage of the mutant gene. Although_mec-4_ and deg-1 are expressed in different neurons, cell death induced by mutant forms of either appears identical. Similarities between the degenerin-induced deaths and the early pathology of dominant myotonias (Engel, 1970; Spier et al., 1990), excitotoxicity (Hajos et al., 1986; Rothman, 1994), and epilepsy (Wasterlain and Shirasaka, 1994) suggest that similar mechanisms underlie other acute cell swellings and necrotic cell deaths.
MATERIALS AND METHODS
Nematode strains. Nematode cultures were grown at 20°C unless otherwise noted, according to standard methods (Brenner, 1974). The mec-4 dominant allele u231 is described in Chalfie and Au (1989). The deg-1(gf) alleles_u38_, u579, and u506 are described inChalfie and Wolinsky (1990) and García-Añoveros et al. (1995). ced-3(n717) and ced-4(n1162) are described by Ellis and Horvitz (1986). Other mutations are described byBrenner (1974). Strain ZB2 harbors an integrated array,bzIs1, that includes a mec-4lacZ fusion gene and the dominant su1006 allele of the rol-6 gene (Mitani et al., 1993).
The otherwise wild-type ZB7 strain contains an array, bzIs3, that is integrated into the X chromosome and has ∼10 copies of plasmid TU#14, which encodes the mec-4(u231) allele (Driscoll and Chalfie, 1991), and several copies of TU#44, a_mec-4lacZ_ fusion gene (Mitani et al., 1993). We generated ZB7 by methods described in Mitani et al. (1993). Copy number of_mec-4(u231)_ was estimated by digesting ZB7 DNA with_Eco_RI, which generates distinct fragments for the genomic and plasmid genes, and by performing Southern blot analysis with a radiolabeled mec-4 probe. The intensity of the hybridization signals from mec-4(u231) plasmid and genomic copies was measured with a Molecular Dynamics PhosphorImager.
We made the deg-1lacZ fusion construct TU#224 by subcloning a 4083 bp Eco_RI–_Msc_I fragment from a 6.7 kb_deg-1 genomic clone (García-Añoveros et al., 1995) into pPD34.11 (Fire et al., 1990). TU#224 has 3 kb of sequence upstream of the deg-1 SL1 _trans_-splice site and DNA to codon 268 in exon 5 (this DNA encodes through the first cysteine-rich domain) fused in frame to the artificial transmembrane coding sequence of the vector and the lacZ gene. TU#148 was created by removing the Kpn_I cassette that encodes the artificial transmembrane domain from TU#224. In all, 100 ng/μl of each construct was coinjected (Fire, 1986) with 100 ng/μl of pRF4, a_rol-6(su1006) marker DNA (Mello et al., 1991), into wild-type animals to produce lines carrying stable extrachromosomal arrays of these DNAs. Roller (Rol) animals were stained for β-galactosidase activity as described by Fire et al. (1990).
We produced strains with integrated arrays of the TU#224_deg-1lacZ_ fusion (uIs5, uIs6, and_uIs7_) as before (Mitani et al., 1993) using a strain with one of the extrachromosomal arrays (uEx141).uIs5 and uIs6 are X-linked; uIs7 is autosomal. To facilitate the identification of_lacZ_-expressing cells, we suppressed the Rol phenotype of_uIs6_ by including the sqt-1(sc103) mutation (Kramer and Johnson, 1993).
Transgenic animals carrying extrachromosomal (uEx141 and_uEx142_) or integrated (uIs5, uIs6, or_uIs7_) arrays of the TU#224 _deg-1lacZ_fusion were stained for β-galactosidase (16 lines; Fire et al., 1990). Because extracellular β-galactosidase is inactive (Fire et al., 1990; Manoil, 1990) and this construct produces a protein in which an artificial transmembrane domain follows the first hydrophobic and first cysteine-rich domains of DEG-1, the first cysteine-rich domain is likely to be extracellular. No staining was detected in animals from any of the five lines carrying a construct without the artificial transmembrane domain (TU#148).
Timing of gene expression and neurodegeneration. mec-4(u231) mutant animals were synchronized to within 3 hr of each other by allowing 100 gravid adults to lay eggs for 1 hr. Larvae were harvested for observation by differential interference contrast microscopy or were stained for β-galactosidase according to the protocol of Fire et al. (1990). At least two different populations were examined at each time point. Individual postembryonic touch receptor neurons could be recognized by their relative positions within the body (Sulston and Horvitz, 1977; White et al., 1986). The onset of_deg-1lacZ_ expression in the PVC neurons was examined in_sc103;uIs6_ animals (n = 565), and the onset of the death of the PVC neurons was examined in deg-1(u38)(n = 1233) and deg-1(u506)(n = 344) animals. These animals were synchronized by collecting animals that had hatched within a 6 hr period. As in_mec-4(u231)_ animals, the onset of cell death was not well synchronized, and dying cells could linger for many hours.
Electron microscopy. ZB7 animals were grown at 25°C and staged under differential interference contrast optics to select cells representing different time points of PVM degeneration. Although PVM is produced at the same time as AVM (Sulston and Horvitz, 1977),mec-4lacZ expression and cell death begin ∼1-2 hr earlier in AVM than in PVM in ZB7 animals. We used AVM swelling as a predictor of the early stages of degeneration of PVM, and we used PVM appearance for the later stages. Selected animals were transferred to primary fixative and cut open on each end with a razor blade to improve penetration. After initial fixation in buffered aldehydes, animals were stained in buffered osmium tetroxide (0.5%) containing 0.5% potassium ferricyanide and embedded in Medcast resin (Ted Pella, Redding, CA) following standard methods (Hall, 1995). Serial thin sections were collected transverse to the body axis, beginning from the gonad and extending posteriorly through the midbody region. In the ZB7 strain, in which cell death occurs during the late L1 stage, the PVM cell body generally was found ∼300 sections behind the center of the developing gonad primordium, where it was embedded in the hypodermis along the left body wall slightly ventral to its sister neuron, SDQL. Electron micrographs were collected by a Philips CM10 electron microscope. In a few cases not documented in detail here, partial series through degenerating neurons gave results consistent with those described here.
Light microscopy also was used to select time points during the death of PVC neurons in deg-1(u38) animals grown at 25°C. We had some difficulty identifying the earliest time points for cell degeneration, because the onset of cell swelling was less predictable than in ZB7 strain animals. After fixation and embedding as above, transverse serial sections were collected through the tail, including the preanal and lumbar ganglia. Complete serial sections were collected for the reported cells.
RESULTS
Several factors influence the onset and extent of degenerin-induced cell death
mec-4 normally is expressed only in a set of six touch receptor neurons (Mitani et al., 1993), and _mec-4(gf)_mutations, such as e1611 and u231, induce the cell-autonomous death of these cells (Chalfie and Sulston, 1981;Herman, 1987; Chalfie and Au, 1989). Two of the cells, AVM and PVM, arise ∼9 hr after hatching at 20°C, so their entire development can be observed easily in the light microscope (Sulston and Horvitz, 1977). By examining the expression pattern of a mec-4lacZ fusion, we found that mec-4 was expressed soon (3–5 hr) after these cells were produced (Fig. 1), with the staining of AVM nearly always occurring first. One-half the AVM cells stained by 12 hr after hatching, whereas one-half of the PVM cells stained by 14 hr after hatching.
Fig. 1.

Summary of the timing of mec-4 and_deg-1_ expression and cell death. Times are given in hours after hatching at 20°C, although the data for the_deg-1_ animals were obtained at 25°C and converted to the equivalent times at 20°C. The production of the cell (PVM for the_mec-4_ animals and PVC for the deg-1_animals) from division of its precursor is shown by open circles. Filled circles indicate the time when the greatest number of cells expresses lacZ from_mec-4 (>50%) or deg-1 (20%) fusions.X indicates the time when the most cell deaths are seen (>50% for both strains). Data for mec-4(e1611) came from Chalfie and Sulston (1981).
The time of onset of PVM degeneration depended on the _mec-4_mutation used and its copy number. The AVM and PVM cells became vacuolated and died in mec-4(e1611) animals when the animals were young adults (at ∼45 hr after hatching; Chalfie and Sulston, 1981; Mitani et al., 1993). In contrast, the PVM cell was noticeably distorted in one-half of the age-synchronized _mec-4(u231)_animals 18 hr after hatching, just 4 hr after mec-4 was first expressed. (Some PVM cells became abnormal later, whereas others never seemed to degenerate.) Increased dosage of _mec-4(u231)in the transgenic line ZB7, which has ∼10 copies of the mutant gene, accelerated the onset of degeneration to shortly after the time that_mec-4 was first expressed.
The rapidly dying cells in the ZB7 animals did not swell as much as the dying cells in u231 animals. In EM reconstructions the diameter of ZB7 cells increased by approximately twofold, increasing the volume ∼10-fold (data not shown). In contrast, the diameters of_u231_ cells, when viewed by differential interference contrast (DIC) optics, increased fivefold to give a volume increase of 100-fold. Presumably, at high gene dosage the toxic product more rapidly inactivated cell functions that contribute to swelling. Consistent with an accelerated time course of cell death, the degenerating cells also disappeared much sooner in ZB7 animals (<6 hr after the onset of visible swelling) than in u231 animals, where the touch cells can persist for 8 hr or longer as distended somata.
Specific gf alleles of deg-1 (the dominant mutations u38 and u529 and the recessive mutation_u506_) cause neuronal degeneration that is morphologically identical to that induced by _mec-4(gf)alleles (Chalfie and Wolinsky, 1990; García-Añoveros et al., 1995). Occasionally, we have found small vacuoles without nuclei anterior to the first bulb of the pharynx and along the body in_deg-1(gf) animals. These empty vacuoles (in distinction to the nucleus-containing neuronal degenerations) may be in neuronal processes, muscles, or epidermal cells.
As predicted for cell-autonomous expression of deg-1, a_deg-1lacZ_ fusion was expressed in many neurons, the positions of which matched those of dying cells in u38 and_u506_ animals (Fig. 2). These cells included the presumptive ASH cells, IL1 cells, AVD cells, the AVG cell, and the PVC cells, although the latter stained weakly. Muscles also expressed the deg-1lacZ fusion. The head muscle quadrants and a muscle near the anus, presumably the anal depressor muscle, stained at all stages, whereas body wall muscles stained most strongly in newly hatched larvae. A similar staining pattern was seen with_deg-1gfp_ fusions (data not shown).
Fig. 2.

Expression of deg-1lacZ. Animals contain the integrated array uIs6 and are homozygous for the sqt-1(sc103) mutation. A, Early L1 larva. B, Late L1 larva. C, L2 larva.D, L4 larva. Note staining (triangles) in head muscles at the front of the L4 larva. Cells are identified by their position within the animals.
As with touch cell death in mec-4 mutants, the onset of PVC death depended on the deg-1 allele used (Fig. 1). Although we were able to see deg-1lacZ expression in PVC cells in only one of our constructs (uIs6), the peak of its expression preceded the peak onset of u38_-induced deaths by ∼6 hr. The PVC deaths induced by the u506 mutation, however, occurred much later at 25°C than those produced by the_u38 mutation. This delay could result because the_u506_ product is either less toxic or less stable than the_u38_ product. With either possibility a greater amount of time would be needed to accumulate a lethal amount of the product. The onset of the cell death also depends on the dosage of the_u38_ mutation, because lower dosage results in a later onset of PVC degeneration (Chalfie and Wolinsky, 1990).
Most of the _deg-1lacZ_-expressing cells, however, did not degenerate in u38 animals (data not shown). Several factors may contribute to cell lethality. First, some cells may make insufficient amounts of the toxic product. Some cell types seem to produce near-threshold amounts of the toxic product, because not all cells of a given type die. For example, variable numbers of IL1 cells (Chalfie and Wolinsky, 1990), ASH neurons (C. Bargmann, personal communication; A. Hart and J. Kaplan, personal communication), and PVC neurons [in deg-1(u506) males] die (data not shown). Second, because alternative splicing occurs at the _deg-1_locus (Chalfie and Wolinsky, 1990; García-Añoveros et al., 1995), a less toxic form of the protein may be made. Third, some cells may compensate better for the toxic product. Fourth, additional proteins needed to form the toxic channel may not be expressed in certain cells. The last three possibilities may explain why the muscle cells in the head, which show the highest level of expression of any cells, survive.
Light microscopy of degenerin-induced cell death
We examined the degeneration of PVM in ∼80 ZB7 animals using DIC microscopy and have divided the process into four periods. The first, or predegeneration, stage lasted 3 hr from the production of the PVM cell. In this initial stage the cells were not detectably different from wild-type cells when viewed with light microscopy (Fig.3A). Obvious pathology is seen after this time (at ∼2 hr before the L1 molt at 25°C). Although the subsequent pattern of degeneration was similar in all animals (Fig. 3), its time course was quite variable, lasting 4–20 hr from the start of the degeneration because of variations in the last of the four periods.
Fig. 3.

Light microscopy of cell death in_mec-4(gf)_ and _deg-1(gf)_animals. A–J, Degeneration of PVM in ZB7.A, A predegeneration normal-looking PVM cell ∼2 hr after its production. This is not the same cell depicted in subsequent panels. The arrowhead points to the cell body.B, Start of degeneration (0 min, early degeneration). A small space (arrow) has begun to appear around the PVM nucleus (arrowhead). C, At 30 min. The cell shows obvious swelling; the nuclear envelope is not easily distinguished. D, At 90 min (middle degeneration). The cytoplasm clears, and the nucleus swells. E, At 120 min. The nucleus has moved to one side of the vacuolated space.F, At 160 min (10 min before the L1 lethargus, late degeneration). The cytoplasmic vacuole and nucleus (arrowhead) are smaller. G, At 190 min (during the L1 lethargus). The vacuole and nucleus are very poorly seen. H, At 7 hr. The vacuole and nucleus (arrowhead) are visible but condensed.I, At 19 hr. The nucleus (arrowhead) has condensed further. J, At 24 hr. Only debris (arrowhead) remains. K–N, Degeneration of PVC in deg-1(u38). K, At ∼0.5 hr after the start of degeneration. L, At 1 hr. The nucleus has enlarged. M, At 1.5 hr. The nucleus begins to condense. N, At 2.2 hr. The nucleus continues to condense, especially at its periphery. The cell disappeared in another 2.2 hr.
The first signs of pathology were detectable in the second, or early degeneration, stage (Fig. 3B,C). Initially, a small space, which was neither clear nor filled with particles displaying Brownian motion, appeared between the plasma membrane and the nucleus. This space enlarged rapidly so that the neuron swelled to its maximal size by the end of this period. The nucleus did not change in size during this period, but its outline was no longer clear (Fig.3C). The early stage was extremely short, lasting ∼15 min.
In the third, or middle degeneration, period, which lasted ∼2 hr (Fig. 3D,E), the space between the plasma membrane and the nucleus cleared and contained particles displaying Brownian motion. The nucleus began to swell to two or three times its normal size, and it was displaced gradually to one side of the cell. The nucleus also contained particles showing Brownian motion.
The onset of the fourth, or late degeneration, stage (Fig.3F–J) came abruptly. The vacuoles in both the cytoplasm and the nucleus shrunk very quickly, and the cell decreased in size. The nucleus gradually condensed and became a small core by the time of the L1 lethargus (the dormancy period preceding the molt). In some animals debris from the PVM cell disappeared in the first hour after the L1 molt. In others the PVM vacuole was more apparent after the molt (Fig. 3H). Sometimes a similar disappearance/reappearance occurred at the L2 lethargus. These apparent changes may be a consequence of the cells being obscured by the molting cuticle or may result from different osmotic conditions during lethargus. All cells had disappeared by the late L3 stage. (The late disappearance of the cells in many observed animals may result from the constant viewing of the cells under a coverslip, because we rarely saw debris from cells 6 hr after the L1 molt in staged animals taken directly from plates.)
The degeneration of the PVC interneurons in deg-1(u38) and_deg-1(u506)_ mutants viewed in the light microscope progressed similarly to the death of PVM cells in ZB7 animals (Fig.3K–N) (data not shown). Cells began to swell and vacuolate before nuclear morphology changed. Nuclei then swelled and began to accumulate refractile material. The cells were not detected 4 or 5 hr after they had begun to vacuolate. The two deg-1_mutants differed, however, in the time of onset of the deaths; the_u506 deaths occurred later, although at a less precise time (see Fig. 1).
In the later stages of the deaths in mec-4(gf) and_deg-1(gf)_ animals, dying cells appeared to condense, a characteristic found in programmed cell death. Although mutations in_ced-3_ and ced-4, genes that are needed for programmed cell death, do not affect the onset of_mec-4(gf)_- and deg-1(gf)-induced cell deaths (Ellis and Horvitz, 1986; Chalfie and Wolinsky, 1990), we tested whether these mutations would affect the cell and nuclear condensation. In neither ced-4;mec-4(e1611) nor_ced-3;deg-1(u38) were cell or nuclear condensation detectably different from that seen in animals lacking the_ced mutations (data not shown).
Ultrastructure of degenerin-induced cell death
We used serial section reconstruction to examine the ultrastructure of a total of seven PVM cells and one AVM cell in the_mec-4(u231)_ overexpression strain ZB7, two at each of the four stages defined by our light microscopic observations. The electron micrographs revealed a reproducible sequence of cellular changes that accompany degeneration.
Predegeneration stage
The two PVM neurons that we sectioned in the predegeneration stage differed from their neighboring lineage sisters, the SDQL cells, by having small membrane whorls at the plasma membrane. In one animal the PVM cell contained a small circle of internalized membrane, which appeared slightly more electron dense than normal plasma membrane or ER and may have contained a second circlet (Fig. 4). In the other animal the PVM cell had five small whorls (nested membranous shells) just inside the plasma membrane and two small circles of dense membrane lying exterior to the cell. Neither cell had vacuoles or cell swelling, and most organelles, including the nucleus, appeared normal. Two mitochondria in the second cell had increased density localized to their outer surface. In each case, as expected for this time in development, a single very short (0.5 and 4 μm) axonal process grew ventrally from the soma toward the ventral nerve cord.
Fig. 4.
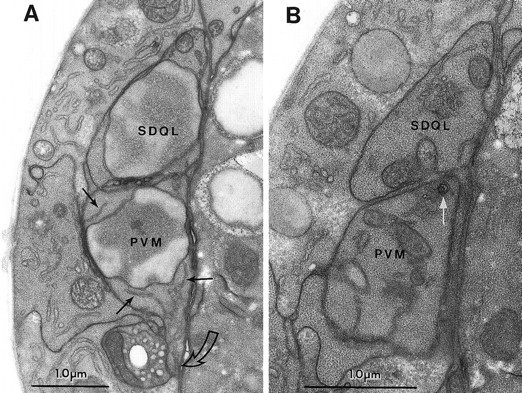
The predegeneration stage. A, Section through the somata of PVM and_SDQL_. In this section the cells look very similar. Note the dispersed chromatin in both nuclei (labeled with the cell names), the strands of ER in the dark cytoplasm of PVM(thin black arrows), and the characteristic ventralward process extending from PVM (open curved arrow).SDQL has a rostral process that begins to extend at this time (not shown). B, A glancing section through a more caudal portion of the same cells as in A. The_thin white arrow_ points to a very small circular set of nested membranes that seems to be the earliest sign of_mec-4(gf)-induced degeneration in the PVM cell. Organelles and dark cytoplasm in PVM and_SDQL look similar.
Early degeneration stage
The two PVM neurons in the early degeneration stage showed many abnormalities (small internal membrane whorls and internal vacuoles) that may have formed from the plasma membrane, because many showed direct connections to it. Other cytoplasmic organelles, however, were still intact and unswollen. One cell had many small whorls surrounding the soma (possibly shed pieces of plasma membrane), three small cytoplasmic vacuoles, and a normal-appearing nucleus (Fig.5A,B). The degeneration of the second cell appeared more advanced in all respects, with more and larger whorls, larger vacuoles, and early signs of nuclear degeneration (chromatin clumping along the inner surface of the nuclear envelope; Fig.5C). A few membrane whorls also were found outside of this cell. Both PVM cells were somewhat swollen by the enlargement of internal vacuoles and membrane whorls. Large vacuoles lay close to the plasma membrane and sometimes appeared to be connected to whorls. Although most ER looked normal in both cells, some was somewhat swollen and more electron dense, suggesting that small vacuoles may occasionally derive directly from the ER. Each cell had a single axon extending toward the ventral nerve cord. This process contained several membrane whorls at the hillock, along its length, and at the growth cone. These axons were shorter than expected for this developmental stage (3 μm long), suggesting that outgrowth may have been disrupted.
Fig. 5.

The early degeneration stage. A,PVM axon hillock in a glancing section. Many small membrane whorls are situated near the plasma membrane.B, A higher magnification view of a whorl from_A_, showing that it consists of onion skin-like concentric layers. C, The PVM soma in a second animal is swollen because of a large membrane-bound vacuole (vac), which may be in direct contact with plasma membrane.Arrows show small nested membrane whorls associated with the plasma membrane. ER, Endoplasmic reticulum;M, mitochondrion.
Middle degeneration stage
The two touch neurons (1 PVM, 1 AVM) at the middle degeneration stage were more swollen by vacuoles and whorls. Their nuclei were distorted and contained large clumps of chromatin. The ER and Golgi bodies were swollen, but the mitochondria were intact (Fig.6). The cytoplasm of these neurons had fewer small whorls but was dominated, instead, by two to four very large vacuoles and/or whorls. The nucleus was displaced and deformed by invagination of a large vacuole in both cells (Fig. 6A,C). The cytoplasm was slightly less electron dense, suggesting that degradation of its contents had begun. No axons were seen in these cells. In addition, the AVM cell, but not the PVM neuron, was wrapped almost completely by a hypodermal process; only a small contact remained with the basal lamina (data not shown).
Fig. 6.
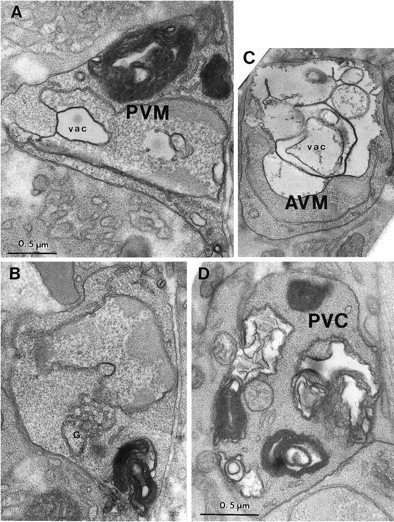
The middle degeneration stage. A, The PVM nucleus (labeled with the cell name) is highly indented, invaded by a cytoplasmic vacuole (vac), and its chromatin is condensed. Several very large whorls of membrane cause cell swelling. The cytoplasm is slightly less electron-dense than that of nearby tissues. B, A different section of the same cell as in A, showing the mild swelling of the Golgi apparatus (G). C, A degenerating_AVM_ cell has a huge vacuole (vac) invading the nucleus (labeled with the cell name), which is pushed to the edge of the soma. Cytoplasm of AVM is lighter than that of surrounding tissues. D, PVC soma in a late L1 stage (12 hr after hatching) deg-1(u38) larva is filled with large and small membrane whorls and vacuoles. The nucleus (not shown) was distorted in shape but otherwise normal in appearance. Magnification is the same in A–C.
Late degeneration stage
The appearance of the two late degeneration stage PVM neurons indicated a complete breakdown of cellular structures. One cell looked similar to the middle degeneration stage PVM cell described above, except for a local failure in the plasma membrane, which allowed some cytoplasmic contents to stream out from the cell (Fig.7A). The nucleus was intact, but somewhat deformed, with very condensed chromatin. The cytoplasm was extremely light, as if degraded. The cytoplasm contained two very large vacuoles with associated whorled membranes that greatly distorted the cell. The cytoplasm also contained several intact mitochondria with moderate cisternal swelling, increased matrix density, and in one case a small outward bleb. Very few small whorls were seen. The second cell, which was large and distended into a lobular shape, had essentially no intact cytoplasmic contents, except for several compact dense bodies (Fig.7B). No other signs of a nucleus remained, nor were any mitochondria present. One of these PVM cells (Fig. 7B) was surrounded completely by a hypodermal arm, which isolated the cell remnants from the basal lamina. The other PVM cell was not engulfed but remained in contact with the basal lamina.
Fig. 7.
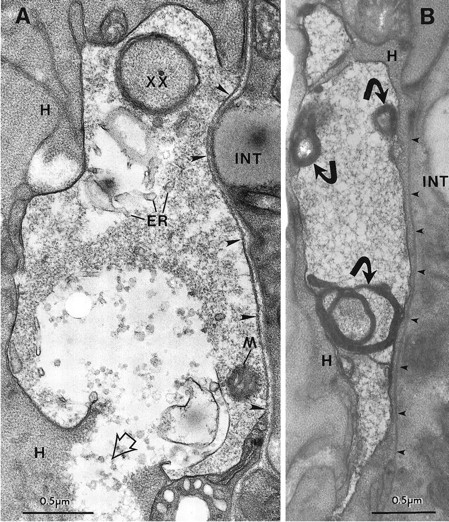
The late degeneration stage. A, PVM soma with a break in the plasma membrane in a ZB7 animal. Note the local damage to the closely apposed hypodermal tissue (H), where the cell contents of PVM have spilled out (open arrowhead). The PVM cytoplasm is very light, and the endoplasmic reticulum (ER) is swollen, but a mitochondrion (M) is still intact. The perimeter of the cell is irregular and is invaded by a finger of hypodermal tissue (xx). The soma is bordered by basal lamina on its right side (large arrowheads) that separates it from the intestine (INT). B, Another PVM soma has highly degraded cytoplasm, but the plasma membrane is intact. Several short projections extend from the swollen, distorted cell. The nucleus was not seen in serial thin sections of this cell. Small dense fragments (curved arrows) are each associated with a large membrane whorl. A thin hypodermal arm projects along the_right_ side of the soma and separates it from the basal lamina (small arrowheads). In serial sections this arm completely engulfed the PVM cell.
The pattern of neuronal death in deg-1(u38) animals, as seen in the degeneration of the PVC interneurons, was similar to that of the touch cells in mec-4 mutants. In an animal selected by light microscopy for having cells in the predegeneration stage, no abnormalities were found in any lumbar neurons, including the bilaterally symmetric PVCL and PVCR cells. In an animal with an early degeneration stage cell, the PVCR appeared normal, but the PVCL was moderately swollen with many large and small membrane whorls and vacuoles in the cytoplasm and a crenated nucleus with normal chromatin (Fig. 6D). In its pathology, PVCL looked intermediate between the early and middle stages of PVM degeneration in ZB7. Most organelles looked normal, although the ER was swollen near a few whorls and vacuoles. The electron density of the cytoplasm was normal. In an animal selected as having a middle degeneration death, the PVCL cell was normal, but the extremely swollen and lobulated PVCR neuron (data not shown) looked similar to a late-stage PVM death in ZB7. The cell had a highly distorted nucleus and a cytoplasm with very low electron density. Most cytoplasmic organelles were absent in PVCR; instead, the cell contained approximately one dozen large, loose membrane circles or whorls and much flocculent material that failed to embed well (in distinction to the surrounding cells that were well embedded). Overall, the progression of degeneration is strikingly similar in ZB7 and_deg-1(u38)_ animals; the same sequence of cellular events is seen.
Other signs of pathology in mec-4(gf) and_deg-1(gf)_ animals
We examined other cells in wild-type, ZB7, and_deg-1(u38)_ animals to determine the specificity of cell damage. Membrane whorls were rarely seen in wild-type animals. We found defects in three of nine animals; specifically, 3 of 120 body wall muscles, 1 of 9 anal depressor muscles, and 2 of 290 neuronal cell bodies had membrane whorls. In a survey of available prints of several hundred fixed animals with various genotypes, only one wild type, an animal incubated in colloidal gold before fixation, showed numerous whorls in muscles, neurons, and other cells in the head.
Cells next to the dying touch cells in ZB7 animals occasionally showed some pathology. In the vicinity of the ruptured late degeneration PVM cell, the hypodermal cytoplasm was damaged where the cytoplasmic contents of PVM had spilled (Fig. 7A).
Defects in neighboring cells also were seen when PVM cells had not lysed. For example, hypodermis, muscle, and neurons near the two early degeneration stage PVM cells showed minor local membrane infoldings or whorls that could be secondary responses to touch cell degeneration. A mild pathology (small membrane whorls and rare small vacuoles), which looked very similar to the earliest signs of toxicity in the touch neurons, was seen in cells that never contacted the touch cells in ZB7 animals. Although these defects may result from ectopic low-level expression of the mec-4(u231) transgene, some or all of this pathology may be attributable to treatment of the animals: 34 of 79 body wall muscles and 29 of 320 neurons in ZB7 animals mounted for light microscopy before fixation had whorls or vacuoles, whereas 4 of 75 body wall muscles and 8 of 200 neuronal cell bodies or axons in animals that were not mounted had them.
Mild pathology, usually membrane whorls and the occasional vacuole, also was seen in electron micrographs of several different cells in_deg-1(u38)_ animals. These cells included neurons (e.g., some of the PHA chemosensory neurons; Fig.8A), muscle cells, hypodermal cells, and other support cells. In the affected muscles, which included the anal depressor muscle (n = 2 of 3), dorsal body wall muscles (n = 5 of 13), and ventral body wall muscles (n = 3 of 12), membrane whorls were concentrated at muscle arms, muscle bellies, and distal extremities (Fig.8B,C). Because these animals also were prepared for light microscopy before fixation, these ectopic signs of degeneration may have resulted from the handling of the animals. However, because empty vacuoles, which are not associated with cell deaths, have been seen by light microscopy throughout the animals (Chalfie and Wolinsky, 1990; García-Añoveros, 1995), some of the whorls and vacuoles probably result from the expression of the toxic_deg-1_ protein.
Fig. 8.
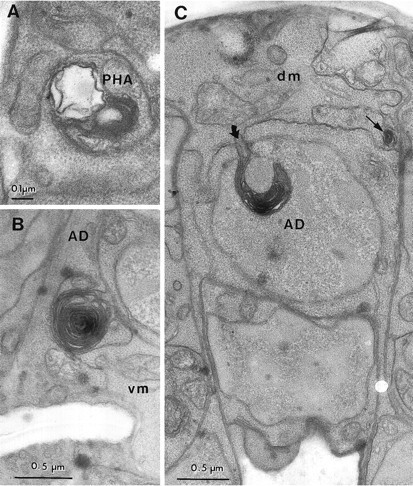
Toxic changes in neurons and muscles in a_deg-1(u38)_ larva. A, A membrane whorl and vacuole were found in a PHA dendrite in the phasmidial nerve. Similar whorls and vacuoles were found along the length of both_PHA_ dendrites in this animal, but not in two other animals scored in serial thin sections. B, A membrane whorl is located at the boundary of the anal depressor muscle (AD) and a ventral body wall muscle (vm).C, Involution of the plasma membrane (curved arrow) in the anal depressor muscle (AD) has caused the insertion of membrane into the muscle cell nucleus (labeled with the cell name). The thin arrow indicates a smaller membrane whorl at the junction of the depressor muscle and a dorsal body wall muscle (dm).
DISCUSSION
The progression of degenerative cell death
The degenerative death induced by gain-of-function alleles of the_C. elegans_ degenerin genes mec-4 and_deg-1_ proceeds in a reproducible series of steps (summarized in Fig. 9). The earliest degenerative events are small infoldings of the plasma membrane. (More rarely, small swellings of the ER also occur.) In both mec-4 and deg-1 mutants the sites of the infoldings are distributed unevenly in the affected cells. In neurons the highest density of the infoldings was on the growing neurites, whereas in muscle cells infoldings were concentrated on the muscle bellies and muscle arms, areas specialized for cell–cell communication. We do not know the subcellular localization of the degenerin channels, but the infoldings could identify the positions of the channels. Alternatively, the high surface-to-volume ratio in these regions of the cell could render them more vulnerable to toxic insult.
Fig. 9.
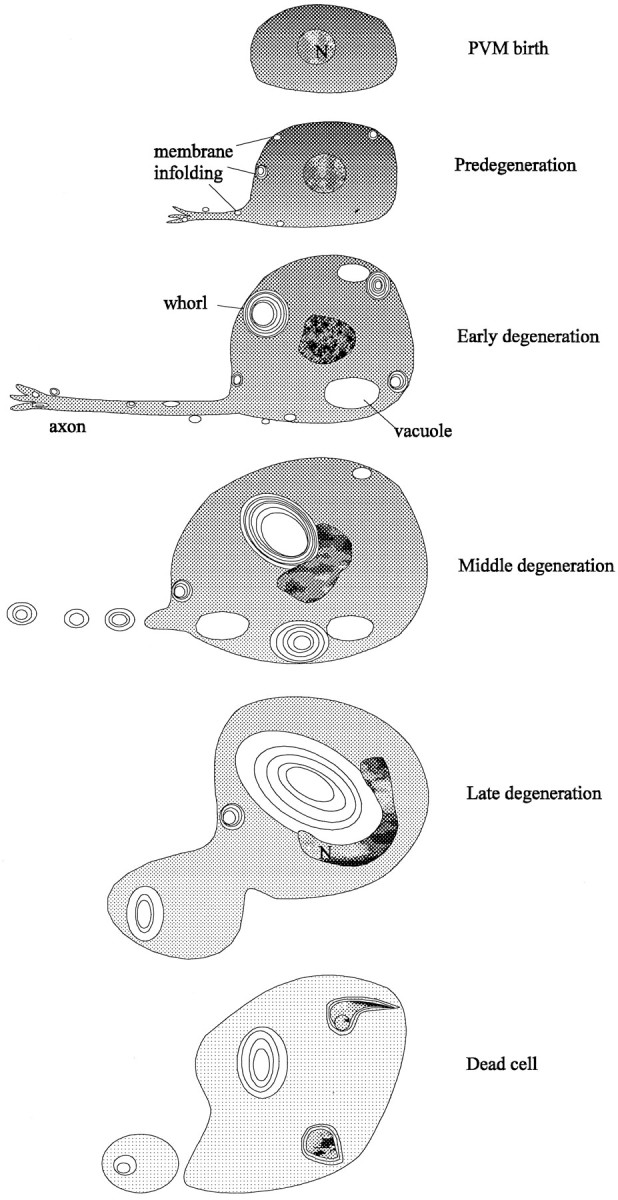
Stages in degenerin-induced cell death. Successive stages in PVM degeneration are depicted from the production of the cell-to-cell lysis. The first signs of pathology are the production of membrane infoldings and whorls. The cytoplasm becomes progressively less electron-dense as whorls and vacuoles accumulate and enlarge. These vacuoles deform the nucleus and dislocate it toward the plasma membrane. The axon seems to be disrupted by the middle degeneration stage. Mitochondria, Golgi, and endoplasmic reticulum, which are unaffected until late stages in degeneration, are not depicted.
The number of active sites of infolding in the PVM cell increases from 2–5 per cell in the predegeneration stage to 15–22 in the early degeneration stage and then falls to 8–10 in the middle degeneration stage and none in the late degeneration stage. Because cells are dominated by a few very large whorls at later times, the infoldings may coalesce into the whorls with increasing numbers of layers. The whorls also may merge with each other, because neighboring whorls are sometimes surrounded by continuous outer shells. Although the layers of the whorls are packed tightly in early degeneration cells, they are packed more loosely as cell death progresses.
The proximity of early vacuoles to the infoldings at the cell surface suggests that vacuoles, like the whorls to which they are connected, originate from infoldings of plasma membrane. Because a few very large vacuoles are found in later stages, they may enlarge or coalesce from smaller vacuoles. If degenerin channels are concentrated in these membranes, as seems likely, the dramatic swelling seen in the dying cells could result from open channels creating osmotic differences between the internal and external compartments. The onion skin structure of the whorls would allow for the formation of an osmotic difference. Growing vacuoles and, perhaps, membrane whorls may contribute to the growing space next to the nucleus observed by light microscopy in early stage degenerations.
Although defects in the plasma and ER membranes are found early, alterations in nuclei and mitochondria occur late. Changes in the nuclei begin at the middle degeneration stage: chromatin progressively condenses as the nucleus invaginates, moves (or is displaced) toward the plasma membrane, and appears to fragment. In several cases, the electron microscopy data suggest that nuclear displacement results from the growth of a very large cytoplasmic vacuole. Mitochondria remain intact through early and middle stages, although occasional mitochondria show increased outer density at the earliest stages. Cristae show some swelling in middle and late-stage cells. Cytoplasmic degradation occurs gradually throughout the period of degeneration, perhaps by the activation of endogenous hydrolases (cf. Clarke, 1990).
In the final stages of cell death organelles disappear, and only a few whorl remnants persist within the intact cell. We do not know whether, as seen with one late-stage PVM cell, the cell membrane usually ruptures as degeneration proceeds, but cell rupture could result in the sudden cell shrinkage that we saw by light microscopy in late-stage degenerations. The damage to surrounding cells seen in the ZB7 animals could be from the spillage of cell contents or from interactions with released membrane whorls. In any event, the dead cells eventually disappear, and serial thin sections of several older larvae show that no cellular debris remains (data not shown). This disappearance seems to require genes that also are needed for the engulfment of programmed cell deaths (Ellis et al., 1991); touch cell corpses persist many hours longer in a_ced-7_;ced-5;mec-4(u231) strain than in a strain with only the mec-4(u231) mutation (S. Chung and M. Driscoll, unpublished observations). Two of the dying cells in our study, a middle degeneration AVM cell and a late degeneration PVM cell, appeared to be engulfed by the surrounding hypodermis.
Degeneration pathology is dependent on degenerin gene dosage
Degenerin-induced cell death can be attributed to the expression of a toxic product. The dosage of the abnormal gene affects the onset of the cell death, e.g., the PVM and AVM deaths occur earlier in ZB7 animals than in mec-4(u231) mutants. Dosage of the toxic product also seems to influence the kinetics of degeneration. In ZB7 animals swollen touch receptor neurons disappear more quickly than they do in mec-4(u231) animals. Dosage also affects the onset of cell death of the PVC cell in deg-1(u38) mutants; cells die earlier in homozygous animals than in heterozygous animals (Chalfie and Wolinsky, 1990). Expression differences also may explain why the PVC cells in deg-1 animals die after many of the other affected cells die, because very low expression of the _deg-1lacZ_reporter fusions was seen in these cells. Although some timing differences distinguish touch receptor degeneration in ZB7 animals from PVC degeneration in deg-1(u38) animals, the same cellular events occur in the same sequence in both.
mec-4 normally is expressed in the six touch receptor neurons, a restricted pattern of expression observed even when a_mec-4lacZ_ fusion gene is present in multiple copies (Mitani et al., 1993). Thus it was surprising that membranous whorls were seen in scattered additional neurons and in hypodermis in the ZB7 strain. These abnormalities may result from leaky expression of the_mec-4(gf)_ array in inappropriate cells or may be a handling artifact. Similarly, several cells appeared abnormal but did not die in the deg-1 animals. Some of these cells may survive because they produce insufficient amounts of the toxic degenerin product. Other cells, however, such as the head muscle cells that expressed high levels of deg-1 reporter fusions (and that appear to accumulate empty vacuoles as seen by light microscopy), may live because they form fewer toxic channels (because they use different combinations of channel subunits) or because they can better compensate for the toxicity.
Similarities and differences with programmed cell death
Programmed cell death (Robertson and Thomson, 1982) and degenerin-induced cell death (this paper) progress in distinctly different ways in C. elegans. In programmed cell death the cell initially condenses, and its cytoplasm becomes very electron dense. Involution of the plasma membrane and the formation of vacuoles, early hallmarks of the degenerin-induced death, do not occur, although some loose whorls of internal and plasma membrane are found in the later stages of programmed cell death. In the late stages of programmed cell death, mitochondria can be distorted and engulfed by vacuoles, but such figures are not seen in the degenerating cells. Finally, the production of a prominent nucleolus seen in programmed cell death is not seen in degenerin-induced death.
Despite many differences, programmed cell death and degenerin-induced death share some features. Both cell deaths are characterized by the distortion of the nucleus, the condensation of chromatin (although condensation occurs less in the degenerin-induced deaths), the invagination of the initially spherical nucleus, and the eventual fragmentation of the nucleus. Both sets of dying cells are engulfed by the surrounding hypodermis. These similarities hint that some steps in both death processes might be shared. As far as the nuclear changes, however, any common mechanisms do not seem to require _ced-3_or ced-4 function.
Similarities also have been seen between vertebrate apoptosis and necrosis: both types of cell death exhibit similar changes in nuclear morphology (chromatin clumping, nuclear fragmentation) (Wyllie, 1981;Clarke, 1990), and cells dying in both ways express common antigens (Fernandez et al., 1994). We also note that the membranous vacuoles we describe here are somewhat similar to the membrane blebbing and prominent vacuolization found in some cases of apoptosis (Martin et al., 1988).
Inappropriate channel activity as a cause of cell pathology and death
Mutations near the putative pore-forming region or in an extracellular regulatory region of C. elegans degenerin proteins cause degenerative cell death (Driscoll and Chalfie, 1991;Huang and Chalfie, 1994; García-Añoveros et al., 1995;Shreffler et al., 1995). In addition, a mutation in the pore-forming region of a C. elegans acetylcholine receptor α subunit, encoded by the gene deg-3, results in a cell death that is morphologically similar to the degenerin-induced death (Treinin and Chalfie, 1995). Because of the location of the degeneration-inducing amino acid substitutions, we have hypothesized that channel hyperactivation or altered ionic conductance leads to cell degeneration by producing inappropriate ion influx or osmotic imbalance. Such changes in channel properties have been seen when the similar mammalian protein, MDEG, is mutated to have the dominant, toxic substitutions found in the C. elegans proteins (Waldmann et al., 1996).
This model for degenerin-induced death shares several features with that proposed for excitotoxic cell death as occurs in ischemia, hypoxia, epilepsy, and, possibly, neurodegenerative disease. In excitotoxic cell death, excess excitatory amino acids binding to glutamate receptors stimulate channel opening and result in excess Na+ and Ca2+ influx. This influx leads to the swelling and death of susceptible neurons (Choi, 1988). Excitotoxic death and degenerin-induced death share some, but not all, ultrastructural features. Acute excitotoxic death in culture and_in vivo_ is accompanied by vacuolation, often from Golgi cisternae or ER (Olney et al., 1974; Hajos et al., 1986), although dramatic plasma membrane infolding is not always seen (although seeChung et al., 1990). As in degenerin-induced deaths, the cytoplasm becomes highly electron-lucent with marked swellings of dendrites and somata, and the plasma membrane ruptures (Olney, 1971; Sperk et al., 1983). Mitochondrial cisternae may swell at advanced stages of degeneration (Olney et al., 1974; Sperk et al., 1983).
Increased ion channel open probability and osmotic imbalance can provoke endocytosis and vacuolation in other systems. For example, Mauthner cell giant synapses accumulate membrane cisternae or whorls and small vacuoles that follow increased endocytosis after direct electrical stimulation, which overwhelms homeostasis during recycling of synaptic vesicle membranes (Model et al., 1975). Treatment with the Ca2+ ionophore A23187 or the Na+ ionophore amphotericin B, which affects membrane permeability, induces “autophagic” cell death characterized by swollen vacuoles, with or without membranous lamellae (Trump and Berezesky, 1985; Clarke, 1990). Infolding of the plasma membrane and formation of vacuoles occur when cultured Aplysia neurons are placed in hypotonic media (Fejtl et al., 1995), and infusion of hypertonic media into the carotid artery can cause necrotic death in rat brains (Salahuddin et al., 1988). Thus, the observed intracellular changes in dying C. elegans cells could reflect a common response to osmotic insult.
Defective ion channels underlie several autosomal dominant muscle diseases in mammals, including hyperkalemic periodic paralysis [HPP(+)] (caused by mutations in the SCNA sodium channel) (Ricker et al., 1994; Sansone et al., 1994; Hudson et al., 1995) and hypokalemic periodic paralysis [HPP(−)] (caused by mutations in CACNL1A3, the dihydropyridine receptor calcium channel) (Boerman et al., 1995; Elbaz et al., 1995). Although the familial myotonias are not characterized by cell death, affected muscle cells swell considerably and, in some instances, exhibit pathology similar to that seen in_mec-4(gf)_ and deg-1(gf) animals. For example, in human HPP(−), membrane infolding leads to endocytosis of extracellular fluid and an applied peroxidase tracer into large intracellular vacuoles and prominent swelling (Engel, 1970). As the disease progresses, small vacuoles continually form at the plasma membrane and fuse into a larger central vacuole within the muscle cell. Vacuolation is restricted to the sarcoplasmic reticulum and possibly the t-tubules. Some larger membrane whorls are associated with the t-tubules. Similar muscle swelling originating at the sarcoplasmic reticulum is found in HPP(+) in humans and in horses (Spier et al., 1990). In addition, mutant Na+ channels from HPP(+) individuals display uncharacteristically long open times (Cannon et al., 1995). The lack of cell death in HPP(+) and HPP(−) suggests that muscle cell homeostasis can override the primary defect, perhaps attributable to the relatively large cell size. In our study of degenerin-induced death, muscles also seem to be less susceptible to swelling deaths than neurons are. As in our study, the degree of pathology correlates with the dosage of the mutant channels (Zhou et al., 1994). Because these conditions involve several different cations, the central problem may be one of osmotic balance rather the nature of the specific ions that flow through the mutant channels.
In weaver mutant mice a defective potassium channel (GIRK2) with altered gating and ion selectivity leads to selective cell death in the cerebellum, dentate gyrus, olfactory bulb, and germ line (Kofugi et al., 1995, 1996; Patil et al., 1995; Verina et al., 1995;Slesinger et al., 1996). Cytologically, the dying cells appear similar to the degenerations we see. Dying granule cells are swollen and vacuolated and sometimes show membranous lamellae within the vacuoles (Rakic and Sidman, 1973a). Dying Bermann glial cells also swell and vacuolate and have electron-lucent cytoplasms (Rakic and Sidman, 1973b). In the mutants, cells die more often and more rapidly with increased gene dosage, i.e., homozygotes are more severely affected than heterozygotes. In addition smaller neurons are more likely to die than larger neurons are. Thus, in many respects, the _weaver_and degenerin mutations produce similar effects.
A common theme in degenerative cell death?
A common feature of degenerin-induced death and the other conditions we have discussed is the production of membrane infoldings, membrane whorls, and vacuoles. These common features suggest that similar mechanisms may underlie several forms of cell pathology. We suggest that, in instances of membrane damage, the cell compensates by enhanced membrane cycling to dilute or to sequester harmful components localized to various membranes and to translocate them to degradative vacuoles. Such a surveillance mechanism may allow a variety of membranes to eliminate inappropriate, malfunctioning, or old components under normal conditions. In the cases of the degenerin-induced deaths, and perhaps other forms of neurodegeneration, this homeostatic mechanism is overwhelmed.
Footnotes
This work was supported by National Institutes of Health Grant GM34775 to M.C. and National Science Foundation Grant IBN 92-0945 to M.D. We thank Jian Xue for determining the copy number of_mec-4(u231)_ in ZB7, Eileen German for photography, and Laura Hall for help with Figure 9.
Correspondence should be addressed to Dr. Martin Chalfie, Department of Biological Sciences, 1012 Fairchild, Columbia University, 1212 Amsterdam Avenue, New York, NY 10027.
Dr. García-Añoveros’ present address: Department of Neurobiology, Harvard Medical School, Massachusetts General Hospital, Wellman 414, Fruit Street, Boston, MA 02114.
REFERENCES
- 1.Boerman RH, Ophoff RA, Links TP, van Eijk R, Sandkuijl LA, Elbaz A, Vale-Santos JE, Wintzen AR, van Deutekom JC, Isles DE, Fontaine B, Padberg GW, Frants RR. Mutation in DHP receptor alpha 1 subunit (CACNL1A3) gene in a Dutch family with hypokalemic periodic paralysis. J Med Genet. 1995;32:44–47. doi: 10.1136/jmg.32.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canessa CM, Horisberger J-D, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature. 1993;361:467–470. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]
- 4.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger J-D, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 5.Cannon SC, Hayward LJ, Beech J, Brown RH., Jr Sodium channel inactivation is impaired in equine hyperkalemic periodic paralysis. J Neurophysiol. 1995;73:1892–1899. doi: 10.1152/jn.1995.73.5.1892. [DOI] [PubMed] [Google Scholar]
- 6.Chalfie M, Au M. Genetic control of differentiation of the Caenorhabditis elegans touch receptor neurons. Science. 1989;243:1027–1033. doi: 10.1126/science.2646709. [DOI] [PubMed] [Google Scholar]
- 7.Chalfie M, Sulston J. Developmental genetics of the mechanosensory neurons of Caenorhabditis elegans. Dev Biol. 1981;82:358–370. doi: 10.1016/0012-1606(81)90459-0. [DOI] [PubMed] [Google Scholar]
- 8.Chalfie M, Wolinsky E. The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature. 1990;345:410–416. doi: 10.1038/345410a0. [DOI] [PubMed] [Google Scholar]
- 9.Chalfie M, Driscoll M, Huang M. Degenerin similarities. Nature. 1993;361:504. doi: 10.1038/361504a0. [DOI] [PubMed] [Google Scholar]
- 10.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 11.Chung SK, Cohen RS, Pfaff DW. Transneuronal degeneration in the midbrain central gray following chemical lesions in the ventromedial nucleus: a qualitative and quantitative analysis. Neuroscience. 1990;38:409–426. doi: 10.1016/0306-4522(90)90038-6. [DOI] [PubMed] [Google Scholar]
- 12.Clarke PGH. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 13.Driscoll M, Chalfie M. The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature. 1991;349:588–593. doi: 10.1038/349588a0. [DOI] [PubMed] [Google Scholar]
- 14.Elbaz A, Vale-Santos J, Jurkatt-Rott K, Lapie P, Ophoff RA, Bady B, Links TP, Piussan C, Vila A, Monnier N, Padberg GW, Abe K, Feingold N, Guimares J, Wintzen AR, van der Hoeven JH, Saudubray JM, Grunfeld JP, Lenoir G, Nivet H, Echenne B, Frants RR, Fardeau M, Lehmann-Horn F, Fontaine B. Hypokalemic periodic paralysis and the dihydropyridine receptor (CACNL1A3): genotype/phenotype correlations for two predominant mutations and evidence for the absence of a founder effect in 16 Caucasian families. Am J Hum Genet. 1995;56:374–380. [PMC free article] [PubMed] [Google Scholar]
- 15.Ellis H, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 16.Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 17.Engel AG. Evolution and content of vacuoles in primary hypokalemic periodic paralysis. Mayo Clin Proc. 1970;45:774–814. [PubMed] [Google Scholar]
- 18.Fejtl M, Szarowski DH, Decker D, Buttle K, Carpenter DO, Turner JN. Three-dimensional imaging and electrophysiology of live Aplysia neurons during volume perturbation: confocal light and high-voltage electron microscopy. J Microsc Soc Am. 1995;1:75–85. [Google Scholar]
- 19.Fernandez PA, Potello R, Rangini Z, Doupe A, Drexler HC, Yuan J. Expression of a specific marker of avian programmed cell death in both apoptosis and necrosis. Proc Natl Acad Sci USA. 1994;91:8641–8645. doi: 10.1073/pnas.91.18.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fire A. Integrative transformation of Caenorhabditis elegans. EMBO J. 1986;5:2673–2680. doi: 10.1002/j.1460-2075.1986.tb04550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fire A, Harrison SW, Dixon D. A modular set of lacZ fusion vectors for studying gene expression in C. elegans. Gene. 1990;93:189–198. doi: 10.1016/0378-1119(90)90224-f. [DOI] [PubMed] [Google Scholar]
- 22.García-Añoveros J (1995) Genetic analysis of degenerin proteins in Caenorhabditis elegans. PhD thesis, Columbia University, New York.
- 23.García-Añoveros J, Ma C, Chalfie M. Regulation of Caenorhabditis elegans degenerin proteins by a putative extracellular domain. Curr Biol. 1995;5:441–448. doi: 10.1016/s0960-9822(95)00085-6. [DOI] [PubMed] [Google Scholar]
- 24.Hajos F, Garthwaite G, Garthwaite JG. Reversible and irreversible neuronal damage caused by excitatory amino acid analogues in rat cerebellar slices. Neuroscience. 1986;18:417–436. doi: 10.1016/0306-4522(86)90163-6. [DOI] [PubMed] [Google Scholar]
- 25.Hall DH. Electron microscopy and 3D image reconstruction. In: Epstein HF, Shakes DC, editors. C. elegans: modern biological analysis of an organism, Vol 48, Methods in cell biology. Academic; San Diego: 1995. pp. 395–436. [DOI] [PubMed] [Google Scholar]
- 26.Hedgecock EM, Sulston JE, Thomson JN. Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science. 1983;220:1277–1279. doi: 10.1126/science.6857247. [DOI] [PubMed] [Google Scholar]
- 27.Hengartner M, Horvitz HR. C. elegans survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 28.Herman RK. Mosaic analysis of two genes that affect nervous system structure in Caenorhabditis elegans. Genetics. 1987;116:377–388. doi: 10.1093/genetics/116.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong K, Driscoll M. A transmembrane domain of the putative channel subunit MEC-4 influences mechanotransduction and neurodegeneration in C. elegans. Nature. 1994;367:470–473. doi: 10.1038/367470a0. [DOI] [PubMed] [Google Scholar]
- 30.Huang M, Chalfie M. Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans. Nature. 1994;367:467–470. doi: 10.1038/367467a0. [DOI] [PubMed] [Google Scholar]
- 31.Hudson AJ, Ebers GC, Bulman DE. The skeletal muscle sodium and chloride channel diseases. Brain. 1995;118:547–563. doi: 10.1093/brain/118.2.547. [DOI] [PubMed] [Google Scholar]
- 32.Kramer JM, Johnson JJ. Analysis of mutations in the sqt-1 and rol-6 collagen genes of Caenorhabditis elegans. Genetics. 1993;135:1035–1045. doi: 10.1093/genetics/135.4.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kofugi P, Davidson N, Lester HA. Evidence that neuronal G-protein-gated inwardly rectifying K+ channels are activated by Gβγ subunits and function as heterodimers. Proc Natl Acad Sci USA. 1995;92:6542–6546. doi: 10.1073/pnas.92.14.6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kofugi P, Hofer M, Millen KJ, Davidson N, Lester HA, Hatten ME. Functional analysis of the weaver mutant GIRK2 K+ channel and rescue of weaver granule cells. Neuron. 1996;16:941–952. doi: 10.1016/s0896-6273(00)80117-8. [DOI] [PubMed] [Google Scholar]
- 35.Manoil C. Analysis of protein localization by use of gene fusion with complementary properties. J Bacteriol. 1990;172:1035–1042. doi: 10.1128/jb.172.2.1035-1042.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mello CC, Kramer JM, Stinchcomb D, Ambros V. Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 1991;10:3959–3970. doi: 10.1002/j.1460-2075.1991.tb04966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitani S, Du H, Hall DH, Driscoll M, Chalfie M. Combinatorial control of touch neuron receptor expression in Caenorhabditis elegans. Development (Camb) 1993;119:773–783. doi: 10.1242/dev.119.3.773. [DOI] [PubMed] [Google Scholar]
- 39.Miura M, Zhu H, Rotello R, Hartweig EA, Yuan J. Induction of apoptosis in fibroblasts by IL-1β-converting enzyme, a mammalian homolog of the C. elegans cell death gene ced-3. Cell. 1993;75:653–660. doi: 10.1016/0092-8674(93)90486-a. [DOI] [PubMed] [Google Scholar]
- 40.Model PG, Highstein SM, Bennett MVL. Depletion of vesicles and fatigue of transmission at a vertebrate central synapse. Brain Res. 1975;98:209–228. doi: 10.1016/0006-8993(75)90002-5. [DOI] [PubMed] [Google Scholar]
- 41.Olney JW. Glutamate-induced neuronal necrosis in the infant mouse hypothalamus. An electron microscopic study. J Neuropathol Exp Neurol. 1971;30:75–90. doi: 10.1097/00005072-197101000-00008. [DOI] [PubMed] [Google Scholar]
- 42.Olney JW, Rhee V, Ho OL. Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 1974;77:507–512. doi: 10.1016/0006-8993(74)90640-4. [DOI] [PubMed] [Google Scholar]
- 43.Patil N, Cox DR, Bhat D, Faham M, Myers RM, Peterson AS. A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat Genet. 1995;11:126–129. doi: 10.1038/ng1095-126. [DOI] [PubMed] [Google Scholar]
- 44.Rakic P, Sidman RL. Sequence of developmental abnormalities leading to granule cell deficit in cerebellar cortex of weaver mutant mice. J Comp Neurol. 1973a;152:103–132. doi: 10.1002/cne.901520202. [DOI] [PubMed] [Google Scholar]
- 45.Rakic P, Sidman RL. weaver mutant mouse cerebellum: defective neuronal migration secondary to specific abnormality of Bergmann glia. Proc Natl Acad Sci USA. 1973b;70:240–244. doi: 10.1073/pnas.70.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ricker K, Moxley RT, III, Heine R, Lehmann-Horn F. Myotonia fluctuans: a third type of muscle sodium channel disease. Arch Neurol. 1994;51:1095–1102. doi: 10.1001/archneur.1994.00540230033009. [DOI] [PubMed] [Google Scholar]
- 47.Robertson AMG, Thomson JN. Morphology of programmed cell death in the ventral nerve cord of Caenorhabditis elegans larvae. J Embryol Exp Morphol. 1982;67:89–100. [Google Scholar]
- 48.Rothman SM. Excitotoxic neuronal death: mechanisms and clinical relevance. Semin Neurosci. 1994;6:315–322. [Google Scholar]
- 49.Salahuddin TS, Johansson BB, Kalimo H, Olsson Y. Structural changes in the rat brain after carotid infusions of hyperosmolar solutions. Acta Neuropathol (Berl) 1988;77:5–13. doi: 10.1007/BF00688236. [DOI] [PubMed] [Google Scholar]
- 50.Sansone V, Rotondo G, Ptacek LJ, Meola G. Mutation in the S4 segment of the adult skeletal sodium channel gene in an Italian paramyotonia congenita (PC) family. Ital J Neurol Sci. 1994;15:473–480. doi: 10.1007/BF02334608. [DOI] [PubMed] [Google Scholar]
- 51.Shreffler W, Margardino T, Shekdar C, Wolinsky E. The unc-8 and sup-40 genes regulate ion channel function in Caenorhabditis elegans motor neurons. Genetics. 1995;139:1261–1272. doi: 10.1093/genetics/139.3.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Slesinger PA, Patil N, Liao J, Jan YN, Jan LY, Cox DR. Functional effects of the mouse weaver mutation on G-protein-gated inwardly rectifying K+ channels. Neuron. 1996;16:321–331. doi: 10.1016/s0896-6273(00)80050-1. [DOI] [PubMed] [Google Scholar]
- 53.Sperk G, Lassmann H, Baran H, Kish SJ, Seitelberger H, Hornytiewícz O. Kainic acid-induced seizures: neurochemical and histopathological changes. Neuroscience. 1983;10:1301–1315. doi: 10.1016/0306-4522(83)90113-6. [DOI] [PubMed] [Google Scholar]
- 54.Spier SJ, Carlson GP, Holliday TA, Cardinet GH, III, Pickar JG. Hyperkalemic periodic paralysis in horses. J Am Vet Med Assoc. 1990;197:1009–1017. [PubMed] [Google Scholar]
- 55.Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- 56.Treinin M, Chalfie M. A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron. 1995;14:871–877. doi: 10.1016/0896-6273(95)90231-7. [DOI] [PubMed] [Google Scholar]
- 57.Trump BF, Berezesky IK. Cellular ion regulation and disease: a hypothesis. Curr Top Membr Transp. 1985;25:279–319. [Google Scholar]
- 58.Vaux DL, Weissman IL, Kim SK. Prevention of programmed cell death in Caenorhabditis elegans by human bcl-2. Science. 1992;258:1955–1957. doi: 10.1126/science.1470921. [DOI] [PubMed] [Google Scholar]
- 59.Verina T, Tang X, Fitzpatrick L, Norton J, Vogelweid C, Ghetti B. Degeneration of Sertoli and spermatogenic cells in homozygous and heterozygous weaver mice. J Neurogenet. 1995;9:251–265. doi: 10.3109/01677069509084161. [DOI] [PubMed] [Google Scholar]
- 60.Waldmann R, Champigny G, Bassilana F, Voilley N, Lazdunski M. Molecular cloning and functional expression of a novel amiloride-sensitive Na+ channel. J Biol Chem. 1995;270:27411–27414. doi: 10.1074/jbc.270.46.27411. [DOI] [PubMed] [Google Scholar]
- 61.Waldmann R, Champigny G, Voilley N, Lauritzen I, Lazdunski M. The mammalian degenerin MDEG, an amiloride-sensitive cation channel activated by mutations causing degeneration in Caenorhabditis elegans. J Biol Chem. 1996;271:10433–10436. doi: 10.1074/jbc.271.18.10433. [DOI] [PubMed] [Google Scholar]
- 62.Wasterlain CG, Shirasaka Y. Seizures, brain damage, and brain development. Brain Dev. 1994;16:279–295. doi: 10.1016/0387-7604(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 63.White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of Caenorhabditis elegans. Philos Trans R Soc Lond [Biol] 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- 64.Wyllie AH. Cell death: a new classification separating apoptosis from necrosis. In: Bowen ID, Lockshin RA, editors. Cell death in biology and pathology. Chapman and Hall; New York: 1981. pp. 9–34. [Google Scholar]
- 65.Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 66.Yuan JY, Horvitz HR. The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-h. [DOI] [PubMed] [Google Scholar]
- 67.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 68.Zhou J, Spier SJ, Beech J, Hoffman EP. Pathophysiology of sodium channelopathies: correlation of normal/mutant mRNA ratios with clinical phenotype in dominantly inherited periodic paralysis. Hum Mol Genet. 1994;3:1599–1603. doi: 10.1093/hmg/3.9.1599. [DOI] [PubMed] [Google Scholar]