Oligonucleotide-Mediated Survival of Motor Neuron Protein Expression in CNS Improves Phenotype in a Mouse Model of Spinal Muscular Atrophy (original) (raw)
Abstract
Spinal muscular atrophy (SMA) is caused by homozygous mutation or deletion of the SMN1 gene encoding survival of motor neuron (SMN) protein, resulting in the selective loss of α-motor neurons. Humans typically have one or more copies of the SMN2 gene, the coding region of which is nearly identical to SMN1, except that a point mutation causes splicing out of exon 7 and production of a largely nonfunctional SMNΔ7 protein. The development of drugs that mitigate aberrant SMN2 splicing is an attractive therapeutic approach for SMA. A steric block antisense oligonucleotide (AO) has recently been developed that blocked an intronic splice suppressor element, and enhanced SMN2 exon 7 inclusion in SMA patient fibroblasts. Here, we show that periodic intracerebroventricular (ICV) delivery of this AO resulted in increased SMN expression in brain and spinal cord to as much as 50% of the level of healthy littermates. Real-time PCR of SMN2 transcripts confirmed the AO-mediated increase in full-length SMN. The AO-derived increase in SMN expression led to a concomitant improvement in bodyweight throughout the lifespan of the SMA animals. Treatment of SMA mice with AO also provided partial correction of motor deficits, manifest as improved righting response. Injections of a scrambled oligonucleotide had no effect on SMN expression or phenotype in the SMA mice. Our results validate that AOs that abrogate aberrant splicing of SMN2 are promising compounds for treating SMA.
Introduction
Spinal muscular atrophy (SMA) is the leading genetic cause of infantile death (Wang and Lunn, 2008) and is mainly caused by mutational loss of the SMN1 gene (Lefebvre et al., 1995), which encodes survival of motor neuron (SMN) protein. Homozygous deletion of SMN1 in SMA individuals results in the progressive loss of spinal α-motor neurons (Wang and Lunn, 2008). Humans also possess the centromeric SMN2 gene. However, a point mutation within exon 7 of SMN2 results in mostly “splicing out” of exon 7 (Lefebvre et al., 1995; Helmken et al., 2003). The resultant SMNΔ7 protein is unstable and hypofunctional (Lorson et al., 1998).
Mice lack the SMN2 gene, and therefore homozygous deletion of Smn is embryonic lethal (Schrank et al., 1997). Embryonic viability is rescued in Smn −/− mice engineered with 1–2 copies of human SMN2 gene, but they die several days after birth (Monani et al., 2000). More recently, these mice were engineered to contain a high copy number of SMN_Δ_7 cDNA, with survival time of the SMNΔ7 SMA mice extended to ∼13 d (Le et al., 2005).
Various approaches have been used to increase the amount of SMN in patient cells and in SMA mice (Wirth et al., 2006). The most effective compounds to date, histone deacetylase (HDAC) inhibitors, promote SMN exon 7 inclusion (Andreassi et al., 2004; Avila et al., 2007) and greatly extended the lifespan of SMNΔ7 SMA mice (Narver et al., 2008).
Several groups have developed steric block RNA oligomer-based compounds that induce SMN2 exon 7 inclusion (Singh et al., 2006; Hua et al., 2007; Baughan et al., 2009). Singh et al. (2006) identified a 2′-_O_-methyl steric block antisense oligonucleotide (AO), which nearly completely reversed the SMN2 splicing pattern toward full-length SMN (FL-SMN) in SMA patient fibroblasts. Increased SMN expression was also recently reported in liver tissue of SMA mice after systemic delivery of steric block 2′-methoxy ethyl oligomers (Dickson et al., 2008; Hua et al., 2008). More recently, SMN expression was reported to be increased in CNS of SMNΔ7 SMA mice after intracerebroventricular (ICV) delivery of 2′-_O_-methyl “bifunctional” oligomers, which contain antisense bases fused to SR binding motifs (Baughan et al., 2009). However, the analysis of SMN expression was limited to 24–72 h after a single injection and phenotypic data were not reported in the SMNΔ7 SMA mice. Importantly, a transient increase in bodyweight and significant improvement in lifespan was found after ICV delivery of this bifunctional oligomer in a more severe SMA mouse strain (∼5 d lifespan), but correlative SMN expression data were not reported (Baughan et al., 2009). Thus, it remains an open question whether AO-mediated induction of SMN expression can be sustained in the CNS of SMNΔ7 SMA mice and improve phenotype.
We show for the first time that ICV injections of an AO targeting SMN2 (Singh et al., 2006) greatly enhanced SMN expression throughout the CNS of SMA mice, resulting in robust and sustained improvements in bodyweight and partial correction of motor deficits, validating this AO as a promising compound for treating SMA.
Materials and Methods
Animals.
Breeding pairs of SMNΔ7 SMA mice homozygous for the two transgenes SMN2 and SMN_Δ_7 and heterozygous for the targeted Smn mutation (SMN_Δ_7 +/+;SMN2 +/+;Smn −/+) were obtained from Jackson Laboratories (see supplemental materials, available at www.jneurosci.org, for genotyping). The SMN_Δ_7 +/+;SMN2 +/+;Smn −/− offspring were designated throughout as SMA mice and the SMN_Δ_7 +/+;SMN2 +/+;Smn +/+ littermates (designated as Smn+/+) were used as healthy controls.
2′-_O_-Methyl antisense oligonucleotides.
A 2′OMe AO (5′-AUUCACUUUCAUAAUGCUGG-3′; Trilink Biotech) was used for SMN induction experiments (Singh et al., 2006). A scrambled 2′OMe oligonucleotide (SCRAM; 5′-UAUCAGUCUAUCGAUGUCUA-3′; Trilink Biotech) containing the same bases as AO was used for control experiments.
ICV injections and tissue harvest.
For SMN expression studies, SMA mice were injected bilaterally with 1 μg of AO or SCRAM-AO into the lateral ventricles on postnatal day 1 (P1), 3, 5, 7, and 10 and harvested 48 h later (see supplemental materials, available at www.jneurosci.org, for details). For AO distribution studies, FVB wild-type mice (background strain; Jackson Laboratories) were injected similarly with FAM-tagged AO (FAM-AO; which contains a 6-FAM fluorophore covalently coupled to the 5′ end of the AO). Brain and spinal cords were harvested on P12 for analysis (see supplemental materials, available at www.jneurosci.org, for details).
Western blot analysis.
Brain and spinal cord samples were extracted and Western blots performed using an SMN antibody (BD Biosciences) and tubulin antibody (Abcam) as a loading control (see supplemental materials, available at www.jneurosci.org, for details).
Real-time PCR of full-length SMN mRNA transcripts.
TaqMan real-time PCR of frozen coronal brain sections directly adjacent to those used for Western blot analysis were used for semiquantitative analysis of AO-mediated changes in SMN2 transcript splicing (see supplemental materials, available at www.jneurosci.org, for details).
Functional assessment.
SMA mice and Smn+/+ littermate controls were evaluated for ambulatory fitness on P12 immediately after body-weight measurement. Individual mice were placed on a flat surface and both right and left side “righting” ability was scrutinized. Mice were scored as either able or unable to right themselves onto all fours within 30 s of being placed on their side (six trials total). We observed that SMA mice that could not right themselves within 5–10 s were unable to right if allowed >30 s, thus we reasoned that prolonging the trial time would not add additional information and could compromise successive trials.
Statistical analysis.
Results are reported as mean values ± SEM. Statistical differences between groups for quantitative measures were determined by Tukey's multiple comparison test. Categorically scored functional results were analyzed using Fisher's exact test.
Results
We opted to deliver AOs to SMNΔ7 SMA mice using ICV injections to maximize speed and extent of exposure to CNS cells, without complications of crossing the blood–brain barrier (BBB). A repeat injection regimen was adopted with ICV injections on P1, 3, 5, 7, and 10; followed by harvest on P12, which is just before the average time of death reported for this strain of SMA mice [13.3 d (Le et al., 2005)]. Accurate free-hand ICV delivery was ensured by first sectioning brains of mice to obtain detailed stereological coordinates and by imaging brains after injections of dye or FAM-AO at various time points. Imaging of whole-brain mounts showed that all ventricles were filled with dye or FAM-AO at 10 min after bilateral ICV injection (Fig. 1 A).
Figure 1.
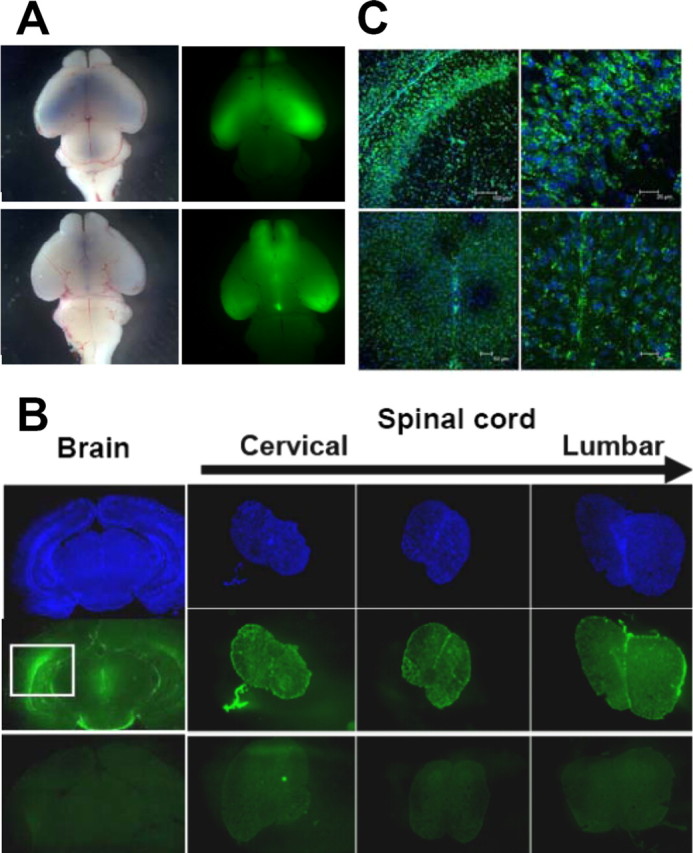
ICV delivery, diffusional distribution, and cellular uptake of AO in CNS of neonatal FVB mice. A, Accurate, free-hand ICV delivery was confirmed using whole mounts of brains harvested 10 min after bilateral ICV injection of either Trypan blue dye (left panels) or FAM-AO (a 6-FAM fluorophore covalently coupled to the 5′ end of the AO; right panels) into lateral ventricles. Both dorsal (top panels) and ventral (bottom panels) views of the whole-brain mounts are shown. AO was well distributed throughout lateral, olfactory, and fourth ventricles. B, C, To evaluate diffusional distribution and cellular uptake of AO in CNS, FVB mice were given bilateral ICV injections of FAM-AO on P1, 3, 5, 7, and 10 and harvested after paraformaldehyde perfusion on P12. B, Coronal sections taken from multiple brain regions (hippocampus shown here) showed FAM-AO was broadly but not uniformly distributed throughout the brain parenchyma after the periodic ICV injections. Transverse sections of spinal cord show that FAM-AO was also distributed along the entire length of the spinal cord after the periodic ICV injections. Images of uninjected brains and spinal cord, obtained using the exact same microscope and camera settings, were used as the negative control (bottom panels). The top panels indicate cell nuclei (DAPI), and middle panels show the FAM-AO (green). The boxed region for FAM-AO in the brain section highlights the extraordinary density of FAM-AO spreading out from the site of injection in the lateral ventricle. C, Confocal images of coronal brain sections (top panels) and transverse spinal cord sections (bottom panels) show cellular uptake, perinuclear localization, and nuclear uptake of FAM-AO. Merged images of FAM-AO (green) and DAPI (blue) are depicted.
To assess AO distribution throughout the CNS, FAM-AO was delivered ICV using the five injection regimen and mice were perfused with paraformaldehyde at P12. Cryosections of brain and spinal cords showed AO was broadly distributed throughout the CNS (Fig. 1 B). Confocal images showed uptake of FAM-AO into cells of the brain and spinal cord with extensive perinuclear localization and pronounced nuclear uptake (Fig. 1 C).
To induce SMN expression, SMA mice were given bilateral ICV injections of AO on P1, 3, 5, 7, and 10 (1 μg per lateral ventricle) and harvested at P12. Western blots showed SMN expression in AO-injected SMA mice was markedly increased in brain sections obtained from the hippocampal region as well as in both cervical and lumbar segments of spinal cord (Fig. 2 A). In contrast, injection of SCRAM, an oligonucleotide containing the same bases and chemistry but scrambled in order, did not increase SMN expression in any area of the CNS (Fig. 2 B). In many cases, we detected two bands for SMN (Fig. 2 A), but similar to many other studies, the doublet was not always resolved (Fig. 2 B). It is not known whether the SMN doublet on Western blots represents the SMNΔ7 and FL-SMN isoforms, or phosphorylation products. Thus, we adopted the method used in all previous publications and quantified SMN expression by summing the upper and lower bands when they appeared as a doublet.
Figure 2.

SMN protein was significantly increased in SMA mice after ICV injections of an AO that causes SMN2 exon 7 inclusion. SMA mice were given bilateral ICV injections of AO or scrambled AO (1 μg per ventricle) on P1, 3, 5, 7, 10, and harvested on P12. A, AO-treated SMA mice showed increased SMN protein expression in hippocampal brain regions (top), cervical spinal cord (middle), and lumbar spinal cord (bottom), relative to uninjected SMA mice. SMN band density was quantified in SMA mice as a percentage of littermate Smn+/+ controls (number in parenthesis below lanes) when normalized to tubulin (T) band density. Samples in each lane are from different mice, with 20 μg of total protein loaded per lane. Note that all samples in the bottom panel were run on the same gel, but the first two lanes were digitally shifted to maintain consistency with the top two panels. Brain, cervical, and lumbar blots are from the same animals. B, SMN protein expression in SCRAM-treated SMA mice was not different from untreated SMA mice in brain (top), cervical spinal cord (middle), or lumbar spinal cord (bottom). Brain, cervical, and lumbar blots are from the same animals. C, Quantitative analysis of SMN protein expression based on Western blots as in A and B. SMN protein expression in AO-injected SMA mice (N = 5) as a percentage of Smn+/+ controls was significantly increased compared with SCRAM-injected SMA mice (N = 4) and uninjected SMA mice (N = 5) in brain (**p < 0.01), cervical spinal cord (**p < 0.01), and lumbar spinal cord (*p < 0.05). There was no significant difference in SMN expression in any region of CNS between SCRAM-injected and uninjected SMA mice. SC, Spinal cord; CSC, cervical SC; LSC, lumbar SC.
As expected, SMN expression in brain and spinal cord of untreated SMA mice ranged from 10 to 20% of the level found in Smn+/+ littermates. On average, SMN expression in the hippocampal region of SMA mice injected with AO was 34.4 ± 1.8% (Fig. 2 C), which was significantly greater than the 15.6 ± 1.9% in uninjected SMA controls (2.2-fold increase; p < 0.001). An even greater increase in SMN expression was observed in cervical spinal cord where SMA mice injected with AO showed SMN expression at 55.4 ± 3.7% of the level in Smn+/+ littermates, which was 3.6-fold greater (p < 0.001) than the 15.4 ± 1.9% found in uninjected SMA controls (Fig. 2 C). Indeed, in a couple of individuals, the level of SMN expression in the cervical spinal cord reached 60–80% of the amount in Smn+/+ littermates. The AO-driven SMN expression in lumbar spinal cord was somewhat less than for cervical segments, reaching only 31.2 ± 2.7% of the level in Smn+/+ littermates, which was 1.9-fold greater (p < 0.05) than the 16.2 ± 3.1% in uninjected SMA controls (Fig. 2 C). In contrast, the average level of SMN expression after ICV injection of SCRAM was not significantly different from uninjected controls in brain or either region of the spinal cord (Fig. 2 C).
Real-time PCR was used to evaluate the level of FL-SMN2 transcripts at P12 in brain sections of SMA mice that were injected ICV with AO. These experiments were performed on serial sections of brains on which the SMN protein levels were previously measured by Western blot analysis. FL-SMN2 mRNA expression was significantly greater by ∼1.4-fold in AO-treated mice than untreated SMA controls (N = 5; p < 0.001).
The high level of SMN expression in brain and spinal cord of SMA mice after ICV injection of AO was accompanied by a significant improvement in bodyweight during postnatal development (P4 through P12; p < 0.05) (Fig. 3 A). In accordance with the SMN expression data, there was no significant difference in bodyweight between uninjected and SCRAM-injected SMA mice.
Figure 3.

Phenotypic improvements in SMA mice after periodic ICV injections of an AO that causes SMN2 exon 7 inclusion. SMA mice received bilateral ICV injections of AO or SCRAM (1 μg per ventricle) on P1, 3, 5, 7, 10, and trial endpoint was P12. A, AO-injected SMA mice (N = 5) showed a significant increase in bodyweight compared with both SCRAM-injected (N = 4) and uninjected (N = 9) SMA mice throughout postnatal development from P4 to P12 (*p < 0.05; **p < 0.01). There was no significant difference in bodyweight at any time point between SCRAM-injected and uninjected SMA mice. B, Righting response measured categorically (able/unable) over a range of stringencies demonstrated that AO-injected SMA mice (N = 5) were significantly better able to right themselves on P12 (*) at three levels of stringency (≥1/6, ≥2/6, ≥3/6) compared with uninjected SMA mice (N = 5; p < 0.05) and SCRAM-injected SMA mice (N = 4; p < 0.01). At the three most conservative stringencies (≥4/6, ≥5/6, 6/6), no statistical difference was observed between AO-injected SMA mice and SCRAM-injected SMA mice. There was no statistically significant difference in righting response between the SCRAM-treated and untreated groups at any stringency level. All mice were subjected to three consecutive righting trials (3 from each side) and were scored as positive if they could right within 30 s of being placed on their side. For each group, the fraction of mice that could right themselves in 6/6, ≥5/6, ≥4/6, ≥3/6, ≥2/6 and ≥1/6 trials was tallied. All Smn+/+ mice could right themselves in 6/6 trials.
We found that AO-treated SMA mice were significantly better at righting themselves than either SCRAM-injected (p < 0.01) or uninjected controls (p < 0.05) at all but the most conservative stringency levels (Fig. 3 B). In addition, the righting response of SCRAM-treated mice was not significantly different from untreated SMA mice at any level of stringency (Fig. 3 B). We further observed that even the strongest of AO-treated SMA mice were still noticeably weaker and physically compromised than their Smn+/+ littermates, which could always right themselves at this age. Thus, ICV injections of AO produced partial correction of the severe motor deficits in SMA mice.
Discussion
SMN2 is present in nearly all SMA individuals, but because of a single point mutation in exon 7 it provides only minor amounts of FL-SMN. However, it represents an attractive target for therapeutic stimulation of FL-SMN expression. Our study provides the first demonstration that splice modulating RNA oligomers can potently and persistently induce SMN expression in the CNS of severely affected SMNΔ7 SMA mice and provide concomitant improvement in the phenotype, including enhanced motor function. ICV injections of AO produced the highest level of SMN expression reported to date in the CNS of severe SMA mice, and demonstrated that the potent AO-mediated conversion from SMNΔ7 to FL-SMN shown previously in vitro (Singh et al., 2006) can be extended to an animal model of SMA and provide functional benefit.
The AO-directed expression of SMN was significantly increased throughout the CNS with the greatest increase in cervical spinal cord (3.6-fold), followed by brain (2.2-fold; hippocampus), and lumbar spinal cord (1.9-fold). In addition, FL-SMN2 transcript levels, determined by real-time PCR, were significantly increased by ∼1.4-fold in brain tissue after ICV injection of AO, confirming that AO significantly increased “splicing in” of exon 7 of SMN2. The increase in SMN expression in AO-treated SMA mice was accompanied by a significant increase in bodyweight relative to controls continuously from P5 to P12, peaking at 60–70% improvement near the trial endpoint at P12. In addition, relative to uninjected or SCRAM-injected mice, AO-injected SMA mice showed increased frequency in ability to right themselves. The fact that there was no difference in righting response between SCRAM-injected and uninjected mice (Fig. 3 B) is consistent with the fact that no significant difference in SMN expression (Fig. 2 C) or bodyweight (Fig. 3 A) was observed between these groups. However, righting responses of SCRAM-treated mice were consistently, if not significantly, less robust than for untreated SMA controls. This trend may indicate deleterious effects of repeat ICV injections to neonatal mice, and that the improved righting response observed in AO-treated SMA mice may be even greater if mechanical damage can be avoided by using either less frequent injections or another mode of AO delivery to the CSF.
Avila et al. (2007) recently showed that daily intraperitoneal injections of the potent HDAC inhibitor trichostatin A (TSA) in SMNΔ7 SMA mice resulted in a 1.8- to 1.9-fold increase in SMN protein expression in the brain and spinal cord at P13. SMN expression in muscles of the TSA-treated SMA mice was ∼3.0-fold higher than controls, but was highly variable and not statistically significant. In comparison, TSA treatment only slightly increased FL-SMN2 transcript levels in the brain and spinal cord, while significantly increasing FL-SMN2 (5-fold) and SMNΔ7 levels (2-fold) in muscle (Avila et al., 2007). Thus, the extent to which TSA improved phenotype of SMA mice (Avila et al., 2007; Narver et al., 2008) by increasing SMN expression in CNS, muscle, or elsewhere remains an intriguing question. Interestingly, mice treated with TSA showed no improvement in bodyweight compared with vehicle-treated controls between P5 and P12 (Avila et al., 2007). In a follow-up study, Narver et al. (2008) used nutritional supplementation during TSA treatment, which resulted in a dramatic increase in lifespan out to P40. However, TSA plus nutritional supplementation also did not noticeably increase bodyweight until after P12; indeed, the predominant bodyweight gain occurred much later (after P30). Our study is the first to demonstrate that drug treatment alone, without external supplementation, can increase bodyweight of SMNΔ7 SMA mice relative to untreated mice throughout postnatal development.
A series of highly engineered splice modulating oligomers that induce SMN2 exon 7 inclusion have been developed in several laboratories (Singh et al., 2006; Hua et al., 2007; Baughan et al., 2009). Steric block oligomers work by interfering with spliceosomal recognition of splicing motifs in target pre-mRNA, a mechanism which distinguishes them from the more commonly recognized classic antisense compounds that function by recruiting RNase-H and cleaving their target mRNA. However, difficulty with delivery of compounds into the CSF of neonatal animals and inability of RNA-based oligomers to cross the BBB has limited the evaluation of these novel compounds in SMA mice. An increase in SMN expression in the CNS of SMNΔ7 SMA mice after ICV delivery of a bifunctional 2′-_O_-methyl splice modulating oligomer was recently reported (Baughan et al., 2009). However, the analysis of SMN expression was restricted to 24–72 h after a single injection on P2, SMN expression did not reliably persist to the 72 h time point, and correlative phenotypic data were not reported in the SMNΔ7 SMA mice. This same study (Baughan et al., 2009) did document phenotypic improvement and enhanced longevity in a more severe SMA mouse model, but without correlative data on SMN expression. In this report, we show that repeated ICV injections in neonatal SMNΔ7 SMA mice is an effective means of evaluating the efficacy of RNA oligomers. The marked increase in SMN expression throughout the CNS demonstrates that AOs display excellent diffusional distribution and cellular uptake properties after direct delivery to CSF. These data are supported by our finding that FAM-AO was broadly distributed throughout the brain and along the full length of the spinal cord after ICV delivery. These results confirm previous reports that showed oligonucleotides of similar chemistry were able to transfect cells throughout the CNS in adult rats and nonhuman primates after ICV delivery (Smith et al., 2006).
A limitation of the present study was that the free-hand ICV delivery did not provide a favorable opportunity to evaluate improvement in lifespan as a consequence of AO injections. Because of the decrease in ventricle volume relative to brain volume and the hardening of the skull with age, it was very difficult to accurately inject the lateral ventricles after P7. Thus, we chose P12 as our endpoint, since we could not reliably continue AO injections. Although our repeat ICV injection regimen provided important biodistribution and efficacy information about AOs in the SMA mice, the question of whether AO-induction improves longevity in SMA mice will be better answered using a mouse model with later onset pathology than the currently available models. In such animals, intrathecal or ICV delivery by cannulation and osmotic pump technology will permit continuous delivery of AOs over extended time frames suitable for longevity studies.
There has been controversy as to whether increasing SMN levels postnatally can cause phenotypic rescue in SMA. A prevailing concern in the field is that most of the damage to motor neurons in SMA individuals may have already occurred during embryonic development, and thus cannot be effectively reversed by postnatal SMN expression. Our data refute this idea and show that sustained delivery of AO to CNS during postnatal development can provide significant phenotypic improvement in SMA mice, including amelioration of bodyweight loss and partial correction of motor deficits. Our study also indicates that the nearly 100% conversion of SMN_Δ_7 to FL-SMN previously observed in vitro can potentially be realized in the CNS of SMA individuals. Peak AO-directed SMN expression in the spinal cord of SMA mice was almost 80% of normal, providing “proof of concept” that with further optimization of dosing and delivery, it may be possible to reach normal levels of SMN expression throughout the CNS. Given that steric block AOs only effect pretranscribed mRNA, there is little concern that AOs would cause toxic or ectopic overexpression of SMN, which may occur with most compounds under development that increase SMN transcription. These results provide compelling evidence that steric block AOs are promising therapeutics that should be further explored and developed with the ultimate goal of clinical trials in patients with SMA.
Footnotes
This work was supported by grants from SMA Foundation and The Commonwealth of Pennsylvania. We acknowledge Cathleen Lutz of Jackson Laboratories for technical advice with genotyping and Mauricio Reginato for assistance with confocal microscopy. We also thank members of the SMA Foundation for critical reading of this manuscript.
References
- Andreassi C, Angelozzi C, Tiziano FD, Vitali T, De Vincenzi E, Boninsegna A, Villanova M, Bertini E, Pini A, Neri G, Brahe C. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12:59–65. doi: 10.1038/sj.ejhg.5201102. [DOI] [PubMed] [Google Scholar]
- Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughan TD, Dickson A, Osman EY, Lorson CL. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum Mol Genet. 2009;18:1600–1611. doi: 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson A, Osman E, Lorson C. A negatively-acting bifunctional RNA increases survival motor neuron in vitro and in vivo. Hum Gene Ther. 2008;19:1307–1316. doi: 10.1089/hum.2008.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmken C, Hofmann Y, Schoenen F, Oprea G, Raschke H, Rudnik-Schöneborn S, Zerres K, Wirth B. Evidence for a modifying pathway in SMA discordant families: reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet. 2003;114:11–21. doi: 10.1007/s00439-003-1025-2. [DOI] [PubMed] [Google Scholar]
- Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5:e73. doi: 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lorson CL, Strasswimmer J, Yao JM, Baleja JD, Hahnen E, Wirth B, Le T, Burghes AH, Androphy EJ. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossoll W, Rossol W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- Narver HL, Kong L, Burnett BG, Choe DW, Bosch-Marcé M, Taye AA, Eckhaus MA, Sumner CJ. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann Neurol. 2008;64:465–670. doi: 10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrank B, Götz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94:9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26:1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, Lobsiger CS, Ward CM, McAlonis-Downes M, Wei H, Wancewicz EV, Bennett CF, Cleveland DW. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116:2290–2296. doi: 10.1172/JCI25424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CH, Lunn MR. Spinal muscular atrophy: advances in research and consensus on care of patients. Curr Treat Options Neurol. 2008;10:420–428. doi: 10.1007/s11940-008-0044-7. [DOI] [PubMed] [Google Scholar]
- Wirth B, Brichta L, Hahnen E. Spinal muscular atrophy: from gene to therapy. Semin Pediatr Neurol. 2006;13:121–131. doi: 10.1016/j.spen.2006.06.008. [DOI] [PubMed] [Google Scholar]