The Actin-Severing Protein Gelsolin Modulates Calcium Channel and NMDA Receptor Activities and Vulnerability to Excitotoxicity in Hippocampal Neurons (original) (raw)
Abstract
Calcium influx through NMDA receptors and voltage-dependent calcium channels (VDCC) mediates an array of physiological processes in neurons and may also contribute to neuronal degeneration and death in neurodegenerative conditions such as stroke and severe epileptic seizures. Gelsolin is a Ca2+-activated actin-severing protein that is expressed in neurons, wherein it may mediate motility responses to Ca2+ influx. Primary hippocampal neurons cultured from mice lacking gelsolin exhibited decreased actin filament depolymerization and enhanced Ca2+ influx after exposure to glutamate. Whole-cell patch-clamp analyses showed that currents through NMDA receptors and VDCC were enhanced in hippocampal neurons lacking gelsolin, as a result of decreased current rundown; kainate-induced currents were similar in neurons containing and lacking gelsolin. Vulnerability of cultured hippocampal neurons to glutamate toxicity was greater in cells lacking gelsolin. Seizure-induced damage to hippocampal pyramidal neurons was exacerbated in adult gelsolin-deficient mice. These findings identify novel roles for gelsolin in controlling actin-mediated feedback regulation of Ca2+ influx and in neuronal injury responses. The data further suggest roles for gelsolin and the actin cytoskeleton in both physiological and pathophysiological events that involve activation of NMDA receptors and VDCC.
Keywords: cytochalasin, cytoskeleton, epileptic seizures, fura-2, knock-out mice, patch-clamp
The calcium ion is a key regulator of a variety of physiological processes in many different cell types, including neurons, in which it controls neurotransmitter release, synaptic plasticity, and growth cone motility (Augustine et al., 1987;Kater et al., 1988; Kennedy, 1989; Miller et al., 1989). Actions of Ca2+ on the polymerization state of microfilaments play important roles in regulating cell motility and vesicle trafficking in each of the just-mentioned processes (Cramer et al., 1994; Neely and Gesemann, 1994). Whereas transient elevations of intracellular Ca2+([Ca2+]i) mediate local modulation of the cytoskeleton, sustained elevations of [Ca2+]i are potentially toxic and lead to disruption of cytoskeletal components including actin filaments (Orrenius et al., 1992; Neely and Gesemann, 1994; Furukawa et al., 1995). Indeed, Ca2+ is implicated as an effector of neurodegeneration and death in disorders ranging from cerebral ischemia to epileptic seizures to Alzheimer’s disease (Mattson et al., 1993a;Wasterlain et al., 1993; Choi, 1995). Recent findings suggest that, in addition to their roles in regulating cell structure and motility, actin filaments may function in the modulation of ion channel function. The evidence is based on pharmacological data showing that the actin filament-disrupting agent cytochalasin D can alter ion flux through Na+ channels (Cantiello et al., 1991; Undrovinas et al., 1995; Berdiev et al., 1996), voltage-dependent calcium channels (VDCC) (Johnson and Byerly, 1993), and NMDA receptor channels (Rosenmund and Westbrook, 1993). Whether actin normally plays roles in regulating Ca2+ influx in physiological and pathophysiological settings is unknown.
The actin cytoskeleton has been most intensively studied in relation to its central roles in modulating cell motility and secretion (Bernstein and Bamburg, 1989; Lee et al., 1993; Sobue, 1993; Condeelis, 1994). An array of proteins that bind actin has been identified (for review, seeHartwig and Kwiatkowski, 1991). One broad class of proteins with F-actin binding and severing activity includes gelsolin (Yin and Stossel, 1979; Yin et al., 1981a), villin (Bretscher and Weber, 1980), and adseverin (Maekawa et al., 1989). Another class of actin-binding proteins that bind actin monomers and exhibit relatively weak actin-severing activity includes actin-depolymerizing factor (Bamburg et al., 1980; Morgan et al., 1993) and cofilin (Yonezawa et al., 1985). Gelsolin is a 93 kDa cytosolic protein that severs actin filaments when it is activated by Ca2+; after cleaving actin filaments, gelsolin remains tightly bound to the actin filament barbed end (Yin et al., 1981b; Cooper et al., 1987; Janmey and Stossel, 1987). Gelsolin is widely expressed in mammalian tissues including the nervous system (Kwiatkowski et al., 1988a,b); in the developing nervous system gelsolin is particularly concentrated in neuronal growth cones (Tanaka et al., 1993). Previous studies have shown that Ca2+influx, including that induced by membrane depolarization, can induce actin depolymerization in neurons (Bernstein and Bamburg, 1985;Furukawa et al., 1995), a process that likely involves gelsolin activity. We recently generated mice genetically deficient in gelsolin and documented enhanced stress fiber formation and altered motility in fibroblasts from these mice (Witke et al., 1995). In the present study we used gelsolin knock-out mice to test the hypothesis that, by inducing actin depolymerization, gelsolin serves as an endogenous modulator of Ca2+ influx through NMDA receptors and VDCC in hippocampal neurons and thereby protects neurons against excitotoxic Ca2+ overload.
MATERIALS AND METHODS
Gelsolin knock-out mice. Methods for generation of mice lacking gelsolin (G−/−) were described previously (Witke et al., 1995). The G−/− mice exhibit no overt phenotype and reproduce normally but exhibit several alterations in rapid motility and structure of platelets, leukocytes, and fibroblasts. Preliminary studies revealed no alterations in brain size or gross or microscopic histological features in Nissl-stained brain sections of G−/− mice. All experiments were performed using littermates from G+/− × G+/− crosses in a mixed Sv129–BALB/c background.
_Hippocampal cell cultures and analysis of neuronal survival._Hippocampal cell cultures were established from 16-d-old embryos using methods essentially identical to those used in our previous studies (Mattson et al., 1988a; Bruce et al., 1996). Hippocampi from each embryo were dissociated by trypsinization and trituration and plated into culture dishes; DNA from the body of each embryo was isolated and used for PCR-based genotyping. Each embryo and the hippocampal cultures from that embryo were given an identification number; experiments were performed without knowledge of genotype, and the code was broken after the experiments. Cells were grown in polyethyleneimine-coated plastic or glass bottom 35 mm culture dishes containing Eagle’s Minimum Essential Medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Life Technologies, Gaithersburg, MD), 20 mmKCl, and 1 mm pyruvate. The atmosphere consisted of 6% CO2/94% room air and was maintained near saturation with water. Experiments were performed in cultures that had been maintained for 8–10 d. Glutamate (Sigma, St. Louis, MO) was prepared as a 200× stock in saline. Neuronal survival was quantified as described previously (Mattson et al., 1989; Furukawa et al., 1995). Briefly, viable neurons in premarked microscope fields (10× objective) were counted before and 24 hr after exposure to vehicle or glutamate. Neuronal viability was assessed by morphological criteria; cells with intact neurites of uniform diameter and a soma with a smooth round appearance were considered viable, whereas neurons with fragmented neurites and a vacuolated or swollen soma were considered nonviable. Analyses were done without knowledge of the treatment history of the cultures. In previous studies of similar hippocampal cultures we found that glutamate induces concentration-dependent (5–200 μm) death of neurons (Mattson et al., 1989), and that cytochalasin D was effective in protecting against cell death across the glutamate concentration range (Furukawa et al., 1995). For most of the experiments in the present study, we therefore used a glutamate concentration of 100 μm.
Phalloidin staining, immunocytochemistry, and Western blot analyses. Methods for phalloidin staining and immunocytochemistry are detailed in our previous studies (Furukawa et al., 1995; Mattson et al., 1997). Briefly, cells were fixed for 30 min in a solution of 4% paraformaldehyde in PBS, and membranes were permeabilized by incubation for 5 min in a solution of 0.2% Triton X-100 in PBS. For phalloidin staining cells were incubated for 30 min in PBS containing 0.005 U/ml fluorescein–phalloidin (Molecular Probes, Eugene, OR) and washed three times with PBS, and an antifade solution containing 100 μm propyl gallate was added. Images of phalloidin fluorescence were acquired using a confocal laser scanning microscope with a 60× oil immersion objective. For immunostaining, cultures were preincubated 10 min in blocking serum (normal horse serum; 15 μl/ml PBS), primary anti-gelsolin antibody (mouse monoclonal generated against a C-terminal domain of human gelsolin; Transduction Laboratories, Lexington, KY) was added to a final dilution of 1:1000, and cells were incubated overnight at 4°C. Cells were then sequentially incubated in solutions of PBS containing biotinylated anti-mouse secondary antibody, avidin–peroxidase complex, and diaminobenzidine (Vector Laboratories, Burlingame, CA) according to the manufacturer’s protocol. Photographs of phase-contrast and bright-field images of immunostained cells were taken using a 40× objective. For Western blot analyses, proteins were separated by SDS-PAGE, transferred to a nitrocellulose sheet, and immunoreacted with primary antibody. The blots were further processed using HRP-conjugated secondary antibody and a chemiluminescence detection method (Amersham, Arlington Heights, IL). Primary antibodies included anti-gelsolin (1:2000 dilution), mouse monoclonal antibody against α-actin (Sigma; 1:500 dilution), and chicken polyclonal antibody against human cofilin (1:700) that was generated in the laboratory of one of the authors (D.J.K.).
Electrophysiological analyses and calcium imaging methods. Whole-cell patch clamp analyses of currents through NMDA receptors and VDCC were performed using a patch-clamp amplifier (Axopatch-1D) and methods similar to those described previously (Hamill et al., 1983; Furukawa et al., 1996). The ionic composition of the external solution for voltage-dependent calcium currents was (in mm): 145 NaCl, 5 CsCl, 8 CaCl2, 10 glucose, 10 HEPES, and 0.0003 tetrodotoxin, pH 7.4. The external solution used for recording NMDA- and kainate-induced currents was (in mm): 150 NaCl, 5 KCl, 2 CaCl2, 10 glucose, 10 HEPES, and 0.01 glycine, pH 7.4. The internal solution for all experiments consisted of (in mm): 90_N_-methyl-d-glucamine, 30 CsCl, 20 tetraethylammonium chloride, 4 MgATP, 10 EGTA, and 10 HEPES pH 7.2. Intracellular free Ca2+ levels were quantified by ratiometric imaging of the fluorescent calcium indicator dye fura-2 (Molecular Probes) as described previously (Mattson et al., 1995). Briefly, cells were loaded with the acetoxymethylester form of fura-2 (30 min incubation in the presence of 10 μm fura-2) and imaged using a Zeiss AttoFluor system with a 40× oil objective. The average [Ca2+]i in individual neuronal cell bodies was determined from the ratio of the fluorescence emissions obtained using two different excitation wavelengths (334 and 380 nm). The system was calibrated using solutions containing either no Ca2+ or a saturating level of Ca2+ (1 mm) using the formula: [Ca2+]i = _K_d[(_R_ −_R_min)/(_R_max −_R_)](_F_0/_F_s).
_Kainate administration and quantification of neuronal injury_in vivo. These methods were essentially identical to those used previously (Bruce et al., 1996; Smith-Swintosky et al., 1996). Briefly, KA (0.3 μg in a volume of 0.5 μl) was injected unilaterally into dorsal hippocampus (dorsoventral, −2.0; mediolateral, +2.4; anteroposterior, −0.8 from bregma) of anesthetized mice. All mice administered KA exhibited seizures within the first hour after injection. Mice were killed 24 hr later and perfused transcardially with 4% paraformaldehyde. Coronal brain sections (30 μm) were cut on a freezing microtome and used for Nissl staining and immunohistochemistry. Nissl-positive undamaged neurons were counted in hippocampal regions CA1, CA3, and CA4/hilus (counts were made in four sections per brain). Cell counts were performed without knowledge of the genotype of the mice.
RESULTS
Reduced actin depolymerization in response to calcium influx in hippocampal neurons lacking gelsolin
Primary cultures of hippocampal neurons were established from embryos of mice lacking gelsolin (G−/−) and their heterozygous (G+/−) and wild-type (G+/+) littermates. Western blot analysis verified lack of gelsolin in G−/− mice and indicated a reduction in gelsolin levels in G+/− mice compared with wild-type littermates (Fig.1A). Immunocytochemistry of cultured embryonic hippocampal neurons showed gelsolin immunoreactivity in the neurites and cell bodies of G+/+ mice; no gelsolin immunoreactivity was present in G−/− neurons in culture (Fig. 1B). The gelsolin appeared to be concentrated in the margins of neuronal somata (Fig. 1B,arrows), suggesting association with the plasma membrane. Exposure of G+/+ hippocampal cultures to glutamate resulted in a decrease in the level of filamentous actin as assessed by confocal laser scanning microscope analysis of cells stained with fluorescent phalloidin (Fig. 2); the actin depolymerization induced by glutamate resulted from Ca2+ influx, because it did not occur in G+/+ neurons incubated in medium lacking Ca2+ (data not shown). In contrast, glutamate did not affect levels of phalloidin fluorescence in G−/− neurons (Fig. 2). Examination of the time course of the glutamate-induced decrease in phalloidin fluorescence in G+/+ neurons indicated that actin depolymerization occurred within 10 min of exposure to glutamate (Fig. 2C).
Fig. 1.
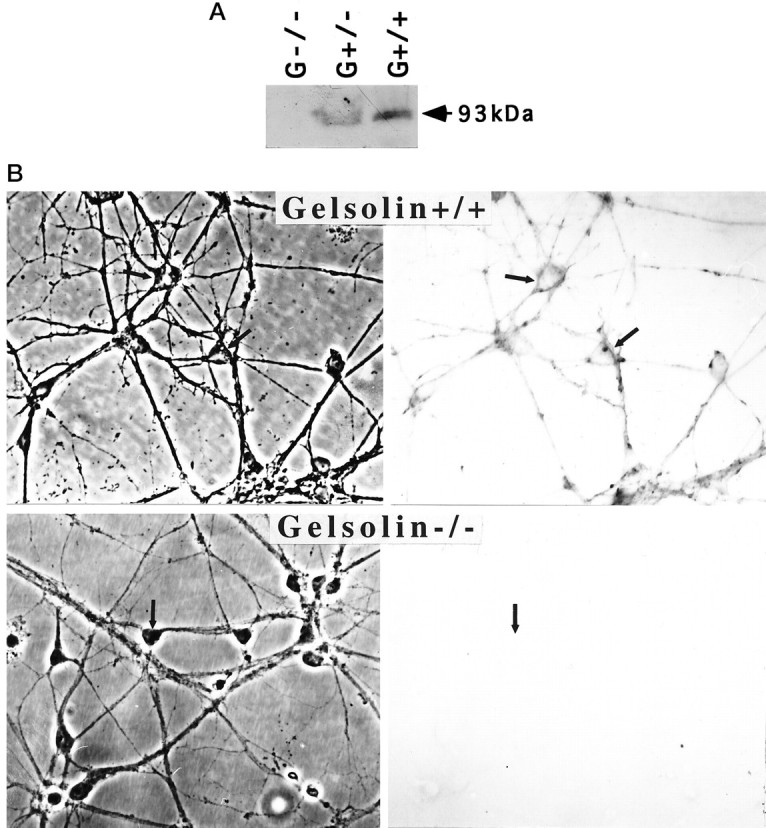
Characterization of gelsolin expression in brain and dissociated hippocampal cell cultures from wild-type and gelsolin knock-out mice. A, Proteins in hippocampal tissue homogenates from G+/+, G+/−, and G−/− mice (100 μg each) were subjected to electrophoresis and Western blot analysis with gelsolin antibody. B, Cultures of hippocampal cells from G+/+ and G−/− mice (9 d in culture) were immunostained with gelsolin antibody. Note immunoreactivity in cell bodies (e.g.,arrows) and neurites of neurons from G+/+ (wild-type) mice and lack of immunoreactivity in neurons from G−/− mice.
Fig. 2.

Neurons lacking gelsolin are resistant to glutamate-induced actin depolymerization. A, Cultures of G+/+ and G−/− hippocampal cells were exposed to either saline (Control) or 100 μm glutamate for 2 hr and were then stained with phalloidin–fluorescein. Confocal laser scanning microscope images of phalloidin fluorescence in hippocampal neurons show decreased levels of filamentous actin after exposure to glutamate in G+/+ cells but not in G−/− cells. B, Levels of phalloidin fluorescence in neuronal cell bodies were quantified in cultures of G+/+, G+/−, and G−/− cells that had been exposed for 2 hr to either saline (Control) or 100 μm glutamate. Values are mean ± SEM of determinations made in four separate cultures (20–30 neurons analyzed per culture). *p < 0.05; **p< 0.01 compared with corresponding control value (ANOVA with Scheffe’s post hoc tests). C, Cultured hippocampal neurons (G+/+) were exposed for increasing periods to 100 μm glutamate, and levels of phalloidin fluorescence in neuronal cell bodies were quantified. Values are the mean and SEM of determinations made in three separate cultures (20–30 neurons analyzed per culture).
The absence of overt alterations in brain development of G−/− mice, and the normal appearance of G−/− hippocampal neurons in culture suggested the possibility that there may be compensatory changes in the levels of other actin-binding proteins such as those in the actin-depolymerizing factor (ADF)–cofilin family (Bamburg et al., 1980; Hayden et al., 1993; Abe et al., 1996; Lappalainen and Drubin, 1997), or in levels of actin itself, in the G−/− mice. Moreover, actin-binding proteins in the ADF–cofilin family can inhibit binding of phalloidin to actin. To address the possibility that compensatory responses influenced the outcomes of our measurements, we performed immunoblot analyses of levels of actin and cofilin in brain tissue homogenates from G+/+, G+/−, and G−/− mice. Levels of actin were similar in brain tissue from mice of each genotype, and levels of cofilin were also unaffected by the absence of gelsolin (Fig.3).
Fig. 3.

Lack of effect of gelsolin genotype on levels of actin and cofilin. Proteins in brain tissue homogenates from G+/+, G+/−, and G−/− mice (50 μg for the actin blot and 100 μg for the cofilin blot) were subjected to SDS-PAGE, transferred to a nitrocellulose sheet, and immunoreacted with either an α-actin antibody (left) or an antibody against cofilin (right). The α-actin immunoreactive band was at ∼42 kDa, and the cofilin immunoreactive band was at ∼27 kDa. The cofilin immunoreactive band comigrated with human recombinant cofilin (data not shown).
Glutamate-induced calcium influx is enhanced in hippocampal neurons lacking gelsolin
Imaging of the calcium indicator dye fura-2 was used to compare [Ca2+]i responses to glutamate in G+/+, G+/−, and G−/− neurons (Fig. 4). The basal [Ca2+]i was essentially identical in neurons of all three gelsolin genotypes (∼120 nm). In G+/+ neurons glutamate induced a rapid elevation of [Ca2+]i to ∼520 nm, which then recovered to ∼300 nm during the subsequent 5–8 min of exposure (Fig. 4A). G+/− and G−/− neurons also showed a rapid rise in [Ca2+]i to levels similar to those seen in G+/+ neurons. However, in contrast to G+/+ neurons, the [Ca2+]i in G−/− neurons remained at ∼600 nm and did not recover (Fig.4A,B). The [Ca2+]iin G+/− neurons exposed to glutamate recovered to a level significantly lower than in G−/− neurons but higher than that of G+/+ neurons. Note that actin depolymerization was largely suppressed in G−/− neurons (Fig. 2) despite a large elevation of [Ca2+]i. The greater elevation of [Ca2+]i in G−/− neurons after exposure to glutamate was observed across a range of glutamate concentrations from 5 to 200 μm (data not shown). When G−/− neurons were pretreated with the actin-depolymerizing agent cytochalasin D before exposure to glutamate, the [Ca2+]i recovery response was similar to that observed in G+/+ neurons (Fig. 4B), indicating that actin depolymerization was sufficient to account for the different [Ca2+]i responses of neurons containing or lacking gelsolin.
Fig. 4.
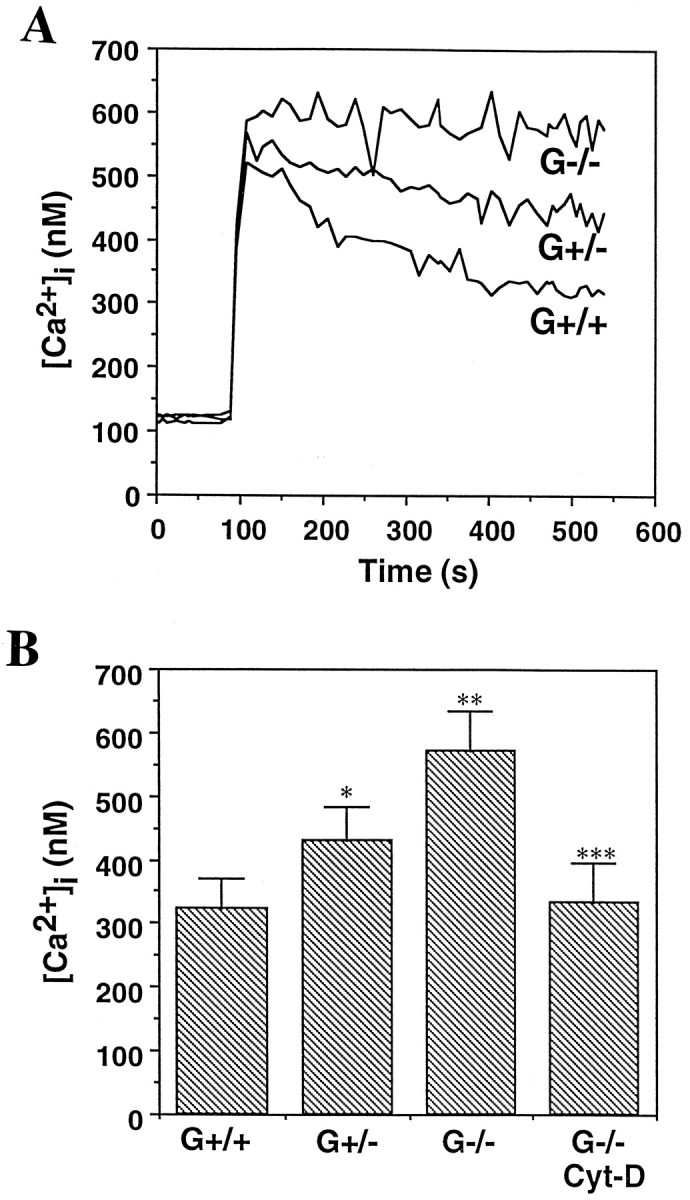
Calcium responses to glutamate are enhanced in hippocampal neurons lacking gelsolin. A, The [Ca2+]i was monitored before and after exposure to 100 μm glutamate in cell bodies of cultured hippocampal neurons from G+/+, G+/−, and G−/− mice.Traces represent the mean [Ca2+]i in 15–20 neurons.B, The sustained [Ca2+]i, measured 5 min after exposure to 100 μm glutamate, was quantified in hippocampal neurons of the different gelsolin genotypes. One set of G−/− cultures was pretreated with 100 nm cytochalasin D 1 hr before exposure to glutamate. Values represent the mean and SEM of determinations made in four to six separate cultures (15–25 neurons per culture). *p < 0.05; **p< 0.01 compared with G+/+ value; ***p < 0.01 compared with G−/− value (ANOVA with Scheffe’s post hoc tests).
Reduced rundown of voltage-dependent calcium current and NMDA current in gelsolin-deficient hippocampal neurons
Whole-cell patch-clamp analyses of voltage-dependent Ca2+ currents showed that both G+/+ and G−/− neurons exhibited currents of similar magnitude (Fig.5A). However, when the time courses of change in current amplitude were examined by applying a depolarizing step pulse every 10 sec for 400 sec, the G−/− neurons exhibited reduced current rundown compared with G+/+ neurons; the difference was highly significant (Fig. 5A). The rate of rundown of current through VDCC in G+/− neurons was intermediate to that of G+/+ and G−/− neurons. NMDA-induced currents also showed more attenuated rundown in G−/− neurons compared with G+/+ neurons when the currents were recorded every 2 min for 30 min; rundown rate of NMDA currents in G+/− neurons was intermediate to that of G+/+ and G−/− neurons (Fig. 5B). In contrast, kainate-induced currents were not different in G+/+, G+/−, and G−/− neurons (Fig.5C). As expected from previous studies (Johnson and Byerly, 1993; Rosenmund and Westbrook, 1993), cytochalasin D accelerated the rate of rundown of currents through both VDCC and NMDA receptors but did not affect kainate-induced currents (data not shown). When taken together with the calcium imaging data, our patch-clamp data indicate that, by inducing actin depolymerization, gelsolin acts to suppress Ca2+ influx through VDCC and NMDA receptors.
Fig. 5.
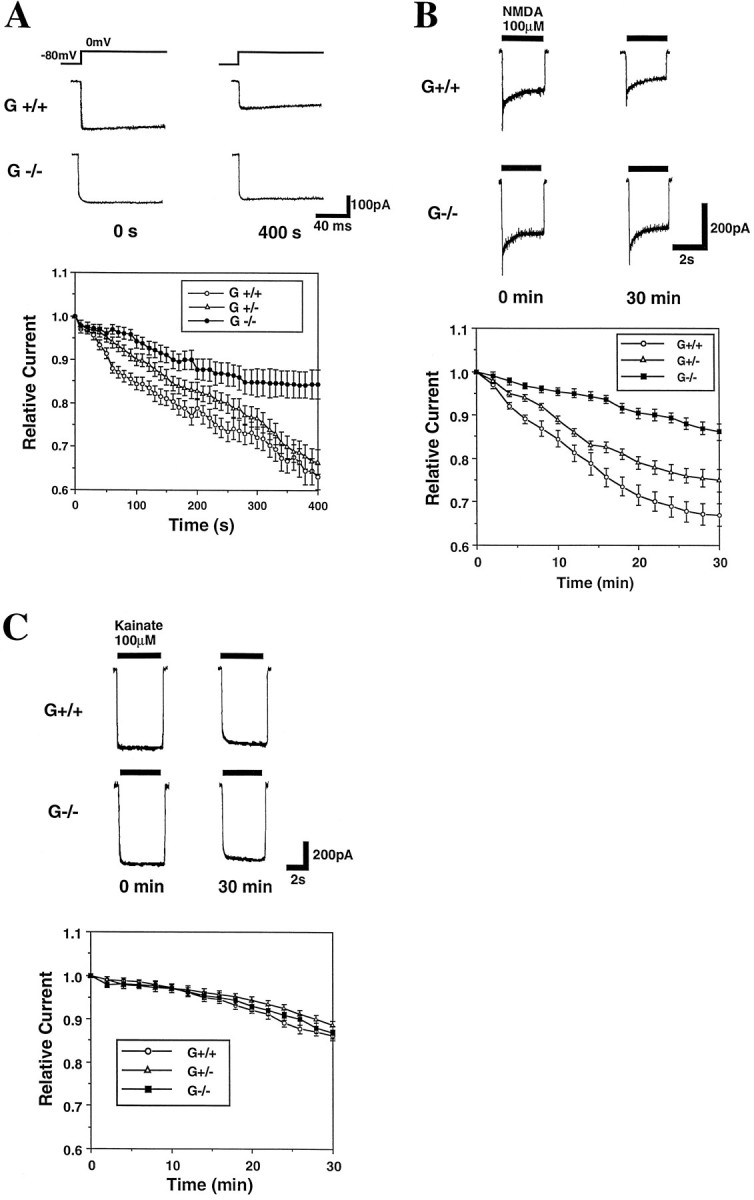
Rate of current rundown through VDCC and NMDA receptor channels is reduced in gelsolin-deficient hippocampal neurons. A, Whole-cell Ca2+ current was recorded in G+/+ and G−/− hippocampal neurons at 10 sec intervals. Top, Representative current recordings taken from a G+/+ neuron and a G−/− neuron at the onset of the experiment and 400 sec later. Bottom, Data from recordings of whole-cell Ca2+ currents in G+/+, G+/−, and G−/− neurons. Values are the ± SEM of determinations made in six to eight neurons. The difference in rate of current rundown was significantly less in the G−/− neurons compared with the G+/+ neurons (p < 0.001 by ANOVA). B, NMDA-induced current was recorded in G+/+ and G−/− hippocampal neurons at 2 min intervals. Top, Representative current recordings taken from a G+/+ neuron and a G−/− neuron at the onset of the experiment and 30 min later. Bottom, Data from recordings of whole-cell NMDA-induced currents in G+/+, G+/−, and G−/− neurons. Values are the mean ± SEM of determinations made in six to nine neurons. The difference in rate of current rundown was significantly less in the G−/− neurons compared with the G+/+ neurons (p < 0.001 by ANOVA). C, Kainate-induced currents were recorded in G+/+ and G−/− hippocampal neurons at 2 min intervals. Top, Representative current recordings taken from a G+/+ neuron and a G−/− neuron at the onset of the experiment and 30 min later. Bottom, Data from recordings of whole-cell kainate-induced currents in G+/+, G+/−, and G−/− neurons. Values are the mean ± SEM of determinations made in six to nine neurons.
Hippocampal neurons lacking gelsolin exhibit increased vulnerability to excitotoxicity
Excitotoxic neuronal death is mediated largely by Ca2+ influx through NMDA receptors and VDCC (Choi, 1995). We found that primary cultured hippocampal neurons from embryos lacking gelsolin are much more vulnerable to glutamate toxicity than are G+/+ neurons, and that G+/− neurons exhibit a level of vulnerability to excititoxicity intermediate to those of G+/+ and G−/− neurons (Fig.6A). Vulnerability of hippocampal neurons to excitotoxicity in vivo was examined using a model of seizure-induced injury in which KA is injected into the dorsal hippocampus (Bruce et al., 1996; Smith-Swintosky et al., 1996). KA primarily damages CA3 and CA4/hilus neurons, but it has little or no effect on CA1 neurons. The dose of KA used (0.3 μg) caused moderate damage to CA3 and CA4/hilus neurons, and no damage to CA1 neurons, in G+/+ mice (Fig. 6A,B). Significantly more neurons in CA3 and CA4/hilus were damaged by KA in G−/− mice compared with G+/+ mice; neuronal damage in G+/− mice was intermediate to that of G+/+ and G−/− mice. Moreover, a significant number of CA1 neurons were damaged by KA in G−/− mice, in contrast to G+/+ and G+/− mice (Fig. 6B).
Fig. 6.
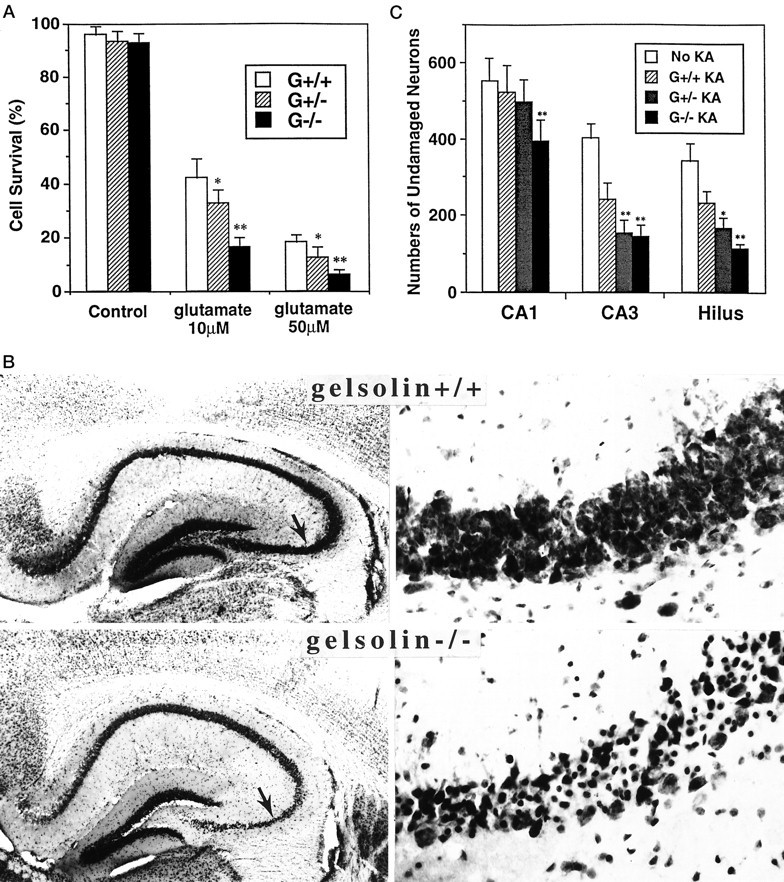
Hippocampal neurons lacking gelsolin exhibit increased vulnerability to excitotoxicity in cell culture and in vivo. A, Hippocampal cultures from G+/+, G+/−, and G−/− mice were exposed for 24 hr to saline (Control) or 100 μm glutamate, and neuronal survival was quantified. Values are the mean ± SEM of determinations made in four separate cultures. *p< 0.05; **p < 0.01 compared with corresponding value for G+/+ cultures (ANOVA with Scheffe’s post hoc_tests). B, Nissl-stained coronal sections of hippocampi from G+/+ and G−/− mice 24 hr after administration of KA into the dorsal hippocampus. The right panels are high-magnification views of the region of CA3 indicated by the_arrow in the corresponding low magnification micrograph. Note greater degeneration of CA3 neurons in G−/− compared with G+/+ mice. C, G+/+, G+/−, and G−/− mice received an injection of KA into the dorsal hippocampus and were killed 24 hr later. Numbers of undamaged neurons in regions CA1, CA3, and CA4/hilus of the injected hippocampus were counted. Additional cell counts were performed in the contralateral uninjected hippocampus (No KA). Values for mice of each genotype were not different and were therefore pooled. Values are the mean ± SEM of determinations made in six to eight mice. *p < 0.05; **p < 0.01 compared with corresponding value for G+/+ mice (ANOVA with Scheffe’s _post hoc_tests).
DISCUSSION
Calcium influx induced by a variety of stimuli, including membrane depolarization and activation of glutamate receptors, can induce actin depolymerization in neurons (Bernstein and Bamburg, 1985; Neely and Gesemann, 1994; Neely et al., 1995). The relative lack of actin depolymerization after exposure to glutamate in cultured hippocampal neurons lacking gelsolin suggests a major role for gelsolin in calcium-induced actin depolymerization in neurons. Previous studies have shown that gelsolin is also the major calcium-activated actin-severing protein involved in regulation of motility and other processes in some types of non-neuronal cells, including platelets, macrophages, and fibroblasts (for review, see Hartwig and Kwiatkowski, 1991). We found that gelsolin immunoreactivity was present in cell bodies and neurites of the cultured embryonic hippocampal neurons. Previous studies of similar cultures have shown that these neuronal compartments contain actin filaments associated with the plasma membrane (Fifkova, 1985; Letourneau and Shattuck, 1989) and also contain NMDA receptors (Mattson et al., 1991, 1993b) and VDCC (Yaari et al., 1987; Westenbroek et al., 1990). Thus, gelsolin, its actin filament substrate, and the ion channels modulated by actin polymerization are colocalized in the same cellular compartments, suggesting the possibility of quite localized regulation of NMDA receptors and VDCC by the actin system. Interestingly, previous studies have provided evidence that gelsolin is associated with plasma membrane in platelets and macrophages, suggesting the possibility that gelsolin acts to promote actin depolymerization in subplasmalemma microdomains (Hartwig et al., 1989). Consistent with the latter possibility, we observed concentrations of gelsolin immunoreactivity in the periphery of neuronal somata, suggesting that gelsolin is also associated with the plasma membrane in neurons.
Several findings in the present study suggest that the alterations in levels of actin depolymerization, ion currents through NMDA receptors and VDCC, and intracellular calcium levels after exposure of G−/− neurons to glutamate were a direct consequence of lack of gelsolin rather than an indirect compensatory response. First, Western blot analysis showed that overall levels of actin and cofilin were essentially identical in G+/+ and G−/− neurons. Second, calcium imaging and patch-clamp analyses showed that responses of G+/− neurons were intermediate to (and significantly different from) responses of G+/+ and G−/− neurons; responses of G+/− neurons might be expected to be similar to G+/+ neurons in a developmental compensation scenario. Third, gelsolin is considered a major calcium-activated actin-depolymerizing factor; glutamate-induced actin depolymerization in G+/+ neurons was mediated by calcium, and the decreased actin depolymerization in G−/− neurons occurred despite a greater elevation of [Ca2+]i in those neurons. Because there are no obvious alterations in normal brain development and function in the G−/− mice, our data showing altered responses of G−/− neurons to excitotoxic levels of calcium influx suggest that gelsolin serves a particularly important function in modulating neuronal calcium homeostasis in pathophysiological conditions such as severe epileptic seizures or cerebral ischemia.
Our calcium imaging data showed that neurons lacking gelsolin exhibit enhanced calcium responses to glutamate. The enhanced response to glutamate was likely the result of decreased actin depolymerization in the G−/− neurons, rather than some other consequence of absence of gelsolin, because the effect was abolished in G−/− neurons treated with the actin-depolymerizing agent cytochalasin D. Previous whole-cell patch-clamp analyses in which actin depolymerization in cultured neurons was induced by pharmacological treatment with cytochalasin D provided evidence that F-actin promotes prolonged opening of NMDA receptor channels (Rosenmund and Westbrook, 1993) and VDCC (Johnson and Byerly, 1993). Our data indicate that calcium-induced, gelsolin-mediated actin depolymerization resulting from physiological stimuli (membrane depolarization and glutamate) promotes rapid rundown of NMDA current and calcium current. Calcium responses to glutamate in G+/− hippocampal neurons were intermediate to those of G+/+ and G−/− neurons. Similarly, rates of rundown of NMDA and calcium currents in the heterozygotes were intermediate to the wild-type and G−/− neurons. In contrast to NMDA current, kainate-induced current was not different in G+/+, G+/−, and G−/− hippocampal neurons, indicating specificity of ion channel regulation by actin.
Although increasing data indicate that actin filaments can influence ion currrents in both non-neuronal cells and neurons, the molecular interactions involved remain to be established. The majority of evidence implicating actin filaments in regulation of ion channel function has come from studies that used cytochalasins. For example, cytochalasin D: decreased sodium current in myocardial cells (Undrovinas et al., 1995); increased the activity of sodium channels in human myeloid leukemia cells (Negulyaev et al., 1996); and enhanced activity of rat epithelial sodium channels (Cantiello et al., 1991). Some data suggest that actin may interact directly with ion channels. For example, short actin filaments enhanced sodium channel activity in planar lipid bilayers, and cytochalasin D blocked the effect of the actin filaments (Berdiev et al., 1996). On the other hand, there are a number of actin-binding proteins that link actin with membranes, and some such proteins (e.g., dystrophin) have been implicated in modulating calcium influx (for review, see Hartwig, 1994). It will be of considerable interest to determine whether prominent neuronal membrane-associated actin-binding proteins such as spectrin play a role in actin-dependent modulation of ion currents through NMDA receptor channels and VDCC.
NMDA receptors and VDCC play fundamental roles in neurotransmission and in developmental and synaptic plasticity. For example, activation of VDCC in presynaptic terminals is a key signal for release of neurotransmitter from synaptic vesicles, and calcium-mediated actin depolmerization may play a role in that process (Kato et al., 1996;Viviani et al., 1996; Iga et al., 1997). Activation of NMDA receptors is involved in regulation of growth cone behaviors and synaptogenesis in visual pathways (Constantine-Paton et al., 1990) and in the hippocampus (Mattson et al., 1988a,b). Calcium influx through NMDA receptors is also involved in long-term depression and long-term potentiation of synaptic transmission in the hippocampus, forms of synaptic plasticity believed to be central to the processes of learning and memory (Collingridge and Bliss, 1987; Bear and Malenka, 1994). Our data suggest that actin depolymerization, effected by gelsolin, results in reduced Ca2+ influx through VDCC and NMDA receptor channels and may thereby play roles in modulating the kinds of calcium-dependent processes just described. Roles for gelsolin in regulating the responses of growth cones to signals that elevate [Ca2+]i are suggested from the presence of gelsolin in growth cones of developing neurons (Tanaka et al., 1993), data demonstrating that calcium and actin regulate growth cone motility (Lankford and Letourneau, 1989; Forscher et al., 1992), and the established role of gelsolin in mediating motility responses to Ca2+ in non-neuronal cells (Hartwig and Kwiatkowski, 1991). However, it should be noted that there are no overt structural or functional alterations in the nervous systems of mice lacking gelsolin, indicating that gelsolin is not required for normal development of the nervous system. Nevertheless, there is ample precedence for more subtle alterations in neuronal physiology resulting from knock-out of certain components of calcium signaling pathways. For example, mice lacking either calcium–calmodulin-dependent protein kinase II (Silva et al., 1992) or the γ subunit of protein kinase C (Abeliovich et al., 1993a,b) exhibit alterations in hippocampal long-term potentiation and spatial learning but, nevertheless, exhibit no structural alterations in the brain and develop and reproduce quite normally. Our data demonstrate clear differences in neuronal calcium signaling and ion channel activity in hippocampal neurons lacking gelsolin; these differences are likely to impact on synaptic plasticity and the associated behaviors subserved by these neurons in vivo.
A striking finding in the present study was that hippocampal neurons lacking gelsolin exhibit increased vulnerability to excitotoxicity, both in cell culture and in vivo. Our data suggest that by promoting actin filament depolymerization and reducing calcium influx through VDCC and NMDA receptors, gelsolin functions to reduce excitotoxic neuronal injury. Moreover, they suggest that gelsolin may have a role in a variety of neurodegenerative conditions that involve excessive Ca2+ influx. Based on the present findings, and a previous study showing that cytochalasin D can protect neurons against excitotoxicity (Furukawa et al., 1995), we propose that gelsolin functions in a feedback pathway that suppresses accumulation of cytotoxic levels of intracellular calcium. Thus, calcium influx activates gelsolin, which, in turn, induces actin filament depolymerization. The loss of F-actin (which normally promotes sustained opening of NMDA receptor channels and VDCC) results in current rundown and reduced calcium influx through NMDA receptors and VDCC. We found that seizure-induced damage to hippocampal neurons is exacerbated in mice lacking gelsolin. It will clearly be of considerable interest to explore further the involvement of gelsolin and the actin cytoskeleton in other neurodegenerative paradigms that involve NMDA receptor activation (e.g., cerebral ischemia and traumatic brain injury). Finally, our data suggest that agents that enhance dynamic changes in the actin filament architecture of neurons may prove effective as therapeutic agents in neurodegenerative conditions such as ischemic stroke, epileptic seizures, and traumatic brain injury.
Footnotes
This work was supported by National Institutes of Health Grants NS29001 and NS30583 to M.P.M., the Alzheimer’s Association, and the Metropolitan Life Foundation. We thank A. Lueck and P. Marks for Western blot analysis of cofilin and R. Pelphrey for technical assistance.
Correspondence should be addressed to Mark P. Mattson, 211 Sanders-Brown Building, University of Kentucky, Lexington, KY 40536-0230.
REFERENCES
- 1.Abe H, Obinata T, Minamide LS, Bamburg JR. Xenopus laevis actin-depolymerizing factor/cofilin: a phosphorylation-regulated protein essential for development. J Cell Biol. 1996;132:871–885. doi: 10.1083/jcb.132.5.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S. Modified hippocampal long-term potentiation in PKCγ-mutant mice. Cell. 1993a;75:1253–1262. doi: 10.1016/0092-8674(93)90613-u. [DOI] [PubMed] [Google Scholar]
- 3.Abeliovich A, Paylor R, Chen C, Kim JJ, Wehner JM, Tonegawa SP. PKCγ mutant mice exhibit mild deficits in spatial and contextual learning. Cell. 1993b;75:1263–1271. doi: 10.1016/0092-8674(93)90614-v. [DOI] [PubMed] [Google Scholar]
- 4.Augustine GJ, Charlton MP, Smith SJ. Calcium action in synaptic transmitter release. Annu Rev Neurosci. 1987;10:633–693. doi: 10.1146/annurev.ne.10.030187.003221. [DOI] [PubMed] [Google Scholar]
- 5.Bamburg JR, Harris HE, Weeds AG. Partial purification and characterization of an actin depolymerizing factor from brain. FEBS Lett. 1980;121:178–181. doi: 10.1016/0014-5793(80)81292-0. [DOI] [PubMed] [Google Scholar]
- 6.Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 7.Berdiev BK, Prat AG, Cantiello HF, Ausiello DA, Fuller CM, Jovov B, Benos DJ, Ismailov Regulation of epithelial sodium channels by short actin filaments. J Biol Chem. 1996;271:17704–17710. doi: 10.1074/jbc.271.30.17704. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein BW, Bamburg JR. Reorganization of actin in depolarized synaptosomes. J Neurosci. 1985;5:2565–2569. doi: 10.1523/JNEUROSCI.05-10-02565.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernstein BW, Bamburg JR. Cycling of actin assembly in synaptosomes and neurotransmitter release. Neuron. 1989;3:257–265. doi: 10.1016/0896-6273(89)90039-1. [DOI] [PubMed] [Google Scholar]
- 10.Bretscher A, Weber K. Villin is a major protein of microvillus cytoskeleton which binds both G and F actin in a calcium-dependent manner. Cell. 1980;20:839–847. doi: 10.1016/0092-8674(80)90330-x. [DOI] [PubMed] [Google Scholar]
- 11.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 12.Cantiello HF, Stow JL, Prat AG, Ausiello DA. Actin filaments regulate epithelial Na+ channel activity. Am J Physiol. 1991;261:882–888. doi: 10.1152/ajpcell.1991.261.5.C882. [DOI] [PubMed] [Google Scholar]
- 13.Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 14.Collingridge GL, Bliss TVP. NMDA receptors: their role in long-term potentiation. Trends Neurosci. 1987;10:288–293. [Google Scholar]
- 15.Condeelis J. Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol. 1994;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- 16.Constantine-Paton M, Cline HT, Debski EA. Patterned activity, synaptic convergence, and the NMDA receptor in developing visual pathways. Annu Rev Neurosci. 1990;13:129–154. doi: 10.1146/annurev.ne.13.030190.001021. [DOI] [PubMed] [Google Scholar]
- 17.Cooper JA, Bryan J, Schwab B, III, Frieden C, Loftus DJ. Microinjection of gelsolin into living cells. J Cell Biol. 1987;104:491–501. doi: 10.1083/jcb.104.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cramer LP, Mitchison TJ, Theriot JA. Actin-dependent motile forces and cell motility. Curr Opin Cell Biol. 1994;6:82–86. doi: 10.1016/0955-0674(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 19.Fifkova E. Actin in the nervous system. Brain Res Rev. 1985;9:187–215. [PubMed] [Google Scholar]
- 20.Forscher P, Lin CH, Thompson C. Novel form of growth cone motility involving site-directed actin filament assembly. Nature. 1992;357:515–518. doi: 10.1038/357515a0. [DOI] [PubMed] [Google Scholar]
- 21.Furukawa K, Smith-Swintosky VL, Mattson MP. Evidence that actin depolymerization protects hippocampal neurons against excitotoxicity by stabilizing [Ca2+]i. Exp Neurol. 1995;133:153–163. doi: 10.1006/exnr.1995.1018. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa K, Barger SW, Blalock E, Mattson MP. Activation of K+ channels and suppression of neuronal activity by secreted β-amyloid precursor protein. Nature. 1996;379:74–78. doi: 10.1038/379074a0. [DOI] [PubMed] [Google Scholar]
- 23.Hamill OP, Bormann J, Sakmann B. Activation of multiple-conductance state chloride channels in spinal neurones by glycine and GABA. Nature. 1983;305:805–808. doi: 10.1038/305805a0. [DOI] [PubMed] [Google Scholar]
- 24.Hartwig JH. Actin-binding proteins 1: spectrin superfamily. Protein Profile. 1994;1:706–778. [PubMed] [Google Scholar]
- 25.Hartwig JH, Kwiatkowski DJ. Actin-binding proteins. Curr Opin Cell Biol. 1991;3:87–97. doi: 10.1016/0955-0674(91)90170-4. [DOI] [PubMed] [Google Scholar]
- 26.Hartwig JH, Chambers KA, Stossel TP. Association of gelsolin with actin filaments and cell membranes of macrophages and platelets. J Cell Biol. 1989;108:467–479. doi: 10.1083/jcb.108.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayden SM, Miller PS, Brauweiler A, Bamburg JR. Analysis of the interactions of actin depolymerizing factor with G- and F-actin. Biochemistry. 1993;32:9994–10004. doi: 10.1021/bi00089a015. [DOI] [PubMed] [Google Scholar]
- 28.Iga M, Inui M, Sobue K. Characterization of the interaction between synapsin I and calspectin (brain spectrin or fodrin). Biochem Biophys Res Commun. 1997;231:852–855. doi: 10.1006/bbrc.1997.6202. [DOI] [PubMed] [Google Scholar]
- 29.Janmey PA, Stossel TP. Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature. 1987;325:362–364. doi: 10.1038/325362a0. [DOI] [PubMed] [Google Scholar]
- 30.Johnson B, Byerly L. A cytoskeletal mechanism for Ca2+ channel metabolic dependence and inactivation by intracellular Ca2+. Neuron. 1993;10:797–804. doi: 10.1016/0896-6273(93)90196-x. [DOI] [PubMed] [Google Scholar]
- 31.Kater SB, Mattson MP, Cohan CS, Connor JA. Calcium regulation of the neuronal growth cone. Trends Neurosci. 1988;11:315–321. doi: 10.1016/0166-2236(88)90094-x. [DOI] [PubMed] [Google Scholar]
- 32.Kato M, Sasaki T, Ohya T, Nakanishi H, Nishioka H, Imamura M, Takai Y. Physical and functional interaction of rabphilin-3A with alpha-actinin. J Biol Chem. 1996;271:31775–31778. doi: 10.1074/jbc.271.50.31775. [DOI] [PubMed] [Google Scholar]
- 33.Kennedy MB. Regulation of neuronal function by calcium. Trends Neurosci. 1989;12:417–420. doi: 10.1016/0166-2236(89)90089-1. [DOI] [PubMed] [Google Scholar]
- 34.Kwiatkowski DJ, Mehl R, Izumo S, Nadal GB, Yin HL. Muscle is the major source of plasma gelsolin. J Biol Chem. 1988a;263:8239–8243. [PubMed] [Google Scholar]
- 35.Kwiatkowski DJ, Mehl R, Yin HL. Genomic organization and biosynthesis of secreted and cytoplasmic forms of gelsolin. J Cell Biol. 1988b;106:375–384. doi: 10.1083/jcb.106.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lankford KL, Letourneau PC. Evidence that calcium may control neurite outgrowth by regulating the stability of actin filaments. J Cell Biol. 1989;109:1229–1243. doi: 10.1083/jcb.109.3.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lappalainen P, Drubin DG. Cofilin promotes rapid actin filament turnover in vivo. Nature. 1997;388:78–82. doi: 10.1038/40418. [DOI] [PubMed] [Google Scholar]
- 38.Lee J, Ishihara A, Theriot JA, Jacobson K. Principles of locomotion for simple-shaped cells. Nature. 1993;362:167–171. doi: 10.1038/362167a0. [DOI] [PubMed] [Google Scholar]
- 39.Letourneau PC, Shattuck TA. Distribution and possible interactions of actin-associated proteins and cell adhesion molecules of nerve growth cones. Development. 1989;105:505–519. doi: 10.1242/dev.105.3.505. [DOI] [PubMed] [Google Scholar]
- 40.Maekawa S, Toriyama M, Hisanaga S, Ynezawa N, Enso S, Hirokawa N, Sakai H. Purification and characterization of a calcium-dependent actin filament severing protein from bovine adrenal medulla. J Biol Chem. 1989;264:7458–7465. [PubMed] [Google Scholar]
- 41.Mattson MP, Dou P, Kater SB. Outgrowth-regulating actions of glutamate in isolated hippocampal pyramidal neurons. J Neurosci. 1988a;8:2087–2100. doi: 10.1523/JNEUROSCI.08-06-02087.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattson MP, Lee RE, Adams ME, Guthrie PB, Kater SB. Interactions between entorhinal axons and target hippocampal neurons: a role for glutamate in the development of hippocampal circuitry. Neuron. 1988b;1:865–876. doi: 10.1016/0896-6273(88)90134-1. [DOI] [PubMed] [Google Scholar]
- 43.Mattson MP, Murrain M, Guthrie PB, Kater SB. Fibroblast growth factor and glutamate: opposing actions in the generation and degeneration of hippocampal neuroarchitecture. J Neurosci. 1989;9:3728–3740. doi: 10.1523/JNEUROSCI.09-11-03728.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mattson MP, Wang H, Michaelis EK. Developmental expression, compartmentalization, and possible role in excitotoxicity of a putative NMDA receptor protein in cultured hippocampal neurons. Brain Res. 1991;565:94–108. doi: 10.1016/0006-8993(91)91740-r. [DOI] [PubMed] [Google Scholar]
- 45.Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, Rydel RE. β-Amyloid precursor protein metabolites and loss of neuronal calcium homeostasis in Alzheimer’s disease. Trends Neurosci. 1993a;16:409–415. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- 46.Mattson MP, Kumar K, Cheng B, Wang H, Michaelis EK. Basic FGF regulates the expression of a functional 71 kDa NMDA receptor protein that mediates calcium influx and neurotoxicity in cultured hippocampal neurons. J Neurosci. 1993b;13:4575–4588. doi: 10.1523/JNEUROSCI.13-11-04575.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- 48.Mattson MP, Fu W, Waeg G, Uchida K. 4-Hydroxynonenal, a product of lipid peroxidation, inhibits dephosphorylation of the microtubule-associated protein tau. NeuroReport. 1997;8:2275–2281. doi: 10.1097/00001756-199707070-00036. [DOI] [PubMed] [Google Scholar]
- 49.Miller RJ, Murphy SN, Glaum SR. Neuronal Ca2+ channels and their regulation by excitatory amino acids. Ann NY Acad Sci. 1989;568:149–158. doi: 10.1111/j.1749-6632.1989.tb12502.x. [DOI] [PubMed] [Google Scholar]
- 50.Morgan TE, Lockerbie RO, Minamide LS, Browning MD, Bamburg JR. Isolation and characterization of a regulated form of actin depolymerizing factor. J Cell Biol. 1993;122:623–633. doi: 10.1083/jcb.122.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neely MD, Gesemann M. Disruption of microfilaments in growth cones following depolarization and calcium influx. J Neurosci. 1994;14:7511–7520. doi: 10.1523/JNEUROSCI.14-12-07511.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neely MD, Nicholls JG. Electrical activity, growth cone motility and the cytoskeleton. J Exp Biol. 1995;198:1433–1446. doi: 10.1242/jeb.198.7.1433. [DOI] [PubMed] [Google Scholar]
- 53.Negulyaev YA, Vedernikova EA, Maximov AV. Disruption of actin filaments increases the activity of sodium-conducting channels in human myeloid leukemia cells. Mol Biol Cell. 1996;7:1857–1864. doi: 10.1091/mbc.7.12.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orrenius S, Burkitt MJ, Kass GEN, Dypbukt JM, Nicotera P. Calcium ions and oxidative cell injury. Ann Neurol. 1992;32:33–42. doi: 10.1002/ana.410320708. [DOI] [PubMed] [Google Scholar]
- 55.Rosenmund C, Westbrook G. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- 56.Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in α-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- 57.Smith-Swintosky VL, Kraemer PJ, Bruce AJ, McCants N, Maki A, Brown RW, Alcala M, Goodman Y, Slevin JT, Mattson MP. Bacterial alkaloids mitigate seizure-induced hippocampal damage and memory deficits. Exp Neurol. 1996;141:287–296. doi: 10.1006/exnr.1996.0163. [DOI] [PubMed] [Google Scholar]
- 58.Sobue K. Actin-based cytoskeleton in growth cone activity. Neurosci Res. 1993;18:91–102. doi: 10.1016/0168-0102(93)90012-f. [DOI] [PubMed] [Google Scholar]
- 59.Tanaka J, Kira M, Sobue K. Gelsolin is localized in neuronal growth cones. Dev Brain Res. 1993;76:268–271. doi: 10.1016/0165-3806(93)90217-x. [DOI] [PubMed] [Google Scholar]
- 60.Undrovinas AI, Shander GS, Makielski JC. Cytoskeleton modulates gating of voltage-dependent sodium channel in heart. Am J Physiol. 1995;269:203–214. doi: 10.1152/ajpheart.1995.269.1.H203. [DOI] [PubMed] [Google Scholar]
- 61.Viviani B, Galli CL, Marinovich M. Is actin polymerization relevant to neurosecretion? A study on neuroblastoma cells. Biochem Biophys Res Commun. 1996;223:712–717. doi: 10.1006/bbrc.1996.0960. [DOI] [PubMed] [Google Scholar]
- 62.Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia. 1993;34:37–53. doi: 10.1111/j.1528-1157.1993.tb05905.x. [DOI] [PubMed] [Google Scholar]
- 63.Westenbroek RE, Ahlijanian MK, Catterall WA. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347:281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- 64.Witke W, Sharpe AH, Hartwig JH, Azuma T, Stossel TP, Kwiatkowski DJ. Hemostatic, inflammatory, and fibroblast responses are blunted in mice lacking gelsolin. Cell. 1995;81:41–51. doi: 10.1016/0092-8674(95)90369-0. [DOI] [PubMed] [Google Scholar]
- 65.Yaari Y, Hamon B, Lux HD. Development of two types of calcium channels in cultured mammalian hippocampal neurons. Science. 1987;235:680–682. doi: 10.1126/science.2433765. [DOI] [PubMed] [Google Scholar]
- 66.Yin HL, Stossel TP. Control of cytoplasmic actin gel-sol transformation by gelsolin, a calcium-dependent regulatory protein. Nature. 1979;281:583–586. doi: 10.1038/281583a0. [DOI] [PubMed] [Google Scholar]
- 67.Yin HL, Albrecht JH, Fattoum A. Identification of gelsolin, a Ca2+-dependent regulatory protein of actin gel-sol transformation and its intracellular distribution in a variety of cells and tissues. J Cell Biol. 1981a;91:901–906. doi: 10.1083/jcb.91.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yin HL, Hartwig JH, Maruyama K, Stossel TP. Ca2+ control of actin filament length. J Biol Chem. 1981b;256:9693–9697. [PubMed] [Google Scholar]
- 69.Yonezawa N, Nishida E, Sakai H. pH control of actin polymerization by cofilin. J Biol Chem. 1985;260:14410–14412. [PubMed] [Google Scholar]