S-Nitrosylation of Divalent Metal Transporter 1 Enhances Iron Uptake to Mediate Loss of Dopaminergic Neurons and Motoric Deficit (original) (raw)
Abstract
Elevated iron deposition has been reported in Parkinson's disease (PD). However, the route of iron uptake leading to high deposition in the substantia nigra is unresolved. Here, we show a mechanism in enhanced Fe2+ uptake via S-nitrosylation of divalent metal transporter 1 (DMT1). While DMT1 could be S-nitrosylated by exogenous nitric oxide donors, in human PD brains, endogenously S-nitrosylated DMT1 was detected in postmortem substantia nigra. Patch-clamp electrophysiological recordings and iron uptake assays confirmed increased Mn2+ or Fe2+ uptake through S-nitrosylated DMT1. We identified two major S-nitrosylation sites, C23 and C540, by mass spectrometry, and DMT1 C23A or C540A substitutions abolished nitric oxide (NO)-mediated DMT1 current increase. To evaluate in vivo significance, lipopolysaccharide (LPS) was stereotaxically injected into the substantia nigra of female and male mice to induce inflammation and production of NO. The intranigral LPS injection resulted in corresponding increase in Fe2+ deposition, JNK activation, dopaminergic neuronal loss and deficit in motoric activity, and these were rescued by the NO synthase inhibitor l-NAME or by the DMT1-selective blocker ebselen. Lentiviral knockdown of DMT1 abolished LPS-induced dopaminergic neuron loss.
SIGNIFICANCE STATEMENT Neuroinflammation and high cytoplasmic Fe2+ levels have been implicated in the initiation and progression of neurodegenerative diseases. Here, we report the unexpected enhancement of the functional activity of transmembrane divalent metal transporter 1 (DMT1) by S-nitrosylation. We demonstrated that S-nitrosylation increased DMT1-mediated Fe2+ uptake, and two cysteines were identified by mass spectrometry to be the sites for S-nitrosylation and for enhanced iron uptake. One conceptual advance is that while DMT1 activity could be increased by external acidification because the gating of the DMT1 transporter is proton motive, we discovered that DMT1 activity could also be enhanced by S-nitrosylation. Significantly, lipopolysaccharide-induced nitric oxide (NO)-mediated neuronal death in the substantia nigra could be ameliorated by using l-NAME, a NO synthase inhibitor, or by ebselen, a DMT1-selective blocker.
Keywords: DMT1, dopaminergic neurons, iron deposition, motoric defects, nitric oxide, S-nitrosylation
Introduction
Iron is an essential cofactor for tyrosine hydroxylase (TH), which is involved in dopamine synthesis (Zecca et al., 2004a). However, Fe2+ is also potentially toxic as a high concentration of unbound Fe2+ induces oxidative stress through the production of highly reactive free radicals, via the Fenton and Haber–Weiss reactions (Kehrer, 2000). Several studies have reported age-related increases in iron deposition in mouse, rat, monkey, and human brains (Hallgren and Sourander, 1958; Massie et al., 1983; Cook and Yu, 1998; Hardy et al., 2005), particularly in regions implicated in neurodegenerative diseases such as Parkinson's disease (PD; Salazar et al., 2008). Interestingly, iron concentration has been shown to increase linearly with age in the substantia nigra (SN), but in contrast, the amount of iron detected remained relatively constant in the locus ceruleus, especially for people >80 years of age (Zecca et al., 2004b). A recent study using whole-brain MRI quantitative susceptibility mapping across the adult lifespan reported that the motor system, chiefly the striatum and midbrain, selectively accumulated iron with age (Acosta-Cabronero et al., 2016). Desferrioxamine, an iron chelator widely used in clinical settings, has been reported to improve motoric functions and dopaminergic neuronal survival in animal models of PD (Guo et al., 2016). A selective iron chelator used in a randomized clinical trial has also been reported to improve PD motor outcomes (Devos et al., 2014).
While the divalent metal transporter 1 (DMT1) is essential for intestinal absorption of Fe2+, in neurons, Fe2+ uptake can be mediated via non-transferrin- or transferrin-dependent pathways (Skjørringe et al., 2015). It has been shown that transferrin-binding sites and transferrin receptor expression were loosely correlated with the final steady-state distributions of Fe (Hill et al., 1985). Moreover, the number of transferrin-binding sites was decreased in the SNc dopaminergic neurons of PD patients, suggesting that transferrin-mediated Fe2+ uptake did not contribute much to the increase in Fe deposition in PD brains (Faucheux et al., 1997). In contrast, the expression of DMT1 with iron response element (IRE) isoform was shown to be increased in the SN of PD patients (Salazar et al., 2008). However, the total DMT1 protein expression seemed unchanged considering that DMT1-IRE protein was decreased in the same PD samples (Salazar et al., 2008). As such, it is still unclear how the higher Fe deposition observed in human PD brains was mediated by DMT1.
On the other hand, we hypothesized that increased Fe deposition was due to enhanced activity of the DMT1. As S-nitrosylation of proteins such as Parkin and X-linked inhibitors of apoptosis (XIAP) were implicated in PD, we wondered first whether DMT1 could be S-nitrosylated and, second, if it did, how would its transport activity be affected. S-nitrosylation is a post-translational modification process that regulates protein function through covalent reaction of nitric oxide (NO) with a cysteine thiol group (Stamler et al., 1992). NO is produced as a coproduct during conversion of l-arginine to citrulline by the enzyme NO synthase (NOS), with Ca2+ and calmodulin as cofactors. Three distinct NOS isoforms have been identified in the brain (Förstermann et al., 1991). The endothelial NOS and neuronal NOS are constitutively expressed in neurons, and their activity is triggered by an increase in intracellular Ca2+. However, inducible NOS (iNOS) is Ca2+ insensitive and contributes to the inflammatory response. Nonetheless, iNOS produces a large amount of NO upon stimulation by proinflammatory cytokines (Green et al., 1994). At low levels, NO supports normal neuronal functions via S-nitrosylation of target proteins. However, when NO is produced in a high concentration under pathological conditions such as inflammation, aberrantly S-nitrosylated proteins can initiate neurodegeneration (Nakamura et al., 2013).
Here, this is the first report to show that DMT1 can be S-nitrosylated, and unexpectedly S-nitrosylation-enhanced DMT1 activity, which is contrary to the compromised functions observed in S-nitrosylated proteins such as Parkin (Chung et al., 2004) or XIAP (Tsang et al., 2009) that is implicated in PD. Endogenous production of NO via lipopolysaccharide (LPS)-induced inflammation in mice could S-nitrosylate DMT1, resulting in increased uptake and deposition of Fe in the SN and in compromised locomotor coordination. Significantly, the application of l-NAME, a NOS inhibitor, or ebselen, a DMT1 inhibitor, reversed dopaminergic neuronal loss and motoric deficit.
Materials and Methods
Human brain tissues.
The postmortem human brain samples for PD were obtained from the National Institute of Child Health and Human Development Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD, and the Harvard Brain Tissue Resource Centre (which is supported in part by United States Public Health Service contract HHSN-270-2013-00030C). The brain tissues were stored at −80°C. The use of the samples had been approved by the SingHealth Centralized Institutional Review Board (2013/1002/A) and the National University of Singapore Institutional Review Board (B-15-032E).
Mice and surgery.
The generation of DMT1-transgenic mice was described in a previous study (Zhang et al., 2017). The cDNA of monkey DMT1 (accession #AF153279) without the 3′ UTR IRE (DMT1B/-IRE) was cloned with a myc-tag at the 3′ end. This DMT1B-myc was then inserted between exons 2 and 3 of the mouse prion promoter MoPrP.Xho expression vector, which can drive high expression in the brain. The transgene was maintained in the heterozygous genotype by crossing heterozygous DMT1 mice with wild-type controls. Mice were maintained on a 12 h light/dark cycle with food and water available ad libitum. C57BL/6 DMT1 heterozygous mice that were 1–14 months old of either sex were used for experiments. Survival surgery was performed on mouse so as to microinject LPS (5 μg/μl, Sigma-Aldrich) or saline into the left SNc to induce inflammation in this region. First, the mouse was anesthetized with a cocktail of ketamine (10 mg/10 g) and xylazine (1 mg/10 g), and then mounted on a stereotaxic frame (Narishige Scientific Instrument Laboratory). An incision was made along the midline, and a burr hole, corresponding to the coordinates of the SNc (anteroposterior, −3.20 mm; mediolateral, −1.5 mm) was then drilled on the left side of the exposed skull. A 33 gauge, stainless steel microinjection needle, attached to a 5 μl Exmire microsyringe (Ito Corporation), was directed into the SNc (dorsoventral, −4.3 mm). A total volume of 1 μl of LPS was administered over a 10 min period, and the microinjection needle was left in place for another 10 min after the final microinjection. Subsequently, the incision site was sutured with nonabsorbable polyamide sutures (metric size, 6.0; B. Braun/Aesculap). The mice were given a single injection of LPS or saline in SN and administered with saline or l-NAME (30 mg/kg) or ebselen (10 mg/kg) by intraperitoneal injection once a day for 2 weeks. After the behavior test, they were transcardially perfused and postfixed with 4% paraformaldehyde for histology analysis or killed for biochemical analysis. All experimental procedures were performed with the approval of and in accordance with the guidelines of the Institutional Animal Care and Use Committee of the National University of Singapore (protocol R14-1566) as well as the National Neuroscience Institute (protocol TNI 14-01-001).
Lentiviral transduction and stereotaxic microinjection.
The shRNA sequence of DMT1 (targeted to both monkey and mouse sequences) or negative control (scrambled sequence) was ligated into the pLVX-shRNA2 plasmid (Clontech). The sequence for DMT1 knockdown was 5′-CTGGCCAGTTTGTCATGGA-3′, whereas the control sequence was 5′-GGAACCGTGCGAAATAATA-3′. All correct insertions were confirmed by DNA sequencing. The lentiviral vector was packaged in HEK293FT cells (a fast-growing, highly transfectable clonal isolate derived from human embryonal kidney cells transformed with the SV40 large T antigen for generating high-titer lentivirus; Thermo Fisher Scientific) and concentrated by centrifugation at 100,000 × g for 90 min. the titers of concentrated viral particles were between 1 × 108 and 1 × 109 transduction units/ml. The concentrated lentivirus was bilaterally microinjected into the SNc of DMT1 transgenic mice. A total volume of 1.5 μl of lentivirus was administered at 0.2 μl/min, and the microinjection needle was left in place for another 10 min after the microinjection. After 7 d, the mice were given a single injection of LPS in SNc, as described above and administered with vehicle or ebselen (10 mg/kg) by intraperitoneal injection once a day for 2 weeks.
Reagents.
S-nitrosocysteine (cysNO) was made freshly by mixing cysteine (200 mm) with sodium nitrite (200 mm) in the presence of HCl (100 mm). After vortexing in the dark at room temperature, NaOH (200 mm) was added to neutralize HCl. The cysNO was exposed in room light for >48 h to prerelease NO and was used as a control (Franklin and Paxinos, 1997). NOC-18 was purchased from Merck Millipore, and a stock solution was kept at −80°C. Thiopropyl Sepharose 6B resin (catalog #17-0420-01, GE Healthcare) was rehydrated with water for 15 min, resuspended, and left for another 10 min at room temperature. Water was removed after centrifugation at 1000 × g for 1 min at room temperature. The resin was rehydrated and washed three times with water and three times with HENS buffer. The 50% resin slurry was kept temporarily at 4°C until use (Guo et al., 2014). Mouse monoclonal antibody against DMT1 (catalog #H00004891-M01, Abnova; RRID:AB_464397), mouse monoclonal antibody against GFP (lot number 10126200 Oct 2015, catalog #11814460001, Roche Applied Science), rabbit polyclonal antibody against DMT1 (catalog #20507-1-AP, Proteintech; RRID:AB_10694284), and rabbit polyclonal antibody against DMT1+IRE (catalog #NRAMP21-A, Alpha Diagnostic International; RRID:AB_1620356) were used in Western blot. Rabbit polyclonal antibody against Iba1 (ionized calcium-binding adapter molecule 1; catalog #019-19741, Wako Chemicals) and rabbit polyclonal antibody against TH (catalog #P40101, Pel-Freez Biologicals) were used for immunostaining. A Quntichrome Iron Detection Kit was purchased from BioAssay Systems (catalog #DIFE-250).
Plasmids, cell cultures, and transfection.
The cDNA of DMT1 (accession #AF153279) without IRE at the 3′ UTR was subcloned into a pEGFPN3 vector. The C23A or C540A substitution of DMT1 was introduced by PCR using pEGFPN3-DMT1 as the template. HEK293 and SH-SY5Y cells were obtained from American Type Culture Collection. The cell lines were not authenticated or tested for mycoplasma contamination after purchase. HEK293 or SH-SY5Y cells were grown in DMEM containing 10% FBS in a 5% CO2 atmosphere at 37°C. The SH-SY5Y cell line overexpressing DMT1 was generated by transfection with a pEGFPN3-DMT1 plasmid construct followed by selection for G418 resistance, and was maintained in 200 μg/ml G418 (Invitrogen)-supplemented DMEM complete medium. HEK293 cells were transfected by the calcium phosphate method and SH-SY5Y cells were transfected using Lipofectamine 2000 reagent (Invitrogen) according to the instructions of the manufacturer. After transfection and culturing for 48 h, the HEK293 cells were lysed, and the cell lysates were treated with 0.2 mm NOC-18 for half an hour or 0.5 mm cysNO for 10 min, and then the cell lysates were processed for resin-assisted capture (RAC) or biotin-switch assay.
Resin-assisted capture.
The RAC protocol is modified from the study by Forrester et al. (2009b). Briefly, brain tissue or cultured cells were lysed in lysis buffer (150 mm NaCl, 30 mm HEPES, 10 mm NaF, 1% v/v Triton X-100, 0.01% w/v SDS, and complete protease inhibitor cocktails, pH 7.5), and samples were adjusted with lysis buffer to achieve an equal concentration of total protein. For experiments using 0.5–2 mg of protein, samples are diluted to 0.9 ml with HEN buffer (100 mm HEPES, 1 mm EDTA, 0.1 mm neocuproine, pH 8.0). Next, 0.1 ml of 25% SDS was added along with 10 μl of 10%& methyl methanethiosulfonate (MMTS; final 1.0 ml volume with 2.5% SDS and 0.1% MMTS). Samples were incubated at 50°C in the dark for 20 min with frequent vortexing. Four volumes of cold acetone (4 ml) were added to each sample. Proteins were precipitated for 20 min at −20°C, and collected by centrifugation at 3000 × g for 5 min. The clear supernatant was aspirated, and the protein pellet was gently washed with 70% acetone (4 × 5 ml). Following suspension in 0.3 ml of HENS buffer (100 mm HEPES, 1 mm EDTA, 0.1 mm neocuproine, and 1% SDS, pH 8.0), the material was added to 50 μl of thiopropyl resin slurry (GE Healthcare) in the presence of sodium ascorbate (final 20 mm unless indicated otherwise), followed by rotation in the dark for 2–4 h. It is critical to avoid any sources of sunlight during this step. The S-nitrosothiol (SNO)-RAC resin was collected by centrifugation at 1000 × g for 1 min, followed by washing with HENS buffer (2 × 1 ml), then HENS/10 (2 × 1 ml). Captured proteins were eluted with 30 μl of HENS/10 (10 mm HEPES, 0.1 mm EDTA, 0.01 mm neocuproine, and 1% SDS, pH 8.0) containing 100 mm 2-mercaptoethanol for 20 min at room temperature, and the eluent was used for SDS-PAGE.
Biotin switch technique.
The biotin switch technique (BST) protocol is modified from the study by Forrester et al. (2009a). Briefly, transfected HEK293 cells were lysed in lysis buffer, and various samples were adjusted to the equal concentration. After treatment with NO donor or control, 0.1 ml of 25% SDS is added along with 10 μl of 10% MMTS (Sigma-Aldrich). Samples were incubated at 50°C in the dark for 20 min with frequent vortexing. Four volumes of cold acetone (4 ml) were added to each sample. Proteins are precipitated for 20 min at −20°C and collected by centrifugation at 3000 × g for 5 min. The clear supernatant was aspirated, and the protein pellet was gently washed with 70% acetone (4 × 5 ml). Following resuspension in 0.24 ml of HENS buffer, the material was transferred to a fresh 1.7 ml Microfuge tube containing 30 μl of biotin-HPDP (N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide; 2.5 mg/ml; Thermo Fisher Scientific). The labeling reaction was initiated by adding 30 μl of 200 mm sodium ascorbate. Alternatively, an equivalent concentration of NaCl was used as an ascorbate-free control. Samples were rotated at room temperature in the dark for 1 h. Three volumes of cold acetone (0.9 ml) were added to each sample. Proteins were precipitated for 20 min at −20°C, and collected by centrifugation at 5000 × g for 5 min. The clear supernatant was aspirated, and the protein pellet was gently washed with 70% acetone (4 × 1 ml). Following complete resuspension in 0.25 ml of HENS/10 buffer, 0.75 ml of neutralization buffer was added. Ten microliters of each sample was removed for analysis of protein “input.” The remaining material was transferred to a fresh 1.7 ml Microfuge tube containing 25–50 μl of prewashed avidin-affinity resin. The samples were gently rotated for 12–18 h at 4°C. Avidin beads were collected by centrifugation at 200 × g for 10 s in a swinging-bucket rotor, followed by washing with wash buffer (4 × 1 ml). After the final wash, the beads were fully dried by gentle aspiration. Thirty to fifty microliters of elution buffer (HENS/10 containing 100 mm 2-mercaptoethanol) was added to each sample. Proteins were eluted at room temperature with frequent shaking, followed by centrifugation at 5000 × g for 30 s. The supernatant was collected without disturbing the pelleted resin, and mixed with 4× SDS loading buffer.
DAN assay.
The method was modified from a previous report (Misko et al., 1993). Briefly, 2,3-diaminonaphthalene (DAN) was dissolved in 0.6 m HCl to make a 3.33 mg/ml stock solution and stored at −20°C. The working solution was freshly made by diluting a DAN stock solution into 50 μg/ml in 0.6 m HCl. Ten microliters of DAN working solution was added to 50 μl samples and mixed. After 15 min of incubation at room temperature in the dark, the reaction was quenched with 60 μl of 0.1 m NaOH. The fluorescence was read by a Tecan infinite M200 Plate Reader (excitation wavelength, 365 nm; emission wavelength, 450 nm).
Western blot.
The samples from RAC or biotin-switch assay were treated in 50°C for 15 min, resolved in acrylamide gels and transferred onto nitrocellulose membranes. After blocking, the membranes were incubated with primary antibodies against target proteins in 4°C overnight. Either anti-mouse IgG or anti-rabbit IgG peroxidase-conjugated secondary antibodies were applied at 1:10,000, and blots were visualized with Luminata Forte ECL Detection Kits (Millipore) in an LAS-4000 Imager (GE Healthcare).
Sliver stain.
An SDS-PAGE gel was fixed for 2 h in a solution containing 50% methanol, 12% acetic acid, and 0.05% formalin under gentle shaking at 55 rpm. Then the gel was washed in 35% ethanol for 20 min, thrice with shaking at 55 rpm, and later sensitized for 2 min in 0.02% sodium thiosulfate while shaking at 80 rpm. After 3 × 5 min washes in water with shaking at 55 rpm, the gel was stained for 20 min in 0.2% AgNO3 and 0.076% formalin while shaking at 80 rpm. After two washes in water for 2 min each at 55 rpm, the gel was developed in a solution containing 6% sodium carbonate, 0.0004% sodium thiosulfate, and 0.05% formalin under vigorous shaking at 110 rpm. Then the solution was changed to 1.46% sodium EDTA to stop the reaction, with shaking at 110 rpm. Finally, the gel was washed in water for 3 × 5 min, gently shaken at 55 rpm, and finally subjected to gel scan.
Immunoprecipitation, Iodoacetyl Tandem Mass Tag switch, and tryptic digestion of DMT1.
HEK293 cells were transfected with pcDNA3-DMT1B-myc using the calcium phosphate method. After 48 h of transfection, the cells were harvested and lysed in lysis buffer for 2 h at 4°C, and centrifuged at 14,000 × g for 10 min. The supernatant was incubated with Anti-Myc Affinity Gel (catalog #B23041, Biotool) overnight at 4°C. After three washings, the gel containing the captured DMT1-myc was treated with cysNO (0.5 mm) or control in lysis buffer for 10 min at room temperature in the dark.
The Iodoacetyl Tandem Mass Tag (iodoTMT; Thermo Fisher Scientific) labeling procedure was modified from a previous protocol (Qu et al., 2014). Free sulfhydryl groups in the protein sample were first blocked with 20 mm MMTS for 30 min at 50°C with frequent vortexing in HEN buffer containing 2.5% (w/v) SDS. To remove excess MMTS, proteins were precipitated using 4× volume of cold acetone for 30 min at −20°C. Proteins were then redissolved in HEN buffer containing 1% (w/v) SDS and labeled with 1 mm iodoTMT Reagent (Thermo Fisher Scientific) in the presence of 5 mm sodium ascorbate for 2 h at room temperature. All of the above procedures were performed with protection from light.
For mass spectrometry (MS) analysis, after acetone precipitation, the iodoTMT-labeled proteins were redissolved with Tris-HCl, pH 8.0, containing 0.1% (w/v) SDS. Protein samples were reduced with 10 mm dithiothreitol at 55°C for 1 h and then alkylated with 25 mm iodoacetamide at 37°C for 1 h in the dark. After acetone precipitation, the protein pellet was resuspended in 100 mm Tris-HCl and 10 mm CaCl2, pH 8.0. Then the protein was digested by chymotrypsin at 25°C for 3 h and then trypsin was added for digestion at 37°C for 16 h. After brief centrifugation, the digested peptides were desalted in a Sep-Pak tC18 96-well plate (Waters) and eluted with 80% acetonitrile. The peptides were then vacuum dried for MS analysis.
S-nitrosylation sites identification by liquid chromatography-tandem MS.
Peptide separation was performed on an Eksigent nanoLC Ultra and ChiPLC-nanoflex (Eksigent) in a Trap Elute configuration. The samples were reconstituted in 2% acetonitrile with 0.1% formic acid before loading on a 200 μm × 0.5 mm column (3 μm; ChromXP C18-CL, SCIEX) and eluted on an analytical 75 μm × 15 cm column (3 μm; ChromXP C18-CL, SCIEX). Peptides were separated by a gradient formed by mobile phase A (2% acetonitrile, 0.1% formic acid) and mobile phase B (98% acetonitrile, 0.1% formic acid) at a flow rate of 3 μl/min. The gradients used for separation were as follows: 5–5% of mobile phase B in 1 min, 5–12% of mobile phase B in 20 min, 12–30% of mobile phase B in 40 min, 30–90% of mobile phase B in 2 min, 90–90% of mobile phase B in 5 min, and 90–5% of mobile phase B in 5 min, and maintained at 5% of mobile phase B for 13 min.
The MS analysis was performed on a TripleTOF 5600 System (SCIEX) in the information-dependent mode. MS spectra were acquired across the mass range of 400–1800 mass/charge ratio (m/z) in high-resolution mode (>30,000) using a 250 ms accumulation time per spectrum. The top 20 precursors per cycle were chosen for fragmentation with 100 ms maximum accumulation time for each precursor and dynamic exclusion of 15 s. Tandem MS (MS/MS) spectra were recorded in high-sensitivity mode (resolution, >15,000) with rolling collision energy on.
Peptide identification was performed with ProteinPilot Software 4.5 (SCIEX) using paragon database search algorithm (4.5.0.0) and integrated false discovery rate (FDR) analysis function. The MS/MS spectra obtained were searched against the UniProtKB database (Macaca fascicularis species, 30,200 protein sequences) with the following search parameters: (1) sample type, identification; (2) cysteine alkylation, carbamidomethylation; (3) digestion, trypsin and chymotrypsin; (4) ID focus, biological modifications; (5) search effort, thorough; and (6) FDR analysis, yes. A reverse database search strategy was adopted to estimate the FDR for peptide identification. For all the shotgun proteomics studies, a strict unused confidence score of ≥1.3 was used as a qualification criterion, corresponding to a peptide confidence level of 95%.
In addition to database searching using ProteinPilot, the same raw data files were also subjected to database search using PEAKS Studio 5.2 (Bioinformatics Solutions). The parameters were error tolerance of 0.1 Da for precursor mass and 0.2 Da for fragment ion, and a maximum of three missed cleavages. The enzymes are trypsin and chymotrypsin. Variable modifications of carbamidomethylation and iodoTMT 6-plex (addition of m/z 329.2266) of cysteine were used, and two combined modifications were allowed for each peptide.
Whole-cell patch-clamp electrophysiology.
HEK293 cells were transfected with wild-type or mutant pEGFPN3-DMT1 using calcium phosphate method. The transfected cells grown on glass coverslips were recorded 48 or 72 h after transfection. The pipette solution contained 138 mm cesium methanesulfonate, 5 mm cesium chloride, 0.5 mm EGTA, 1 mm MgCl2, 2 mg/ml Mg-ATP, and 10 mm HEPES, pH 7.3 and 290–300 mOsm). The bath solution contained 140 mm TEA methanesulfonate, 100 μm MnCl2, 10 μm CaCl2, and 10 mm HEPES, pH 5.5 and 300–310 mOsm. The cells was perfused in a bath solution containing NO donor or control using a gravity perfusion system. Data were collected using an Axopatch 200B patch-clamp amplifier, a Digidata 1320, and pClamp 8.0 software (Molecular Devices). Whole-cell currents were digitized at 10 kHz and filtered at 2 kHz.
DAB-enhanced Perl's stain.
The diaminobenzidine (DAB)-enhanced Perl's stain was performed as previously reported (Moos and Møllgård, 1993). Briefly, 40-μm-thick free-floating sections were immersed in Perls' solution that contained equal amounts of freshly prepared potassium ferrocyanide (2%) and hydrochloric acid (2%) for 30 min. After washing, the sections were intensified with 0.025% DAB plus 0.0033% H2O2 in TBS for 10 min. The staining conditions were kept the same for the parallel experiments. After several washes with TBS, the slides were mounted onto the gelatin-coated slides and dried for 3 h. Then the slices were dehydrated, covered with the mounting medium, and examined with a light microscope (model BX51, Olympus) equipped with a digital camera (model DP70, Olympus).
Immunohistochemistry and stereological assessments of TH+ neurons
Stereological assessment was performed as previously described (Schmitz and Hof, 2005). Briefly, fixed mouse brain was sectioned into 40-μm-thick slices spanning the rostral-caudal extent of the SNc using a CM3050S Cryostat (Leica Biosystems). Every fourth section was collected and kept in PBS. Endogenous peroxidase activity of each section was quenched with 0.3% H2O2 (Sigma-Aldrich) for 30 min, followed by washing with TNT buffer (Tris-Cl 0.1 m, NaCl 150 mm, Tween 0.1% v/v, pH 7.5). Brain sections were then blocked with TNT blocking buffer containing 5% normal goat serum (Jackson ImmunoResearch) for another 30 min. Subsequently, sections were incubated in rabbit polyclonal anti-TH antibody (Pel-Freez Biologicals) in 5% normal goat serum TNT blocking buffer overnight at 4°C and washed with TNT buffer the following day. Brain sections were then incubated in secondary biotinylated antibody (Vector Laboratories) for 1 h before incubation with ABC reagent kit (Vector Laboratories) for 30 min at room temperature. TH+ cells were made visible via DAB staining before the sections were mounted on glass slides for visualization. To analyze the TH+ neurons in the SNc region of each brain section, a design-based stereological methodology was used with a computer-assisted system consisting of an Axio Imager 2 Microscope (Carl Zeiss), equipped with a motorized ASI MS-2000 stage (Applied Scientific Instrumentation) and an IEEE 1394 camera (Imi Tech) interfaced with Stereologer Software (Stereology Resource Centre). To count the SNc TH+ neurons, a 2.5× contour objective, a 63×/1.40 oil-immersion counting objective, and optical dissectors with a counting frame measuring 60 × 60 μm and a height of 20 μm (guard zones, 2 μm) were used. Cell counts were performed on every fourth evenly spaced sections from each brain. The cell counts were expressed as the mean number of TH+ cells in SNc region of the injection side. The cell count number were obtained using Stereologer Software and further analyzed by Excel 2010 (Microsoft) and GraphPad Prism 7 for group comparisons.
Rotarod test.
Motor performance assays were performed using a rotarod (Ugo Basile). Briefly, the mice were placed on the rotarod with a rotating speed of 4 rpm. Once the mice were ready on the beam, the speed was accelerated to 40 rpm within 4 min and the time spent on the rotarod was recorded. The mice were trained for 2 d before the testing day. Each mouse was given three trials with 10–15 min of rest after every trial. We excluded the mice whose latency to fall was <40 s because of overweight (Balkaya et al., 2013).
qRT-PCR.
Total RNA was isolated from the saline or LPS-injected side of mouse SN, using the TRIzol method (Invitrogen). The RNA samples were treated with RNase-free DNase (Ambion) before reverse transcription. The first strand of cDNA was synthesized by the Superscript III RT (Invitrogen) from 1 μg of total RNA with oligo (dT)18 primers according to the instructions of the manufacturer. Negative controls were performed without reverse transcriptase to exclude contamination by genomic DNA. The primers specific for detection of the mouse DMT1+IRE transcript (forward 5′-ATACCAGCGGTGGCAACATT, reverse 5′-TCCTAAATGCAGTCTGTGCAAC), both mouse and transgenic monkey DMT1 (panDMT1; forward 5′-AGCGTTTTTTGGCTTTCTC-ATCA, reverse 5′-CTAAGGCAGAATGCAGGTACAT), and mouse GAPDH (forward 5′-CCCC-AGCAAGGACACTGAGCAA, reverse 5′-GTGGGTGCAGCGAACTTTATTGATG) were used to detect the respective mRNA levels. Normalization was performed against GAPDH levels. The reaction mixes were assembled using SYBR Green Master Mix (Kapa Biosystems). Real-time PCRs were performed on an ABI7500 System (Applied Biosystems) using the following cycling parameters: 95°C for 10 min, and then 40 cycles of 95°C (10 s), 60°C (10 s), and 72°C (10 s) followed by a melting curve analysis. All reactions were performed with two technical replicates, and at least three biological repeats were performed. The data were analyzed by ABI7500 Software version 2.0.5, Excel 2010, and GraphPad Prism 7.
Experiment design and statistical analysis.
Both male and female mice were used in this study. LPS, l-NAME, and ebselen injections were randomized across cages (not all animals in one cage received the same drug injection). We did not notice significant difference in response to these drugs between sexes. Researchers were blinded to drug treatments in the rotarod test. The sample size for each experiment was based on our previous study (Zhang et al., 2017), as well as published literature on similar experiments (Herrera et al., 2000; Salazar et al., 2008). All data were presented as the mean ± SEM, and individual experimental points are depicted in scatter plots. Statistical analyses were performed with GraphPad Prism 7 with statistical significance set at p < 0.05. The Student's two-tailed t test was used for two-group comparisons, and one-way ANOVA was used for multigroup comparisons, followed by post hoc Bonferroni tests (the p values reported in the Bonferroni test were adjusted for pairwise group comparisons). The n value stands for biological repeat. Each n value is indicated in the column graph and the results description. Details for the statistical tests used were provided within figure legends or the relative results description.
Results
DMT1 could be S-nitrosylated in vitro and in vivo
To determine whether DMT1 could be S-nitrosylated, we used two NO donors, NOC-18 (Chung et al., 2004) and cysNO (Forrester et al., 2009b), to S-nitrosylate DMT1 transfected into SH-SY5Y or HEK293 cells. S-nitrosylation of DMT1 were examined by the biotin switch (Forrester et al., 2009a) or RAC (Forrester et al., 2009b) methods, and omission of ascorbate was used as a negative control. Our results show that DMT1 could be robustly S-nitrosylated by both NO donors that were administered exogenously (Fig. 1A,B). Next, we assessed whether DMT1 could be S-nitrosylated in vivo. Because Fe deposition is age dependent (Zecca et al., 2001) and have been detected in the SN, we therefore isolated the SN region of 1-, 2-, 6-, or 14-month-old DMT1 transgenic mice and subjected the lysates to RAC and Western blot analyses (Fig. 1C). The results showed that there is a clear age-dependent increase in S-nitrosylation of DMT1 in the mouse SN (SNO-DMT1/total DMT1: 1 month, 1.00 ± 0.084, n = 6; 2 months, 2.12 ± 0.12, n = 6; 6 months, 3.49 ± 0.056, n = 6; 14 months, 3.66 ± 0.080, n = 6; one-way ANOVA, F(3,20) = 202.8, p < 0.0001), while the total protein levels were similar (normalized total DMT1: 1 month, 1.00 ± 0.052, n = 6; 2 months, 1.06 ± 0.073, n = 6; 6 months, 0.98 ± 0.050, n = 6; 14 months, 0.99 ± 0.059, n = 6; one-way ANOVA, F(3,20) = 0.55, p = 0.65). For clinical relevance, we investigated the S-nitrosylation of DMT1 in human brains of PD patients (Salazar et al., 2008). Our data showed that DMT1 S-nitrosylation in PD brains was significantly higher than in age-matched controls (SNO-DMT1/total DMT1: control, 0.32 ± 0.044, n = 6; PD, 1.00 ± 0.15, n = 6; Student's t test, t(10) = 4.34, p = 0.0015), while the total protein levels were similar (Fig. 1D; total DMT1/GAPDH: control, 0.93 ± 0.19, n = 6; PD, 1.00 ± 0.14, n = 6; Student's t test, t(10) = 0.31, p = 0.77). Together, we showed that DMT1 could be robustly S-nitrosylated by the addition of exogenous NO in vitro, and, importantly in human PD brains, after an endogenous S-nitrosylation process, enough DMT1 remained S-nitrosylated for detection in archived postmortem tissues.
Figure 1.

DMT1 was S-nitrosylated both in vitro and in vivo. A, Biotin-switch assay showed that DMT1 was S-nitrosylated in the presence of NO donor NOC-18. The lysate of DMT1 stably transfected SH-SY5Y cells were treated with NOC-18 (200 μm) or control for 1 h, and then subjected to biotin-switch assay. The DMT1-GFP fusion protein was detected by anti-GPF antibody. Top, S-nitrosylation fraction. Bottom, Input of the cell lysate. Omission of ascorbate resulted in the almost complete loss of biotinylation and served as negative control. The location of molecular weight markers is shown in kilodaltons on the right. B, RAC assay showed that DMT1 was S-nitrosylated in the presence of cysNO (500 μm) in pEGFPN3-DMT1-transfected HEK293 cells. The DMT1-GFP fusion protein was detected by anti-GPF antibody. C, Representative Western blot showing the age-dependent DMT1 S-nitrosylation in the SN of DMT1-transgenic mouse. SN from 1-, 2-, 6-, or 14-month-old mice was subjected to RAC assay and Western blot. Pooled: the mixture of equal amount protein lysate from each sample. The DMT1-myc fusion protein was detected by anti-DMT1 antibody. Bar chart and scatter plots show the statistics of SNO-DMT1/total DMT1 ratio and normalized total DMT1 protein level normalized to 1-month-old mice. One-way ANOVA: p values are indicated above the bar chart. D, RAC assay shows the S-nitrosylation of DMT1 in human PD brains, as well as age-matched controls. Postmortem SNs of PD patients were used for RAC assay. The endogenous DMT1 was detected by anti-DMT1 antibody. Bar chart and scatter plots show the statistics of SNO-DMT1/total DMT1 or total DMT1/GAPDH ratio. Two-tailed Student's t test: p values are indicated above the bar chart. The n value is indicated in the column graph. Error bars represent the SEM.
S-nitrosylation of DMT1 increased its transporter activity
We next examined whether the post-translational modification of DMT1 by NO could change DMT1 transporter activity. Whole-cell patch-clamp electrophysiological recordings were used to record proton-driven Mn2+ currents of DMT1 (Xu et al., 2004) heterologously expressed in HEK293 cells. The DMT1 currents were evoked by repeated voltage ramps from −140 to +60 mV with a holding potential of +20 mV in a bath solution maintained at pH 5.5 (Fig. 2A), an acidic pH environment similar to that found inside the endosome, where endosomal membrane-bound DMT1 plays a critical role for transferrin-dependent Fe2+ flux into the cytoplasm (Zecca et al., 2004a). The current amplitudes evoked at −130 or +50 mV were used to plot the I–t curves. As reported, DMT1 could produce an inward-rectifying current (Xu et al., 2004) in which the current amplitude at −130 mV was used to evaluate DMT1 function, while the current amplitude at +50 mV was used to monitor the leaky current (Fig. 2A). We observed that the DMT1 currents were increased during 50 μm cysNO perfusion compared with control perfusion, which contained cysNO that was exposed to room light for >48 h to prerelease NO (Lei et al., 1992; Fig. 2A,B). While the vector transfected HEK293 cells only exhibited very small currents at pH 5.5 (Fig. 2C), we observed that the small current evoked in vector-transfected HEK293 cells was also increased by cysNO perfusion and this increase may be due to activation of the endogenous DMT1. The leaky currents were very small and stable when compared with the large inward currents at −130 mV (Fig. 2A), providing assurance that the seal was good during whole-cell patch-clamp recordings.
Figure 2.
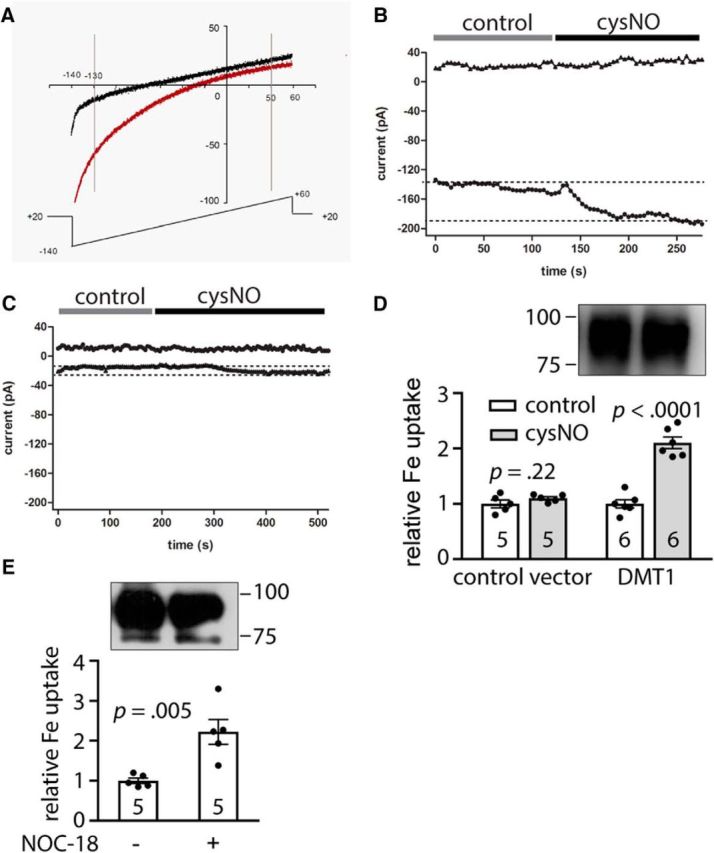
The transporter function of DMT1 was enhanced by S-nitrosylation. A, Whole-cell currents were elicited by repeated voltage ramps (−140 to +60 mV, 1 s), with a 4 s interval between ramps. The holding potential was +20 mV. Representative current traces elicited by protons, pH 5.5, and Mn2+ (100 μm) in wild-type DMT1-transfected HEK293 cells are shown: black trace, control; red trace, perfused with cysNO. A–C, The current amplitude elicited at +50 mV (triangles) and −130 mV (circles) was plotted against time (A) with control or cysNO perfusion, pH 5.5, in a DMT1-transfected (B) or a vector (pEGFPN3)-transfected (C) HEK293 cell. The dotted line shows the baseline or steady-state amplitude with cysNO perfusion. D, E, Quantichrome iron assay shows that the iron uptake was increased in the NO donor-treated group vs control, although the protein expression levels were comparable. SHSY5Y cells were transfected with pEGFPN3-DMT1 plasmid for 48 h, and then treated with cysNO (D), NOC-18 (E), or control with 10 μm FeCl2 for 1 h. After thorough washing, the cells were lysed and subjected to iron assay. Insets are the representative Western blot image showing the DMT1 expression in the cells used for iron assay. Bar chart and scatter plots show the statistics of iron uptake in the NO donor-treated group vs the control group. The iron concentration was normalized to total protein concentration and then normalized to the control group. Student's t test: p values are indicated above the bar chart. The n value is indicated inside the column. Error bars represent the SEM.
In addition to electrophysiological recordings, we also used an Fe uptake assay to detect Fe2+ transport through S-nitrosylated DMT1. SH-SY5Y cells were used because of the low background in Fe uptake under control conditions. However, SH-SY5Y cells transiently transfected with DMT1, treated with a NO donor (cysNO or NOC-18) and 10 μm FeCl2 for 1 h, were found to increase Fe2+ uptake by at least twofold, compared with controls (Fig. 2D: pEGFPN3-DMT1B transfection: control, 1.00 ± 0.074, n = 6; cysNO treatment, 2.10 ± 0.10, n = 6; Student's t test, t(10) = 8.59, p < 0.0001; Fig. 2E: pEGFPN3-DMT1B transfection: control, 1.00 ± 0.068, n = 5; cysNO treatment, 2.24 ± 0.31, n = 5; Student's t test, t(8) = 3.83, p = 0.005). Notably, the DMT1 proteins were expressed at comparable levels (Fig. 2D,E, insets). The cysNO treatment, however, did not increase Fe2+ uptake in vector-transfected SH-SY5Y cells (Fig. 2D; pEGFPN3 transfection: control, 1.00 ± 0.071, n = 5; cysNO treatment, 1.10 ± 0.030, n = 5; Student's t test, t(8) = 1.33, p = 0.22), indicating that Fe2+ uptake was through the DMT1. Together, we demonstrated that S-nitrosylation of DMT1 increased its transporter activity, akin to a gain in function.
Identifying DMT1 S-nitrosylation sites by MS
DMT1 has 12 cysteinyl residues in its protein sequence, and we used an unbiased iodoTMT switch and mass spectrometry method to identify the residues that could be S-nitrosylated (Qu et al., 2014). HEK293 cells were transfected with pcDNA3-DMT1-myc, and the resin-captured DMT1-myc protein was harvested after extensive washing, and its relative purity was examined by detection on a silver-stained gel (Fig. 3A). The resin-captured DMT1-myc was then subjected to iodoTMT MS analysis. As shown in the MS spectrum, the m/z value of C23 cysteine from cysNO-treated sample was 432 (Fig. 3C), which is the sum of cysteine (103) and iodoTMT labeling (329). In contrast, the m/z of the C23 from the control-treated sample was only 160 (Fig. 3B), which is the sum of cysteine (103) and iodoacetamide labeling (57). The m/z value of DC540 residues from the cysNO-treated sample was 548 (Fig. 3E), which is the sum of DC540 (219) and iodoTMT labeling (329). The DC540 residues from the control-treated sample was only 276 (Fig. 3D), which is the sum of cysteine (219) and iodoacetamide labeling (57). Thus, we have identified two major S-nitrosylation sites of DMT1: C23 and C540 (Fig. 3B–E).
Figure 3.

Identification of the DMT1 S-nitrosylation sites by mass spectrometry. A, Silver-stained gel image showing the purification of DMT1-myc from DMT1-myc transfected HEK293 cells. The resin-captured DMT1-myc was subjected to iodoTMT-switch and tryptic digestion, and then to MS analysis. Those fragment peaks that appear to extend from the amino terminus are termed “b ions.” Similarly, groups of peptide fragment ions appear to extend from the C terminus; these peaks are termed “y ions.” B, C, The MS spectrum of control-treated (B) or cysNO-treated (C) C23-containing peptides are shown. The m/z of C23 was calculated by b16 − b15. A larger shift was detected in nitrosylated C23, showing the iodoTMT modification of the cysteine residue. D, E, Similarly, the MS spectrum of control-treated (D) or cysNO-treated (E) C540-containing peptides is shown at the bottom. The m/z of DC(540) was identified by b2. A larger shift was detected in DC(540), showing the Iodo-TMT modification of C540.
C23A or C540A substitutions abolished cysNO-induced DMT1 current increase
To validate the MS screening results, we determined the S-nitosylation levels of DMT1 C23A and C540A substitutions versus wild type. We found that the C23A or C540A substitutions significantly decreased the S-nitrosylation of DMT1 (Fig. 4A,B; SNO-DMT1/total DMT1: wild type, 1.00 ± 0.064, n = 5; C23A, 0.44 ± 0.051, n = 5; C540A, 0.53 ± 0.071, n = 5; one-way ANOVA, F(2,12) = 21.67, p = 0.0001; post hoc Bonferroni test: wild-type vs C23A, p = 0.0001; wild-type vs C540A, p = 0.0005). Next, we investigated whether the C23A or C540A substitutions influenced NO effects on DMT1 currents. Our results showed that the C23A or C540A substitutions abolished cysNO-induced DMT1 current increase (Fig. 4C,D), consistent with the RAC data that showed the lack of S-nitrosylation in DMT1-C23A or DMT1-C540A. The DMT1 current returned to a very small amplitude when perfused with a pH 7.4 bath solution (Fig. 4C,D), indicating that the cells were healthy. Thus, we have shown functionally that the C23 or C540 residues are major sites for S-nitrosylation of DMT1.
Figure 4.

C23A and C540A substitutions abolished S-nitrosylation and NO-enhanced transporter function of DMT1. A, RAC assay shows that the S-nitrosylation of DMT1 was abolished by the C23A or C540A substitutions in the presence of 0.5 mm cysNO. The DMT1-transfected HEK293 cell lysates were treated with cysNO for 10 min and subjected to RAC assay. Omission of ascorbate served as a negative control. B, Bar chart and scatter plots showing the statistics of normalized SNO-DMT1/total DMT1 ratio for wild-type, C23A, or C540A substitutions. One-way ANOVA followed by post hoc Bonferroni test: p values are indicated above the bar chart. The n value is indicated inside the column. Error bars represent the SEM. C, D, The response of C23A and C540A currents to cysNO treatment. Whole-cell currents were elicited by repeated voltage ramps (−140 to +60 mV, 1 s). Holding potential was +20 mV. C, D, The current amplitude elicited at +50 mV (open triangles) and −130 mV (open circles) were plotted against time with control or cysNO perfusion, pH 5.5, in a DMT1C23A-transfected (C) or a DMT1C540A-transfected (D) HEK293 cell. The HEK293 cell was perfused with pH 7.4 external solution in the last stage of recording to ensure the cell viability.
S-nitrosylation of DMT1 was enhanced in SN after LPS injection
For physiological or pathophysiological relevance, we used a single intranigral injection of LPS to evoke NO production by NOS in glia cells or neurons of the SNc (Nakamura et al., 2013). The dosage of LPS used has been reported for selective effects of inflammatory reactions on the dopaminergic system (Herrera et al., 2000). The SNc has been reported to be far more sensitive than the striatum to the inflammatory stimulus, and importantly, only the dopaminergic neurons of the SN were affected, with no detectable damage to either the GABAergic or the serotoninergic neurons (Herrera et al., 2000). Therefore, the LPS inflammatory model is a good in vivo model to test the effects of endogenously produced NO on DMT1 activity. The NOS inhibitor l-NAME was used to inhibit NO production to rescue the mice from an NO-mediated effect. DMT1 transgenic mice were given a single injection of 5 μg of LPS or saline in the left side of the SNc and administered with saline or l-NAME (30 mg/kg) by intraperitoneal injection once a day for 2 weeks as reported previously (Singh et al., 2005). After 2 weeks, we examined the activation of microglia using the microglia activation marker Iba1 (Ito et al., 1998) and found that Iba1 immunoreactivity in the SNc of the LPS-injected mice was much stronger than in the control saline-injected mice (Fig. 5A). We also examined the relative NO concentrations in SN using the DAN assay and found that the relative NO concentration in LPS-injected mice was significantly increased compared with the saline-injected control. Moreover, application of the NOS inhibitor l-NAME decreased the LPS-elevated NO concentration (Fig. 5B; NO concentration: saline, 1.38 ± 0.41, n = 6; LPS 6.24 ± 0.52, n = 6; LPS plus l-NAME, 2.37 ± 0.69, n = 6; one-way ANOVA: F(2,15) = 21.48, p < 0.0001; post hoc Bonferroni test: control vs LPS, p < 0.0001; LPS vs LPS plus l-NAME, p = 0.0004). We further used the RAC method to evaluate S-nitrosylation of DMT1 in LPS-injected mouse brain, and our results showed that the level of S-nitrosylation of DMT1 were significantly higher in LPS-injected mouse than in the saline-injected control. Notably, l-NAME blocked LPS-induced S-nitrosylation of DMT1, which led to reversal to low level of S-nitrosylated DMT1 (Fig. 5C,D; SNO-DMT1/total DMT1: saline, 1.00 ± 0.10, n = 6; LPS, 2.51 ± 0.41, n = 6; LPS plus l-NAME, 0.90 ± 0.26, n = 6; one-way ANOVA: F(2,15) = 10.00, p = 0.0017; post hoc Bonferroni test: control vs LPS, p = 0.0039; LPS vs LPS plus l-NAME, p = 0.0024), while the total DMT1 protein levels were similar (Fig. 5C,D; normalized total DMT1: saline, 1.00 ± 0.070, n = 6; LPS, 1.02 ± 0.094, n = 6; LPS plus l-NAME, 1.01 ± 0.13, n = 6; one-way ANOVA: F(2,15) = 0.0089, p = 0.99). Together, our data showed that LPS-induced production of endogenous NO was sufficient to S-nitrosylate DMT1 expressed in the SN, and the blockade of NO production by l-NAME, an inhibitor of NOS, abolished this effect.
Figure 5.

LPS-evoked inflammation increased S-nitrosylation of DMT1 in mouse SN. A, Immunofluorescence staining shows the activation of microglia in SN (boxed by dotted line) of the LPS or saline-injected mice. The mouse brains slices were stained with rabbit anti-Iba1 antibody. Scale bar, 200 μm. B, DAN assay shows the NO concentration in SN lysate after injection with saline, LPS, or LPS plus l-NAME. One-way ANOVA followed by post hoc Bonferroni test: p values are indicated above the bar chart. C, Representative RAC assay shows that DMT1 S-nitrosylation was enhanced by LPS-induced inflammation and blocked by l-NAME in SN of DMT1 transgenic mice. D, Bar chart and scatter plots show the statistics of SNO-DMT1/total DMT1 ratio and normalized total DMT1 protein level. One-way ANOVA followed by post hoc Bonferroni test: p values are indicated above the bar chart. The n value is indicated in the column graph. Error bars represent the SEM.
LPS injection increased iron deposition in SN of DMT1 transgenic mouse
We have shown above that intranigral injection of LPS increased NO production and DMT1 S-nitrosylation in DMT1 transgenic mice. Next, we wondered whether the LPS-mediated NO production might enhance iron uptake into the brain via DMT1 (Skjørringe et al., 2015). Using DAB-enhanced Perls' stain (Moos and Møllgård, 1993), we detected enhanced deposition of Fe in the SN. To exclude the confounding effects of other proteins subject to S-nitrosylation, such as NMDA receptor (Shi et al., 2013), we used the DMT1 inhibitor ebselen to directly block DMT1 transport. DMT1 transgenic mice were given a single injection of 5 μg of LPS or saline in the left side of SN and administered saline, l-NAME (30 mg/kg), or ebselen (10 mg/kg; Takasago et al., 1997) by intraperitoneal injection once a day for 2 weeks. Our results showed that Fe2+ deposition in the SN region was significantly greater in LPS-injected mice than in saline-injected controls (Fig. 6A,B). Moreover, both the NOS inhibitor l-NAME and the DMT1 inhibitor ebselen blocked the SNO-DMT1-mediated increase in iron deposition (Fig. 6A,B; Perl's DAB intensity: saline, 10.81 ± 1.78, n = 8; LPS, 45.35 ± 3.88, n = 6; LPS plus l-NAME, 11.80 ± 2.23, n = 6; LPS plus ebselen, 10.61 ± 2.06, n = 7; one-way ANOVA, F(3,23) = 44.07, p < 0.0001; post hoc Bonferroni test: saline vs LPS, p < 0.0001; LPS vs LPS plus l-NAME, p < 0.0001; LPS vs LPS plus ebselen, p < 0.0001). As ferritin is important in iron storage and can be induced by Fe2+, we examined ferritin heavy chain (FTH1) expression, but was surprised to discover that the FTH1 level did not change significantly after LPS injection (Fig. 6C; FTH1/GAPDH: saline, 1.00 ± 0.083, n = 5; LPS, 0.99 ± 0.073, n = 5; LPS plus l-NAME, 1.05 ± 0.13, n = 5; one-way ANOVA: F(2,13) = 0.10, p = 0.90). Ferritin contains an IRE in the 5′ UTR, and increased iron uptake activates translational upregulation of FTH1 to enhance iron storage (Pantopoulos, 2004), while a NO-mediated increase in IRP1-IRE binding could potentially inhibit transcription to decrease the mRNA level (Weiss et al., 1993; Jaffrey et al., 1994). In summation, the level of FTH1 might remain unchanged. Our results therefore indicated that the failure to upregulate FTH1 level to buffer the labile iron pool might contribute to increased oxidative stress and cell death. As iron overload can activate the JNK-mediated cell death pathway (Kim and Choi, 2010; Hua et al., 2017), we examined and found a significant increase in phospho-JNK (pJNK) in LPS-injected DMT1 transgenic mice. These results revealed that LPS upregulated pJNK level while l-NAME or ebselen restored it to a normal level, and the total JNK levels remained unchanged (Fig. 6D; pJNK/total JNK: saline, 1.00 ± 0.10, n = 6; LPS, 1.81 ± 0.16, n = 6; LPS plus l-NAME, 1.13 ± 0.094, n = 6; one-way ANOVA: F(2,15) = 12.25, p = 0.0007; post hoc Bonferroni test: saline vs LPS, p = 0.0007; LPS vs LPS plus l-NAME, p = 0.003; normalized total JNK: saline, 1.00 ± 0.038, n = 6; LPS, 0.98 ± 0.080, n = 6; LPS plus l-NAME, 0.95 ± 0.096, n = 6; one-way ANOVA: F(2,15) = 0.11, p = 0.89; Fig. 6E; pJNK/total JNK: saline, 1.00 ± 0.11, n = 6; LPS, 1.78 ± 0.066, n = 6; LPS plus ebselen, 1.11 ± 0.13, n = 6; one-way ANOVA, F(2,15) = 15.92, p = 0.0002; post hoc Bonferroni test: saline vs LPS, p = 0.0002; LPS vs LPS plus ebselen, p = 0.0009; normalized total JNK: saline, 1.00 ± 0.011, n = 6; LPS, 1.00 ± 0.094, n = 6; LPS plus ebselen, 1.01 ± 0.080, n = 6; one-way ANOVA: F(2,15) = 0.0021, p = 0.99). As ebselen is a broad antioxidant and anti-inflammatory compound, we needed to separate these effects from its DMT1 inhibitor activity. Here, we found that adding ebselen with LPS did not alter the level of S-nitrosylated DMT1 in SN (Fig. 6F; SNO-DMT1/total DMT1: saline, 1.00 ± 0.10, n = 6; LPS, 2.40 ± 0.32, n = 6; LPS plus ebselen, 2.30 ± 0.22, n = 6; one-way ANOVA: F(2,15) = 10.00, p = 0.0017; post hoc Bonferroni test: saline vs LPS, p = 0.0012; LPS vs LPS plus ebselen, p = 0.93; normalized total DMT1: saline, 1.00 ± 0.10, n = 6; LPS, 1.03 ± 0.073, n = 6; LPS plus ebselen, 1.07 ± 0.080, n = 6; one-way ANOVA: F(2,15) = 0.14, p = 0.87). Thus, the protective effect of ebselen is through directly inhibiting DMT1 activity in the LPS-injected mice.
Figure 6.

LPS-evoked inflammation enhanced iron deposition and JNK activation in mouse SN. A, Enhanced Perl's stain shows increased iron deposition in SN (boxed by dotted line) after injection with saline, LPS, LPS plus l-NAME, or LPS plus ebselen (Eb). Scale bar, 200 μm. B, Bar chart and scatter plots show the population data of Perl's DAB intensities. One-way ANOVA followed by post hoc Bonferroni test: p values are indicated above the bar chart. C, Representative Western blots show that FTH1 expression was not significantly changed by LPS or l-NAME injection in SN of DMT1 transgenic mice. D, E, Representative Western blots show that pJNK level was upregulated by LPS-induced inflammation and was blocked by l-NAME or ebselen in SN of DMT1 transgenic mice. F, Representative RAC assay shows that the LPS-enhanced DMT1 S-nitrosylation was not significantly changed by ebselen in SN of DMT1 transgenic mice. Bar charts and scatter plots show the statistics of FTH1/GAPDH, pJNK/total JNK ratio, total JNK, SNO-DMT1/total DMT1 or total DMT1 shown in C–F. One-way ANOVA followed by post hoc Bonferroni test: p values are indicated above the bar chart. The n value is indicated in the column graph. Error bars represent the SEM.
Blockade of DMT1 reversed LPS-mediated dopaminergic neuronal loss and motoric deficit
Because NO could S-nitrosylate multiple proteins, we needed to clarify whether the enhanced uptake of Fe2+ due to SNO-DMT1 might be deleterious to dopaminergic neurons of the SNc, and whether DMT1 played a major contributory role to cell death. To enumerate the surviving neurons after Fe-mediated cell death, we used the design-based stereological method to count TH+ neurons. The coefficient of error is between 0.06 and 0.09 in each unilateral SNc counting. Our results showed that intranigral LPS injection decreased dopaminergic neuron number significantly compared with the saline control, while the application of the NOS inhibitor l-NAME prevented dopaminergic neuron loss (Fig. 7A,B; dopaminergic cell number: saline, 7289 ± 231, n = 7; LPS, 4432 ± 195, n = 8; LPS plus l-NAME, 6537 ± 386, n = 6; one-way ANOVA: F(3,23) = 16.82, p < 0.0001; post hoc Bonferroni test: saline vs LPS, p < 0.0001; LPS vs LPS plus l-NAME, p = 0.0004). This result indicates that inhibiting NO production and therefore S-nitrosylation rescued dopaminergic neuronal loss. To exclude the secondary effects due to other S-nitrosylated proteins, we used the DMT1-selective inhibitor ebselen to block DMT1 function. Significantly, a similar protection by ebselen was observed (Fig. 7A,B; dopaminergic cell number: saline, 7289 ± 231, n = 7; LPS, 4432 ± 195, n = 8; LPS plus ebselen, 6912 ± 517, n = 6; one-way ANOVA: F(3,23) = 16.82, p < 0.0001; post hoc Bonferroni test: LPS vs LPS plus ebselen, p < 0.0001), suggesting that LPS-induced dopaminergic neuron loss was mediated via Fe2+ uptake by DMT1.
Figure 7.

S-nitrosylation of DMT1 contributes to LPS-induced dopaminergic neuronal loss and behavioral deficit. A, Immunohistochemistry images (2.5×) show the TH+ neuron staining in SNc region (boxed by dotted line). The insets are images under 63× objective for stereological counting. DMT transgenic mice were given a single intranigral injection of LPS or saline and then administered with saline, l-NAME, or ebselen (Eb). The fixed mouse brains were cut into 40-μm-thick slices and stained by anti-TH antibody. Scale bar, 200 μm. B, Bar chart and scatter plots show the stereological analysis of the TH+ neurons in SNc. The sections in A were counted using the design-based stereological method. Every fourth slice was take for analysis throughout the SNc and counted under a 63× oil lens. The statistical chart and scatter plots show the TH+ neuron number on the injection side. C, Rotarod behavior test shows that l-NAME or ebselen rescued the LPS-induced motoric deficiency in DMT1 transgenic mice. The bar chart and scatter plots show statistical analysis of time to fall on rotarod. One-way ANOVA followed by post hoc Bonferroni test: p values are indicated above the bar chart. D, Representative Western blot shows the expression of DMT1 in SN of scrambled shRNA or DMT1-specific shRNA-expressing mice. The bar chart and scatter plots show statistical analysis of normalized DMT1 expression. Two-tailed Student's t test: p values are indicated above the bar chart. E, Stereological analysis of the TH+ neuron number in the injection-side SNc of LPS injection, or LPS plus ebselen injection in control or DMT1 knock-down mice. One-way ANOVA followed by post hoc Bonferroni test: p values are indicated in the bar chart. F, Real-time PCR results showing the expression of mouse DMT1+IRE (left) or mouse and transgenic monkey DMT1 (panDMT1, right) normalized against GAPDH. Two-tailed Student's t test: p values are indicated above the bar chart. G, Representative Western blot shows the expression of DMT1+IRE in SN of saline or the LPS-injected side. The bar chart and scatter plots show statistical analysis of normalized DMT1+IRE expression. Two-tailed Student's t test: p values are indicated above the bar chart. The n value is indicated inside the column. Error bars represent the SEM.
As dopaminergic neurons in SNc play an important role in motor function (Hodge and Butcher, 1980), we assessed the effect of the S-nitrosylation of DMT1 on locomotor activity. The rotarod is a suitable test for the evaluation of motoric deficit in rodents (Lalonde et al., 1995), and we found that LPS injection decreased the latency to fall significantly, and critically both l-NAME and ebselen rescued LPS-mediated locomotor deficit (Fig. 7C; latency to fall: saline, 214.9 ± 4.42 s, n = 15; LPS, 132.2 ± 7.29 s, n = 15; LPS plus l-NAME, 197.8 ± 5.83 s, n = 15; LPS plus ebselen, 200.7 ± 9.15 s, n = 15; one-way ANOVA, F(3,56) = 28.61, p < 0.0001; _post hoc_ Bonferroni test: control vs LPS, _p_ < 0.0001; LPS vs LPS plus l-NAME, _p_ < 0.0001; LPS vs LPS plus ebselen, _p_ < 0.0001). Gratifyingly, the behavioral results corresponded well with the restoration of TH+ neurons. In addition, to further support the critical role of DMT1, we used lentiviral delivery of specific shRNA against DMTI to knock down DMT1 expression, and tested its effect on LPS-induced loss and the protective effect of ebselen on dopaminergic neurons in DMT1 transgenic mice. The protein level of DMT1 in shRNA-injected SN was decreased to ∼40% compared with scrambled shRNA-injected control (Fig. 7_D_; DMT1/GAPDH: scrambled, 1.00 ± 0.13, _n_ = 6; shRNA, 0.38 ± 0.052, _n_ = 6; Student's _t_ test, _t_(10) = 4.56, _p_ = 0.0001). The knock-down effect should be more potent within the injection sites as the SN area dissected for Western blot was larger than the lentiviral transduced area. Nonetheless, our results revealed that DMT1 knockdown abolished LPS-induced dopaminergic neuronal death, while the coapplication of ebselen did not further protect against neuronal death (Fig. 7_E_; dopaminergic cell number: scrambled, LPS, 4107 ± 332, _n_ = 7; scrambled, LPS plus ebselen, 5941 ± 186, _n_ = 8; DMT1 knockdown, LPS, 6540 ± 372, _n_ = 6; DMT1 knockdown, LPS plus ebselen, 6874 ± 177, _n_ = 6; one-way ANOVA: _F_(3,23) = 20, _p_ < 0.0001; _post hoc_ Bonferroni test: scrambled, LPS vs scrambled, LPS plus ebselen, _p_ = 0.0001; scrambled, LPS vs DMT1 knockdown, LPS, _p_ < 0.0001; DMT1 knockdown, LPS vs DMT1 knockdown, LPS plus ebselen, _p_ > 0.99). This knock-down experiment strongly supports the notion that enhanced DMT1 activity via S-nitrosylation contributes to dopaminergic cell death, and the major protective effect of ebselen was via its role in blocking DMT1 transport.
To further exclude the possibility that the LPS-induced DMT1 transporter activity increase was due to an elevation of DMT1 expression, we performed qRT-PCR and Western blot analysis to detect the expression of DMT+IRE and panDMT1. The results showed that neither the mRNA level of endogenous DMT1+IRE (Fig. 7F, left; DMT1+IRE/GAPDH: saline, 2.25 ± 0.15 × 10−3, n = 8; LPS, 2.24 ± 0.22 × 10−3, n = 6; Student's t test: t(12) = 0.024, p = 0.98) nor panDMT1 (Fig. 7F, right; panDMT1/GAPDH: saline, 8.28 ± 0.95 × 10−2, n = 6; LPS, 8.28 ± 0.99 × 10−2, n = 6; Student's t test: t(10) = 0.00080, p = 0.999) were significantly changed after LPS injection. We also detected the protein level with a specific rabbit polyclonal antibody, which recognize the 90 kDa glycosylated DMT1+IRE (Roth et al., 2000; Salazar et al., 2008), and found there was no significant difference after LPS injection (Fig. 7G; DMT1+IRE/GAPDH: saline, 1.00 ± 0.11, n = 6; LPS, 1.07 ± 0.09, n = 6; Student's t test: t(10) = 0.024, p = 0.64). Thus, the increased transporter activity is due to S-nitrosylation of DMT1. DMT1+IRE contains an IRE in the 3′ UTR. The increased iron level would decrease the DMT1+IRE expression (Hubert and Hentze, 2002), while NO could potentially increase DMT1 expression through enhancing IRP1-IRE binding (Weiss et al., 1993; Jaffrey et al., 1994). In summation, the level of DMT1+IRE might remain unchanged. Together, we have demonstrated that S-nitrosylation of DMT1 contributed majorly to LPS-mediated motoric deficit.
Discussion
During aging, progressive elevation of Fe in the brain poses a threat to cell survival as Fe in an oxygen-rich environment facilitates the generation of oxidative stress and reactive oxygen species (Zecca et al., 2004a). Increasing evidence supports the hypothesis that Fe accumulation in the brain plays a role in the pathogenesis of age-related PD, with iron-induced oxidative stress as the underlying mechanism (Kumar et al., 2012). Here, we provided evidence for the unexpected enhancement of DMT1 activity via S-nitrosylation, and that endogenous production of NO via LPS-induced inflammatory response resulted in enhanced Fe2+ uptake in the SN and the associated dopaminergic neuronal loss and motoric deficit. Significantly, dopaminergic neuronal loss and motoric deficit could be reversed by the application of either l-NAME, a NOS inhibitor, or ebelsen, a DMT1 blocker.
The Fe2+ uptake is regulated through DMT1 in several ways. Two regulatory regions, the 5′ promoter exon 1A region and the IRE-containing terminal exon, participate in iron regulation of DMT1 mRNA in response to cytoplasmic Fe2+ levels (Hubert and Hentze, 2002). NO could act on DMT1 by increasing IRP1 binding to the IRE sequence located at the 3′ UTR, and to stabilize and increase the DMT1 transcript level, akin to cells exposed to low iron condition (Weiss et al., 1993; Jaffrey et al., 1994; Wallander et al., 2006; Skjørringe et al., 2015). Another pathway to increase iron uptake via DMT1 is by S-nitrosylation of Dexras 1 to indirectly regulate DMT1 function and enhanced Fe2+ uptake, as reported in PC12 and cortical neurons (Cheah et al., 2006). A homolog of Dexras1, Rhes, which is enriched in the striatum, also increased Fe2+ uptake via DMTI. However, in contrast to Dexras1, Rhes is not S-nitrosylated, but instead protein kinase A-mediated phosphorylation of Rhes enhanced Fe2+ influx via DMT1 (Choi et al., 2013). Another report (Ayton et al., 2015) also showed that NO can act indirectly to enhance iron deposition in the brain via decreasing APP expression. By contrast, we showed that NO can directly modulate DMT1 and enhanced its function via S-nitrosylation. This is unexpected as the S-nitrosylation of proteins important in PD, such as Parkin and XIAP, resulted in compromised functions. In addition, many S-nitrosylated proteins have been identified in the past decade, and, of note, those that have been functionally characterized have a loss of function (Nakamura et al., 2013). In this regard, enhanced DMT1 activity arising from S-nitrosylation would therefore present a mechanism by which Fe2+ could be accumulated over aging and contribute to age-dependent neurodegeneration.
Due to the low abundance and labile nature of the S-nitrosothiol bond, detection of the cysteine residue site for S-nitrosylation has been a challenging task. Over the past 2 decades, a variety of strategies have been established, including both direct and indirect methods of detection. A BST was developed and widely used, in which S-nitrosylated cysteine (SNO-Cys) is indirectly detected by switch labeling with a reversible biotin-HPDP reagent (Forrester et al., 2009a). Subsequently, several modifications were introduced with improved specificity, sensitivity, and throughput, including the RAC method (Forrester et al., 2011). To specifically assess SNO-Cys modification status and increase the throughput of detection, several MS-based methods have been developed, including SNO-Cys site identification, SNO-RAC, S-alkylating labeling strategy, and cysTMT switch assay. Recently, Qu et al. (2014) presented a novel MS-based modified BST, the iodoTMT switch assay (ISA), using Cys thiol-reactive iodoTMT sixplex reagents to identify and quantify protein S-nitrosylation. During MS/MS analysis of the eluted peptides, SNO-Cys sites are indicated on the MS spectra by a mass increment of 329 Da from the iodoTMT tag. The iodoTMT switch allows for specific MS detection and SNO-Cys site mapping (Qu et al., 2014). Here, we used the ISA and MS to screen for SNO-Cys sites in the full-length DMT1 protein and have identified two cysteine residues that were S-nitrosylated. To the best of our knowledge, this could be the first report in which S-nitrosylation sites of a specific full-length membrane protein were identified using MS.
Inflammation is commonly observed in the brains of animal models of or patients with neurodegenerative diseases like PD (Block and Hong, 2005). We mimicked the inflammatory response by using intranigral LPS injection to induce NO production and to model hypernitrosative stress in vivo. SN contains 4.5 times as many microglia compared with the cortex and other regions of the brain, and as such this region is especially responsive to LPS (Kim et al., 2000). This notion is borne by the report that only the application of LPS into the SN resulted in neurodegeneration, while injections of LPS into the hippocampus or cortex of adult rats did not (Kim et al., 2000). Such results suggest that dopaminergic neurons are more vulnerable to oxidative stress and microglial activation relative to other cell types (Loeffler et al., 1994). While MPP+ or 6-OHDA acts directly on dopaminergic neurons to cause death, LPS causes neuronal death indirectly via the inflammatory response. Therefore, LPS injection within the SNc is a suitable model for investigating the selective effects of inflammation on the dopaminergic system and hence for studying PD (Herrera et al., 2000). LPS induced microglial activation and activated microglia release proinflammatory and cytotoxic factors such as IL-1β, TNF-α, and NO, which leads to dopaminergic neuronal damage (Arimoto and Bing, 2003). Here we successfully used this model to test how NO produced by inflammation S-nitrosylated DMT1 to increase its transporter activity and enhanced Fe2+ uptake, thereby resulting in the loss of dopaminergic neurons and compromised motoric function.
Our results would highly suggest that high iron uptake via S-nitrosylated DMT1 could potentially play a role and contribute toward the development of PD, a disease in which the pathogenesis could be multifactorial.
Footnotes
This work was supported by the National Medical Research Council (NMRC), Ministry of Health, Singapore, with Grants NMRC/CBRG12nov078 and NMRC-TCR/013-NNI/2014 (to T.W.S.); and NMRC-EDG 10nov065 and NMRC-TCR/013-NNI/2014 (to K.-L.L.). We thank Tan Fong Yong for excellent technical assistance, and Dr. Chan Yiong Huak for the helpful comments on statistics.
The authors declare no competing financial interests.
References
- Acosta-Cabronero J, Betts MJ, Cardenas-Blanco A, Yang S, Nestor PJ (2016) In vivo MRI mapping of brain iron deposition across the adult lifespan. J Neurosci 36:364–374. 10.1523/JNEUROSCI.1907-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimoto T, Bing G (2003) Up-regulation of inducible nitric oxide synthase in the substantia nigra by lipopolysaccharide causes microglial activation and neurodegeneration. Neurobiol Dis 12:35–45. 10.1016/S0969-9961(02)00017-7 [DOI] [PubMed] [Google Scholar]
- Ayton S, Lei P, Hare DJ, Duce JA, George JL, Adlard PA, McLean C, Rogers JT, Cherny RA, Finkelstein DI, Bush AI (2015) Parkinson's disease iron deposition caused by nitric oxide-induced loss of β-amyloid precursor protein. J Neurosci 35:3591–3597. 10.1523/JNEUROSCI.3439-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkaya M, Kröber JM, Rex A, Endres M (2013) Assessing post-stroke behavior in mouse models of focal ischemia. J Cereb Blood Flow Metab 33:330–338. 10.1038/jcbfm.2012.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76:77–98. 10.1016/j.pneurobio.2005.06.004 [DOI] [PubMed] [Google Scholar]
- Cheah JH, Kim SF, Hester LD, Clancy KW, Patterson SE 3rd, Papadopoulos V, Snyder SH (2006) NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1. Neuron 51:431–440. 10.1016/j.neuron.2006.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BR, Bang S, Chen Y, Cheah JH, Kim SF (2013) PKA modulates iron trafficking in the striatum via small GTPase, Rhes. Neuroscience 253:214–220. 10.1016/j.neuroscience.2013.08.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM (2004) S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science 304:1328–1331. 10.1126/science.1093891 [DOI] [PubMed] [Google Scholar]
- Cook CI, Yu BP (1998) Iron accumulation in aging: modulation by dietary restriction. Mech Ageing Dev 102:1–13. 10.1016/S0047-6374(98)00005-0 [DOI] [PubMed] [Google Scholar]
- Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C, Jonneaux A, Ryckewaert G, Garçon G, Rouaix N, Duhamel A, Jissendi P, Dujardin K, Auger F, Ravasi L, Hopes L, Grolez G, Firdaus W, Sablonnière B, Strubi-Vuillaume I, et al. (2014) Targeting chelatable iron as a therapeutic modality in Parkinson's disease. Antioxid Redox Signal 21:195–210. 10.1089/ars.2013.5593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faucheux BA, Hauw JJ, Agid Y, Hirsch EC (1997) The density of [125I]-transferrin binding sites on perikarya of melanized neurons of the substantia nigra is decreased in Parkinson's disease. Brain Res 749:170–174. 10.1016/S0006-8993(96)01412-6 [DOI] [PubMed] [Google Scholar]
- Forrester MT, Foster MW, Benhar M, Stamler JS (2009a) Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic Biol Med 46:119–126. 10.1016/j.freeradbiomed.2008.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS (2009b) Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol 27:557–559. 10.1038/nbt.1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester MT, Hess DT, Thompson JW, Hultman R, Moseley MA, Stamler JS, Casey PJ (2011) Site-specific analysis of protein S-acylation by resin-assisted capture. J Lipid Res 52:393–398. 10.1194/jlr.D011106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förstermann U, Schmidt HH, Pollock JS, Sheng H, Mitchell JA, Warner TD, Nakane M, Murad F (1991) Isoforms of nitric oxide synthase. characterization and purification from different cell types. Biochem Pharmacol 42:1849–1857. 10.1016/0006-2952(91)90581-O [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G (1997) The mouse brain in stereotaxic coordinates. San Diego, London: Academic. [Google Scholar]
- Green SJ, Scheller LF, Marletta MA, Seguin MC, Klotz FW, Slayter M, Nelson BJ, Nacy CA (1994) Nitric oxide: cytokine-regulation of nitric oxide in host resistance to intracellular pathogens. Immunol Lett 43:87–94. 10.1016/0165-2478(94)00158-8 [DOI] [PubMed] [Google Scholar]
- Guo C, Hao LJ, Yang ZH, Chai R, Zhang S, Gu Y, Gao HL, Zhong ML, Wang T, Li JY, Wang ZY (2016) Deferoxamine-mediated up-regulation of HIF-1alpha prevents dopaminergic neuronal death via the activation of MAPK family proteins in MPTP-treated mice. Exp Neurol 280:13–23. 10.1016/j.expneurol.2016.03.016 [DOI] [PubMed] [Google Scholar]
- Guo J, Gaffrey MJ, Su D, Liu T, Camp DG 2nd, Smith RD, Qian WJ (2014) Resin-assisted enrichment of thiols as a general strategy for proteomic profiling of cysteine-based reversible modifications. Nat Protoc 9:64–75. 10.1038/nprot.2013.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallgren B, Sourander P (1958) The effect of age on the non-haemin iron in the human brain. J Neurochem 3:41–51. 10.1111/j.1471-4159.1958.tb12607.x [DOI] [PubMed] [Google Scholar]
- Hardy PA, Gash D, Yokel R, Andersen A, Ai Y, Zhang Z (2005) Correlation of R2 with total iron concentration in the brains of rhesus monkeys. J Magn Reson Imaging 21:118–127. 10.1002/jmri.20244 [DOI] [PubMed] [Google Scholar]
- Herrera AJ, Castaño A, Venero JL, Cano J, Machado A (2000) The single intranigral injection of LPS as a new model for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis 7:429–447. 10.1006/nbdi.2000.0289 [DOI] [PubMed] [Google Scholar]
- Hill JM, Ruff MR, Weber RJ, Pert CB (1985) Transferrin receptors in rat brain: neuropeptide-like pattern and relationship to iron distribution. Proc Natl Acad Sci U S A 82:4553–4557. 10.1073/pnas.82.13.4553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge GK, Butcher LL (1980) Pars compacta of the substantia nigra modulates motor activity but is not involved importantly in regulating food and water intake. Naunyn Schmiedebergs Arch Pharmacol 313:51–67. 10.1007/BF00505805 [DOI] [PubMed] [Google Scholar]
- Hua Y, Wang C, Jiang H, Wang Y, Liu C, Li L, Liu H, Shao Z, Fu R (2017) Iron overload may promote alteration of NK cells and hematopoietic stem/progenitor cells by JNK and P38 pathway in myelodysplastic syndromes. Int J Hematol 106:248–257. 10.1007/s12185-017-2237-x [DOI] [PubMed] [Google Scholar]
- Hubert N, Hentze MW (2002) Previously uncharacterized isoforms of divalent metal transporter (DMT)-1: implications for regulation and cellular function. Proc Natl Acad Sci U S A 99:12345–12350. 10.1073/pnas.192423399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998) Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 57:1–9. 10.1016/S0169-328X(98)00040-0 [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Cohen NA, Rouault TA, Klausner RD, Snyder SH (1994) The iron-responsive element binding protein: a target for synaptic actions of nitric oxide. Proc Natl Acad Sci U S A 91:12994–12998. 10.1073/pnas.91.26.12994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrer JP. (2000) The haber-weiss reaction and mechanisms of toxicity. Toxicology 149:43–50. 10.1016/S0300-483X(00)00231-6 [DOI] [PubMed] [Google Scholar]
- Kim EK, Choi EJ (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802:396–405. 10.1016/j.bbadis.2009.12.009 [DOI] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS (2000) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci 20:6309–6316. 10.1523/JNEUROSCI.20-16-06309.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Lim HW, More SV, Kim BW, Koppula S, Kim IS, Choi DK (2012) The role of free radicals in the aging brain and Parkinson's disease: convergence and parallelism. Int J Mol Sci 13:10478–10504. 10.3390/ijms130810478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R, Bensoula AN, Filali M (1995) Rotorod sensorimotor learning in cerebellar mutant mice. Neurosci Res 22:423–426. 10.1016/0168-0102(95)00916-H [DOI] [PubMed] [Google Scholar]
- Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, Sucher NJ, Lipton SA (1992) Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron 8:1087–1099. 10.1016/0896-6273(92)90130-6 [DOI] [PubMed] [Google Scholar]
- Loeffler DA, DeMaggio AJ, Juneau PL, Havaich MK, LeWitt PA (1994) Effects of enhanced striatal dopamine turnover in vivo on glutathione oxidation. Clin Neuropharmacol 17:370–379. 10.1097/00002826-199408000-00009 [DOI] [PubMed] [Google Scholar]
- Massie HR, Aiello VR, Banziger V (1983) Iron accumulation and lipid peroxidation in aging C57BL/6J mice. Exp Gerontol 18:277–285. 10.1016/0531-5565(83)90038-4 [DOI] [PubMed] [Google Scholar]
- Misko TP, Schilling RJ, Salvemini D, Moore WM, Currie MG (1993) A fluorometric assay for the measurement of nitrite in biological samples. Anal Biochem 214:11–16. 10.1006/abio.1993.1449 [DOI] [PubMed] [Google Scholar]
- Moos T, Møllgård K (1993) A sensitive post-DAB enhancement technique for demonstration of iron in the central nervous system. Histochemistry 99:471–475. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA (2013) Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 78:596–614. 10.1016/j.neuron.2013.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantopoulos K. (2004) Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci 1012:1–13. 10.1196/annals.1306.001 [DOI] [PubMed] [Google Scholar]
- Qu Z, Meng F, Bomgarden RD, Viner RI, Li J, Rogers JC, Cheng J, Greenlief CM, Cui J, Lubahn DB, Sun GY, Gu Z (2014) Proteomic quantification and site-mapping of S-nitrosylated proteins using isobaric iodoTMT reagents. J Proteome Res 13:3200–3211. 10.1021/pr401179v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JA, Horbinski C, Feng L, Dolan KG, Higgins D, Garrick MD (2000) Differential localization of divalent metal transporter 1 with and without iron response element in rat PC12 and sympathetic neuronal cells. J Neurosci 20:7595–7601. 10.1523/JNEUROSCI.20-20-07595.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nuñez MT, Garrick MD, Raisman-Vozari R, Hirsch EC (2008) Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson's disease. Proc Natl Acad Sci U S A 105:18578–18583. 10.1073/pnas.0804373105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz C, Hof PR (2005) Design-based stereology in neuroscience. Neuroscience 130:813–831. 10.1016/j.neuroscience.2004.08.050 [DOI] [PubMed] [Google Scholar]
- Shi ZQ, Sunico CR, McKercher SR, Cui J, Feng GS, Nakamura T, Lipton SA (2013) S-nitrosylated SHP-2 contributes to NMDA receptor-mediated excitotoxicity in acute ischemic stroke. Proc Natl Acad Sci U S A 110:3137–3142. 10.1073/pnas.1215501110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Das T, Ravindran A, Chaturvedi RK, Shukla Y, Agarwal AK, Dikshit M (2005) Involvement of nitric oxide in neurodegeneration: a study on the experimental models of Parkinson's disease. Redox Rep 10:103–109. 10.1179/135100005X38842 [DOI] [PubMed] [Google Scholar]
- Skjørringe T, Burkhart A, Johnsen KB, Moos T (2015) Divalent metal transporter 1 (DMT1) in the brain: implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front Mol Neurosci 8:19. 10.3389/fnmol.2015.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel DJ, Loscalzo J (1992) S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc Natl Acad Sci U S A 89:444–448. 10.1073/pnas.89.1.444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasago T, Peters EE, Graham DI, Masayasu H, Macrae IM (1997) Neuroprotective efficacy of ebselen, an anti-oxidant with anti-inflammatory actions, in a rodent model of permanent middle cerebral artery occlusion. Br J Pharmacol 122:1251–1256. 10.1038/sj.bjp.0701426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang AH, Lee YI, Ko HS, Savitt JM, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM, Chung KK (2009) S-nitrosylation of XIAP compromises neuronal survival in Parkinson's disease. Proc Natl Acad Sci U S A 106:4900–4905. 10.1073/pnas.0810595106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallander ML, Leibold EA, Eisenstein RS (2006) Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim Biophys Acta 1763:668–689. 10.1016/j.bbamcr.2006.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G, Goossen B, Doppler W, Fuchs D, Pantopoulos K, Werner-Felmayer G, Wachter H, Hentze MW (1993) Translational regulation via iron-responsive elements by the nitric oxide/NO-synthase pathway. EMBO J 12:3651–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Jin J, DeFelice LJ, Andrews NC, Clapham DE (2004) A spontaneous, recurrent mutation in divalent metal transporter-1 exposes a calcium entry pathway. PLoS Biol 2:E50. 10.1371/journal.pbio.0020050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Gallorini M, Schünemann V, Trautwein AX, Gerlach M, Riederer P, Vezzoni P, Tampellini D (2001) Iron, neuromelanin and ferritin content in the substantia nigra of normal subjects at different ages: consequences for iron storage and neurodegenerative processes. J Neurochem 76:1766–1773. 10.1046/j.1471-4159.2001.00186.x [DOI] [PubMed] [Google Scholar]
- Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR (2004a) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5:863–873. 10.1038/nrn1537 [DOI] [PubMed] [Google Scholar]
- Zecca L, Stroppolo A, Gatti A, Tampellini D, Toscani M, Gallorini M, Giaveri G, Arosio P, Santambrogio P, Fariello RG, Karatekin E, Kleinman MH, Turro N, Hornykiewicz O, Zucca FA (2004b) The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc Natl Acad Sci U S A 101:9843–9848. 10.1073/pnas.0403495101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CW, Tai YK, Chai BH, Chew KCM, Ang ET, Tsang F, Tan BWQ, Hong ETE, Asad ABA, Chuang KH, Lim KL, Soong TW (2017) Transgenic mice overexpressing the divalent metal transporter 1 exhibit iron accumulation and enhanced parkin expression in the brain. Neuromolecular Med 19:375–386. 10.1007/s12017-017-8451-0 [DOI] [PMC free article] [PubMed] [Google Scholar]