Leptin Signaling in GABA Neurons, But Not Glutamate Neurons, Is Required for Reproductive Function (original) (raw)
Abstract
The adipocyte-derived hormone leptin acts in the brain to modulate the central driver of fertility: the gonadotropin releasing hormone (GnRH) neuronal system. This effect is indirect, as GnRH neurons do not express leptin receptors (LEPRs). Here we test whether GABAergic or glutamatergic neurons provide the intermediate pathway between the site of leptin action and the GnRH neurons. Leptin receptors were deleted from GABA and glutamate neurons using Cre-Lox transgenics, and the downstream effects on puberty onset and reproduction were examined. Both mouse lines displayed the expected increase in body weight and region-specific loss of leptin signaling in the hypothalamus. The GABA neuron-specific LEPR knock-out females and males showed significantly delayed puberty onset. Adult fertility observations revealed that these knock-out animals have decreased fecundity. In contrast, glutamate neuron-specific LEPR knock-out mice displayed normal fertility. Assessment of the estrogenic hypothalamic-pituitary-gonadal axis regulation in females showed that leptin action on GABA neurons is not necessary for estradiol-mediated suppression of tonic luteinizing hormone secretion (an indirect measure of GnRH neuron activity) but is required for regulation of a full preovulatory-like luteinizing hormone surge. In conclusion, leptin signaling in GABAergic (but not glutamatergic neurons) plays a critical role in the timing of puberty onset and is involved in fertility regulation throughout adulthood in both sexes. These results form an important step in explaining the role of central leptin signaling in the reproductive system. Limiting the leptin-to-GnRH mediators to GABAergic cells will enable future research to focus on a few specific types of neurons.
Introduction
Circulating leptin is an important metabolic signal regulating food intake and energy expenditure. Leptin also forms a permissive modulator of fertility (Ahima et al., 1997; Nagatani et al., 1998). Reproduction is centrally regulated by the drivers of the hypothalamic-pituitary-gonadal (HPG) axis: the gonadotropin releasing hormone (GnRH) neurons. A significant body of research has been devoted to trying to understand how leptin's effects are relayed to this reproductive axis. Leptin acts directly on the brain as transgenic removal of leptin receptors (LEPRs) from forebrain neurons results in the same infertile and obese phenotype as does global mutation in the leptin gene itself (Ingalls et al., 1950; Swerdloff et al., 1975; Quennell et al., 2009). However, leptin affects GnRH neurons indirectly as they do not express LEPRs (Quennell et al., 2009). To elucidate the identity of the neuronal network that must exist between leptin-responsive neurons and GnRH neurons, we investigated leptin signaling in the major inhibitory and excitatory neuronal cell populations: GABA and glutamate neurons.
The GnRH neurons express both GABAA and GABAB receptor isoforms, and binding of GABA to the GABAB receptor causes an inhibition of pulsatile GnRH release (Todman et al., 2005; Zhang et al., 2009). Although GABA is generally an inhibitory neurotransmitter, the effect of GABAA receptor activation in GnRH neurons can be excitatory or inhibitory depending on the chloride concentration within the neuron (Krnjevic, 1974; Herbison and Moenter, 2011). Hypothalamic locations where GABAergic LEPR neurons are concentrated include the arcuate nucleus (Arc), dorsomedial nucleus (DMN), and lateral hypothalamus (Ovesjö et al., 2001; Vong et al., 2011). Functionally, acute fasting (i.e., reducing leptin levels) alters GABAergic transmission to GnRH neurons and decreases GnRH neuronal activity (Sullivan et al., 2003; Sullivan and Moenter, 2004), suggesting that leptin sensing GABAergic afferents integrate metabolic cues to modulate GnRH release.
Glutamate is the main excitatory neurotransmitter of the CNS. The cell bodies of GnRH neurons are innervated by glutamatergic terminals that stimulate GnRH release (Bourguignon et al., 1995; Kiss et al., 2003). Hypothalamic glutamatergic LEPR neurons can be found in the Arc, the ventral medial nucleus (VMN), and the ventral premammilary nucleus (PMV) (Kocsis, 2003; Vong et al., 2011). Lesion experiments indicate that PMV LEPR neurons are involved in leptin's effects on fertility (Donato et al., 2009; Leshan et al., 2009). Hence, glutamatergic LEPR neurons form a likely leptin-to-GnRH candidate.
To specifically delete LEPRs from either vesicular GABA transporter (Vgat) or vesicular glutamate transporter 2 (Vglut2) expressing neurons, targeting GABA and glutamate neurons, respectively, Cre-Lox transgenics was used. The effects of these deletions on fertility were studied in female and male animals. Specifically, puberty onset, adult fertility, and estradiol feedback on the HPG axis were assessed to determine which of these two major neuronal populations is primarily responsible for leptin's effects on reproductive function.
Materials and Methods
Animals.
Female and male transgenic mice with Vglut2 (_Vglut2_-ires-Cre) or Vgat (_Vgat_-ires-Cre) specific LEPR deletions were bred from transgenic lines as described by Vong et al. (2011) (on a mixed FVB and C57BL/6J background). These animals are hereafter referred to as GABA- and glutamate-specific LEPR knock-outs. Homozygous LEPR floxed (_Lepr_flox/flox) littermates were used as controls. Animals were group housed or paired with an animal of the opposite sex (for fertility assessment). Mice were kept on a 12 h:12 h light and dark cycle (lights on at 0800 h), at constant room temperature (21 ± 1°C), with ad libitum access to water and standard rodent chow unless otherwise noted. The University of Otago Animal Ethics Committee approved all experimental animal protocols.
Tissue collection and preparation.
For tissue collection animals were fasted overnight and injected subcutaneously with recombinant leptin (5 mg/kg; National Hormone and Peptide Program) or vehicle (0.01 m PBS, pH 7.8). Two hours after injection, animals were sedated and blood collected from the inferior vena cava before transcardial perfusion with 4% paraformaldehyde in 0.1 m PBS, pH 7.4. Brain tissue was prepared for immunohistochemistry, as previously described by Quennell et al. (2011).
Immunohistochemistry and image analysis.
Immunohistochemical labeling of leptin-induced phosphorylation of the leptin signaling molecule signal transducer and activator of transcription 3 (pSTAT3) was performed to determine loss of hypothalamic leptin-responsiveness in the knock-out mice. To do so, we performed an antigen retrieval step (15 min; 1 mm EDTA, pH 8.0, at 90°C), and used polyclonal rabbit anti-pSTAT3 as the primary antibody (1:3000; Tyr705; #9145, Cell Signaling Technology). This was followed by a biotinylated goat anti-rabbit secondary antibody (1:1000; Vector Laboratories), and labeling was visualized with 3,3′-diaminobenzidine (Quennell et al., 2011). To analyze the effects of LEPR knock-out on the number of pSTAT3-positive neurons, stained cells were counted in hypothalamic nuclei [median preoptic area (MnPO), rostral preoptic ares (rPOA), medial preoptic area (MPA), rostral Arc (bregma −0.94 to −1.34 mm), medial Arc (bregma −1.46 to −1.94 mm), caudal Arc (bregma −2.06 to −2.54 mm), VMN, lateral hypothalamus, DMN, PMV] and the nucleus of the solitary tract (NTS). Positive leptin-responsive cells were counted when a clear circular and darkly stained nucleus was present. Counts were performed in photomicrographs of at least three tissue sections per area from each animal. The three Arc subregions were later merged to form a single bar on the graph (see Fig. 1J).
Figure 1.

Characterization of the knock-out models by labeling leptin-induced phosphorylated STAT3. A–I, Representative photomicrographs of staining in different hypothalamic areas counted. A–C, MnPO and rPOA. D–F, Arc, VMN, LH, and DMN. G–I, Arc and PMV. The first column shows leptin-induced (5 mg/kg) pSTAT3 labeling in control animals. Second and third columns show pSTAT3 labeling in GABA- and glutamate-specific LEPR knock-out animals, respectively. J, Quantification of immunohistochemical staining (number of positive labeled cells per section) in control animals (black bars; n = 11), GABA-specific (gray bars; n = 10), and glutamate-specific (white bars, n = 8; n = 2 for NTS) knock-outs. There was no difference between the pSTAT3 counts of the two control groups (n = 6 and n = 5) that are derived from the two different Cre lines; therefore, they have been pooled in this graph. In all regions, experimental groups are compared with the control animals to show significant differences. MnPO includes the region of the organum vasculosum of the lamina terminalis (OVLT); rPOA, ventral part. Scale bar, 100 μm. *p < 0.05.
Fluorescent double-label immunohistochemistry of GnRH and vGAT was performed in control and GABA-specific LEPR knock-out male animals. Brain sections containing GnRH neurons were incubated in polyclonal guinea pig anti-GnRH (1:2000; #GA02, in-house) and rabbit anti-vGAT (1:750; #5062P, Millipore) primary antibodies. This was followed by a direct labeling with fluorescent secondary antibodies, using goat anti-guinea pig AlexaFluor-488 (Invitrogen) to visualize GnRH and goat anti-rabbit AlexaFluor-568 (Invitrogen) to label vGAT. Sections were mounted on microscope slides with Vectashield (Vector Laboratories) for confocal microscopy (Zeiss LSM 710). Using an argon laser exciting at 488 nm (GnRH), a helium neon laser exciting at 543 nm (vGAT) and a ×40 PlanApochromat objective lens, a _z_-stack (0.5 μm optical slices) was made of each individual GnRH soma and proximal projections. Omission of the primary vGAT antibody resulted in an absence of stained terminals. Twenty GnRH neurons per animal were photographed and used for counting (10 within the medial septum population and 10 in the rPOA). Zeiss LSM image browser software was used to count the number of appositions (where red pixels touched the green, for at least two consecutive optical slices) on the GnRH soma and in three 10 μm segments progressing distally along the projections. This tool was also used to measure the circumference of the soma. Where data were obtained from two projections per GnRH neuron, these were averaged.
Leptin ELISA.
Plasma leptin concentration of control and knock-out animals was measured by ELISA (Mouse Leptin ELISA kit, Chrystal Chem). The average coefficient of variation was 4.1%, and the sensitivity of the assay was 0.04 ng/ml. All samples were repeated in duplicate on one single plate.
Fertility experiments.
In female animals, puberty onset was measured by vaginal opening along with the age at first estrus (based on vaginal cytology). From 26 d of age, experimental (GABA- and glutamate-specific LEPR knock-out) and control mice were checked daily. Once vaginal opening was established, vaginal cytology was studied to assess for signs of first estrus. Male mice were mated with adult wild-type C57BL/6J females from 36 d of age. Male puberty onset was calculated by subtracting 21 d (gestation period) from the date when their first litter was born. Males were kept in their breeding pairs to further assess adult fertility. To determine adult female estrous cyclicity, vaginal smears were taken for 10 (glutamate-specific knock-outs and controls) or 28 (GABA-specific knock-outs and controls) consecutive days starting at least 14 d after first estrus. Estrus cycle duration was calculated as the average time between two proestrus phases. Afterward, female knock-out and control animals of both experimental groups were paired with adult wild-type C57BL/6J males to study fecundity.
Estimation of daily sperm production.
After transcardial perfusion and brain collection, one testis from control or knock-out male mice was removed, wet weight recorded, and stored at −80 C. Daily sperm production was subsequently determined as described previously with slight modifications (Robb et al., 1978; Singireddy et al., 2013). Briefly, testis fragments (25 mg) were homogenized in a tissue lyser (TissueLyser II, QIAGEN) for 10 min. Spermatids in stages 14–16 of spermatogenesis (Leblond and Clermont, 1952) are resistant to homogenization, and their nuclei were counted in 10 μl aliquots of homogenate using a hemocytometer. Total counted nuclei were divided by 4.84 because developing spermatids spend 4.84 d in stages 14–16 during spermatogenesis in mice.
Assessment of HPG axis regulation by estradiol.
Negative feedback assessment of estradiol was based on the experiments described by Moore et al. (2012). Briefly, GABA-specific LEPR knock-out and control mice were anesthetized with isoflurane and tail blood samples taken at baseline (day 0; intact). Ovariectomies were performed, and 14 d later another blood sample was taken (OVX). Animals were subsequently implanted with a chronic slow-release 17β-estradiol subcutaneous implant (50 μg/kg; 11–26 mm long depending on body weight); 8 d later, another blood sample was taken (OVX+implant). Subsequently, preovulatory-like luteinizing hormone (LH) surge induction was performed as described by Quennell et al. (2009). On day 22, all mice received a bolus injection of estradiol benzoate (50 μg/kg, s.c.) 9 h before lights out. At lights out on day 23, final trunk blood samples were taken. All blood samples were processed within 10 min of collection. Plasma was separated by centrifugation, immediately frozen, and stored at −20°C. The concentration of LH in the plasma was measured by radioimmunoassay, as described by Quennell et al. (2009). The sensitivity of the assay (95% confidence interval at 0 ng/ml) was 0.14 ng/ml, and the intra-assay coefficient of variation was 5.5%. All samples were analyzed in triplicate within the same assay.
Statistical analysis.
All results are presented as mean ± SEM. Student's t tests were used to identify significant statistical differences (p < 0.05) between control and knock-out mice in the pSTAT3 cell counts, plasma leptin concentrations, daily sperm production, and other measures of fertility. The body weight and negative feedback data were compared using a two-way ANOVA with repeated measures and a Bonferroni multiple-comparison post hoc test. Pooling of _Lepr_flox/flox control groups was used in immunohistochemical, plasma leptin concentration and body weight comparisons because the two experiments were run concurrently and controls were identically produced.
Results
Loss of leptin signaling in GABA- and glutamate-specific LEPR knock-out neurons
To validate our transgenic knock-out models, we identified the loss of leptin-responsive cells using pSTAT3 immunohistochemistry. The Cre-Lox animal models used in this study disrupt leptin signaling by deletion of exon 17 of the Lepr gene. This exon codes for the part of the receptor responsible for JAK-STAT signaling (McMinn et al., 2004); therefore, loss of phosphorylated STAT3 provides an accurate representation of where LEPR was deleted. The transgenic disruption of LEPR from either GABA or glutamate neurons resulted in a marked reduction of leptin-induced pSTAT3 immunoreactivity in the hypothalamic nuclei where these neurotransmitters are known to be strongly expressed (Vong et al., 2011). Removing LEPRs from GABA neurons caused a 42% reduction of leptin-induced pSTAT3 in the Arc (particularly in the medial and caudal Arc sections; the effect was not seen in the most rostral sections; Fig. 1E,H,J). Similar reductions were seen in the lateral hypothalamus (49%) and the DMN (31%; Fig. 1E,J). Deletion of LEPRs from glutamate neurons caused a different pattern of pSTAT3 staining. Strikingly, pSTAT3 immunoreactivity was 80–97% reduced in the PMV and VMN, respectively (Fig. 1F,I,J). Significant decreases were also evident in the ventral rPOA (55%; Fig. 1C,J) and MPA (47%; Fig. 1J) as well as the lateral hypothalamus (30%; Fig. 1F,J) when glutamate-specific LEPR knock-outs were compared with controls. Extra-hypothalamic LEPR deletion was addressed by counting pSTAT3-positive nuclei in the NTS. No significant differences were found between the GABA-specific knock-outs and controls (Fig. 1J); this was expected because several studies have shown that the NTS LEPR neurons are not GABAergic (Vong et al., 2011; Garfield et al., 2012). The number of animals in the glutamate-specific LEPR knock-out group was low; nevertheless, a trend toward a reduced pSTAT3 response (albeit nonsignificant; n = 2; t(8) = 1.261, p = 0.24) was observed similar to what has been reported (Vong et al., 2011).
In vehicle-treated animals, the overnight fast was unable to reduce pSTAT3 immunoreactivity to the same extent in the GABA-specific LEPR knock-outs compared with the controls. This phenomenon is likely the result of the high circulating leptin found in these very obese animals (Fig. 2C).
Figure 2.
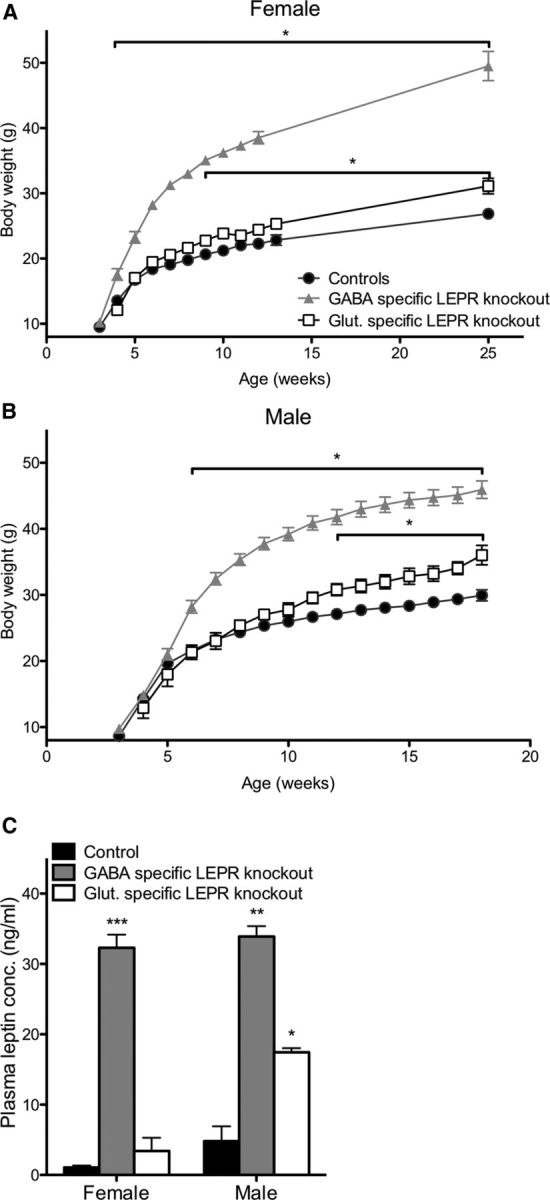
Effects of LEPR knock-outs on body weight and plasma leptin concentration. A, B, Female and male GABA- and glutamate-specific LEPR knock-out and control animals body weights. Female GABA-specific knock-out animals (gray triangles; n = 10) are significantly heavier than controls (black circles; n = 19) at 4 weeks of age; glutamate-specific LEPR knock-out animals (white squares; n = 9) are significantly heavier than controls (black circles) at 9 weeks of age. Male GABA-specific LEPR knock-out animals (gray triangles; n = 9) had an increase in body weight from 6 weeks onwards and glutamate-specific knock-out males (white squares; n = 6) from 12 weeks onwards compared with controls (black circles; n = 15). We were unable to gather body weight data for the female groups throughout the breeding study because females were at different stages of pregnancy. C, Plasma leptin concentrations of control animals (black bars; female n = 8, male n = 3) are significantly lower than GABA-specific knock-out animals (gray bars; female n = 8, male n = 2). In glutamate-specific LEPR knock-outs, male animals show significantly higher leptin than the controls (white bars; female n = 4, male n = 2). *p < 0.05. **p < 0.01. ***p < 0.001.
The above-mentioned findings agree with previously described distributions of Vgat and Vglut2 mRNA in the hypothalamus (Vong et al., 2011), confirming that the LEPR deletions were indeed targeted to neurons expressing Vgat and Vglut2. There appeared to be very little overlap between the regions affected by GABA and glutamate LEPR knock-out. The lateral hypothalamus was an exception; both GABA- and glutamate-specific LEPR deletion reduced the numbers of neurons able to respond to leptin here.
Disrupted body weight regulation in both GABA- and glutamate-specific LEPR knock-out animals
Body weights of all knock-out and control groups were measured fortnightly. The genotype of the knock-outs had a significant effect on body weight in females (two-way repeated-measures ANOVA: F(2,32) = 247.06, p < 0.001) and in males (two-way repeated-measures ANOVA: F(2,26) = 69.50, p < 0.001) compared with controls. As previously described, LEPR knock-out from GABA neurons caused an obese phenotype (Vong et al., 2011). Post hoc testing revealed that female GABA-specific LEPR knock-out animals were significantly heavier than littermate controls from the age of 4 weeks onwards (Fig. 2A), whereas for males a significantly greater body weight was reached at 6 weeks of age (Fig. 2B). Glutamate-specific LEPR knock-out caused a milder metabolic phenotype. Females were significantly heavier than their littermate controls by 9 weeks of age (Fig. 2A). The males showed a significant weight difference from 12 weeks of age compared with control animals (Fig. 2B). These results are similar to the body weights described by Vong et al. (2011), with the GABA-specific knock-outs being notably heavier than the glutamate-specific knock-out animals. Blood collected at the end of the breeding studies showed that plasma leptin concentrations were significantly elevated in both female and male GABA-specific LEPR knock-outs (female: t(14) = 16.46, p < 0.0001 and male: t(3) = 9.85, p = 0.0022; Fig. 2C). In glutamate-specific knock-outs, a significant increase in leptin concentration was only evident in male animals (t(3) = 4.56, p = 0.02; Fig. 2C).
Delayed puberty onset in GABA-specific LEPR knock-out animals
To assess puberty onset, date of vaginal opening and first estrus in females were measured, whereas for males the age of first fertile mating was determined. Female mice with LEPR deleted from GABAergic neurons showed a significant, 7 d delay in vaginal opening (t(18) = 2.40, p = 0.027), coupled with a 20 d delay in first estrus (t(16) = 10.31, p < 0.0001; Fig. 3A,C). When male GABA-specific LEPR knock-out animals and controls were mated with inbred wild-type C57BL/6J females of reproductive age, they showed a significant delay in puberty onset. When the age at puberty was backdated, it revealed that the knock-out animals went through puberty ∼13 d later than the controls (t(12) = 3.40, p = 0.005; Fig. 3A,D).
Figure 3.

Puberty onset in GABA- and glutamate-specific LEPR knock-out animals. A, In GABA-specific LEPR knock-out animals (gray bars; n = 7–10), vaginal opening, first estrus, and male puberty onset were all significantly delayed compared with their control littermates (black bars; n = 7–10). B, Vaginal opening, first estrus, and male puberty onset all occurred at the same time in glutamate-specific knock-out animals (white bars; n = 6–9) compared with their control littermates (black bars; n = 8–9). C, Survival profiles showing puberty onset of female GABA-specific LEPR knock-out (gray lines) and control animals (black lines). The percentage of mice showing vaginal opening (intermittent lines) and first estrus (continuous lines) is plotted for each time point. D, Puberty onset (date of first successful mating) profiles of male GABA-specific knock-outs (gray line) and controls (black line) over time is plotted. VO, Vaginal opening. *p < 0.05. **p < 0.01. ***p < 0.001.
Puberty onset of glutamate-specific LEPR knock-out females and males did not differ from controls (Fig. 3B). Both female knock-out and control animals showed vaginal opening at 30.2 ± 0.7 d. In these animals, first estrus was evident ∼3–4 d later. Male glutamate-specific LEPR knock-out animals did not show any significant difference in the timing of puberty onset either (Fig. 3B).
Reduced adult fertility in GABA-specific LEPR knock-out animals
To assess adult fertility, we investigated female estrous cycles and measured fecundity in both sexes. Vaginal cytology was determined for 28 consecutive days beginning at day 60 in female GABA-specific LEPR knock-out animals; significantly less time was spent in proestrus (t(17) = 7.32, p < 0.0001; Fig. 4A) and more time in estrus compared with controls (t(17) = 2.28, p = 0.036). The time spent in met- or diestrus was not different between groups (t(17) = 0.63, p = 0.54; Fig. 4A). Estrus cycle duration averaged 13.6 ± 1.8 d for knock-outs compared with 4.9 ± 0.2 d for controls, showing that GABA-specific LEPR knock-out resulted in significantly prolonged cycles (t(17) = 5.02, p = 0.0001). For glutamate-specific LEPR knock-out females, there was no difference in the frequency of cycle stage compared with controls. On average, one day was spent in each of the pro- and estrus phases and 2 d in met- and diestrus per cycle (Fig. 4C).
Figure 4.

Adult fertility in knock-out and control groups. A, Frequency of estrous cycle stage was monitored for 28 d in GABA-specific LEPR knock-out females (gray bars; n = 9) and controls (black bars; n = 10). A significant decrease in time spent in proestrus was seen in the knock-out females. B, The time from pairing with a wild-type male to first litter in GABA knock-out females (gray bar; n = 10) was longer than in littermate controls (black bar; n = 10). Number of days between litters was significantly increased in male GABA-specific knock-out animals (n = 7 in both groups). The same trend was seen in surviving females, but low animal number limited statistical comparison (n = 3). C, Frequency of estrous cycle stage was monitored for 10 d in glutamate-specific knock-out mice (white bars; n = 9) and control littermates (black bars; n = 9). No significant differences were seen between the groups. D, Time to first litter and number of days between litters were the same for all glutamate-specific LEPR knock-out females and males (white bars; n = 6 and 9) compared with littermate controls (black bars; n = 8 and 9). *p < 0.05. ***p < 0.001.
Fecundity of adult mice was assessed by litter size and frequency after pairing with wild-type mates. In the GABA-specific LEPR knock-out females, three animals had to be killed because of dystocia complications; and because of these welfare considerations, the experiment had to be terminated prematurely (after 38 d of mating). Only 30% (3/10) of GABA-specific LEPR knock-out females produced two litters within this time frame, a further four of the knock-out females delivered successfully but only had one litter. In contrast, all control females produced two litters after 38 d of mating. The interlitter interval for the multiparous three knock-outs was not significantly different compared with controls (t(11) = 1.12, p = 0.29; Fig. 4B). However, there was a significant delay in the time from pairing to first delivery for knock-out females (t(18) = 4.21, p = 0.0005; Fig. 4B). The average number of pups in the litters that were born did not differ between groups (knock-out: 6.2 ± 0.4 and control 7.6 ± 0.6 pups per litter; t(17) = 1.79, p = 0.09). Male knock-out and control animals were kept in their breeding pairs for 100 d after their first litter was born. The time between litters over this period was significantly greater in the knock-out group compared with the control animals (t(12) = 2.74, p = 0.0178; Fig. 4B). The average number of pups per litter was not different between groups (knock-out: 7.8 ± 0.7 and control 7.8 ± 0.5 pups per litter; t(12) = 0.11, p = 0.91). To assess whether this reduced fertility in the male animals was the result of lower sperm counts, daily sperm production was measured in GABA-specific LEPR knock-out males and controls. This revealed no significant differences between the two groups (control: 1.2 × 106 ± 0.3 × 106; knock-out: 1.4 × 106 ± 0.5 × 106 spermatocytes per testis per day; t(8) = 0.32, p = 0.76). Glutamate-specific LEPR knock-outs and their controls were also assessed for litter frequency and time to first litter. After pairing with an inbred wild-type mate, both female and male knock-outs and controls were proven to be equally fertile (Fig. 4D).
These results, together with the delay in puberty onset, indicate a critical role for GABAergic leptin-sensing neurons in the onset and maintenance of male and female fertility. Interestingly, the results show that LEPR signaling in glutamate (vGLUT2) neurons is not required for normal reproductive function.
GABA-specific LEPR knock-out animals have an impaired response to estradiol in a positive feedback paradigm
The experiments described above have demonstrated a significant delay in puberty onset and a reduction in fertility when LEPR signaling is disrupted in GABA neurons. To explore the underlying mechanism of this decrease in fertility, we assessed the hypothalamic response to estradiol. Measuring the circulating concentration of LH in different estrogenic states provides a direct index of GnRH neuronal activity because GnRH is a potent stimulator of LH release. Estrogens inhibit GnRH/LH secretion at most times during the female cycle (negative feedback); however, on the day before ovulation, a rising estradiol concentration triggers a massive preovulatory GnRH/LH surge (positive feedback), eventually causing ovulation (Herbison, 1998).
Intact control and GABA-specific LEPR knock-out mice had similar LH levels that increased significantly after OVX (controls t(16) = 6.12; knock-outs t(16) = 5.70; p < 0.0001 for both; Fig. 5A). The ability of estradiol to suppress LH levels was undiminished in GABA-specific LEPR knock-out compared with control animals (controls t(16) = 5.01; knock-outs t(16) = 5.27; p < 0.0001 for both; Fig. 5A), showing that estrogenic negative feedback remains functional in these mice despite their fertility defects and high circulating leptin concentration. To test whether leptin signaling in GABA neurons is required for the positive estradiol feedback, we used an ovariectomized plus high-dose estradiol model. In this model, a bolus injection of estradiol benzoate results in an artificially induced preovulatory-like LH surge the following day at the time of lights out (Wintermantel et al., 2006). When our animals were subjected to this treatment, the knock-out animals had a 46% lower plasma LH concentration at the time of the preovulatory-like surge compared with controls (t(16) = 2.49, p = 0.025; Fig. 5B). These data suggest that an impaired positive feedback mechanism may underlie the subfertile phenotype seen in the GABA-specific LEPR knock-out mice.
Figure 5.
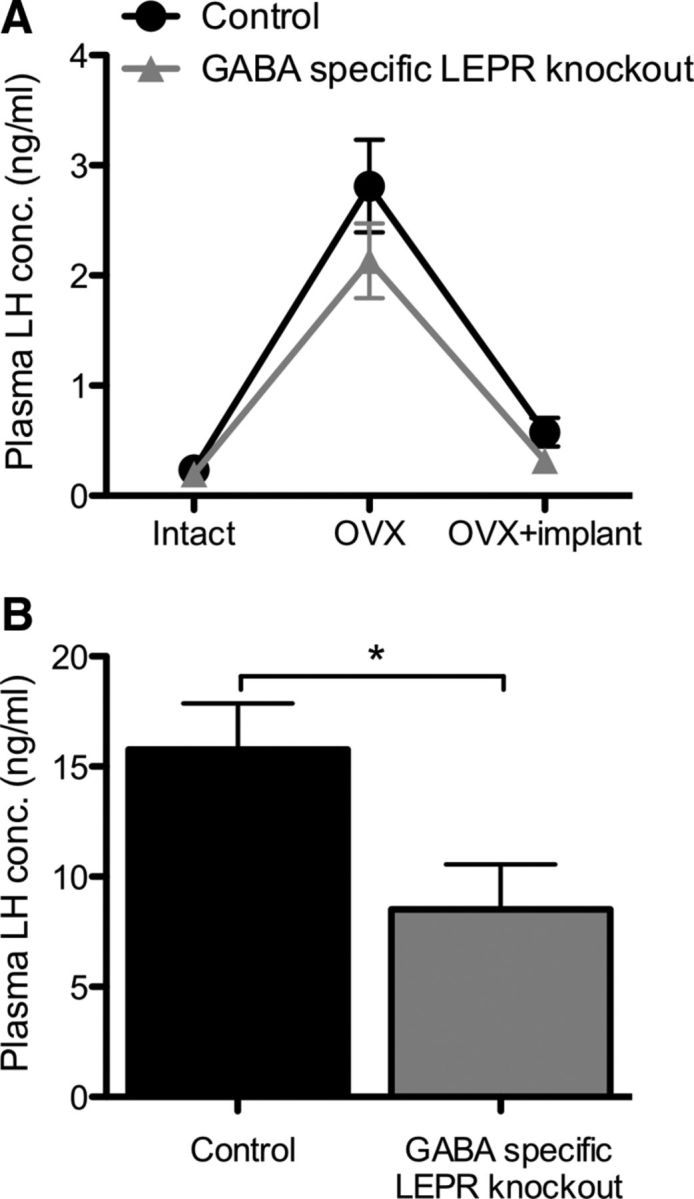
The effects of estradiol on plasma LH concentration in GABA-specific LEPR knock-outs and controls. A, Estradiol-negative feedback was assessed by measuring plasma LH concentration in serial blood samples. Intact plasma samples were taken on day 0, day 14 (OVX), and day 22 (OVX+implant). The negative feedback actions of estradiol remained intact in GABA-specific LEPR knock-out animals (gray triangles; n = 9) compared with littermate controls (black circles; n = 9). B, Estradiol-positive feedback was assessed based on plasma LH concentration in trunk bloods taken at the time of an estradiol-induced preovulatory-like surge. There was a significant suppression of LH concentration in GABA-specific knock-out female animals (gray bar; n = 9) compared with controls (black bar; n = 9). *p < 0.05.
The reduction in fertility of our GABA-specific LEPR knock-out animals could be a result of a difference in GABAergic wiring onto GnRH neurons. To investigate this, we counted GABAergic appositions (vGAT-positive) on GnRH soma and the proximal projections in both groups. Double-label immunohistochemistry and confocal imaging were used to visualize GnRH neurons and surrounding vGAT-positive terminals. We found that the circumference (knock-out: 31.5 ± 0.78 μm and controls: 32.3 ± 1.83 μm; t(8) = 0.85, p = 0.42) and number (knock-out: 5.9 ± 1.32 GnRH neurons per section and controls: 5.4 ± 0.95 GnRH neurons per section; t(8) = 0.35, p = 0.74) of GnRH soma did not differ between knock-out and control animals. When the number of GABAergic appositions onto the GnRH soma and projections were counted, there were no differences between the groups (Fig. 6), indicating that there are no detectable deficits in GABAergic GnRH inputs in the GABA-specific LEPR knock-out mice.
Figure 6.

GABAergic (vGAT-positive) appositions on GnRH neurons in male GABA-specific knock-outs and controls. A, Representative image of a GnRH neuron (green) with vGAT (red) appositions on the soma and proximal projection (white arrowheads). White lines indicate 10 μm segments of the neuronal projection. B, Number of vGAT-positive appositions per 10 μm of GnRH soma membrane and the first three 10 μm segments of the projection, in control (black bars; n = 5) and GABA-specific LEPR knock-out animals (gray bars; n = 5). No significant differences were found between groups. Scale bar, 10 μm.
Discussion
Neuronal pathways that communicate leptin and other metabolic signals to the HPG axis are poorly understood. Here, LEPRs were selectively deleted from the principal inhibitory (GABA) and excitatory (glutamate) neuronal populations to identify whether these neurons mediate interactions between leptin and the reproductive axis. Using these validated mouse models, we conclude that leptin signaling in GABAergic neurons is critical for HPG axis functioning in both sexes, and surprisingly glutamatergic LEPR neurons are not.
Leptin signaling in the PMV is thought to play a major role in regulating fertility rather than metabolism. Lesion studies of this nucleus cause a disruption of estrous cyclicity in rats (Donato et al., 2009), and selective PMV LEPR reexpression in LEPR-null mice induces female puberty onset and some pregnancies (Donato et al., 2011). Additionally, PMV LEPR neurons (of which >80% are glutamatergic) are known to project to GnRH cell bodies, AVPV kisspeptin neurons, and GnRH terminals in the median eminence (Rondini et al., 2004; Leshan et al., 2009; Donato et al., 2011; Louis et al., 2011). Although the present study showed a reduction of ∼80% in PMV leptin signaling in the glutamate-specific LEPR knock-outs, it was unexpected to see that these neurons are not required for fertility. However, they may well participate in this role under normal conditions and even be sufficient to permit fertility in the absence of other brain LEPRs (Donato et al., 2011). Alternatively, the remaining ∼20% of LEPR neurons in the PMV are not glutamatergic and may fulfill the leptin-to-GnRH role. In a recent paper by Leshan et al. (2012), removal of LEPR from neuronal nitric oxide synthase (nNOS) neurons, which are strongly represented in the PMV, caused a delay in puberty onset. Thus, PMV nitric oxide neurons might mediate leptin's effects to the HPG axis. Interestingly, these nNOS LEPR knock-out mice were considerably more obese than our glutamate-specific LEPR knock-outs. This is probably because the nNOS LEPR knock-out targets Arc neurons in addition to those in the PMV (Leshan et al., 2012).
In contrast to the fully fertile glutamate-specific LEPR knock-outs, male and female GABA-specific LEPR knock-outs exhibited markedly reduced fertility and the females had impaired estrous cycles. To try to identify the cause of subfertility in the GABA-specific LEPR knock-out mice, we assessed estradiol feedback in females and daily sperm production in males. The results of our estrogenic feedback experiment indicate that knock-out mice exhibit normal negative feedback regulation of LH but are impaired in their ability to mount a full preovulatory-like LH surge. This conclusion is consistent with the reduced occurrence of proestrus in these mice. Similar findings with regards to the LH surge have been reported in LEPR-deficient (in all forebrain neurons) and high fat diet-fed mice (Quennell et al., 2009; Sharma et al., 2013). As with the fertility data in intact animals, the concurrence of severe obesity with this effect means that caution must be exercised in attributing the disrupted preovulatory-like LH surge specifically to deficient leptin signaling in GABA neurons. Male ob/ob mice completely lacking leptin produce relatively few mature spermatozoa, but this deficit appears to be the result of apoptotic effects of leptin deficiency at the gonadal level (Bhat et al., 2006; Zhu et al., 2013). Daily sperm production in our animals, which were not leptin deficient, did not differ between GABA-specific LEPR knock-out mice and controls, suggesting that our male mice had no such apoptotic effects. Perhaps their subfertility was instead the result of reduced sperm efficacy or to mating behavior deficits.
GABA is well known to be an important modulator of GnRH neurons, and the present results are consistent with the idea that leptin may communicate with GnRH through GABA. It is known that fasting alters GABAA receptor-mediated transmission to GnRH neurons, and leptin is able to modulate this change (Sullivan et al., 2003; Sullivan and Moenter, 2004). Similarly, GABA-specific LEPR knock-out mice in the study of Vong et al. (2011) were characterized by an increased inhibitory GABAergic tone upon Arc pro-opiomelanocortin neurons. Therefore, GABAergic input upon GnRH neurons may also be compromised in our GABA-specific LEPR knock-out mice, causing the subfertile phenotype.
There are a few other candidates for providing the leptin-to-GnRH intermediate signal, in the form of neuropeptides that are potentially coreleased from GABAergic LEPR neurons. First, the neuropeptide kisspeptin is the most potent known stimulator of GnRH release and regulates the timing of puberty onset (Han et al., 2005; Clarkson and Herbison, 2006). Additionally, leptin is able to regulate kisspeptin expression (Quennell et al., 2011). Disruption of the function of these cells would provide a convenient explanation for the inability of GABA-specific LEPR knock-out mice to mount a full LH surge in this study. However, conditional LEPR deletion in kisspeptin neurons or even reexpression in LEPR-null mice has revealed that a direct action is neither required nor sufficient for fertility (Donato et al., 2011; Cravo et al., 2013). Therefore, if kisspeptin function is reduced in these mice, it probably does not occur as a direct response to LEPR deficiency. Second, agouti-related peptide (AgRP)/neuropeptide Y (NPY) neurons in the Arc are affected by the knock-out of LEPR because they are GABAergic (Horvath et al., 1997). Both the AgRP and NPY neuropeptides have been linked to metabolic control of reproduction by direct actions onto GnRH neurons (Klenke et al., 2010; Roa and Herbison, 2012) and indirectly by inhibiting the stimulatory α melanocyte-stimulating hormone produced from the Pomc gene (Israel et al., 2012). In both leptin-deficient ob/ob and LEPR-deficient db/db animals, ablation of AgRP neurons restores normal puberty onset and fertility (Israel et al., 2012; Wu et al., 2012). These experiments suggest that leptin-responsive AgRP/NPY/GABA neurons are likely to act as intermediates between metabolism and GnRH neurons, and thus suppress the HPG axis in absence of leptin or leptin signaling. We suggest that these LEPR/AgRP/NPY/GABA neurons are likely to be at least partly responsible for the subfertile phenotype observed in our GABA-specific LEPR knock-out mice. Last, there are galanin-like peptide (GALP) neurons in the Arc and cocaine and amphetamine-regulated transcript (CART) neurons in the DMN that express LEPR and project to the area of the GnRH neurons (Lebrethon et al., 2000; Takatsu, 2001; Rondini et al., 2004). Whether GALP and CART neurons corelease GABA has not been identified; therefore, it is unclear whether they have been targeted in our knock-out model.
Regardless of whether leptin's permissive actions on the HPG axis occur via GABA inhibition or other predominantly inhibitory peptidergic neurotransmitters, such as NPY, deletion of LEPRs from GABA neurons might be predicted to cause upregulation of GABA/NPY function leading to an increased inhibitory tone upon GnRH neurons. Alternatively, if leptin's signals are relayed by stimulatory neuropeptides (i.e., GALP and CART), and knowing that GABA has the ability to stimulate GnRH neuronal activity (Herbison and Moenter, 2011), then a reduction in the tone of these pathways upon GnRH neurons would be predicted in our knock-out model. It would be interesting to determine the nature of such changes in our GABA-specific LEPR knock-out mice.
The knock-out animals in which we observed a delay in puberty onset were also characterized by obesity and had high circulating leptin concentrations. Transgenic removal of exon 17 from the Lepr gene happens at early embryonic stages because Vgat expression is detectable from mid-gestation (Oh et al., 2005). Therefore, leptin receptor deletion in GABA neurons would have encompassed the second half of gestational development in all GABA-specific LEPR knock-out animals. The reproductive phenotype we observed in these animals could be an effect of defective wiring or the resulting obesity. However, early obesity or high circulating leptin is known to cause an advance in puberty onset rather than a delay (Ahima et al., 1997; Li et al., 2012). Additionally, HPG axis functioning can be successfully stimulated by exogenous administration of gonadotrophins and progesterone, or of leptin in ob/ob animals (Smithberg and Runner, 1957; Chehab et al., 1996). These studies indicate that neurotropic deficits resulting from deficient leptin signaling throughout development are relatively minor, at least as far as fertility is concerned. In support of this, no deficits in GABAergic GnRH inputs were apparent in the GABA-specific LEPR knock-out mice because no detectable differences were noted in the number of vGAT-positive appositions on the GnRH neurons. Collectively, these results suggest that the obesity or any differences in neuronal wiring in our GABA-specific LEPR knock-outs are unlikely to be primarily responsible for their reduced fertility.
Here, we have limited the first-order leptin-to-GnRH mediators to GABAergic neurons. These experiments show that there is a pivotal role for LEPRs in GABA neurons, but surprisingly not in glutamate neurons, in metabolic regulation of male and female fertility. This serves to focus attention on GABA or a few known peptide cotransmitters, specifically NPY, AgRP, GALP, and CART as critical neurotransmitters for the control of reproduction. Future studies should determine which neurons are GABAergic, leptin-responsive, and send inputs to GnRH neurons to control fertility.
Footnotes
This work was supported by the Health Research Council of New Zealand. We thank S. Chua (Institute of Human Nutrition, Columbia University College of Physicians and Surgeons, New York) for Leprflox/flox transgenic mice and Bradford B. Lowell (Department of Medicine, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston) for _Vgat_-ires-Cre and _Vglut2_-ires-Cre mice.
The authors declare no competing financial interests.
References
- Ahima RS, Dushay J, Flier SN, Prabhakaran D, Flier JS. Leptin accelerates the onset of puberty in normal female mice. J Clin Invest. 1997;99:391–395. doi: 10.1172/JCI119172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat GK, Sea TL, Olatinwo MO, Simorangkir D, Ford GD, Ford BD, Mann DR. Influence of a leptin deficiency on testicular morphology, germ cell apoptosis, and expression levels of apoptosis-related genes in the mouse. J Androl. 2006;27:302–310. doi: 10.2164/jandrol.05133. [DOI] [PubMed] [Google Scholar]
- Bourguignon JP, Gerard A, Alvarez Gonzalez ML, Purnelle G, Franchimont P. Endogenous glutamate involvement in pulsatile secretion of gonadotropin-releasing hormone: evidence from effect of glutamine and developmental changes. Endocrinology. 1995;136:911–916. doi: 10.1210/en.136.3.911. [DOI] [PubMed] [Google Scholar]
- Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet. 1996;12:318–320. doi: 10.1038/ng0396-318. [DOI] [PubMed] [Google Scholar]
- Clarkson J, Herbison AE. Postnatal development of kisspeptin neurons in mouse hypothalamus; sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology. 2006;147:5817–5825. doi: 10.1210/en.2006-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravo RM, Frazao R, Perello M, Osborne-Lawrence S, Williams KW, Zigman JM, Vianna C, Elias CF. Leptin signaling in Kiss1 neurons arises after pubertal development. PLoS One. 2013;8:e58698. doi: 10.1371/journal.pone.0058698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato J, Jr, Silva RJ, Sita LV, Lee S, Lee C, Lacchini S, Bittencourt JC, Franci CR, Canteras NS, Elias CF. The ventral premammillary nucleus links fasting-induced changes in leptin levels and coordinated luteinizing hormone secretion. J Neurosci. 2009;29:5240–5250. doi: 10.1523/JNEUROSCI.0405-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato J, Jr, Cravo RM, Frazão R, Gautron L, Scott MM, Lachey J, Castro IA, Margatho LO, Lee S, Lee C, Richardson JA, Friedman J, Chua S, Jr, Coppari R, Zigman JM, Elmquist JK, Elias CF. Leptin's effect on puberty in mice is relayed by the ventral premammillary nucleus and does not require signaling in Kiss1 neurons. J Clin Invest. 2011;121:355–368. doi: 10.1172/JCI45106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfield AS, Patterson C, Skora S, Gribble FM, Reimann F, Evans ML, Myers MG, Jr, Heisler LK. Neurochemical characterization of body weight-regulating leptin receptor neurons in the nucleus of the solitary tract. Endocrinology. 2012;153:4600–4607. doi: 10.1210/en.2012-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SK, Gottsch ML, Lee KJ, Popa SM, Smith JT, Jakawich SK, Clifton DK, Steiner RA, Herbison AE. Activation of gonadotropin-releasing hormone neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci. 2005;25:11349–11356. doi: 10.1523/JNEUROSCI.3328-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbison AE. Multimodal influence of estrogen upon gonadotropin-releasing hormone neurons. Endocr Rev. 1998;19:302–330. doi: 10.1210/er.19.3.302. [DOI] [PubMed] [Google Scholar]
- Herbison AE, Moenter SM. Depolarising and hyperpolarising actions of GABA(A) receptor activation on gonadotrophin-releasing hormone neurones: towards an emerging consensus. J Neuroendocrinol. 2011;23:557–569. doi: 10.1111/j.1365-2826.2011.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Bechmann I, Naftolin F, Kalra SP, Leranth C. Heterogeneity in the neuropeptide Y-containing neurons of the rat arcuate nucleus: GABAergic and non-GABAergic subpopulations. Brain Res. 1997;756:283–286. doi: 10.1016/S0006-8993(97)00184-4. [DOI] [PubMed] [Google Scholar]
- Ingalls AM, Dickie MM, Snell GD. Obese, a new mutation in the house mouse. J Hered. 1950;41:317–318. doi: 10.1093/oxfordjournals.jhered.a106073. [DOI] [PubMed] [Google Scholar]
- Israel DD, Sheffer-Babila S, de Luca C, Jo YH, Liu SM, Xia Q, Spergel DJ, Dun SL, Dun NJ, Chua SC., Jr Effects of leptin and melanocortin signaling interactions on pubertal development and reproduction. Endocrinology. 2012;153:2408–2419. doi: 10.1210/en.2011-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss J, Kocsis K, Csáki A, Halász B. Evidence for vesicular glutamate transporter synapses onto gonadotropin-releasing hormone and other neurons in the rat medial preoptic area. Eur J Neurosci. 2003;18:3267–3278. doi: 10.1111/j.1460-9568.2003.03085.x. [DOI] [PubMed] [Google Scholar]
- Klenke U, Constantin S, Wray S. Neuropeptide Y directly inhibits neuronal activity in a subpopulation of gonadotropin-releasing hormone-1 neurons via Y1 receptors. Endocrinology. 2010;151:2736–2746. doi: 10.1210/en.2009-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocsis K, Kiss J, Csáki A, Halász B. Location of putative glutamatergic neurons projecting to the medial preoptic area of the rat hypothalamus. Brain Res Bull. 2003;61:459–468. doi: 10.1016/S0361-9230(03)00180-1. [DOI] [PubMed] [Google Scholar]
- Krnjevic K. Chemical nature of synaptic transmission in vertebrates. Physiol Rev. 1974;54:418–540. [Google Scholar]
- Leblond CP, Clermont Y. Spermiogenesis of rat, mouse, hamster and guinea pig as revealed by the “periodic acid-fuchsin sulfurous acid” technique. J Anat. 1952;90:167–215. doi: 10.1002/aja.1000900202. [DOI] [PubMed] [Google Scholar]
- Lebrethon MC, Vandersmissen E, Gérard A, Parent AS, Junien JL, Bourguignon JP. In vitro stimulation of the prepubertal rat gonadotropin-releasing hormone pulse generator by leptin and neuropeptide Y through distinct mechanisms. Endocrinology. 2000;141:1464–1469. doi: 10.1210/en.141.4.1464. [DOI] [PubMed] [Google Scholar]
- Leshan RL, Louis GW, Jo YH, Rhodes CJ, Münzberg H, Myers MG., Jr Direct innervation of GnRH neurons by metabolic- and sexual odorant-sensing leptin receptor neurons in the hypothalamic ventral premammillary nucleus. J Neurosci. 2009;29:3138–3147. doi: 10.1523/JNEUROSCI.0155-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leshan RL, Greenwald-Yarnell M, Patterson CM, Gonzalez IE, Myers MG., Jr Leptin action through hypothalamic nitric oxide synthase-1-expressing neurons controls energy balance. Nat Med. 2012;18:820–823. doi: 10.1038/nm.2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XF, Lin YS, Kinsey-Jones JS, O'Byrne KT. High-fat diet increases LH pulse frequency and kisspeptin-neurokinin B expression in puberty-advanced female rats. Endocrinology. 2012;153:4422–4431. doi: 10.1210/en.2012-1223. [DOI] [PubMed] [Google Scholar]
- Louis GW, Greenwald-Yarnell M, Phillips R, Coolen LM, Lehman MN, Myers MG., Jr Molecular mapping of the neural pathways linking leptin to the neuroendocrine reproductive axis. Endocrinology. 2011;152:2302–2310. doi: 10.1210/en.2011-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMinn JE, Liu SM, Dragatsis I, Dietrich P, Ludwig T, Eiden S, Chua SC., Jr An allelic series for the leptin receptor gene generated by CRE and FLP recombinase. Mamm Genome. 2004;15:677–685. doi: 10.1007/s00335-004-2340-1. [DOI] [PubMed] [Google Scholar]
- Moore AM, Prescott M, Campbell RE. Estradiol negative and positive feedback in a prenatal androgen-induced mouse model of polycystic ovarian syndrome. Endocrinology. 2012;1543:796–806. doi: 10.1210/en.2012-1954. [DOI] [PubMed] [Google Scholar]
- Nagatani S, Guthikonda P, Thompson RC, Tsukamura H, Maeda KI, Foster DL. Evidence for GnRH regulation by leptin: leptin administration prevents reduced pulsatile LH secretion during fasting. Neuroendocrinology. 1998;67:370–376. doi: 10.1159/000054335. [DOI] [PubMed] [Google Scholar]
- Oh WJ, Noggle SA, Maddox DM, Condie BG. The mouse vesicular inhibitory amino acid transporter gene: expression during embryogenesis, analysis of its core promoter in neural stem cells and a reconsideration of its alternate splicing. Gene. 2005;351:39–49. doi: 10.1016/j.gene.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Ovesjö M, Gamstedt M, Collin M, Meister B. GABAergic nature of hypothalamic leptin target neurons in the ventromedial arcuate nucleus. J Neuroendocrinol. 2001;13:505–516. doi: 10.1046/j.1365-2826.2001.00662.x. [DOI] [PubMed] [Google Scholar]
- Quennell JH, Mulligan AC, Tups A, Liu X, Phipps SJ, Kemp CJ, Herbison AE, Grattan DR, Anderson GM. Leptin indirectly regulates gonadotropin-releasing hormone neuronal function. Endocrinology. 2009;150:2805–2812. doi: 10.1210/en.2008-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quennell JH, Howell CS, Roa J, Augustine RA, Grattan DR, Anderson GM. Leptin deficiency and diet-induced obesity reduce hypothalamic kisspeptin expression in mice. Endocrinology. 2011;152:1541–1550. doi: 10.1210/en.2010-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roa J, Herbison AE. Direct regulation of GnRH neuron excitability by arcuate nucleus POMC and NPY neuron neuropeptides in female mice. Endocrinology. 2012;153:5587–5599. doi: 10.1210/en.2012-1470. [DOI] [PubMed] [Google Scholar]
- Robb GW, Amann RP, Killian GJ. Daily sperm production and epididymal sperm reserves of pubertal and adult rats. J Reprod Fertil. 1978;54:103–107. doi: 10.1530/jrf.0.0540103. [DOI] [PubMed] [Google Scholar]
- Rondini TA, Baddini SP, Sousa LF, Bittencourt JC, Elias CF. Hypothalamic cocaine- and amphetamine-regulated transcript neurons project to areas expressing gonadotropin releasing hormone immunoreactivity and to the anteroventral periventricular nucleus in male and female rats. Neuroscience. 2004;125:735–748. doi: 10.1016/j.neuroscience.2003.12.045. [DOI] [PubMed] [Google Scholar]
- Sharma S, Morinaga H, Hwang V, Fan W, Fernandez MO, Varki N, Olefsky JM, Webster NJ. Free fatty acids induce Lhb mRNA but suppress Fshb mRNA in pituitary LbetaT2 gonadotropes and diet-induced obesity reduces FSH levels in male mice and disrupts the proestrous LH/FSH surge in female mice. Endocrinology. 2013;154:2188–2199. doi: 10.1210/en.2012-2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singireddy AV, Inglis MA, Zuure WA, Kim JS, Anderson GM. Neither signal transducer and activator of transcription 3 (STAT3) or STAT5 signaling pathways are required for leptin's effects on fertility in mice. Endocrinology. 2013;154:2434–2445. doi: 10.1210/en.2013-1109. [DOI] [PubMed] [Google Scholar]
- Smithberg M, Runner MN. Pregnancy induced in genetically sterile mice. J Hered. 1957;48:97–100. [Google Scholar]
- Sullivan SD, Moenter SM. Gamma-aminobutyric acid neurons integrate and rapidly transmit permissive and inhibitory metabolic cues to gonadotropin-releasing hormone neurons. Endocrinology. 2004;145:1194–1202. doi: 10.1210/en.2003-1374. [DOI] [PubMed] [Google Scholar]
- Sullivan SD, DeFazio RA, Moenter SM. Metabolic regulation of fertility through presynaptic and postsynaptic signaling to gonadotropin-releasing hormone neurons. J Neurosci. 2003;23:8578–8585. doi: 10.1523/JNEUROSCI.23-24-08578.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdloff RS, Batt RA, Brau GA. Reproductive hormonal function in the genetically obese (ob/ob) mouse. Endocrinology. 1975;98:1359–1364. doi: 10.1210/endo-98-6-1359. [DOI] [PubMed] [Google Scholar]
- Takatsu Y, Matsumoto H, Ohtaki T, Kumano S, Kitada C, Onda H, Nishimura O, Fujino M. Distribution of galanin-like peptide in the rat brain. Endocrinology. 2001;142:1626–1634. doi: 10.1210/en.142.4.1626. [DOI] [PubMed] [Google Scholar]
- Todman MG, Han SK, Herbison AE. Profiling neurotransmitter receptor expression in mouse gonadotropin-releasing hormone neurons using green fluorescent protein-promoter transgenics and microarrays. Neuroscience. 2005;132:703–712. doi: 10.1016/j.neuroscience.2005.01.035. [DOI] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. doi: 10.1016/j.neuron.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wintermantel TM, Campbell RE, Porteous R, Bock D, Grone HJ, Todman MG, Korach KS, Greiner E, Perez CA, Schutz G, Herbison AE. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron. 2006;52:271–280. doi: 10.1016/j.neuron.2006.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Whiddon BB, Palmiter RD. Ablation of neurons expressing agouti-related protein, but not melanin concentrating hormone, in leptin-deficient mice restores metabolic functions and fertility. Proc Natl Acad Sci U S A. 2012;109:3155–3160. doi: 10.1073/pnas.1120501109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Bosch MA, Rønnekleiv OK, Kelly MJ. Gamma-aminobutyric acid B receptor mediated inhibition of gonadotropin-releasing hormone neurons is suppressed by kisspeptin-G-protein -coupled receptor 54 signaling. Endocrinology. 2009;150:2388–2394. doi: 10.1210/en.2008-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu CC, Zhang H, Zhang JS, Li Z, Zhao J, Li W, Zhang YQ. Inhibition of ghrelin signaling improves the reproductive phenotype of male ob/ob mouse. Fertil Steril. 2013;99:918–926. doi: 10.1016/j.fertnstert.2012.11.022. [DOI] [PubMed] [Google Scholar]