Study of Receptor-Mediated Neurotoxins Released by HIV-1-Infected Mononuclear Phagocytes Found in Human Brain (original) (raw)
Abstract
Although there is growing evidence that neurotoxic molecules produced by HIV-1-infected mononuclear phagocytes damage neurons, the precise mechanisms of neuronal attack remain uncertain. One class of cytotoxin involves neuronal injury mediated via the NMDA receptor. We examined blood monocytes and brain mononuclear cells isolated at autopsy from HIV-1-infected individuals for the ability to release NMDA-like neuron-killing factors. We found that a neurotoxic amine, NTox, was produced by blood monocytes and by brain mononuclear phagocytes infected with retrovirus. In vivo injections of minute quantities of NTox produced selective damage to hippocampal pyramidal neurons. NTox can be extracted directly from brain tissues infected with HIV-1 and showed structural features similar to wasp and spider venoms. In contrast to NTox, HIV-1 infection did not increase the release of the NMDA excitotoxin quinolinic acid (QUIN) from mononuclear cells. Although we found modest elevations of QUIN in the CSF of HIV-1-infected individuals, the increases were likely attributable to entry through damaged blood–brain barrier. Taken together, our data pinpoint NTox, rather than QUIN, as a major NMDA receptor-directed toxin associated with neuro-AIDS.
Keywords: AIDS, neurotoxin, gp120, microglia, macrophage, brain, dementia, HIV-1, NMDA, quinolinic acid
Clinical studies have shown that individuals with AIDS often suffer cognitive dysfunction (Snider et al., 1983;Faulstich, 1986; Navia et al., 1986a; Gabuzda et al., 1987; Mangos et al., 1989), which can range from distractibility and delirium to impaired memory and dementia. At autopsy, the brains of AIDS patients revealed cortical atrophy, invasion of macrophages, nodules of reactive microglia, and giant cell formation (Navia et al., 1986b; Budka, 1989;Gelman, 1993), as well as loss of large neurons in frontal, temporal, and parietal regions (Navia et al., 1986b; Ketz-ler et al., 1990;Wiley et al., 1991). More recently, neuronal pathology in the hippocampus has been linked to HIV-1 dementia (Masliah et al., 1992). Because neuronal and synaptic loss in the CNS are likely to be responsible for impaired cognition, much attention has focused on the mechanisms to account for retrovirus-induced neuron pathology (Brenneman et al., 1988; Heyes et al., 1989; Giulian et al., 1990;Dreyer et al., 1990).
It is now widely believed that HIV-1 attacks neurons through stimulated production of neurotoxic molecules (Giulian, 1992a; Lipton, 1992b). The likely cellular sources of brain-derived neurotoxins are CD4(+) mononuclear phagocytes such as blood-borne monocytes, resident microglia, and multinucleated giant cells (Giulian et al., 1993a;Gendelman et al., 1994). The first evidence that HIV-1-infected mononuclear cells injured neurons came from in vitro work using human cell lines (Giulian et al., 1990) or human blood monocytes (Pulliam et al., 1991). These studies showed that infected mononuclear phagocytes, but not lymphocytes, release neuron-killing factors. Subsequent reports demonstrated that mononuclear phagocytes were a source of cytotoxic agents when activated not only by viral infection but also by other immune stimuli, including the viral envelope protein gp120 (Colton and Gilbert, 1987; Thery et al., 1991; Boje and Arora, 1992; Lipton, 1992a; Giulian et al., 1993a). Although a number of candidate poisons—cytokines, free radicals, nitric oxide, platelet activating factor—were derived from monocytes, there has been uncertainty as to which of these agents is relevant to neuro-AIDS (Gendelman et al., 1994). We and other investigators have suggested that neurons bearing NMDA receptors are sensitive targets of cytotoxins generated during HIV-1 infection (Giulian et al., 1990; Lipton, 1992a). Two pertinent neurotoxins with NMDA-like properties are quinolinic acid (QUIN; Schwarcz et al., 1983), found elevated in the CSF of HIV-1-infected individuals (Heyes et al., 1989), and a neurotoxic amine (referred to here as NTox) released by HIV-1-infected monocytic cell lines (Giulian et al., 1990, 1993a). Although both of these toxins may be brain-damaging agents in neuro-AIDS, no direct evidence exists that HIV-1 infection actually drives mononuclear phagocytes to release QUIN or that NTox exists within cells or tissues of infected individuals. In this report, we compare the capacities of blood and brain mononuclear phagocytes to produce factors toxic to NMDA receptor-bearing neurons. Although elevated QUIN levels appear in CSF of HIV-1(+) patients, no increased production by infected mononuclear cells was detected. In contrast, NTox was found to be released by blood- and brain-derived mononuclear phagocytes recovered from HIV-1-infected individuals. Moreover, NTox was a potent neuron poison in vivo, sharing some structural features with the polyamine amide class of wasp and spider venoms.
MATERIALS AND METHODS
Culture of mononuclear cells. Cell lines were cultured in RPMI-1640 with l-glutamine supplemented with 10% fetal bovine serum (FBS) or in N2 media (Bottenstein and Sato, 1979) and maintained in suspension culture at 37°C in a humidified atmosphere of 95% air/5% CO2. The continuous human T lymphocyte line (H9) was obtained from Dr. R. Gallo through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health. The human monocyte cell line THP-1 (ATCC TIB 202) was obtained from the American Type Culture Collection and infected with the HIV-1IIIB at a multiplicity of infection of 5 × 105TCID50/106 cells. The cells were passaged and monitored periodically for HIV antigens by indirect immunofluorescence using a polyclonal anti-HIV-1 serum. At 15–20 d postinfection, the cells were >80% HIV antigen-positive. The cell line (THP-1/HIV) produced a virus that was cytopathic in the MT-4 cell line (∼104 TCID50/ml culture fluid) and released p24/p25 antigen (>103 pg/ml culture fluid). Electron microscopy of THP-1/HIV revealed HIV-1-like retrovirus particles. All the cell lines used grew well under standard culture conditions (without the addition of cytokines) and were free of mycoplasma contamination as determined by a ribosomal RNA detection assay (Mycoplasma Rapid Detection System, Gen-Probe, San Diego, CA).
Buffy coats from HIV-1(+) and HIV-1(−) individuals were obtained through the Baylor Center for AIDS Research and the Gulf Coast Regional Blood Center (Houston, TX). Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation on a ficol/sodium diatrizoate gradient (Histopaque 1077; Sigma, St. Louis, MO). The PBMCs were washed four times with PBS, pH 7.4, resuspended in RPMI-1640 medium with l-glutamine (Gibco) or N2 media containing 10% fetal bovine serum and 5% IL-2 (Cellular Products, Buffalo, NY), seeded into 100 mm plastic culture dishes at 20 × 106 cells/10 ml. After a 1 hr incubation at 37°C in an atmosphere of 95% air/5% CO2, nonadherent cells were removed from culture dishes by gentle washing with three changes of warmed medium and replated at a density of 2 × 106 cells/ml in plastic culture dishes for 24 hr. Flow cytometric analyses showed that >90% of these nonadherent cells contained the lymphocyte marker CD45, whereas <1% were CD14(+) monocytes. Adherent cells were recovered by washes with ice-chilled Ca2+- and Mg2+-free PBS and plated at a density of 1 × 106/ml. After a 24 hr incubation, >95% of the adhering cells were monocytes as indicated by the presence of nonspecific esterase activity, acetylated low-density lipoprotein (LDL) receptors, and the CD14 surface antigen. Isolated lymphocytes or monocytes were maintained for 48–72 hr in RPMI-1640 or N2 medium containing 10% FBS, 5% IL-2, and 10% media conditioned by GCT cell line (ATTC TIB 223). Productive infection was monitored by immunoassay for p24/25 antigen released into culture media (Coulter HIV-1 Antigen Assay Kit, Hileah, FL).
Acquisition of CNS tissues and cells. Human brain tissue was obtained within a 6 hr postmortem interval through the Rapid Autopsy Program of the Department of Pathology, Methodist Hospital, Houston, TX, under the direction of Dr. J. Kirkpatrick as described previously (Giulian et al., 1995a). Normal control brains were from adult patients with no brain pathology. HIV-1(+) brain tissues were acquired from infected adults showing microglial nodules and giant cells but lacking evidence of other pathology, including parasitic infection, neoplasm, infarction, or abscess. Coded serum and CSF samples were acquired through the Neurological Research Specimen Bank at the Veterans Administrative Medical Center, Los Angeles, CA, from volunteers who were either seropositive for HIV-1(+) (n = 63) or who had a clinical diagnosis of multiple sclerosis (n = 28). Neurological assessments and other laboratory data, including serum albumin concentrations, were provided by Dr. W. Tourtellotte and colleagues.
Viable adult human microglia were isolated from neocortical gray matter within a postmortem interval of <6 hr (Giulian et al., 1995a). Tissues were dissociated by trituration in 0.15% trypsin, placed in N2 media with 10% FBS, centrifuged through 35% sucrose, and separated by a ficol/sodium diatrizoate gradient (Histopaque 1077; Sigma). Cells were placed in N2 media containing gentamycin (48 μg/ml) and recovered by selective adhesion to plastic for 12 hr (Giulian and Baker, 1986). We typically recover 1 to 3 × 105 cells/gram wet weight of tissue. Viable cells were identified by the >95% presence of phagocytic activity and by endocytosis of the scavenger receptor ligand acetylated LDL (ac-LDL) bound to the fluorescent probe 1,1′-dioctadecyl-1,3,3,3′,3-tetramethylindocarbocyanine (DiI-ac-LDL; Biomedical Technologies, Stoughton, MA). Cells were also identified by immunostaining for CD4 (1:80 dilution, BioGenex, San Ramon, CA) and for HLA-DR (1:80; BioGenex). In vitro infection of mononuclear cells involved exposure to HIV-1IIIB stock (1 × 107TCID50/106 cells) for 24 hr. Cells were washed with fresh N2 media and monitored for viability, neurotoxin production, and p24/25 production. Mock controls were prepared in an identical manner using virus-free stock solutions.
Cells adhering to glass coverslips were fixed in 2% glutaraldehyde in 50 mm cacodylate buffer, pH 7.4, containing 0.1 m sucrose for 30 min at 37°C. After several rinses with PBS, the cells were post-fixed in 2% OsO4 in 0.1 m phosphate buffer, pH 7.4, for 1 hr at 4°C. After dehydration with a graded series of ethanol, the cells were washed three times with 100% acetone, dried under CO2 in a critical point drying apparatus (Denton Vacuum, Cherry Hill, NJ), and coated with gold (Denton Sputter Etch Unit). Samples were mounted on copper specimen boats with conductive colloidal silver and viewed with a JEOL JEM-100 CX electron microscope between 500 and 10,000× magnification. Cell surface features used to distinguish microglia from macrophages were described previously (Giulian et al., 1995b).
Neurotoxicity assays. Ciliary neurons from embryonic day 9 chick embryos were plated onto poly-l-lysine-coated coverslips in 24-well plates at two ganglia per well in N2 media (diluted to 90%) and supplemented with 30 mm KCl plus 0.6% horse serum [modified from Giulian et al. (1993b)]. Cultures consisted of ∼50% neurofilament(+) neurons mixed with Schwann cells and were free of mononuclear phagocytes. Ciliary neurons were sensitive to the toxic effects of NMDA, QUIN, and α-amino-3-hydroxy-5-methyl-4-isoxazoleacetic acid zwitterion (Giulian et al., 1993a,b). Neurotoxic activity was measured after 48 hr incubations by viewing cells with phase microscopy at 250× magnification, with untreated cultures providing internal controls for each experiment. For cell counts, we defined a healthy, surviving neuron as one that exhibits a distinct nuclear membrane with characteristic nucleoli and a cytoplasm free of large vacuoles. The percent neuron kill score was calculated as [1 − (neurons per field in treated group/neurons per field in the untreated control group)] × 100%. Data were expressed as mean values ± SE, with each value obtained from 18 fields per coverslip using at least six coverslips per group. NMDAR stain was a gift from Dr. Robert Wenthold, National Institutes of Health (Petralia et al., 1994).
Rat hippocampal neurons (fetal day 18) were plated onto poly-l-lysine-coated coverslips in 24-well plates at 250,000 cells/well in N2 culture media and 5% FBS. Gradual reduction of serum began on day 7 in vitro by 1:1 volume replacement with chemically defined media. These cultures consisted of process-bearing neurons (10–20% of total cell population) atop a bed of astroglia (>70%) mixed with microglia (5–10%). Microglia-free cultures were produced by a treatment with saporin (sap; a ribosome-inactivating protein) coupled to ac-LDL. Sap-ac-LDL (10 μg/ml for 18 hr) selectively bound to microglial scavenger receptors, thereby specifically depleting cultures of these cells to <0.1% of the total population, with no effect on numbers or viability of either neurons or astroglia in long-term cultures. After 11 d in vitro, cultures (with a final concentration of 0.6% serum) were exposed to test substances in the presence or absence of exogenous microglia for 72 hr. At the end of this time, the cultures were fixed in 3% paraformaldehyde at room temperature for 12 hr and immunostained by overnight incubation with a mixture of anti-neurofilament antibodies (SMI-311, 1:500; RT-97, 1:150; Sternberger Monoclonals) plus anti-MAP-2 (Boehringer Mannheim, 184959; 1:500) at 4°C in the presence of 2% horse serum and 0.3% Triton X-100 to delineate both cell bodies and neurites. Indirect immunofluorescence labeling used biotinylated horse anti-mouse IgG (1:1000; Vector Laboratories) and rhodamine streptavidin (Jackson ImmunoResearch, 1:1200) in buffered saline, pH 7.6. Neuron-killing scores were calculated as described above by determining the number of NF(+) MAP-2(+) cells in nine randomly selected fields per coverslip at 200× magnification. Glutamate receptor antagonists, includingd(−)-2-amino-5-phosphonopentanoic acid (AP5),d(−)-2-amino-phosphonoheptanoic acid (AP7), 6-cyano-7-nitroquinoline-2,3-dione,d-γ-glutamylaminomethanesulfonic acid, ifenprodil, 5,7-dichlorokynurenic acid, and MK-801, were obtained from Research Biochemicals (Natick, MA).
Isolation and biochemical study of neurotoxin. Sonicates in dH2O (10 vol sterile distilled water per tissue weight) from minced gray matter of frozen human brain were centrifuged at 20,000 × g for 15 min and separated by ultrafiltration through YM-30 membrane followed by YM-1 membrane; conditioned media were passed only through YM-1 membranes (Amicon; estimated molecular mass cutoff = 1000 Da). The ultrafiltrates were then washed with equal volumes of ethyl acetate, pH 4.0, and extracted into ethyl acetate under alkaline conditions, pH 10.5. Material was re-extracted into an acidic aqueous phase, pH 2.0, and dried under vacuum. Neurotoxin was flushed with nitrogen gas, sealed under vacuum, and subjected to acid hydrolysis (in 6N HCl for 24 hr at 105°C). Hydrolysate was then extracted into basic ethyl acetate and eluted twice from C18 reverse-phase high-pressure liquid chromatography (HPLC) (3.9 × 150 mm, Nova-Pak, Waters, Milford, MA) with a 0–20% acetonitrile gradient developed over 35 min (solvent A, 0.1% trifluoroacetic acid in dH2O; solvent B, 0.1% trifluoroacetic acid in dH2O:acetonitrile 5:95, v/v). Phenolyic and amine content were used to estimate concentrations of NTox found within highly purified HPLC fractions. Assigning a UVmaxof 265 nm (0.1% trifluoroacetic acid in 14% acetonitrile in dH2O), peaks of NTox eluted from C18-HPLC were compared with a standard curve of tyramine eluted under identical conditions measured with a multiple wavelength detector (Rainin Dynamax UV-M). Amine content was determined by the fluorescamine method using tyramine as a standard. These detection methods gave similar values for a given NTox preparation; the estimates of toxin concentration assumed one amine and phenolic ring per molecule.
Acid-catalyzed esterification of neurotoxins was performed with 3N HCl in _n_-butanol (Regis Chemical, Morton Grove, IL) for 60 min at 80°C; short acetylation was performed in acetic anhydride in methanol (1:3 v/v; Sigma) for 1 min at 25°C and the reaction was terminated by addition of excess glycine at room temperature. Toxin was modified by excess pentafluoropropionic anhydride (PFPA; Fluka Chemie, Switzerland) at 60°C for 60 min. For strong reducing conditions, toxins were resuspended in 100% ethanol containing 5 mg of rhodium (5% on carbon particles; Aldrich Chemical, Milwaukee, WI). Each sample was then flushed with hydrogen gas and incubated at 40°C for 24 hr under 275 kpa. For weak reduction, samples were reconstituted in 2.0 ml of 4.4% formic acid in methanol and loaded on a 0.5 × 2.0 cm2 column packed with palladium PEI powder (Pierce Chemical, Rockford, IL). The column was washed with 1 ml of methanol and 1 ml of 4.4% formic acid in methanol. Methylation of toxins was performed with diazomethane/ether in tetrahydrofuran for 5 min at room temperature. (Diazomethane/ether was prepared from Diazald according to the procedures in Aldrich Technical Information Bulletin No. AL-180.)
Neurotoxins were also treated with 100 units/ml of plasma amine oxidase (amine:oxygenoxidoreductase; 1.4.3.6; Worthington Biochemical, Freehold, NJ) at 25°C in 1 ml of 10 mm PBS, pH 7.0, for 4 hr or 390 units of polyphenol oxidase (monophenol, dihydroxyphenylalanine:oxygenoxidoreductase; 1.14.18.1, Worthington) at 25°C in 2 ml of 10 mm PBS, pH 7.0, for 2 hr. Other enzymatic treatments included exposure to 100 units lipase (3.1.1.3; Worthington) in 1 ml of 10 mm Tris buffer, pH 8.0, at room temperature for 5 hr or to 2 units ofl-amino acid oxidase (1.4.3.2; Worthington) in 1 ml of 10 mm Tris buffer containing 150 mm KCl, pH 7.4, at 37°C for 1 hr or at 25°C for 4 hr. In all cases, enzymatic reactions were terminated by boiling for 15 min. Before incubation with the neurotoxin, inactivated-enzyme controls were prepared by boiling.
Measurement of quinolinic acid. Quantitative measurements of QUIN levels found in CSF and in cell-conditioned media were performed directly by a radioenzymatic assay (Foster et al., 1986) using 50 μl samples in quadruplicate with internal standards of QUIN ranging from 2 nm to 2 μm. Determinations of QUIN in cell-conditioned media were also made by gas chromatography/mass spectrometry (GC/MS) using a Finningan MAT Incos 50 GC/MS. The GC column was a 15 m DB-5 (film thickness 0.25 μm) fused silica capillary column, programmed from 200 to 300°C at 10°C/min. Samples (3 ml of culture media conditioned by cells) were mixed with 100 ng dipicolinic acid (pyridine, 2,6-dicarboxylic acid, DPIC) as an internal standard, loaded onto a glass column packed with DOWEX-1X2, and eluted with 5 ml 6N formic acid. Lyophilized eluants were then mixed with 25 μl of pyridine and 75 μl of_N_-methyl-_N_-tert-butyldimethylsilyl-trifluoroacetamide (Pierce, Rockford, IL) and heated at 60°C for 30 min. Two microliters of derivatized material were injected for analysis by electron impact ionization mass spectrometry. Although the molecular ion at mass/charge (m/z) 395 was not observed, a fragment ion at m/z 338 (M+-tert-butyl) was the most abundant ion observed in the mass spectra of both silylated QUIN and DPIC. This ion was selected for specific molecular detection and quantitation of the compounds recovered from biological samples. To estimate recovery of QUIN from culture media by GC/MS, 3 ml of culture media were mixed with 3, 10, 30, and 100 ng of QUIN plus 100 ng DPIC in each sample to serve as the internal standard. After column fractionation and lyophilization, samples then received 100 ng of pyridine 2,4-dicarboxylic acid as an _external_standard. Both results showed good linear correlation with the actual amounts of QUIN added (correlation coefficients of 0.996 and 0.987, respectively). The percent recovery of QUIN for the entire protocol, including column fractionation, was estimated at 83 ± 11% (determined by dividing amounts of QUIN measured by the external standard method by those measured by the internal standard method), which was in good agreement with the 85 ± 5% recovery estimated by3H-QUIN.
Separation of QUIN from NTox was readily achieved by exploiting differences in charge or hydrophobicity (QUIN at physiological pH is an anion with avid binding to the anion exchanger, whereas NTox is a cation). Passing 3H-QUIN through tandem anion and cationic exchanger allowed ∼96% recovery of QUIN bound to the DOWEX-1X2 resin with <0.5% eluted from the secondary SP-Sephadex C25 column. In contrast, we found that >95% NTox bound to the secondary cation exchanger. Under basic conditions (pH 10.5), NTox could be extracted into ethyl acetate and then recovered in acidified aqueous phase (pH 4.0) with >90% recovery; under identical conditions, only 0.5% of 3H-QUIN was recovered from the two-step extraction procedure. In this way, separation of QUIN from NTox could be achieved by either tandem ion exchange chromatography or by two-step aqueous/ethyl acetate extractions.
Infusion of neurotoxin into rat brain. Adult albino rats (250–300 gm) were anesthetized by intraperitoneal injection of a combination anesthetic (0.86 mg xylazine, 4.30 mg ketamine, and 0.14 mg acepromazine per 100 gm body weight) and placed in a stereotaxic device. Using aseptic technique, the scalp was reflected and burr holes were placed lateral to the midline. A microsyringe with a 26 gauge needle was then inserted down 2.9 mm from the brain’s surface, at a distance of 4.5 mm caudal to bregma, 3.0 mm lateral to the midline into the hippocampus; concentrated fractions reconstituted in artificial CSF, pH 7.4, were infused in 1.0 μl over a period of 4 min. The scalp was closed with wound clips, and recovery was monitored under a heat lamp for 60 min before returning to a home cage.
Histology. Five days after brain injections, animals received overdoses of intraperitoneal anesthetic and were perfused with 60 ml of PBS containing 2 U/ml of heparin followed by 60 ml of 3% formaldehyde in PBS. Brain tissues were then placed in buffered 3% formaldehyde at 4°C overnight and processed for histology. Paraffin-embedded rat brains were cut into coronal serial sections (8 μm thickness) and stained using a modified degenerative stain (Fink and Heimer, 1967) or with cresyl violet. Quantitative measures of neuron injury were performed with cresyl violet-stained CA3 cells obtained from sections 100 μm rostral and caudal to the needle tract. Because the sizes of healthy and pyknotic cells differed significantly (30.8 vs 19.3 μm mean diameters, respectively, at 5 d postinfusion), correction factors were applied for split elements (Abercrombie, 1946) to the raw counts (section thickness/section thickness + mean cell diameter; the correction factors at 5 d postinfusion were 0.206 for healthy neurons and 0.293 for pyknotic neurons. These corrected counts were then used to derive the percent neuron damage with damage scores defined as: corrected number of pyknotic neurons/corrected number of pyknotic + corrected number of healthy neurons × 100%.
RESULTS
QUIN, a dicarboxylic acid, and NTox, a neurotoxic amine, are two agents thought to contribute to the neuronal pathology observed during HIV-1 infection. Although both of these factors readily destroyed ciliary or hippocampal neurons in vitro, culture conditions did influence neurotoxic potencies in different ways. QUIN, for example, showed an ED50 of ∼25 nm with ciliary neurons grown in chemically defined culture media that increased to 30 μmwhen medium was supplemented with dialyzed 10% newborn calf serum. NTox, in contrast, was a more potent agent with an ED50 ∼20 pm, and remained unaffected by the presence of serum (Fig.1A). QUIN killing of hippocampal neurons (grown atop a feeder layer of astroglia) required micromolar concentrations (ED50 = 10 μm), whereas NTox readily destroyed neurofilament(+) MAP-2(+) hippocampal cells in the low picomolar range (ED50 = 50 pm; Fig.1B). Neuron killing by both QUIN and NTox was blocked by 10 μm of AP7 or MK-801, antagonists to the NMDA receptor channel complex (Fig. 1C,D).
Fig. 1.
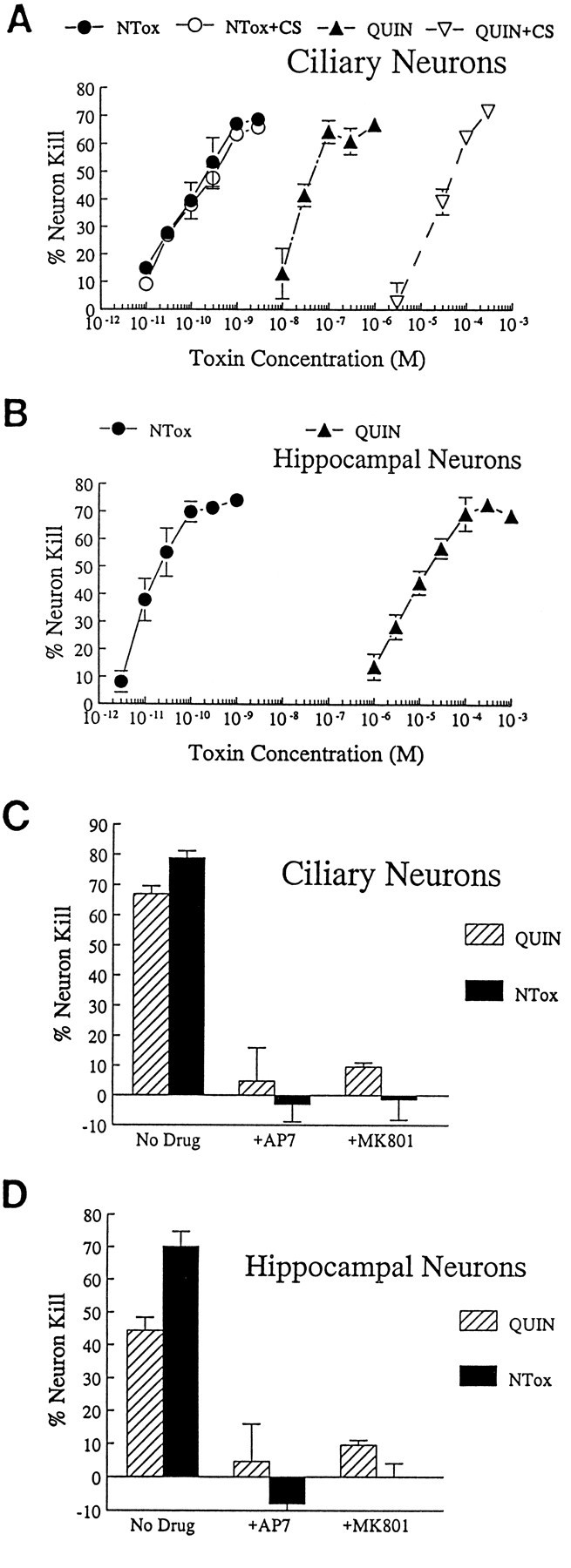
A, Dose–response curves showing relative potency of NTox and quinolinic acid (QUIN) after exposure to ciliary neuron cultures for 48 hr. NTox demonstrates greater neuron-killing capacity with ED50 values estimated in <100 pm. In contrast, quinolinic acid shows similar effects at ∼20 nm. QUIN toxicity was further affected by the presence of 10% dialyzed newborn calf serum (CS), which decreased its ED50 to ∼30 μm. Similar effects were not observed for NTox. B, Dose–response curves show ∼100,000-fold difference in potency for NTox and QUIN using embryonic hippocampal neurons as targets.C, D, Both neurotoxins are inhibited in either culture system by 10 μm of the NMDA receptor blockers AP7 or MK-801 (MK801). Concentrations of 1 μm QUIN and 1 nm of NTox were used.
Neurotoxins released by HIV-1-infected cell lines
Earlier studies (Giulian et al., 1990, 1993a) have shown that human monocytic cell lines infected with HIV-1 released NTox (Fig.2A). To determine whether these cells secreted other neurotoxic agents, we monitored QUIN in conditioned media from HIV-1(+) THP-1 and U937 cells. Using both radioenzymatic and GC/MS detection methods (Fig. 3), we found that there were no significant differences in the amount of QUIN released by infected and noninfected cells (Table 1). There was no correlation between the presence of neurotoxic activity in culture media and the measured levels of QUIN (Fig. 2B). Because QUIN bound to strong anionic exchangers such as DOWEX-2, and NTox bound to strong cationic exchangers such as SP-Sephadex, it was possible to show that neurotoxic activity in culture media was entirely attributable to NTox (Fig. 2A).
Fig. 2.
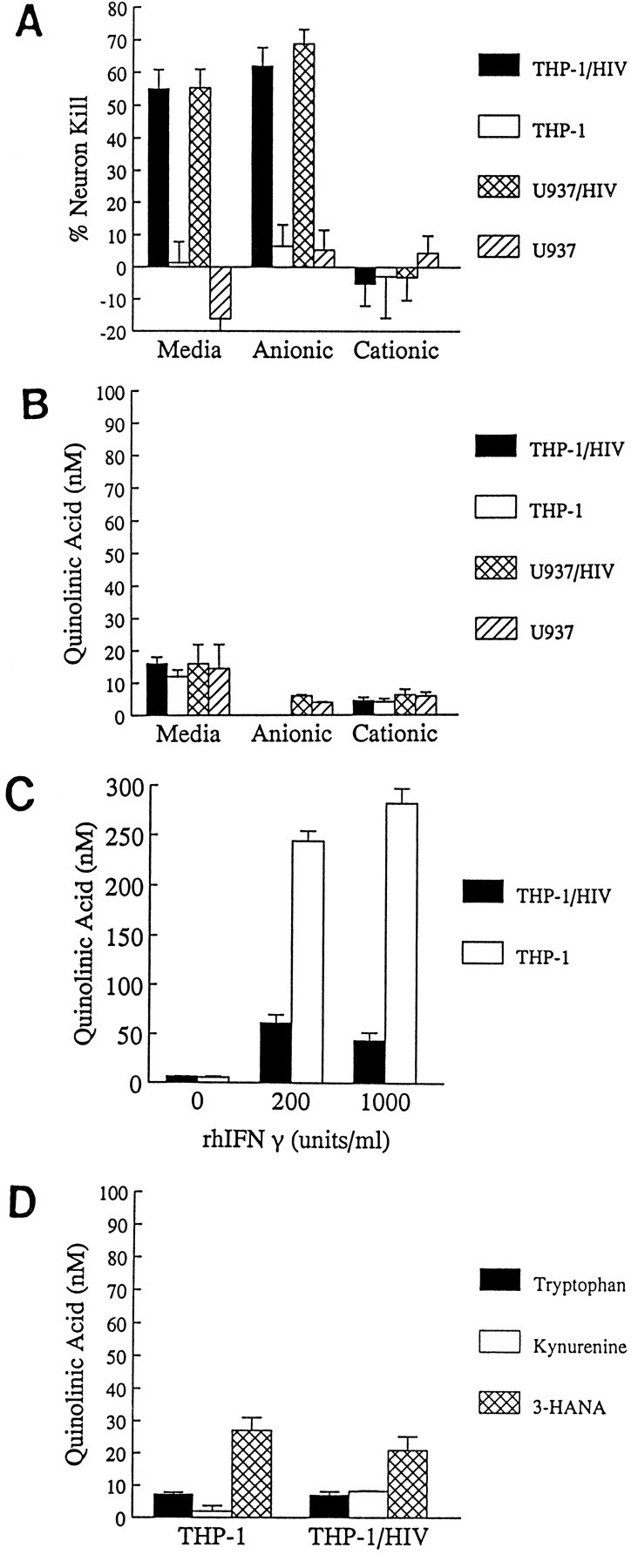
A, B, Ionic exchange chromatographic fractionation of neurotoxins found in cell culture media. Media conditioned by THP-1 or U937 cells (106 cells/ml) for 48 hr with or without HIV-1 infection were fractionated by strong anionic (DOWEX-2) or cationic (SP-Sephadex) exchangers to separate NTox from quinolinic acid. Nearly all the neurotoxic activity recovered from infected cells is NTox, which binds to the cationic exchange resin but not to the anionic exchanger. The same amounts of QUIN found in these fractions do not correlate to measured neurotoxic activity. C, THP-1 exposed to high concentrations of IFNγ to induce indoleamine 2,3-dioxygenase show production of modest levels of quinolinate, whereas cells infected with retrovirus show far less of a response. D, QUIN production by cells in the presence of 2.5 mmtryptophan (t = 24 hr exposure), 50 μm kynurenine (t = 24 hr), or 10 μm 3-HANA (t = 2 hr) remains unaffected by HIV-1 infection.
Fig. 3.
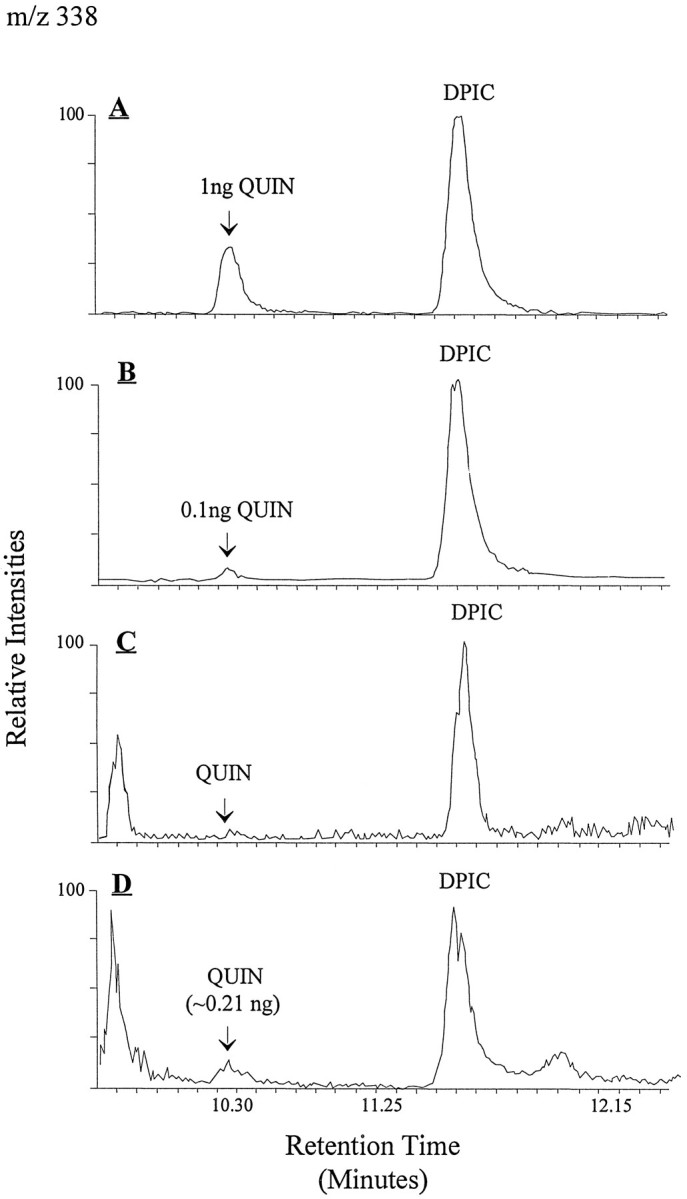
To estimate recovery of QUIN from culture media by GC/MS, 3 ml of culture media were mixed with 3, 10, 30, and 100 ng of_QUIN_ plus 100 ng dipicolinic acid (DPIC) in each sample to serve as the internal standard. After column fractionation and lyophilization, samples then received 100 ng of pyridine 2,4-dicarboxylic acid as an external standard. Both results showed good linear correlation with the actual amounts of QUIN added (correlation coefficients of 0.996 and 0.987, respectively). The percent recovery of QUIN for the entire protocol including column fractionation was estimated at 83 ± 11% (determined by dividing amounts of QUIN measured by the external standard method by those measured by the internal standard method), which was in good agreement with the 85 ± 5% recovery estimated by 3H-QUIN. A, Chromatographic separation of silylated QUIN and of DPIC from standard solutions (1 ng QUIN and 2 ng DPIC in 2 μl injected volume).B, A similar chromatogram shows the sensitivity of the GC/MS at 0.1 ng QUIN with a signal-to-noise ratio of >10. C, Control sample from 3 ml of N2 culture media with no detectable QUIN.D, N2 media conditioned by THP-1 cells infected by HIV-1 for 48 hr (106 cells/ml).
Table 1.
Quinolinic acid release by HIV-1 infected cells
| Radioenzymatic detection | GC/mass spectrometric detection | |
|---|---|---|
| THP-1 | 13.0 ± 3.7 nm (n = 6) | 13.9 ± 0.9 (3) |
| THP-1/HIV | 13.6 ± 2.3 nm (7) | 15.9 ± 2.3 (3) |
| H9 | 7.7 ± 2.1 nm (4) | 17.9 ± 1.7 (2) |
| H9/HIV | 6.2 ± 0.9 nm (4) | 10.8 ± 2.0 (2) |
| U937 | 8.8 ± 3.6 nm (5) | 16.8 ± 9.4 (2) |
| U937/HIV | 9.0 ± 2.0 nm (7) | 13.8 ± 1.8 (2) |
QUIN is a metabolite of tryptophan and appears as an intermediary product of the kynurenine degradation pathway. It has been proposed that HIV-1 infection of mononuclear cells might alter tryptophan or kynurenine metabolism to increase QUIN production (Heyes et al., 1992). Although we did not detect elevations of QUIN in mononuclear cells infected with virus, it remained possible that alternative culture conditions might boost the biosynthetic capacity by infected cells. An initial step in the QUIN pathway involves the flavin-dependent enzyme indoleamine 2,3-dioxygenase (IDO), which converts tryptophan to_N_-formylkynurenine. Because the cytokine interferon γ (IFNγ) can induce IDO, it might also accelerate production of QUIN (Saito et al., 1993). We found that concentrations of IFNγ in the physiological range (<100 units/ml) did not stimulate THP-1 or U937 cells to produce QUIN. Very high cytokine concentrations (≥200 units/ml of IFNγ), however, did elicit QUIN (Fig. 2C), but not from HIV-1-infected cells (Fig. 2C). We also incubated infected and noninfected monocytic cells with various QUIN precursors, including tryptophan, kynurenine, and 3-hydroxyanthranilic acid (3-HANA). Media supplementation of tryptophan or its metabolites did not alter the ability of HIV-1-infected cells to produce QUIN (Fig.2D). Overall, these experiments showed that QUIN production by infected mononuclear phagocytes remained low despite IFNγ stimulation or excess concentrations of biosynthetic precursors.
Infected blood cells release neurotoxins
Although cultured monocytic cell lines released neuron poisons after exposure to HIV-1 in vitro, it remained uncertain whether mononuclear cells actually produced neuron-killing agents within HIV-1-infected individuals. To address this question, we monitored freshly isolated blood cells collected from volunteers with and without HIV-1 infection for the ability to produce neurotoxins. Enriched preparations of monocytes or lymphocytes were coded and placed in culture for 72 hr (106 cells/ml); media conditioned by these cell preparations were then screened for neurotoxic activity using cultured neurons as targets. Monocytes from the blood of HIV-1-infected adults produced neuron-killing activity that was dose-dependent and was not found among blood cells of normal adult volunteers (Fig. 4A–C). From randomized, coded samples, we found AP5-sensitive neurotoxic activity in 16 of 16 blood monocyte cultures from HIV-1-infected individuals but none in lymphocyte cultures prepared from the same individuals. Importantly, coded monocytes isolated from normal individuals (0/6) were free of neuron poisons (Fig. 4A,C). In all samples tested, the neurotoxic activity released by HIV-1-infected monocytes was identified as NTox by ion exchange chromatography and could be separated from QUIN. The estimated ED50 for media conditioned by monocytes of HIV-1(+) individuals (106 cells/ml for 72 hr) was 3% ± 0.5% volume in the ciliary neuron assay with neurons grown in the presence of serum.
Fig. 4.

A, Blood monocytes from HIV-1-infected donors release neurotoxic factors. Photomicrographs of E18 rat hippocampal neurons (immunolabeled for neurofilament and MAP-2) show that neurons thrive in the presence of conditioned media from monocytes of normal volunteers (A; 10% by volume), whereas a significant loss of neurons occurred in cultures exposed to media from monocytes of an HIV-1-infected individual (B; Scale bar, 20 μm). C, Graph shows the dose-dependent killing of neurons by media conditioned by monocytes isolated from infected [HIV(+)] or uninfected [HIV(−)] individuals. D, Blood cells isolated from an HIV-1-infected donor show that increasing cell number increases the amount of NTox produced, but has no effect on the measured levels of QUIN. Blood cells from a normal donor [HIV(−)] have no significant neurotoxic activity. The high basal levels of QUIN found in these culture media come from the supplements (fetal bovine sera, giant cell derived growth factors) used to maintain productive HIV-1 infection among human blood cells.
To enhance cell survival and to promote productive viral infection, supplements of fetal bovine sera and GCT cell-derived growth factors variably raised the basal level of QUIN in leukocyte culture media between 1 and 5 μm. In general, QUIN production by these human blood preparations were below QUIN levels intrinsic to the culture media. Increasing the number of HIV-1(+) mononuclear cells in culture increased the amount of NTox released without elevating QUIN concentrations above background media levels (Fig. 4D). Based on these experiments, we concluded that essentially all NMDA-like neurotoxic activity released by blood cells cultured from HIV-1(+) individuals was attributable to NTox.
Neurotoxic agents released by HIV-1-infected brain cells
To determine whether mononuclear brain cells also had the ability to secrete neurotoxins, we isolated microglia from human neocortex. As reported (Giulian et al., 1995a), gray matter from normal adult human brain provided highly enriched preparations of viable microglia (>98% homogeneity) if obtained within a 6 hr post mortem interval. These cells, when isolated from normal brain, were CD4(+) and covered with spinous projections (Fig. 5A,D). Although normal brain-derived microglia did not show spontaneous release of neuron poisons (Fig. 5B), these cells become neurotoxic after infection with HIV-1 in vitro or after exposure to 1 nm gp120 (Fig. 5B). No QUIN levels above basal concentrations were found in culture media conditioned by infected or gp120-stimulated human microglia (Fig. 5C). As recently described (Giulian et al., 1995b), mononuclear phagocytes isolated from brains of HIV-1-infected individuals consisted of mixed-cell populations, including both microglia with spinous morphology (Fig. 5D) and blood-borne macrophages with ruffled surfaces (Fig. 5E). [These ruffled cells were estimated to make up >5% of the total brain mononuclear phagocyte population recovered from infected CNS (Giulian et al., 1995b).] Unlike mononuclear phagocytes from normal brain, cells derived from HIV-1-infected brain released NTox, which was detected within 3 d of culture (Fig. 5F). There was no significant QUIN production by these same brain-cell preparations.
Fig. 5.

A, Photomicrograph of human microglia isolated from normal adult human brain demonstrates the presence of CD4 surface receptor by immunoperoxidase staining. CD4 receptor provides the site for HIV-1 infection of mononuclear cells. B, The neurotoxic activity of human microglia isolated from normal brain can be elicited by exposure to 1 nm gp120 or by_in vitro_ infection with HIV-1. The mock infection control (HIV-1 control) or gp120 alone (gp120 only) do not cause neuron damage. Fractionation by ion exchange chromatography confirmed that such neurotoxic activity is entirely attributable to NTox. C, In contrast to NTox, quinolinate acid production is not altered in human microglia by exposure to gp120 or HIV-1.D, Electron photomicrograph of scanning EM that shows a microglial cell isolated from human brains after a rapid autopsy. Human microglia are process-bearing and covered with spines (2500× magnification). E, In addition to microglia, HIV-1-infected brains also contain invading macrophages, which can be identified by ruffled surfaces (2500× magnification). F, Mononuclear phagocytes isolated from HIV-1-infected brain (106 cells/ml) show increasing amounts of neurotoxic activity over time. Under identical conditions, microglia from normal brains did not release neuron-killing factors (data not shown).
Neurotoxins found within HIV-1-infected brain tissues
Because brain cells released NTox after HIV-1 infection, we anticipated that this same neurotoxic factor could be recovered from HIV-1-infected brain tissues. As shown in Figure 6, ultrafiltrates from viral-infected, but not normal, gray matter contained NTox. Biochemical properties, including stability after acid hydrolysis, binding to cationic exchange resins, pH-dependent extraction into ethyl acetate, and copurification using RP-HPLC, confirmed that NTox released by infected cells in culture was indistinguishable from the toxin extracted from the CNS (Fig.6C; see below).
Fig. 6.
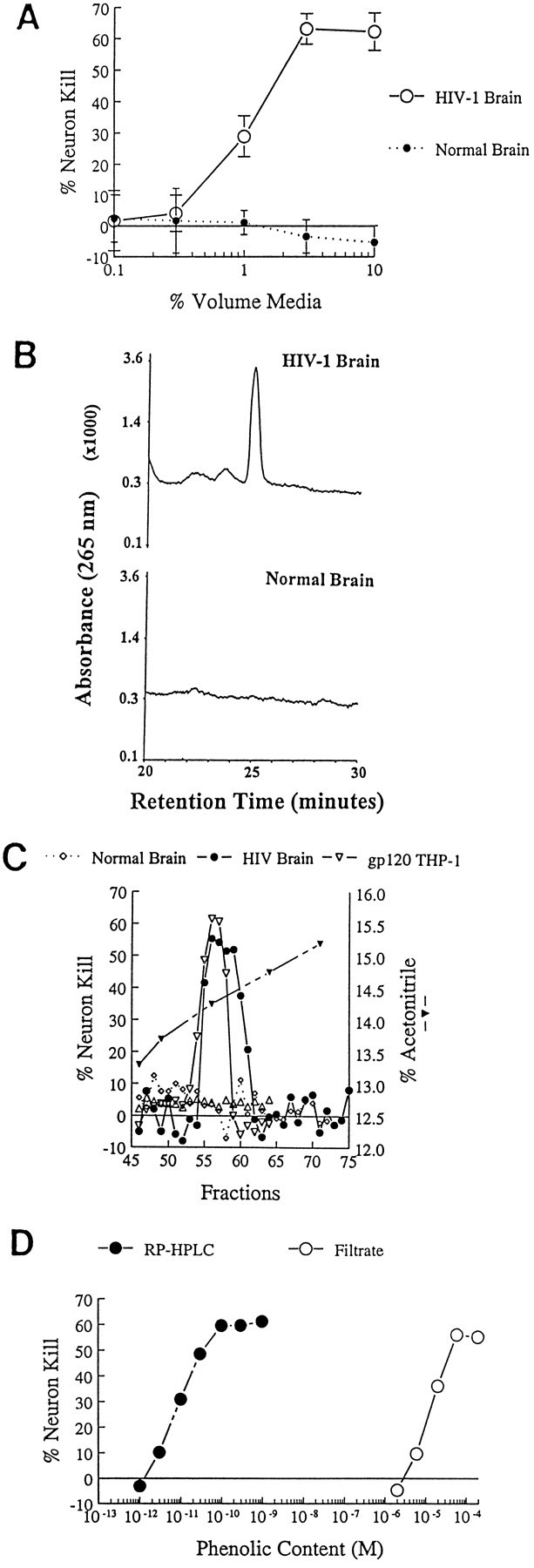
A, Characterization of neurotoxic activity found in ultrafiltrates from HIV-1-infected brain tissue. Aqueous extracts (10 vol of H20 per gram of tissue) of HIV-1-infected gray matter obtained at autopsy were separated by ultrafiltration (cutoff 1000 Da). Biological assays show that ultrafiltrates from HIV-1-infected (HIV-1 Brain), but not from normal control brains (Normal Brain), contain neurotoxic activity that destroys rat hippocampal neurons in a dose-dependent manner. B, Pooled neocortical gray matter from HIV-1-infected brain (HIV-1 Brain) or Normal Brain were processed as described in Materials and Methods using pH-dependent organic extractions, acid hydrolysis, and sequential C18 RP-HPLC. As shown here, a distinctive peak at ∼25 min retention time (UVmax of 265) is found in the HIV-1-infected, but not control, tissues. C, The UV peak shown in_B_ was fractionated further by RP-HPLC using a gradient of acetonitrile. As shown, microfractions (100 μl each) of highly purified NTox found in HIV-1-infected brain coeluted with NTox released by gp120-stimulated THP-1 cells (gp120 THP-1). No biological activity was recovered from identical fractions of normal brain.D, Dose–response curves show that the ED50 = 10 μm in the ultrafiltrate (Filtrate) compared with 10 pm for highly purified NTox after C18 RP-HPLC. From such preparations, we estimate >100,000-fold purification. The phenolic content was estimated by UVmax at 265 nm against a standard curve of tyramine. A similar result was obtained when values were normalized to amine content measured against a tyramine curve using the fluorescamine method.
As noted, HIV-1-infected human cell lines, blood monocytes, or brain microglia did not release significant amounts of QUIN. Moreover, brain tissue levels of QUIN were nearly identical in samples tested (infected brain ultrafiltrates containing 30 ± 5 nm QUIN and normal brain ultrafiltrates containing 25 ± 4 nm). Such results were puzzling in view of the numerous reports describing an association between QUIN and AIDS dementia (Heyes et al., 1989; Achim et al., 1993). To address this problem, we returned to the original observation that HIV-1 infection caused QUIN elevations in CSF. We found significantly higher mean concentrations of CSF QUIN (208 ± 31 nm;n = 66) for HIV-1(+) donors when compared with donors with multiple sclerosis (MS; 73 ± 6 nm QUIN;n = 20; Student’s t test; t = 2.31; df = 84; p = 0.023). Further analysis revealed that HIV-1(+) individuals with or without neurological dysfunction had QUIN concentrations in the CSF above those associated with MS (Fig.7A). In a similar way, plasma of donors infected with HIV-1 contained 1406 ± 127 nm QUIN (n = 63), which was significantly higher than levels found in MS donors (677 ± 89; n = 20; Student’s _t_test; t = 3.14; df = 81; p = 0.002; Fig.7B). Notably, there was a striking correlation between CSF elevations of QUIN and CSF elevations of albumin (Fig. 7C;r = 0.722; n = 63; p < 0.0001), i.e., the highest concentrations of CSF QUIN were found in those HIV-1-infected individuals with the greatest CSF elevations in a serum protein. The abnormally high concentrations of CSF albumin found in HIV-1(+) donors indicated defects in the blood–brain barrier as noted by other investigators (Petito and Cash, 1992). Taken together, our observations suggest that QUIN elevations in CSF are the result of leakage across the blood–brain barrier, and not enhanced production by HIV-1-infected mononuclear phagocytes.
Fig. 7.
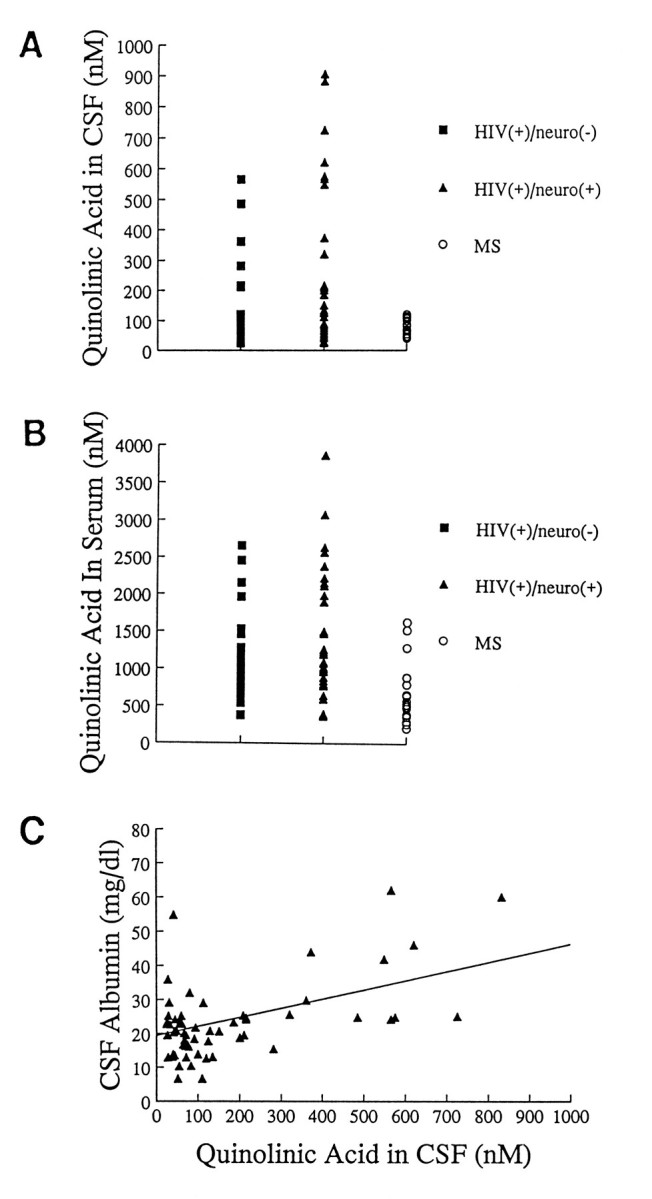
Measurement of quinolinic acid in donors with HIV-1 infection or multiple sclerosis (MS).A, Individual concentrations of QUIN found in CSF of HIV-1(+) donors with [_neuro(+)_] or without [_neuro(−)_] neurological abnormalities. As shown, higher values are noted in both categories of infected donors when compared with MS controls. Mean CSF QUIN concentrations in HIV-1 groups were 126 ± 26 nm for neuro(−) and 268 ± 50 nm for neuro(+) compared with 74 ± 6 nm for the MS group. B, Mean serum QUIN concentrations are higher for HIV-1-infected donors than for those measured in the MS group. [mean scores for serum QUIN were 1249 ± 170 nm for neuro(−); 1530 ± 180 for neuro(+); and 677 ± 90 nm for MS]. C, Correlation of mean CSF albumin concentration with mean QUIN concentrations found in HIV-1-infected donors. As shown, increasing amounts of QUIN correspond to increasing amounts of albumin (r = 0.722;n = 63; p < 0.0001). Such data suggest that defects in blood–brain barrier account for elevations of both QUIN and a serum protein.
Properties of brain-derived neurotoxin
To characterize NTox found in HIV-1-infected CNS further, we dispersed frozen gray matter by sonication (1:10 wt/vol in dH2O) and removed particulate material by centrifugation. Bulk purification was then performed by ultrafiltration of the soluble fraction, by extractions into ethyl acetate at pH 10.5, by acid hydrolysis, and by sequential gradient chromatography using RP-HPLC. A peak detected by UV at 265 nm eluted at ∼14% acetonitrile from C18 RP-HPLC and contained all recovered neurotoxic activity (Fig.6B,C). Importantly, this peak of biological activity was estimated to be >100,000-fold enriched when compared with brain ultrafiltrates (Fig. 6D) and was not detected in identical fractions recovered from normal brain (Fig. 6B,C). Dose–response curves indicated that brain-derived NTox was capable of destroying cultured neurons in low picomolar concentrations (ED50=20 pm; Fig.6D). The recovery of ∼10 pmol of NTox per gram of HIV-1-infected gray matter suggested that cell-damaging levels of this agent might be achieved within brain tissues during the course of viral infection.
NTox recovered from human brain has a mass <1000 Da and a lipophilic quality that is pH-sensitive (extraction into ethyl acetate at increasingly alkaline pH); step-gradient extractions with pH increasing from 8.0 to 11.0 suggested an isoelectric point between 9.5 and 10.5. Copurification studies confirmed that NTox from HIV-1-infected human brain was identical to that from HIV-1-infected cell lines (Fig. 6C). Acetylation with acetic anhydride or derivatization with PFPA indicated the presence of -NH3 groups, whereas inactivation by plasma amine oxidase uncovered terminal amines (Fig.8A,B). There were no apparent -COOH groups, because NTox was resistant to esterification with acidified butanol (Fig. 8A); NTox was not sensitive tol-amino acid oxidase or lipase (Fig.8B) arguing against simple amino acids or lipids as the toxic factor. Resistance to acid hydrolysis (24 hr, 105°C in 6N HCl) ruled out a number of molecules, including proteins and peptides, as the neuron-killing agent. Although unaltered by mild reduction with palladium, NTox did lose activity after hydrogenation by rhodium, which suggested the presence of double-bond structures such as phenolic or pyridine rings (Fig. 8A). Inactivation of toxic activity by both polyphenol oxidase (pH 6.5, 37°C, 30 min) and methylation with diazomethane favored the presence of phenols (Fig. 8B). Although a class of potent polyamine amide neurotoxins (Fig.8C) has some biological and structural properties in common with NTox, these venoms produced by wasps and spiders require peptide bonds for biological activity (Asami et al., 1989). Despite the fact that chemical and enzymatic modifications indicated NTox to be a phenolic amine with lipophilic properties (Fig. 8C), additional work will be needed to determine whether NTox, in fact, represents a new class of mammalian-derived neurotoxin.
Fig. 8.
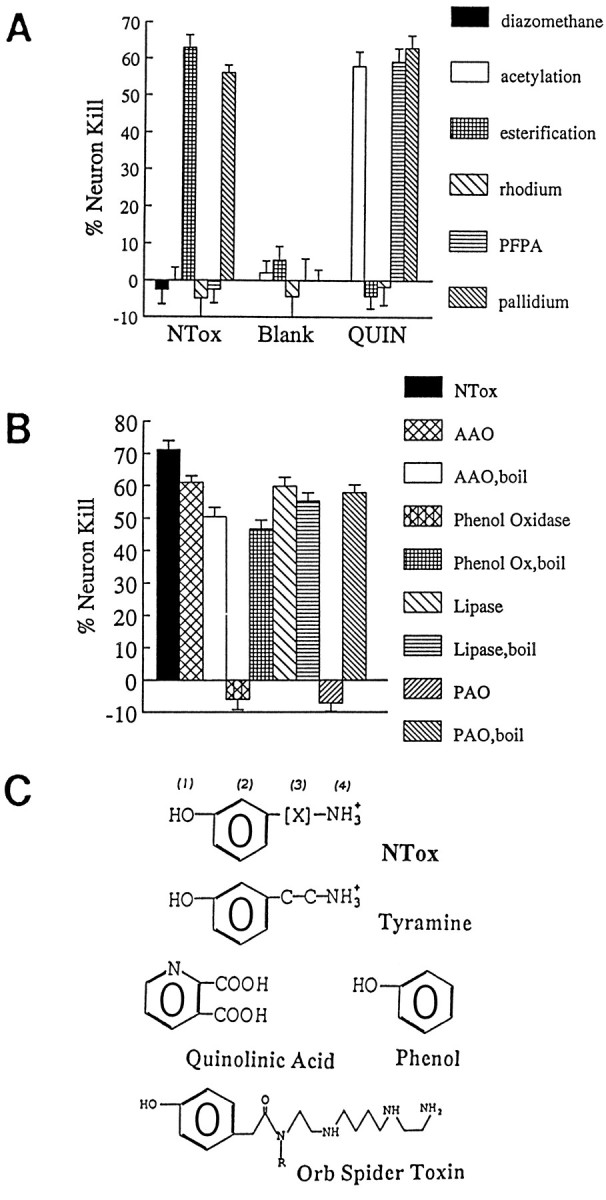
A, Chemical modifications of NTox indicate that amino groups sensitive to acetylation by acetic anhydride or derivatization by PFPA are present, whereas carboxyl groups sensitive to butanol esterification are not. Weak reduction by palladium did not alter neurotoxic activity, whereas strong reducing conditions with rhodium or methylation by diazomethane eliminated neuron killing. These data suggested the presence of double-bonded ring structures. B, Enzymatic treatments showed NTox sensitivity to polyphenol oxidase and plasma amine oxidase (PAO) but not to l-amino acid oxidase (AAO) or lipase. These data suggest that a phenolic ring and terminal amine are required for toxic activity. As a control, enzymatic activity was eliminated by immediate boiling (boil) to confirm specificity of action on the neurotoxin. C, Structural features of NTox. Important features that distinguish NTox from other potent neu-rotoxins include the lack of carboxyl groups (as found in quinolinic acid), the presence of amine (lacking in phenol), and the lack of peptide bonds (as found in orb spider venom). (1), Hydroxyl group on ring structure suggested by sensitivity to polyphenol oxidase and diazomethane; (2), ring structure suggested by rhodium reduction, UVmax, and polyphenol oxidase inactivation; (3), lipophilic region indicated elution with RP-HPLC and organic extractions; (4), amine group indicated by inactivation with acetylation, PFPA, and plasma amine oxidase.
Brain-derived neurotoxin acts on NMDA receptor-bearing neurons
NTox destroyed cultured neurons bearing the NMDA receptor (Fig. 9A) which, as noted above, could be blocked by NMDA antagonists. Because of the potential for therapeutic interventions, we sought to define the relationship of NTox to the NMDA receptor by comparing the ability of specific drugs, which act at select sites of the receptor complex, with block neurotoxicity. Highly purified NTox was applied to neuron cultures to establish a dose curve with a saturating response; we then examined the effects of increasing concentrations (ranging from 1 nm to 10 μm) of AP5 (NMDA receptor site) and MK-801 (NMDA channel site) on the general shape of the established toxicity response curve. As shown in Figure 9, AP5 shifted the ED50 of NTox, whereas MK-801 did not. Such data suggested that the neurotoxin competed with the NMDA receptor site as shown by a shift of the neuron-killing response to the right; in contrast, noncompetitive interactions between the neurotoxin and MK-801 altered saturation of responses (Goldstein et al., 1974). A plot of ratios of ED50 values (Arunlakshana and Schild, 1959) showed a linear relationship for AP5, again implicating a direct competition between NTox and AP5 at a common site of binding (Fig.9E).
Fig. 9.

A, NMDA receptor(+) ciliary neurons survive when grown in the presence of media conditioned by monocytes (10% volume) from a normal volunteer; in contrast, there is a marked loss of NMDAR1(+) neurons (B) when exposed to media from an HIV-1-infected individual. Scale bar, 7 μm. C, D, NTox interactions with the NMDA receptor. Using ultrafiltrate from HIV-infected brain, we examined its toxic effects on ciliary neurons with increasing concentrations of AP5, an NMDA receptor antagonist, or MK-801 (MK801), an NMDA channel blocker. As shown, increasing amounts of AP5 shift dose responses to the right, producing ever larger ED50 values with no change in saturating responses. Such a pattern suggests there is a direct competition between NTox and AP5 to elicit cell death. Although increasing concentrations of MK-801 block toxicity, the degree of the responses are increasingly reduced, suggesting a noncompetitive relationship between NTox and MK-801. E, A Schild analysis for AP5 curves indicates a linear relationship, again indicative of competition between neurotoxin and AP5. (The Schild analysis assumes an equilibrium is reached among agonists and receptors that might not occur in a toxicity assay with ongoing loss of cells.)
Actions of HIV-1 neurotoxin in vivo
If NTox, in fact, played a role in the pathogenesis of human brain injury, it should also demonstrate biological action when infused into the brain. The rationale for hippocampal injections into rats was based on our observations that NMDA receptor(+) hippocampal neurons were particularly sensitive to the toxin in vitro. After infusion of NTox (derived from gp120-stimulated THP-1 cells), large numbers of pyknotic and dying pyramidal neurons were apparent 5 d later in the CA1 and CA3 regions of the hippocampus (Fig. 10). Little cell damage in the hippocampus was produced by injection of an identical, highly purified fraction from unstimulated THP-1 cells (Fig.10). Study of animals infused with NTox (100 pmol) showed a uniform pattern of pyknotic neurons (Fig. 10E) within 100 μm (rostral to caudal) to either side of the injection site. Damage occurred primarily among pyramidal neurons in the CA1 and CA3 regions with sparing of the dentate granule cells and CA2. By contrast, a 60 nmol infusion of QUIN showed total loss of pyramidal cells in CA1 and CA3 without pyknosis. No seizure activity was noted after NTox infusion. We estimated that 100 pmol of NTox produced >65% loss of CA3 neurons 5 d after stereotaxic infusion compared with about a 10% cell loss after injections of identical nontoxic fraction (Fig.11). One nmol of AP5 infused before NTox protected CA3 neurons (Fig. 11), confirming that NTox targeted NMDA receptor-bearing neurons in vivo.
Fig. 10.
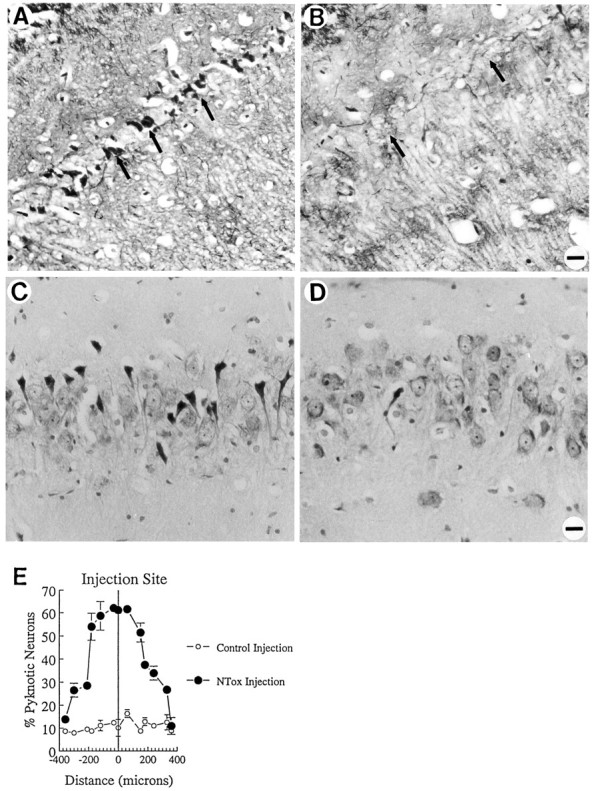
NTox infused directly into rat hippocampus kills neurons in vivo. Five days after injections, neurons most sensitive to NTox included the pyramidal cells of the CA1 and CA3 regions. A, Fink–Heimer degeneration stain of CA1 neurons after NTox infusion (purified from media conditioned by gp120-stimulated THP-1 cells). B, In contrast, contralateral hippocampus infused with a nontoxic control fraction (recovered from media conditioned by unstimulated THP-1 cells) showed little cell damage. C, Cresyl violet-stained rat hippocampus (CA3 region) after stereotaxic injections also showed far more dying, pyknotic neurons after infusion of NTox when compared with the contralateral hippocampus control infusion (D; Scale bar, 40 μm). E, Measurement of cell loss in the CA3 region after infusion of NTox in serial sections (8 μm thick) rostral and caudal to the site of injection. As shown, ∼60% neuronal loss appeared within 150 μm of the site of toxin infusion. One microliter of artificial CSF with or without NTox was injected over a 4 min period at 2.9 mm from the brain’s surface, at a distance of 4.5 mm caudal to bregma, 3.0 mm lateral to the midline into the hippocampus. We estimate ∼100 pmol of NTox were contained in the 1 μl fluid volume injected into the hippocampus.
Fig. 11.

The in vivo action of NTox can be blocked by preinfusion of 1 nmol of AP5 prepared in 1 μl artificial CSF (A) before NTox injection. In contrast, a control preinfusion with 1 μl artificial CSF did not block hippocampal injury as shown in B. Quantitative measure of CA3 neuronal damage (C) shows a 60–70% after-NTox infusion with ∼10% pyramidal cell loss seen in control injections (Student’s _t_test; p < 0.0001). Blockade with AP5 reduces cell loss to control levels. Cell counts were obtained at 100 μm rostral and caudal to the site of injection from 8-μm-thick sections stained with cresyl violet.
DISCUSSION
The mechanisms by which mononuclear phagocytes attack neurons after HIV-1 infection are thought to involve neuron-selective toxins (Giulian, 1990; Gendelman et al., 1994). Because NMDA receptor-bearing neurons play a role in cognition and memory, it has been tempting to connect perturbations in NMDA neurotransmission with dementia (Lipton, 1992b). Two NMDA receptor-mediated neurotoxic agents, QUIN and NTox, have been linked to HIV-1 infection through clinical observations (Heyes et al., 1989, 1991) or in vitro modeling (Giulian et al., 1990, 1993a). To explore the role of these two toxic molecules in neuro-AIDS, we determined whether their stimulated production occurred in mononuclear cells isolated from HIV-1-infected individuals. Although blood-derived or brain-derived infected mononuclear phagocytes did not produce excessive amounts of QUIN, we did find marked production of NTox by both blood monocytes and microglia recovered from infected donors. Biochemical studies revealed NTox to be a phenolic amine lacking both peptide bonds and carboxyl groups. Overall, NTox found in HIV-1-infected brain was indistinguishable from the microglia-derived or monocyte-derived NTox recovered from culture media as indicated by identical elution profiles with ultrafiltration, ion chromatography, organic extractions, and reverse-phase HPLC. Importantly, NTox concentrations found in HIV-1-infected CNS were similar to those levels capable of inflicting hippocampal damage when infused into animals.
As described here, NMDA receptor blocking agents have neuroprotective effects against NTox in vitro, and an NMDA receptor antagonist prevented NTox-induced damage to pyramidal hippocampal neurons in vivo. A comparison between AP5 and MK-801 suggested further that the neuronal injury brought about by NTox in culture involved a direct receptor interaction at the level of the NMDA binding site. These observations, together with dose–response comparisons with QUIN, suggest that NTox is among the most potent of the known NMDA receptor-dependent neuron poisons. Caution, however, must be applied to this interpretation because the experiments reported here involved a multicellular assay system in which the fate of the toxic agent (uptake vs metabolism) was unknown. The structural model for NTox based on functional group analyses by chemical and enzymatic modifications indicated some similarities between NTox and polyamine amides found in orb spider and wasp venoms. Although NTox has functional dependent phenolic groups and a terminal amine, it lacked peptide bonds found in all known polyamine amide venoms. Thus, although similarities exist between the polyamine amides and NTox, the agents were not identical. Other differences between NTox and polyamine amides might exist. For example, current literature indicates that polyamine-containing agents act on the NMDA channel (Asamin et al., 1989; Jasys et al., 1990; Williams et al., 1990) and not the NMDA binding site as proposed for NTox. A more detailed modeling of NTox/receptor interactions awaits further investigation by direct binding assays; structural studies currently under way will help to determine whether NTox, in fact, represents a new class of mammalian-derived neuron poison.
Other investigators have proposed that HIV-1 infection stimulated release of such mononuclear cell products as nitric oxide, cytokines, platelet activating factor, eicosanoids, and free radicals (Genis et al., 1992; Epstein and Gendelman, 1993). It has not been clear, however, which of these factors were produced in significant amounts in the CNS during HIV-1 infection or which actually contributed to neuronal pathology. Human mononuclear cells, for example, have little capacity to synthesize nitric oxide (Denis, 1994). Although tumor necrosis factor α was readily secreted by activated microglia and was found in HIV-1-infected brains, this cytokine has no capacity to kill neurons in vitro (Giulian et al., 1994). Platelet activating factor (PAF), another potential neurotoxin released by activated monocytes (Kornecki and Ehrlich, 1988), has been found in low concentrations in CSF of a variety of neurological disorders including patients with HIV-1 dementia (Gelbard et al., 1994); it remains uncertain, however, whether neuron-damaging concentrations of PAF were produced in brain tissues. Alternatively, some viral proteins have also been described as neuron poisons. Brenneman et al. (1988) have reported that the HIV-1 envelop glycoprotein gp120 acted as a direct neuronal toxin, a finding later supported by Dreyer et al. (1990). However, because cell culture systems used in these experiments were mixtures of neurons and glia, it was not possible to demonstrate direct toxic effects on neurons independent of microglia. Subsequent work using CNS cultures lacking mononuclear phagocytes showed that gp120 was not toxic to neurons but rather served as a stimulus to induce NTox release from mononuclear cells (Giulian et al., 1993a). Similarly, Lipton (1992a)found that destruction of microglia in retinal cell cultures eliminated the ability of gp120 to kill ganglion cells. With the exception of detergent-like cytotoxic effects of tat at high concentrations (Sabatier et al., 1991), other HIV-1 proteins have no neuron-killing capacity (Giulian et al., 1993a).
The question of QUIN production during HIV-1 infection was addressed here using two different approaches. First, we showed that HIV-1-infected mononuclear phagocytes or microglia did not have an enhanced capacity to produce or release QUIN. In the case of HIV-1-infected THP-1 cells, we also demonstrated that exposure to three bioprecursors of QUIN, i.e., tryptophan, kynurenine, and 3-hydroxyanthranilate, did not cause synthesis beyond control levels. Similar data were also obtained from infected blood monocytes (data not shown). These results must be contrasted with several reports by Heyes and coworkers (1989, 1991), who had provided indirect evidence favoring infiltrating HIV-1-infected macrophages as the source of the often dramatic increases in CSF and brain tissue QUIN levels in AIDS (Heyes et al., 1992). We did confirm elevations in CSF QUIN concentrations during HIV-1 infections, although the increases were far lower in magnitude than those reported previously (Heyes et al., 1989). It should be noted, however, that the HIV-1(+) patients assessed here were volunteer outpatients, and not the severely demented, hospitalized population used in other studies (Heyes et al., 1989, 1992). Importantly, the highest levels of CSF QUIN described here were in those samples with the highest levels of serum albumin. The most likely explanation for this correlation was that both QUIN and albumin entered the CSF through defects in the blood–brain barrier, perhaps the result of previous CNS pathologies such as bacterial infections, parasitic abscesses, or neoplasms. Such defects have been suggested by Petito and Cash (1992) based on increased serum protein deposits in brain tissues of patients with AIDS dementia. The idea that changes in cerebral QUIN concentrations reflect abnormal blood–brain barrier is further supported by the fact that disease states with the highest reported levels of CSF QUIN involved meningeal pathology, such as bacterial, carcinomatous, or fungal meningitides (Heyes and Markey, 1988; Heyes and Lackner, 1990; Heyes et al., 1989, 1992). Although our data do not provide a satisfactory answer regarding the possible participation of raised QUIN levels in the pathophysiology of AIDS, steady exposure to elevated QUIN concentrations could lead to cognitive decline. Moreover, sustained hyperphysiological levels of QUIN (Whetsell and Schwarcz, 1989) might result in enhanced vulnerability of NMDA receptor-bearing cells to indirect excitotoxic insults (Beal et al., 1991). If this were true, it follows that the use of pharmacological agents that inhibit peripheral QUIN synthesis (such as halogenated 3-HANA analogs) would offer a strategy to protect against QUIN elevations during HIV-1 infection (Walsh et al., 1991).
Because clusters of reactive mononuclear phagocytes are considered a hallmark abnormality of HIV-1-infected brains, the release of poisons from such cells would likely be important in the pathogenesis of AIDS dementia. We believe that neurotoxic agents acting on NMDAR(+) cells contribute to the genesis of HIV-1 neuronal pathology. One of these toxins, QUIN, although not produced by HIV-1-infected mononuclear cells in blood or brain, appeared to penetrate the CNS as the result of alterations in the blood–brain barrier. Such defects in blood–brain barrier might be cumulative and reflect recurrent CNS injury over the course of chronic immune suppression and cryptogenic infections (as well as toxoplasmosis, overt bacterial infections, and neoplasms). We believe that the second toxic factor, NTox, is the principal neurotoxic agent released by HIV-1-infected mononuclear phagocytes. Importantly, NTox was extracted directly from HIV-1-infected brain cells and brain tissues. With a neuron-killing potency of >500-fold above that of QUIN (for example, in vitro ED50 = 10 pm vs ED50 = 20 nm), NTox is likely to be a dominant CNS poison. As noted here, both blood monocytes and brain microglia exposed to virus released NTox. Because HIV-1-infected brains are populated by both invading macrophages and reactive microglia (Giulian et al., 1995b), it is likely that both classes of cells elicit neuronal pathology through the chronic release of NTox. What might distinguish HIV-1 infection from other inflammatory brain injuries is the persistence of neurotoxic mononuclear cells among vulnerable neurons.
Footnotes
FNa
We note the loss of a friend and colleague; Dr. Noonan died on Feb. 28, 1995.
The work was supported by Grant MH48652 from the National Institute of Mental Health, by Grant NS25637 from National Institutes of Health, and by the core facilities of the Center for AIDS Research at Baylor College of Medicine.
Correspondence should be addressed to Dr. Dana Giulian, Department of Neurology, Baylor College of Medicine, Houston, TX 77030.
REFERENCES
- 1.Abercrombie M. Estimation of nuclear population from microtome sections. Anat Rec. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- 2.Achim CL, Heyes MP, Wiley CA. Quantitation of human immunodeficiency virus, immune activation factors, and quinolinic acid in AIDS brain. J Clin Invest. 1993;91:2769–2775. doi: 10.1172/JCI116518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol. 1959;14:48–57. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asami T, Kagechika H, Hashimoto Y, Shudo K, Miwa A, Kawai N, Nakajima T. Acylpolyamines mimic the action of Joro spider toxin (JSTX) on crustacean muscle glutamate receptors. Biomed Res. 1989;10:185–189. [Google Scholar]
- 5.Beal MF, Swartz KJ, Hyman BT, Storey E, Finn SF. Aminooxyacetic acid results in excitotoxin lesions by a novel indirect mechanism. J Neurochem. 1991;57:1068–1073. doi: 10.1111/j.1471-4159.1991.tb08258.x. [DOI] [PubMed] [Google Scholar]
- 6.Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- 7.Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenneman DE, Westbrook GL, Fitzgerald SP, Ennist DL, Elkius KL, Ruff MR, Pert PC. Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature. 1988;335:639–642. doi: 10.1038/335639a0. [DOI] [PubMed] [Google Scholar]
- 9.Budka H. Human immunodeficiency virus (HIV)-induced disease of the central nervous system: pathology and implications for pathogenesis. Acta Neuropathol. 1989;77:225–236. doi: 10.1007/BF00687573. [DOI] [PubMed] [Google Scholar]
- 10.Colton CA, Gilbert DL. Production of superoxide anion by a CNS macrophage, the microglia. FEBS Lett. 1987;223:284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- 11.Denis M. Human monocytes/macrophages: NO or no NO? J Leukoc Biol. 1994;55:682–684. doi: 10.1002/jlb.55.5.682. [DOI] [PubMed] [Google Scholar]
- 12.Dreyer EB, Kaiser PK, Offermann JT, Lipton SA. HIV-1 coat protein neurotoxicity prevented by calcium channel antagonists. Science. 1990;248:364–367. doi: 10.1126/science.2326646. [DOI] [PubMed] [Google Scholar]
- 13.Epstein LG, Gendelman HE. HIV-1 infection of the nervous system: pathogenetic mechanisms. Ann Neurol. 1993;33:429–436. doi: 10.1002/ana.410330502. [DOI] [PubMed] [Google Scholar]
- 14.Faulstich ME. AIDS: an overview of CNS complications and neuropsychological sequelae. Int J Neurosci. 1986;30:249–254. doi: 10.3109/00207458608985675. [DOI] [PubMed] [Google Scholar]
- 15.Fink RP, Heimer L. Two methods for selective silver impregnation of degenerating axons and their synaptic endings in the central nervous system. Brain Res. 1967;4:369–374. doi: 10.1016/0006-8993(67)90166-7. [DOI] [PubMed] [Google Scholar]
- 16.Foster A, Okuno E, Brougher D, Schwarcz R. A radioenzymatic assay for quinolinic acid. Anal Biochem. 1986;158:98–103. doi: 10.1016/0003-2697(86)90595-6. [DOI] [PubMed] [Google Scholar]
- 17.Gabuzda DH, Hirsch MS. Neurologic manifestations of infection with human immunodeficiency virus. Ann Intern Med. 1987;107:383–391. doi: 10.7326/0003-4819-107-2-383. [DOI] [PubMed] [Google Scholar]
- 18.Gelbard HA, Nottet HS, Swindells S, Jett M, Dzenko KA, Genis P, White R, Wang L, Choi YB, Zhang D. Platelet-activating factor: a candidate human HIV-1-induced neurotoxin. J Virol. 1994;68:4628–4635. doi: 10.1128/jvi.68.7.4628-4635.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gelman BB. Diffuse microgliosis associated with cerebral atrophy in the acquired immunodeficiency syndrome. Ann Neurol. 1993;34:65–70. doi: 10.1002/ana.410340112. [DOI] [PubMed] [Google Scholar]
- 20.Gendelman HE, Lipton SA, Tardieu M, Epstein L. The neuropathogenesis of HIV-1 infection. J Leukoc Biol. 1994;56:389–398. doi: 10.1002/jlb.56.3.389. [DOI] [PubMed] [Google Scholar]
- 21.Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, Keane RW, Resnick L, Mizrachi Y, Volsky DJ. Cytokines and arachidonic metabolites produced during HIV-infected macrophage-astroglia interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–1718. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giulian D. Brain inflammatory cells, neurotoxins, and acquired immunodeficiency syndrome. Fidia Res Found Symp Ser. 1992a;9:229–234. [Google Scholar]
- 23.Giulian D. Microglia and diseases of the nervous system. Curr Neurol. 1992b;12:23–54. [Google Scholar]
- 24.Giulian D. Microglia and neuron dysfunction. In: Kettenmann H, Ransom B, editors. Neuroglia. Oxford UP; Oxford: 1995. pp. 671–684. [Google Scholar]
- 25.Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci. 1986;6:2163–2178. doi: 10.1523/JNEUROSCI.06-08-02163.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giulian D, Noonan C, Vaca K. HIV-1-infected mononuclear phagocytes release neurotoxins. Science. 1990;250:1593–1595. doi: 10.1126/science.2148832. [DOI] [PubMed] [Google Scholar]
- 27.Giulian D, Wendt E, Vaca K, Noonan C. The envelope glycoprotein of HIV-1 stimulates monocyte release of neurotoxins. Proc Natl Acad Sci USA. 1993a;90:2769–2773. doi: 10.1073/pnas.90.7.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giulian D, Vaca K, Corpuz M. Brain glia release factors with opposing actions upon neuronal survival. J Neurosci. 1993b;13:29–37. doi: 10.1523/JNEUROSCI.13-01-00029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giulian D, Leara B, Keenan C, Li J. Phagocytic microglia release cytokines and cytotoxins that regulate survival of astrocytes and neurons in culture. Neurochem Int. 1994;25:227–233. doi: 10.1016/0197-0186(94)90066-3. [DOI] [PubMed] [Google Scholar]
- 30.Giulian D, Haverkamp LJ, Li J, Karshin WL, Yu J, Tom D, Li X, Kirkpatrick J. Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem Int. 1995a;27:119–137. doi: 10.1016/0197-0186(95)00067-i. [DOI] [PubMed] [Google Scholar]
- 31.Giulian D, Li J, Bartel S, Broker J, Li X. Cell surface morphology identifies microglia to be a distinct class of mononuclear phagocytes. J Neurosci. 1995b;15:7712–7726. doi: 10.1523/JNEUROSCI.15-11-07712.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldstein A, Aronow L, Kalman SM. Wiley; New York: 1974. Principles of drug action. Chap 4, 2nd Ed, pp 301–355. . [Google Scholar]
- 33.Heyes MP, Lackner A. Increased cerebrospinal fluid quinolinic acid, kynurenic acid, and l-kynurenine in acute septicemia. J Neurochem. 1990;55:338–341. doi: 10.1111/j.1471-4159.1990.tb08857.x. [DOI] [PubMed] [Google Scholar]
- 34.Heyes MP, Markey SP. Quantification of quinolinic acid in rat brain, whole blood, and plasma by gas chromatography and negative chemical ionization mass spectrometry: effects of systemicl-tryptophan administration on brain and blood quinolinic acid concentrations. Anal Biochem. 1988;174:349–359. doi: 10.1016/0003-2697(88)90556-8. [DOI] [PubMed] [Google Scholar]
- 35.Heyes MP, Rubinow D, Lane C, Markey SP. Cerebrospinal fluid quinolinic acid concentrations are increased in acquired immune deficiency syndrome. Ann Neurol. 1989;26:275–277. doi: 10.1002/ana.410260215. [DOI] [PubMed] [Google Scholar]
- 36.Heyes MP, Brew BJ, Martin A, Price RW, Salazar AM, Sidtis JJ, Yergey JA, Mouradian MM, Sadler AR, Keilp J. Quinolinic acid in cerebrospinal fluid and serum in HIV-1 infection: relationship to clinical and neurological status. Ann Neurol. 1991;29:202–229. doi: 10.1002/ana.410290215. [DOI] [PubMed] [Google Scholar]
- 37.Heyes MP, Saito K, Crowley JS, Davis LE, Demitrack MA, Der M, Dilling LA, Elia J, Kruesi MJ, Lackner A. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain. 1992;115:1249–1273. doi: 10.1093/brain/115.5.1249. [DOI] [PubMed] [Google Scholar]
- 38.Ketzler S, Weis S, Haug H, Budka H. Loss of neurons in the frontal cortex in AIDS brains. Acta Neuropathol (Berl) 1990;80:92–94. doi: 10.1007/BF00294228. [DOI] [PubMed] [Google Scholar]
- 39.Kornecki E, Ehrlich YH. Neuroregulatory and neuropathological actions of the ether-phospholipid platelet-activating factor. Science. 1988;240:1792–1794. doi: 10.1126/science.3381103. [DOI] [PubMed] [Google Scholar]
- 40.Jasys VJ, Kelbaugh PR, Nason DM, Phillips D, Rosnack KJ, Saccomano NA, Stroh JG, Volkmann RA. Isolation, structure elucidation, and synthesis of novel hydroxylamine-containing polyamines from the venom of the Agelenopsis aperta spider. J Am Chem Soc. 1990;112:6696–6704. [Google Scholar]
- 41.Lipton SA. Requirement for macrophages in neuronal injury induced by HIV envelope protein gp120. NeuroReport. 1992a;3:913–915. doi: 10.1097/00001756-199210000-00023. [DOI] [PubMed] [Google Scholar]
- 42.Lipton SA. Models of neuronal injury in AIDS: another role for the NMDA receptor? Trends Neurosci. 1992b;15:75–79. doi: 10.1016/0166-2236(92)90013-x. [DOI] [PubMed] [Google Scholar]
- 43.Mangos JA, Doran T, Aranda-Naranjo B, Rodriguez-Escobar Y, Scott A. Pediatric AIDS: clinical presentation and diagnosis. Tex Med. 1989;85:32–34. [PubMed] [Google Scholar]
- 44.Masliah E, Ge N, Achim CL, Hansen LA, Wiley CA. Selective neuronal vulnerability in HIV encephalitis. J Neuropathol Exp Neurol. 1992;51:585–593. doi: 10.1097/00005072-199211000-00003. [DOI] [PubMed] [Google Scholar]
- 45.Navia BA, Cho E-S, Petito CK, Price RW. The AIDS dementia complex II: neuropathology. Ann Neurol. 1986a;19:525–535. doi: 10.1002/ana.410190603. [DOI] [PubMed] [Google Scholar]
- 46.Navia BA, Jordan BD, Price RW. The AIDS dementia complex. I. Clinical features. Ann Neurol. 1986b;19:517–524. doi: 10.1002/ana.410190602. [DOI] [PubMed] [Google Scholar]
- 47.Petito CK, Cash KS. Blood-brain barrier abnormalities in the acquired immunodeficiency syndrome: immunohistochemical localization of serum proteins in postmortem brain. Ann Neurol. 1992;32:658–666. doi: 10.1002/ana.410320509. [DOI] [PubMed] [Google Scholar]
- 48.Petralia RS, Yokotani N, Wenthold RJ. Light and electron microscope distribution of the NMDA receptor subunit NMDAR1 in the rat nervous system using a selective anti-peptide antibody. J Neurosci. 1994;14:667–696. doi: 10.1523/JNEUROSCI.14-02-00667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pulliam L, Herndier BG, Tang NM, McGrath MS. HIV-1 infected macrophages produce soluble factors that cause histological and neurochemical alterations in cultured human brains. J Clin Invest. 1991;87:503–512. doi: 10.1172/JCI115024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sabatier JM, Vives E, Mabrouk K, Benjouad A, Rochat H, Duval A, Hue B, Bahraoui E. Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J Virol. 1991;65:961–967. doi: 10.1128/jvi.65.2.961-967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saito K, Chen CY, Masana M, Crowley JS, Markey SP, Heyes MP. 4-Chloro-3-hydroxyanthranilate, 6-chlorotryptophan, and norharmane attenuate quinolinic acid formation by interferon gamma-stimulated monocytes (THP-1 cells). Biochem J. 1993;291:11–14. doi: 10.1042/bj2910011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwarcz R, Whetsell WO, Mangano RM. Quinolinic acid: an endogenous metabolite that produces axon-sparing lesions in rat brain. Science. 1983;219:316–318. doi: 10.1126/science.6849138. [DOI] [PubMed] [Google Scholar]
- 53.Snider W, Simpson DM, Nielsen S, Gold JW, Metroka CW, Posner JB. Neurological complications of AIDS: analysis of 50 patients. Ann Neurol. 1983;14:403–418. doi: 10.1002/ana.410140404. [DOI] [PubMed] [Google Scholar]
- 54.Thery C, Chamak B, Mallat M. Cytotoxic effect of brain macrophages on developing neurons. Eur J Neurosci. 1991;3:1155–1164. doi: 10.1111/j.1460-9568.1991.tb00050.x. [DOI] [PubMed] [Google Scholar]
- 55.Walsh J, Todd WP, Carpenter BK, Schwarcz R. 4-Halo-3-hydroxyanthroanilic acids: potent competitive inhibitors of 2-hydroxyanthranilic acid oxygenase in vitro. Biochem Pharmacol. 1991;42:985–990. doi: 10.1016/0006-2952(91)90279-e. [DOI] [PubMed] [Google Scholar]
- 56.Whetsell WO, Schwarcz R. Prolonged exposure to submicromolar concentrations of quinolinic acid causes excitotoxic damage in organotypic cultures of rat corticostriatal system. Neurosci Lett. 1989;97:271–275. doi: 10.1016/0304-3940(89)90609-5. [DOI] [PubMed] [Google Scholar]
- 57.Wiley CA, Masliah E, Morey M, Lemere C, DeTeresa R, Grafe M, Hansen L, Terry R. Neocortical damage during HIV infection. Ann Neurol. 1991;29:651–657. doi: 10.1002/ana.410290613. [DOI] [PubMed] [Google Scholar]
- 58.Williams K, Dawson VL, Romano C, Dichter MA, Molinoff PB (1990) Characterization of polyamines having agonist, antagonist, an inverse agonist effects at the polyamine recognition site of the NMDA receptor. Neuron 5:199–208. [DOI] [PubMed]