Phosphorylation of SNAP-25 at Ser187 Mediates Enhancement of Exocytosis by a Phorbol Ester in INS-1 Cells (original) (raw)
Abstract
Activation of diacylglycerol (DAG) signaling pathways with phorbol esters dramatically enhances Ca2+-triggered exocytosis from both endocrine cells and neurons, however the relevant targets of DAG are controversial. A possible effector mechanism for this signaling pathway is phosphorylation of SNAP-25 (25 kDa synaptosome-associated protein) at Ser187 by PKC. Here, we investigated the role of Ser187 in the enhancement of exocytosis by the phorbol ester PMA (phorbol 12-myristate 13-acetate). We used patch-clamp measurements of membrane capacitance together with photorelease of caged-Ca2+ and membrane depolarization to study exocytosis. Expression of the nonphosphorylatable S187C SNAP-25 mutant did not attenuate the enhancement of exocytosis by PMA in either bovine chromaffin cells or the INS-1 insulin-secreting cell line. To test the effects of Ser187 mutations under conditions in which the endogenous SNAP-25 is disabled, we expressed botulinum toxin serotype E to cleave SNAP-25 in INS-1 cells. Coexpression of a toxin-resistant mutant (TR), but not wild-type SNAP-25, was able to rescue PMA-modulated exocytosis. Coexpression of the toxin with the TR-S187C SNAP-25 mutant was able to completely block the enhancement of exocytosis by PMA in response to photoelevation of [Ca2+]i to low μm levels or to a depolarizing train. The phospho-mimetic S187E mutation enhanced the small, fast burst of exocytosis evoked by photelevation of Ca2+, but, like PMA, had smaller effects on exocytosis evoked by a depolarizing train. This work supports the hypothesis that phosphorylation of Ser187 of SNAP-25 by PKC is a key step in the enhancement of exocytosis by DAG.
Keywords: phorbol esters, PKC, SNAP-25, exocytosis, BoNT/E, phosphomimetic mutations
Introduction
Phorbol esters are plant-derived tumor-promoting organic compounds that structurally resemble the second-messenger diacylglycerol (DAG). They are commonly used as stable and potent DAG mimics to study the role of DAG signaling in cellular processes. Whereas most attention has focused on the ability of these compounds to activate PKC, it has recently become clear that DAG and phorbol esters have additional cellular targets (for review, see Brose and Rosenmund, 2002). For example, experiments in cultured hippocampal neurons suggest that phorbol ester enhancement of Ca2+-triggered exocytosis is attributable to activation of the DAG/phorbol ester binding protein munc-13 rather than PKC (Rhee et al., 2002). However, the enhancement of exocytosis resulting from short-term exposure to phorbol esters can be mostly or completely blocked by PKC inhibitors that act independently of DAG binding in endocrine cells (Terbush and Holz, 1990; Gillis et al., 1996; Zhu et al., 2002; Wan et al., 2004; Yang and Gillis, 2004) and neurons (Braha et al., 1990; Yawo, 1999; Wu and Wu, 2001), therefore activation of PKC is, indeed, necessary for the enhancement of exocytosis by phorbol ester in a number of preparations.
A possible target of PKC phosphorylation in modulating exocytosis is SNAP-25. SNAP-25 is one of three proteins that form the SNARE core complex that plays an important, although not fully defined, role in exocytosis. One of the key observations of the essential role of SNAP-25 is that botulinum neurotoxins (BoNTs) of serotype ‘A’ and ‘E’ inhibit exocytosis by selectively cleaving SNAP-25 near the C terminus (Blasi et al., 1993; Schiavo et al., 1993). Within the C terminus of SNAP-25, Ser187 is the only known PKC phosphorylation site (Shimazaki et al., 1996). Ser187 is phosphorylated by PKC in vitro (Shimazaki et al., 1996; Yang et al., 2007) and in vivo in response to phorbol ester application and depolarizing stimuli in endocrine cells (Shimazaki et al., 1996; Gonelle-Gispert et al., 2002; Nagy et al., 2002) and hippocampal organotypic cultures (Genoud et al., 1999).
The role of Ser187 phosphorylation in regulating exocytosis is controversial because Ser187 phosphorylation levels do not always correlate with modulation of exocytosis (Iwasaki et al., 2000; Gonelle-Gispert et al., 2002) and mutation of this residue has been reported to either have no effect (Gonelle-Gispert et al., 2002; Finley et al., 2003) or somewhat subtle effects on exocytosis (Nagy et al., 2002; Yang et al., 2007). Two recent studies report that phosphomimetic Ser187 mutations (to Asp or Glu) partially mimic phorbol ester effects in enhancing vesicle recruitment (Nagy et al., 2002) and highly Ca2+-sensitive exocytosis (Yang et al., 2007).
An important test of whether phosphorylation of Ser187 of SNAP-25 mediates effects of PKC on exocytosis is whether mutation of this residue can block some or all of the effects of phorbol esters on exocytosis. We used photorelease of caged Ca2+ together with patch-clamp capacitance measurements to directly study the effects of Ser187 mutants and phorbol esters on the Ca2+ sensitivity of exocytosis in the insulin-secreting INS-1 cell line. Here we present experimental evidence that phorbol-ester enhancement of exocytosis triggered by photo-elevation of [Ca2+]i to the low μm range or by a depolarizing train is blocked by expressing Ser187 mutations. However, this block requires coexpression of botulinum neurotoxin E to cleave the endogenous SNAP-25 to enable functional substitution of transfected toxin-resistant SNAP-25 mutants.
Materials and Methods
Materials.
All cell culture reagents were from Invitrogen (Carlsbad, CA), whereas all other reagents were from Sigma (St. Louis, MO) unless otherwise indicated. The rabbit antibody, which selectively recognizes SNAP-25 phosphorylation at Ser187 was described by Kataoka et al. (2000). All other antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell culture.
Chromaffin cells were isolated from freshly collected bovine adrenal glands as previously described (Yang et al., 2007) and seeded onto 25 mm diameter round glass coverslips coated with poly(d)-lysine in DMEM supplemented with 10% (v/v) FBS and 1% penicillin/streptomycin. Cells were kept in a 37°C incubator with 5% CO2 and used 1–3 d after preparation.
Rat insulinoma INS-1 cells were cultured as previously described (Asfari et al., 1992; Yang and Gillis, 2004). Briefly, the cells were maintained in culture media consisting of RPMI 1640 medium supplemented with 50 μm 2-mercaptoethanol, 2 mm l-glutamine, 10 mm HEPES, 100 U/ml penicillin, 100 mg/ml streptomycin, 1 mm sodium pyruvate, and 10% FBS. Cells were kept in a humidified 37°C incubator with 5% CO2 and were subcultured once per week. For patch clamp experiments, INS-1 cells were plated onto 25 mm round glass coverslips in 35 mm dishes at a density of 5 × 105 cells per dish. In immunoblot experiments, INS-1 cells were plated into 60 mm dishes at a density of 2.5 × 106 cells per dish.
Generation of Semliki Forest virus.
A DNA-based Semliki Forest virus (SFV) vector (DiCiommo and Bremner, 1998) and an RNA-based SFV vector (Invitrogen) (Ashery et al., 1999; Duncan et al., 1999) were used to produce SFV. Both methods expressed a construct with green fluorescent protein (GFP) fused to the N terminus of SNAP-25a as previously described (Yang et al., 2007).
Chromaffin cells were infected the day after isolation. Thawed SFV-containing media was diluted 1:1 with DMEM media without FBS. α-chymotrypsin (0.2 mg/ml in HBS solution) was added and the solution was allowed to incubate for 50 min at room temperature to activate the virus. Then aprotinin (0.6 mg/ml in HBS solution) was added and allowed to incubate for an additional 5 min to inactivate α-chymotrypsin. Media was removed from the chromaffin cells and replaced with the activated SFV solution. After allowing 12 h for infection, the virus-containing media was replaced with fresh chromaffin cell media. Cells were used for experiments 1–2 d after infection. Cells expressing GFP–SNAP-25 and GFP–SNAP-25 mutants were identified by fluorescence microscopy using 480 nm excitation and used for patch-clamp experiments. Cells were selected with similar total fluorescence intensity (approximately ±25%) as measured by a photodiode so that expression levels of GFP–SNAP-25 and the various mutants were comparable.
DNA constructs.
The constructs of BoNT/E light chain and the toxin-resistant human SNAP-25a (TR SNAP-25a) were kindly provided by the laboratory of Dr. Philippe Halban (University of Geneva, Geneva, Switzerland) (Gonelle-Gispert et al., 1999, 2002). The cDNAs of BoNT/E light chain and TR SNAP-25 were cloned into the pCMV5 vector and pcDNA3 vector, respectively. The cDNA coding green fluorescent protein (GFP) was fused to the N terminus of the SNAP-25. The pcDNA3–GFP–TR–SNAP-25 was used as a template for both a phosphomimetic mutation (S187E) and a non-phosphomimetic mutation (S187C) with the QuickChange Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA), according to the manufacturer's instructions (Gonelle-Gispert et al., 2002). The sequences of all constructs were confirmed by DNA-sequencing.
Transfection of INS-1 cells.
The INS-1 cells were transfected with SNAP-25 DNA using LIPOFECTAMINE 2000 (Invitrogen) according to the manufacturer's instructions. Transfection medium was changed 6 h later to fresh cell culture medium. Cells were used for experiments 2 d after transfection. In the experiments of cotransfection of BoNT/E with SNAP-25 constructs, the ratio of BoNT/E and SNAP-25 was 3:1 to make it more likely that cells identified from fluorescence of GFP–SNAP-25 also express the toxin. The transfection efficiency of coexpression experiments was estimated to be ∼5–8% from GFP fluorescence measurements. The transfection efficiency during transfection with a single plasmid was ∼15%.
[Ca2+]i calibration.
A combination of the low affinity Ca2+ indicator fura2-ff (100 μm; Teflabs, Austin, TX) and the higher affinity indicator bisfura-2 (100 μm; Invitrogen) was used to measure [Ca2+]i over a wide dynamic range. The Ca2+ indicator combination was calibrated in cells as described before (Yang and Gillis, 2004). Briefly, eight solutions with free [Ca2+]i of, 0.31, 1.2, 3.6, 7.2, 28, 127 μm and 10 mm were prepared using Ca2+ buffers EGTA (_K_D = 150 nm at pH 7.2, total concentration 10 mm), _N_-hydroxyethylethylenediaminetriacetic acid (KD = 3.6 μm, concentration 10 mm), or 1,3-diaminopropan-2-ol-tetraacetic acid (KD = 81 μm, concentration 30 mm). Dissociation constants for the buffers were obtained from (Martell and Smith, 1974–1989). Whole-cell recordings were made using each of the calibration solutions in the pipette, and the ratio of fluorescence emission (535 ± 25 nm) for 340 and 365 nm excitations were recorded after allowing 2 min for the pipette solution to diffuse into the cell. Three to five recordings were made for each calibration solution. The following equation was fit to the eight measured ratio values (R) to find the five unknown parameters (Voets, 2000):

Electrophysiology.
All experiments were performed at room temperature (∼22°C). Whole-cell patch-clamp measurements were performed using an EPC-9 patch-clamp amplifier and PULSE acquisition software (HEKA Elektronik, Lambrecht/Pfalz, Germany). Pipettes (2–4 MΩ resistance) were pulled from Kimax glass capillaries, coated with dental wax at the tips, and fire polished. The pipette potential was held at a dc value of −70 mV. Capacitance measurements were performed using the “sine + dc” software lock-in amplifier method implemented in PULSE software (Pusch and Neher, 1988; Gillis, 2000). The assumed reversal potential was 0 mV and the sinusoid had an amplitude of 25 mV and a frequency of 1.5 kHz.
A monochromator (Polychrome 4; TILL Photonics, Graefelfing, Germany) coupled to the epifluorescence port of an Olympus Optical (Tokyo, Japan) IX-50 microscope with a fiber-optic cable were used to excite the Ca2+ indicators at 340 and 365 nm. A two-port condenser (TILL/ASI) combined the monochromator excitation path with that of a flash lamp (TILL/ASI). A 40× 1.15 NA water-immersion lens (U-APO; Olympus Optical) focused the excitation light and collects fluorescent light. The fluorescent light (535 ± 25 nm) was measured using a photodiode mounted in a viewfinder (TILL/ASI).
The standard bath solution consisted of (in mm) 140 NaCl, 5.5 KCl, 1 MgCl2, 5 CaCl2, 10 HEPES titrated to pH 7.2 with NaOH. The bath solution also contained 2.6 mm glucose. Standard pipette solution consisted of (in mm) 130 l-glutamic acid, 1 MgCl2, 2 Na2-ATP, and 40 HEPES titrated to pH 7.2 with CsOH. In caged-Ca2+ experiments, NP-EGTA (Ellis-Davies and Kaplan, 1994), ∼80% loaded with CaCl2 was added to the pipette solution. Basal [Ca2+]i was set to ∼500 nm before the flash.
Immunocytochemistry.
INS-1 cells plated on glass coverslips in 35 mm dishes were transfected with GFP or SNAP-25 mutants plus or minus BoNT/E according to the protocol described above. Two days after transfection, the culture medium was removed. Cells were then washed with bath solution. After treatment with 100 nm PMA for 30 min, the cells were fixed with 4% paraformaldehyde (PFA) in PBS for 20 min, and permeabilized with 0.25% Triton X-100 in PBS for 6 min. After washing three times with PBS and blocking with 2% BSA in PBS twice, the cells were incubated with primary antibodies. Primary antibodies were either mouse anti-BoNT/E, 1:50 (Abcam, Cambridge, MA), rabbit antiphospho-Ser187 SNAP-25 antibody, 1:200, or goat anti-SNAP-25 (N-19), 1:200 (Santa Cruz Biotechnology) for 2 h. They were then incubated with rhodamine-conjugated secondary antibodies for 1 h. Secondary antibodies were either goat anti-mouse IgG, 1:50 (Biomeda, Foster City, CA), goat anti-rabbit IgG, 1:500 (Chemicon, Temecula, CA), or TRITC rabbit anti-goat IgG, 1:200 (Biomeda). Glass coverslips were mounted with antifade Clarion Mounting Medium (M54; Biomeda), followed by fluorescence image acquisition and quantitative analysis using an Olympus Optical Fluoview FV1000 Confocal Laser Scanning Biological Microscope. All the images were acquired at room temperature. GFP–SNAP-25 fluorescence was detected during excitation with the 488 nm line from an Argon laser, whereas rhodamine immunofluorescence was detected during excitation with a 559 nm diode laser.
Immunoblot analysis.
The INS-1 cells were lysed with ice-cold RIPA buffer (50 mm Tris HCl, pH 8.0; 150 mm NaCl; 1% NP-40; 0.5% sodium deoxycholate; 0.1% SDS; 2 mm EDTA). Protease inhibitors (protease inhibitor cocktail, Sigma catalog number P8340; 1 mm PMSF; 1 mm Na3VO4) were added freshly. The lysates were harvested and centrifuged at 12,000 rpm for 20 min at 4°C. The supernatants were collected and the protein concentrations were measured by using the DCA method (Bio-Rad, Hercules, CA). Equal amounts of samples (15 μg of protein unless otherwise stated) were separated electrophoretically on a 12% SDS-polyacrylamide gel and then transferred onto nitrocellulose membranes. The membranes were incubated with primary antibodies (rabbit antiphospho-Ser187 SNAP-25 antibody, 1:250; goat anti-SNAP-25 antibody, N-terminal, 1:250) for 2 h at room temperature, followed by 1 h incubation with corresponding secondary antibodies (goat anti-rabbit IgG-HRP and bovine anti-goat IgG-HRP, 1:2000, respectively) for 1 h at room temperature. ECL Western blotting detection reagents (Pierce, Rockford, IL) were used to develop the membranes and the proteins bands were visualized with Hyperfilm ECL (GE Healthcare, Little Chalfont, UK). To normalize the signals in some experiments, the membranes were stripped in stripping buffer (1.5% glycine (W/V), 0.1% SDS (W/V), 1% Tween20 (V/V), pH 2.0) for 30 min at room temperature. After being washed with PBS, the membranes were blotted with another pair of primary antibody and secondary antibody (e.g., mouse anti-actin, 1:250; bovine anti-mouse IgG-HRP, 1:2000) according to the procedure described above. For the quantification of the Western blot data, the developed films were scanned, the immunoreactive bands were digitized, and the densitometry was performed using Scion Image for Windows (Scion, Frederick, MD).
Data analysis.
Data analysis and curve fitting were performed using Igor software (WaveMetrics, Lake Oswego, OR). Results and histograms were expressed as mean ± SEM except for Figure 7B, which is expressed as mean ± SD. Statistical comparisons between groups were performed using Student's t test. A single asterisk is used to indicate p < 0.05, whereas double asterisks indicate p < 0.01.
Figure 7.
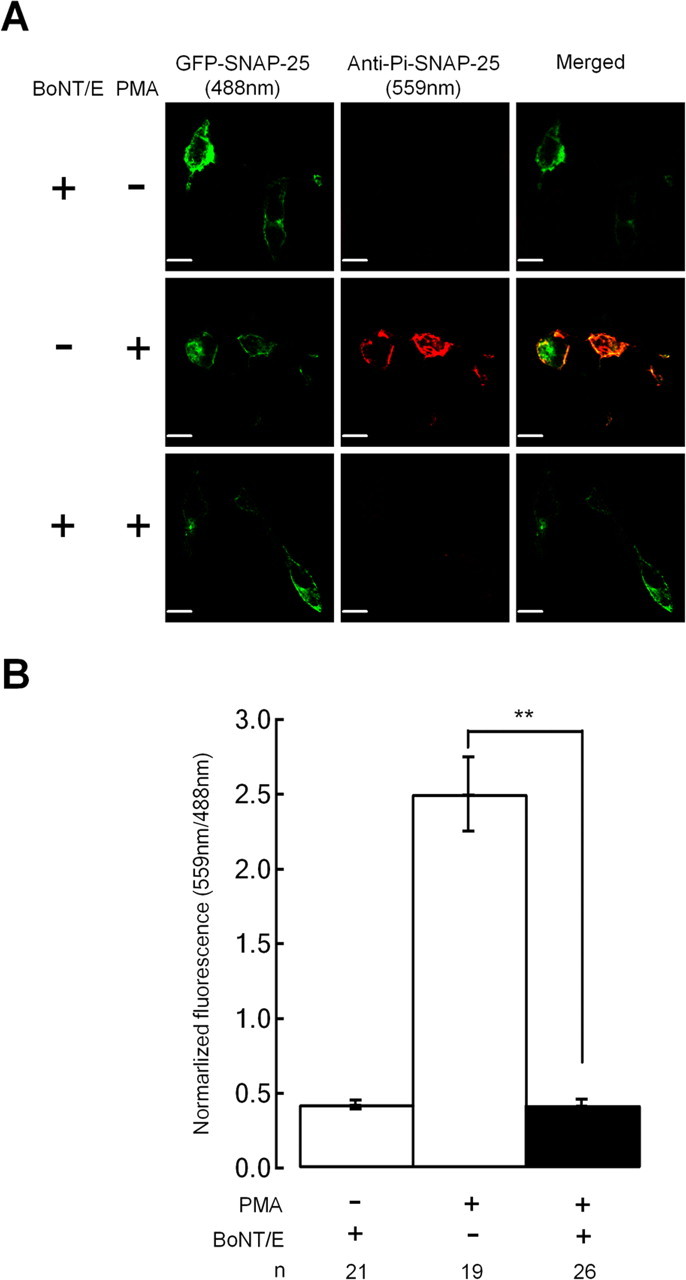
Single cell immunofluorescence measurements demonstrate that expression of TR SNAP-25 does not block cleavage of endogenous SNAP-25 by BoNT/E. INS-1 cells were transfected with the TR/S187C SNAP-25 mutant plus or minus BoNT/E as indicated. PMA treatment for 30 min was used to induce phosphorylation of endogenous SNAP-25. Cells were fixed and probed with the pSer187 phospho-specific SNAP-25 antibody. The cells were excited with a 488 nm laser line to image exogenously expressed GFP–SNAP-25 mutant (TR/S187C; green pseudocolor), whereas a 559 nm laser line was used to identify endogenous Pi–SNAP-25 immunoreactivity (red pseudocolor). Coexpression of BoNT/E leads to a loss of Pi–SNAP-25 immunoreactivity presumably because the C terminus containing pSer187 is cleaved by the toxin. Scale bars, 10 μm. A, Sample cell images. B, Bar graph summarizing the fluorescent ratio (pSer187/GFP–SNAP-25) for the indicated number of cells.
Results
Overexpressing a nonphosphorylatable mutant of SNAP-25 does not attenuate the enhancement of Ca2+-triggered exocytosis by PMA in chromaffin cells or INS-1 cells
We expressed GFP-tagged mutants of SNAP-25a using the SFV expression system in bovine chromaffin cells and assayed Ca2+-triggered exocytosis using membrane capacitance (_C_m) measurements while stimulating cells with photorelease of caged Ca2+ and membrane depolarization. Fusion of GFP to the N terminus of SNAP-25 allows fluorescent selection of cells expressing the mutant protein at levels many-fold higher than the endogenous protein, yet viral expression of the fusion protein has no obvious effects on Ca2+-triggered exocytosis (Wei et al., 2000).
Chromaffin and other endocrine cells respond with a small but rapid burst of exocytosis in response to photoelevation of [Ca2+]i into the low μm range that has been attributed to release of a highly Ca2+ sensitive pool (HCSP) of granules, whereas elevation of [Ca2+]i into the tens of μm is required to elicit fast release from the larger readily releasable pool (RRP) of granules (Yang et al., 2002, 2007; Zhu et al., 2002; Wan et al., 2004; Yang and Gillis, 2004). Figure 1A, top, depicts a protocol we developed to estimate the size of the HCSP and RRP in a single sweep (Yang and Gillis, 2004; Yang et al., 2007). The Ca2+ cage NP-EGTA together with the Ca2+ indicator dyes Fura2-FF and bisFura2 are introduced into cells through the patch pipette during whole-cell recording. A flash of UV light to photolyze the cage (arrow) is followed by 10 depolarizing pulses, 30 ms in duration, to + 20 mV. We fit the initial Δ_C_m burst to an exponential and define the size of the HCSP as the amplitude of this exponential fit (Fig. 1A, top). The subsequent train of depolarizing pulses leads to Ca2+ influx through voltage-gated Ca2+ channels (_I_Ca) (also depicted in Fig. 1) and exocytosis of the RRP. We define the Δ_C_m from the 10 depolarizing pulses as Δ_C_10 and use this as an index of the size of the RRP (Fig. 1A, top). Δ_C_10 is comparable in size with the RRP defined by the _C_m burst that results from photo-elevation of [Ca2+]i to several tens of μm (Voets, 2000).
Figure 1.
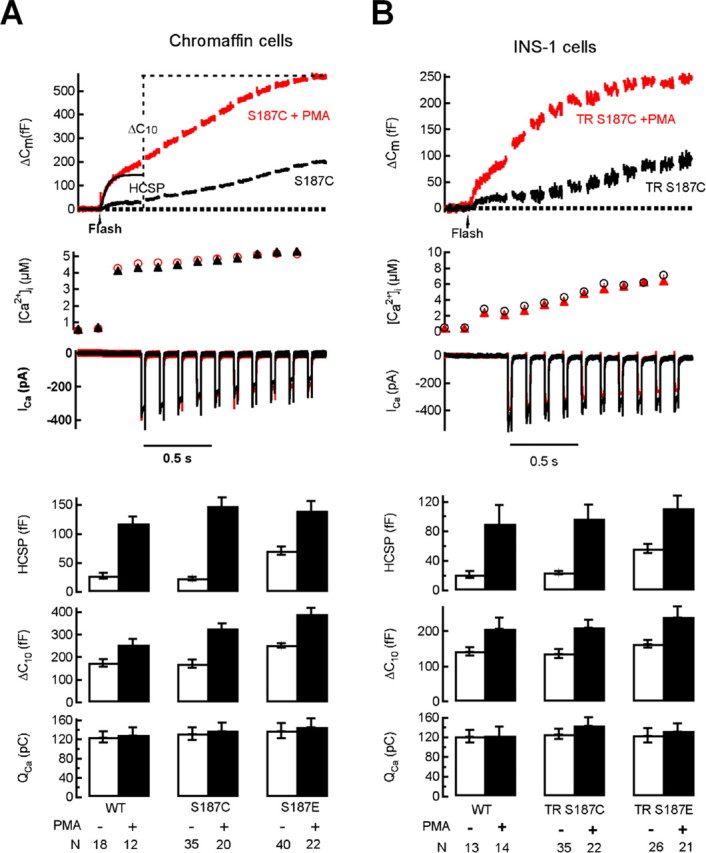
Expression of Ser187 SNAP-25 mutants does not attenuate the enhancement of exocytosis by PMA in chromaffin cells or INS-1 cells. Whole-cell recordings were made from GFP-positive cells. Flash photolysis of caged Ca2+ at the time indicated by the arrow leads to a burst of exocytosis (Δ_C_m) from the HCSP. This is followed 300 ms later by a train of depolarizing pulses to trigger Ca2+ influx (_I_Ca) and release of granules that are less responsive to Ca2+ (the RRP). A, The top shows sample responses from chromaffin cells expressing GFP–SNAP-25 with the S187C mutation using the Semliki Forest virus expression system in either the absence or presence of 100 nm PMA. The bottom summarizes data obtained from chromaffin cells with the number of cells (N) indicated below each experimental condition. _Q_Ca is the integral of the cumulative _I_Ca for the 10 depolarizing pulses, whereas Δ_C_10 is the total increase in capacitance in response to the depolarizing pulses. The data in the absence of PMA are from the study of Yang et al. (2007). B, The top shows sample responses from INS-1 cells expressing GFP–SNAP-25 with the TR and S187C mutations in either the absence or presence of 100 nm PMA. The bottom summarizes data obtained from INS-1 cells.
SFV expression of phosphomimetic Ser187 mutations (to Asp or Glu) partially mimic phorbol ester effects in enhancing vesicle recruitment (Nagy et al., 2002) and highly Ca2+ sensitive exocytosis (Yang et al., 2007) in chromaffin cells (Fig. 1A). If phosphorylation of Ser187 by PKC is an important player in phorbol ester-mediated enhancement of exocytosis, then mutation of Ser187 to a residue incapable of being phosphorylated should block phorbol ester effects. Whereas Ala substitution is a commonly used control mutation, expressing the S187A mutant results in a general inhibition of Ca2+-triggered exocytosis compared with expression of wild-type (WT) SNAP-25 (Nagy et al., 2002; Yang et al., 2007), therefore we tested the Cys mutation (S187C) which structurally resembles Ser and produces responses indistinguishable from cells transfected with WT GFP–SNAP-25 (Nagy et al., 2002; Yang et al., 2007) (Fig. 1). Figure 1A, top, depicts the response of 2 cells expressing the S187C mutant in either the absence or presence of the phorbol ester PMA. Results from a number of cells expressing either WT GFP–SNAP-25 or the S187C or S187E mutants in the absence or presence of PMA are summarized in the bottom of Figure 1A. These results clearly demonstrate that expressing Ser187 mutants (either S187C or S187E) have little or no effect on the enhancement of exocytosis by PMA.
We explored the universality of this result by performing similar experiments in the INS-1 insulin-secreting cell line (Asfari et al., 1992). INS-1 cells exhibit Ca2+-triggered exocytosis with an HCSP and RRP that are regulated in a manner comparable with bovine chromaffin cells or primary rat pancreatic β cells (Wan et al., 2004; Yang and Gillis, 2004). Just as in these primary endocrine cells, the HCSP in INS-1 cells is increased many fold in response to PMA, whereas the RRP is increased by ∼50%. We expressed S187C and S187E mutants in INS-1 cells using cationic lipid transfection and found virtually identical results as for chromaffin cells, i.e., mutating Ser187 of SNAP-25 has no effect on the enhancement of Ca2+-triggered exocytosis by PMA (Fig. 1B). Nevertheless, similar to chromaffin cells, the phosphomimetic S187E mutation partially mimics PMA treatment in selectively enhancing the size of the HCSP. The similar results between chromaffin cells and INS-1 cells demonstrate that these observations are independent of endocrine cell type, transfection method, or expression vector.
A strategy to disable endogenous SNAP-25 by expressing BoNT/E together with rescue of exocytosis with coexpression of a toxin-resistant (TR) SNAP-25 mutant
One possible explanation for the data of Figure 1 is that Ser187 phosphorylation by PKC is either unimportant for phorbol ester effects or that other pathways activated by the drug act redundantly with Ser187 phosphorylation to enhance Ca2+-triggered exocytosis. However, another possible explanation is a confounding role of the endogenous SNAP-25 in these expression studies. It is usually assumed that overexpressed SNAP-25 dominates interaction with potential binding partners such as the other members of the core SNARE complex (synaptobrevin/VAMP and syntaxin) because the transfected SNAP-25 is expressed at levels many-fold higher than that of the endogenous protein (supplemental data, available at www.jneurosci.org as supplemental material) (Wei et al., 2000). However, certain mutants of SNAP-25, although highly expressed (evidenced from GFP fluorescence), may not effectively functionally compete with endogenous SNAP-25. One approach to this problem is to express SNAP-25 in cells obtained from SNAP-25 knock-out mice (Sorensen et al., 2003), but it is possible that compensatory changes in expression of functionally related proteins may occur during development.
We adopted a strategy to disable endogenous SNAP-25 by expressing BoNT/E light chain while simultaneously expressing a toxin-resistant (TR) SNAP-25 mutant to functionally substitute for the endogenous protein (Gonelle-Gispert et al., 2002; Finley et al., 2003). The mutation strategy is depicted in Figure 2A. BoNT/E cleaves SNAP-25 between residues R180 and I181 whereas the toxin is ineffective on TR SNAP-25 mutant which contains three substitutions at Arg176, Asp179 and Met182 (Sadoul et al., 1997; Gonelle-Gispert et al., 1999). To increase the probability that the cells expressing exogenous SNAP-25 were also transfected with BoNT/E, we used a threefold higher concentration of BoNT/E DNA than the GFP–SNAP-25 DNA.
Figure 2.
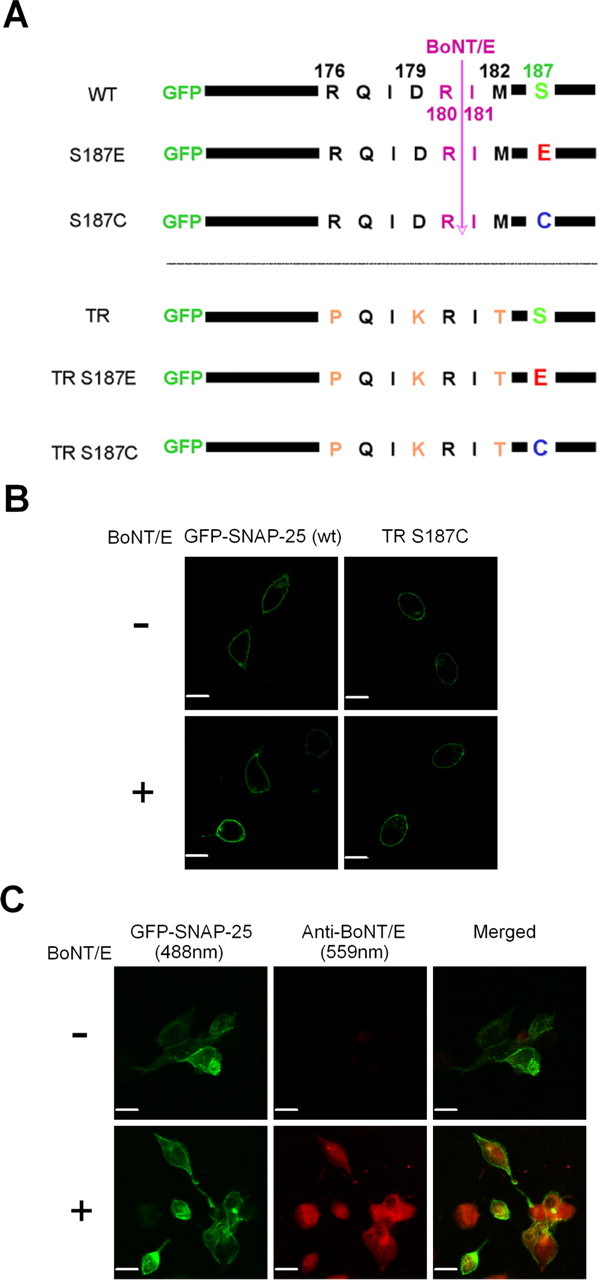
A strategy for expressing TR mutations of SNAP-25 that are resistant to BoNT/E cleavage. A, Schematic representation of the SNAP-25 mutants used in this study. GFP is fused to the N terminus of SNAP-25 to serve as a reporter of protein expression. BoNT/E cleaves SNAP-25 between Arg180 and Ile181, but toxin resistance can be conferred with a TR set of mutations (R176P, D179K, M182T). Finally, mutation of Ser187 to Cys precludes phosphorylation by PKC, whereas the Glu mutation is phosphomimetic. B, Four micrographs of confocal fluorescence from live cells demonstrate that expressed GFP–SNAP-25 is primarily localized to the plasma membrane of INS-1 cells. The top left micrograph depicts expression pattern of WT GFP–SNAP-25, whereas the top right micrograph depicts INS-1 cells cotransfected with BoNT/E and GFP–SNAP-25. The bottom micrographs depict expression of GFP–SNAP-25 with the TR/S187C mutations with (bottom right) and without (bottom left) cotransfection with BoNT/E. Scale bar, 10 μm. C, Sample confocal immunofluorescence images demonstrate that TR SNAP-25 and BoNT/E are coexpressed in the same cells. The cells were treated with anti-BoNT/E antibody and excited with a 488 nm laser line to image GFP–SNAP-25 (green pseudocolor), whereas a 559 nm laser line was used to identify BoNT/E immunoreactivity (red pseudocolor). The images were merged to show the coexpression of BoNT/E and TR SNAP-25 in the same cells. Scale bars, 10 μm.
To confirm that expressed SNAP-25 in our study is targeted properly to the surface membrane, we transfected INS-1 cells with GFP–SNAP-25 and acquired images from live cells using confocal microscopy. Figure 2B clearly shows that the expressed SNAP-25 is primarily localized in the plasma membrane, which is consistent with a previous study (Gonelle-Gispert et al., 2002). The lower right panel of Figure 2B depicts localization of TR/S187C SNAP-25 in cells cotransfected with BoNT/E. Thus this SNAP-25 mutant shows apparently normal membrane targeting under the transfection conditions of our functional assays. We used single-cell immunofluorescence with a SNAP-25 antibody to quantify that exogenous TR SNAP-25 is expressed at levels approximately sevenfold higher than endogenous SNAP-25 (supplemental data, available at www.jneurosci.org as supplemental material). The other mutants of SNAP-25 we tested expressed at comparable levels because we selected cells with similar GFP fluorescent intensities.
We also used single-cell immunofluorescent imaging to confirm that most cells expressing GFP–SNAP-25 also express BoNT/E in cotransfection experiments. Figure 2C presents sample images of cells transfected with TR SNAP-25 with (bottom row) or without (top row) cotransfection with BoNT/E. The left column depicts the fluorescence of GFP–SNAP-25 (488 nm excitation) whereas the middle column depicts immunofluorescence of BoNT/E (559 nm excitation) from the same field. We found that 11 of 13 GFP-positive cells we quantified exhibited BoNT/E immunofluorescence at levels at least 2 SDs above background levels. It is worth noting, however, that the percentage of cells successfully transfected was significantly lower (∼5–8%) in coexpression experiments compared with cells transfected with only SNAP-25 mutants (∼15%). This might be attributable to the larger amount of DNA used in the cotransfection experiments compared with experiments in which a single plasmid was used for transfection.
BoNT/E blocks exocytosis in cells expressing WT GFP–SNAP-25
We cotransfected INS-1 cells with the light chain of BoNT/E and WT GFP–SNAP-25 and assayed exocytosis using the stimulus protocol presented in Figure 1. A sample response is depicted in Figure 3A, whereas summarized results are presented in Figure 3B. Expression of BoNT/E dramatically inhibited Ca2+-triggered exocytosis with almost complete inhibition of exocytosis observed in 31 of 37 cells. Expression of GFP–SNAP-25 was ineffective at negating the effect of the toxin, presumably because it was cleaved along with the endogenous protein (also see Fig. 6). Conversely, coexpression of TR SNAP-25 was able to completely rescue Ca2+-triggered exocytosis (Fig. 3). Thus the TR mutation, although resistant to cleavage by BoNT/E, has no apparent deleterious effects on exocytosis under our stimulus conditions (also see Fig. 6) (Gonelle-Gispert et al., 2000).
Figure 3.

Expression of TR, but not WT, GFP–SNAP-25 can rescue exocytosis in INS-1 cells coexpressing BoNT/E. A, Sample traces from INS-1 cells cotransfected with BoNT/E and WT (GFP–SNAP-25; black trace) or TR SNAP-25 (red trace). B, Bar graph summary of data obtained from cells expressing WT or TR SNAP-25 with or without coexpression of BoNT/E.
Figure 6.
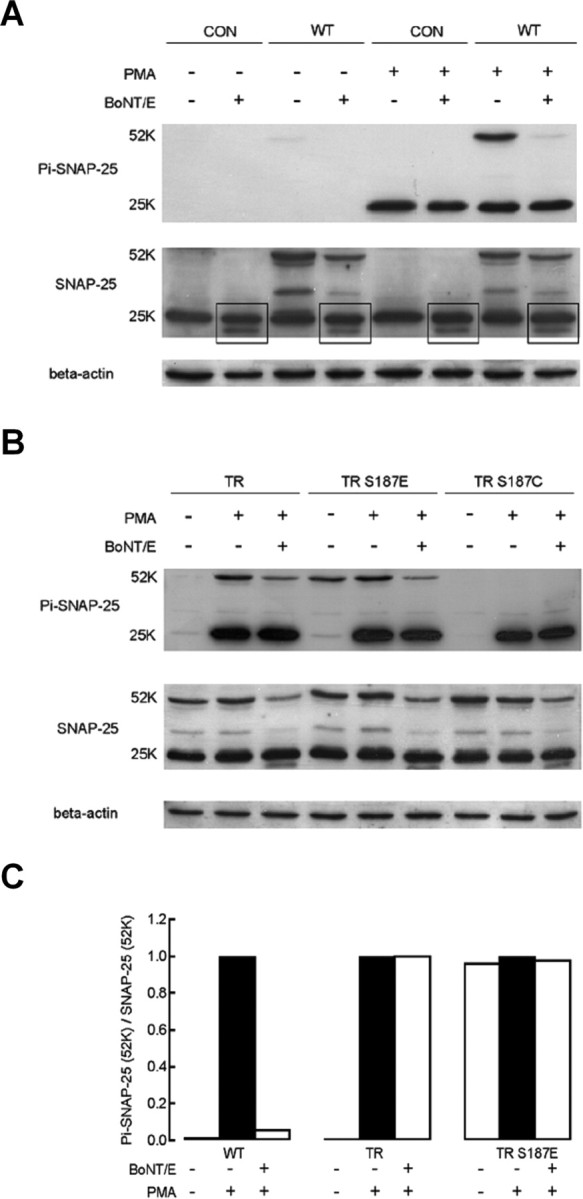
Immunoblots demonstrate that BoNT/E transfection cleaves transfected WT GFP–SNAP-25 and endogenous SNAP-25 but not TR SNAP-25. Antibodies used for immunoblots are the same as described for Figure 4. Expressed GFP–SNAP-25 is identified as a 52 kDa band, whereas endogenous SNAP-25 is ∼25 kDa. A, BoNT/E transfection abolishes the 52 kDa band recognized by the Ser187 phospho-specific antibody in GFP–SNAP-25 transfected cells. Transfection with BoNT/E leads to a second, lower-molecular-weight SNAP-25 band (highlighted by boxes) that presumably indicates truncated SNAP-25. CON refers to nontransfected cells, whereas WT refers to transfection with GFP–SNAP-25. B, BoNT/E cotransfection does not affect the relative intensity of the 52 kDa band recognized by the Ser187 phospho-specific antibody in TR SNAP-25 or TR/S187E transfected cells. C, Quantification of the relative intensity of the 52 kDa band representing phosphorylated GFP–SNAP-25. The intensity of the 52 kDa band recognized by the Ser187 phospho-specific antibody was divided by the intensity of the 52 kDa band recognized by the N-terminal SNAP-25 antibody. The relative intensity was then normalized to a value of 1.0 under the condition of exposure to PMA without cotransfection with BoNT/E (filled bars).
Note that coexpression of BoNT/E and WT GFP–SNAP-25 results in a ∼50% reduction in the size of the depolarization-evoked Ca2+ current (_I_Ca) or Ca2+ charge entry (_Q_Ca, the integral of _I_Ca) (Fig. 3), suggesting that the C terminus of SNAP-25 is linked with Ca2+ channel function, as previously reported (Wiser et al., 1996; Rettig et al., 1997; Andres-Mateos et al., 2005). Thus cleavage of SNAP-25 by BoNT/E can inhibit exocytosis both by reducing Ca2+ influx and by directly blocking Ca2+-triggered exocytosis (Xu et al., 1998).
PMA rapidly induces phosphorylation of Ser187 in INS-1 cells and the phosphomimetic S187E mutant is recognized by a Ser187 phosphorylation-specific antibody
We used a SNAP-25 Ser187 phosphorylation-specific antibody (Shimazaki et al., 1996; Kataoka et al., 2000) for immunoblot experiments. Application of PMA 100 nm leads to rapid phosphorylation of SNAP-25 at Ser187 in INS-1 cells (Fig. 4A) similar to what has been previously demonstrated (Gonelle-Gispert et al., 2002). Note that robust phosphorylation is evident within 5 min of PMA application and appears to be maximal within ∼30 min. We also used an antibody that recognizes the N terminus of SNAP-25 to probe total SNAP-25. This antibody detected both the endogenous SNAP-25 (∼25 kDa) and the expressed GFP–SNAP-25 (∼52 kDa) (Fig. 4B). Note that expression of the SNAP-25 mutants did not lead to any obvious change in the level of the endogenous SNAP-25.
Figure 4.

Incubation of INS-1 cells in PMA leads to rapid phosphorylation of SNAP-25 at Ser187. The S187E phosphomimetic mutant is detected by a Ser187 phospho-specific antibody. A, The time course of SNAP-25 phosphorylation during incubation of INS-1 cells in 100 nm PMA for the indicated period of time. Control lanes include sham incubations (Bath solution) and incubation in the vehicle DMSO for 30 min. Two groups of samples were prepared in parallel and separated using electrophoresis by SDS-PAGE gels. After transfer to membranes, they were probed with either the Ser187 phospho-specific antibody (Pi–SNAP-25; top) or an antibody that recognized the N terminus of SNAP-25 (SNAP-25; middle), respectively. One of the membranes was then stripped and probed with a β-actin antibody (bottom). B, Detection of expressed SNAP-25 mutants in INS-1 cells. INS-1 cells either incubated in PMA (100 nm) or standard bath solution for 30 min before lysis. CON refers to nontransfected cells, whereas WT refers to transfection with GFP–SNAP-25.
We also found that the Ser187 phospho-specific antibody recognized expression of the S187E GFP–SNAP-25 mutant (Fig. 4B), but not TR SNAP-25 or the S187C mutants, similar to what has been recently reported for the bacterially expressed protein (Yang et al., 2007). This result indicates that the TR/S187E protein is indeed phosphomimetic as demonstrated by an increased affinity for a Ser187-specific binding partner. Conversely, although the expression level of the 52 kDa TR/S187E protein (Fig. 4B, lane 5) is approximately sevenfold higher (supplemental data, available at www.jneurosci.org as supplemental material) than the endogenous 25 kDa SNAP-25 (Fig. 4B, lane 2), the 52 kDa SNAP-25-Pi band from the TR/S187E-expressing cells is much lighter than the 25 kDa SNAP-25-Pi band from the PMA treated control cells. This suggests that this SNAP-25 Ser187 phosphorylation-specific antibody does not bind to TR/S187E with as high an affinity as bona-fide phosphorylated Ser187. Thus the S187E SNAP-25 mutant may not perfectly mimic Ser187 phosphorylation.
Mutations of Ser187 block the enhancement of exocytosis by PMA if (and only if) endogenous SNAP-25 is disabled by coexpression of BoNT/E
Having established that expression of TR SNAP-25 can fully restore exocytosis after cleavage of endogenous SNAP-25 with coexpression of BoNT/E, we tested Ser187 mutations in the TR SNAP-25/BoNT/E background. Figure 5, A and C, demonstrates that PMA can still potently enhance exocytosis in cells coexpressing BoNT/E and TR SNAP-25. Conversely, the TR/S187C mutation completely blocked the enhancement of exocytosis by PMA under this experimental condition (Fig. 5B,C). This contrasts with the lack of effect of expression of TR/S187C SNAP-25 on the enhancement of Ca2+-triggered exocytosis by PMA in cells in which the endogenous SNAP-25 remained intact (Fig. 1). Expression of the phosphomimetic TR/S187E mutant together with BoNT/E resulted in a preferential enhancement of the HCSP (Fig. 5C) similar to what we observed without BoNT/E (Fig. 1). Addition of PMA caused no additional increase in Ca2+-triggered exocytosis during expression of the TR/S187E mutant in cells coexpressing BoNT/E (Fig. 5C), in contrast to the enhancement by PMA observed without the toxin (Fig. 1). These results suggest that Ser187 of SNAP-25 is essential for PMA to enhance Ca2+-triggered exocytosis under our stimulus conditions.
Figure 5.
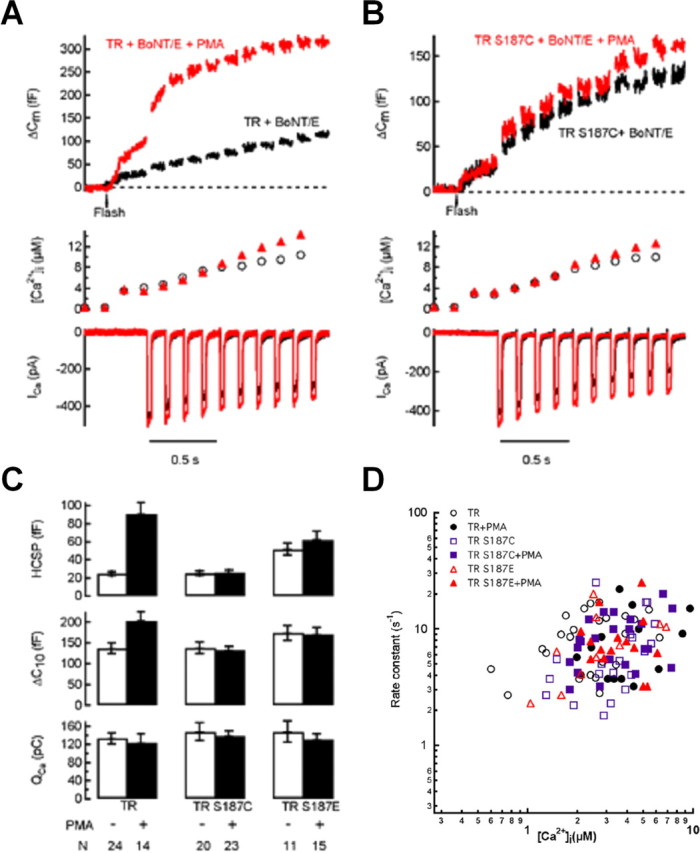
Mutations of Ser187 blocks the enhancement of exocytosis by PMA if endogenous SNAP-25 is disabled by coexpression of BoNT/E. The kinetics of exocytosis from the HCSP are not affected by Ser187 mutations or PMA incubation. All experiments were performed in fluorescent-positive cells cotransfected with BoNT/E and the indicated GFP–SNAP-25 mutant. Cells were used within 1 h after addition of 100 nm PMA. A, Sample traces from cells expressing TR SNAP-25 and BoNT/E incubated in the absence (black trace) or presence (red trace) of PMA. B, Sample traces from cells expressing TR/S187C and BoNT/E incubated in the absence (black trace) or presence (red trace) of PMA. C, Bar graph summarizing data from the indicated number of cells. D, Plot of the rate constant of exocytosis from the HCSP plotted against the post-flash [Ca2+]i for cells expressing either TR (circles), TR/S187C (squares), or TR/S187E (triangles). Filled symbols represent cells exposed to 100 nm PMA. The rate constant was obtained from an exponential fit to the _C_m trace after flash photolysis of caged Ca2+.
We also examined the Ca2+-dependent kinetics of exocytosis from the HCSP under conditions in which the endogenous SNAP-25 was disabled by expression of BoNT/E. The rate constants obtained from exponential fits of the _C_m time course to photorelease of caged Ca2+ are plotted versus post-flash [Ca2+]i in Figure 5D. Neither Ser187 mutation nor PMA incubation affected the rate constant of exocytosis from the HCSP. This is consistent with previous reports demonstrating that the amplitudes, but not the rate constants, of Ca2+-triggered exocytosis are modulated by phorbol esters (Gillis et al., 1996; Wan et al., 2004; Yang and Gillis, 2004).
Immunoblot analysis demonstrates that expression of BoNT/E efficiently cleaves WT SNAP-25, but not TR SNAP-25; expression of TR SNAP-25 does not block cleavage of endogenous SNAP-25 by BoNT/E
We performed immunoblot experiments in INS-1 cells to confirm that expression of BoNT/E cleaves endogenous, but not TR SNAP-25. Ser187 is part of the 26-residue C-terminal fragment that is released from the SNAP-25 protein by BoNT/E cleavage, therefore loss of immunolabeling by the Ser187 phospho-specific antibody can be used to assay toxin action. Figure 6A presents an immunoblot demonstrating that the phospho-specific antibody labels both endogenous SNAP-25 (∼25 kDa) and expressed GFP–SNAP-25 (∼52 kDa) when cells are incubated in 100 nm PMA for 30 min. Coexpression of BoNT/E leads to almost complete loss of the phospho-specific band for the expressed 52 kDa protein whereas the endogenous 25 kDa protein band is essentially intact. The mild attenuation of the endogenous band is presumably because a smaller percentage of cells express GFP–SNAP-25 when cells are cotransfected with the toxin. The near complete loss of the 52 kDa band suggests that the great majority of cells that express GFP–SNAP-25 also express highly functional BoNT/E in these cotransfection experiments and is consistent with our immunofluorescence experiments (Fig. 2C) and the nearly complete loss of exocytosis from these cells (Fig. 3). We stripped the phospho-specific antibody and probed the same membrane with an antibody that recognizes the N terminus of SNAP-25. This blot (Fig. 6A) demonstrates that cotransfected cells still express GFP–SNAP-25 (52 kDa band), therefore loss of labeling by the phospho-specific antibody likely represents cleavage by the toxin. Figure 6C presents a quantitative analysis of the band intensity for phospho-specific antibody normalized to the N-terminal SNAP-25 band to demonstrate nearly total cleavage of the 52 kDa protein by coexpression of BoNT/E.
Close inspection of immunoblots with the N-terminal SNAP-25 antibody reveals that there are two endogenous SNAP-25 bands evident in all samples with BoNT/E transfection (highlighted by boxes in Fig. 6A). The lower band is likely the truncated endogenous SNAP-25 and provides additional evidence for the effectiveness of BoNT/E expression.
We next tested whether the TR mutation can protect SNAP-25 from cleavage by BoNT/E (Fig. 6B). In TR SNAP-25 transfected cells, both the endogenous 25 kDa SNAP-25 and the expressed 52 kDa TR SNAP-25 are phosphorylated during treatment with PMA (Fig. 6B). In TR/S187E transfected cells, PMA phosphorylates the endogenous 25 kDa SNAP-25 but has no effect on the immunoreactivity of the expressed 52 kDa protein. During expression of either TR SNAP-25 or TR/S187E, BoNT/E coexpression does not change the phospho-specific immunoreactivity of the 52 kDa SNAP-25 mutant relative to the immunoreactivity probed with the N-terminal SNAP-25 antibody (Fig. 6B,C).
It is not straightforward to determine the extent that the endogenous SNAP-25 is cleaved in cells expressing BoNT/E using immunoblots of the entire cell population because only ∼5–8% of cells express the transfected proteins. Therefore we used single-cell immunofluorescence with the Ser187 phospho-specific antibody to assay cleavage of endogenous SNAP-25. A question of particular relevance is whether expression of TR SNAP-25 can block BoNT/E from cleaving the endogenous protein, therefore we expressed the TR/S187C mutant with or without cotransfection with BoNT/E. Because the S187C mutant cannot be phosphorylated by PKC, the phospho-specific antibody will not recognize this exogenous form of SNAP-25 even in cells treated with PMA (Fig. 6B). Pi–SNAP-25 immunoreactivity, originating from endogenous SNAP-25, is noted only if cells are incubated in PMA (Fig. 7). Pi–SNAP-25 immunofluorescence is equivalent to background levels in PMA-treated cells that coexpress TR/S187C and BoNT/E, presumably because the toxin cleaves the endogenous SNAP-25. Sample images are presented in Figure 7A, whereas normalized fluorescent data are summarized in Figure 7B.
Discussion
Phosphorylation of Ser187 of SNAP-25 by PKC is necessary for phorbol ester enhancement of Ca2+-triggered exocytosis
Our results demonstrate that expression of nonphosphorylatable Ser187 mutants of SNAP-25 can almost totally block enhancement of exocytosis by the DAG analog PMA if, and only if, endogenous SNAP-25 is disabled by expressing BoNT/E. This contrasts with a study by Finley et al. (2003) that reported little effect of Ser187 mutations on enhancement of exocytosis by a phorbol ester in CA3 hippocampal neurons. This study was performed using a similar strategy of rescuing BoNT/E intoxication with a toxin-resistant SNAP-25 mutant, although it is not clear in this study how extensively the endogenous SNAP-25 was cleaved by the toxin. Nevertheless, it is likely that phorbol esters have effects on exocytosis that are not mediated by PKC and the relative importance of the PKC-mediated effect likely depends on the cell type and stimulus conditions. For example, we have observed that the effects of PMA on exocytosis in chromaffin cells are not always completely blocked by the PKC inhibitor bisindolylmaleimide I if cells are treated with the phorbol ester for greater than ∼30 min (data not shown) (Shoji-Kasai et al., 2002). A very recent study in cultured hippocampal neurons found that PKC inhibitors are less effective at blocking the enhancement of exocytosis elicited by a second exposure to the phorbol ester PDBu (Wierda et al., 2007).
Our conclusions are based on the assumption that endogenous SNAP-25 is disabled and functionally replaced with the expressed TR SNAP-25 mutants. We found that BoNT/E and TR SNAP-25 are indeed expressed in the same cells (Fig. 2C) and BoNT/E expression cleaves GFP–SNAP-25, but not TR SNAP-25 (Fig. 6). Expression of BoNT/E almost completely abolishes exocytosis when expressed together with GFP–SNAP-25, but coexpression of TR SNAP-25 completely rescues exocytosis (Fig. 3). In addition, our immunofluorescence experiments suggest that expression of TR SNAP-25 does not block the ability of coexpressed BoNT/E to cleave endogenous SNAP-25 (Fig. 7). We have not investigated possible complications of this approach introduced by the presence of cleavage products of SNAP-25. In addition, our approach to assay SNAP-25 cleavage with the phospho-specific antibody in Figures 6 and 7 is indirect, although the double bands depicted in Figure 6A strongly support the hypothesis that expression of BoNT/E indeed cleaves endogenous SNAP-25.
A likely alternative target of phorbol esters to enhance exocytosis is munc-13. A study by (Rhee et al., 2002) suggested that munc-13 is the only important target of phorbol esters in enhancing exocytosis because mutation of the phorbol ester binding site of this protein was able to completely block phorbol ester effects. However, our results and those of others published within the last year suggest that other proteins in addition to munc-13 play important roles in phorbol-ester mediated enhancement of exocytosis in both neurons and endocrine cells. While preparing this manuscript, a study by (Wierda et al., 2007) reports that mutating the PKC phosphorylation sites of munc-18 can completely block the enhancement of EPSC amplitude by phorbol esters in cultured hippocampal neurons. Another recent study reports that mutation of the PKC phosphorylation site in Aplysia SNAP-25 blocks the ability of a phorbol ester to reduce the level of depression observed in response to repetitive stimulation of sensory-to-motor synapses (Houeland et al., 2007). An important future area of study is to further delineate how these three proteins coordinate to mediate effects of the DAG signaling pathway on exocytosis.
How does phosphorylation of Ser187 affect exocytosis?
We found that expression of the phosphomimetic S187E SNAP-25 mutant increases exocytosis elicited by low [Ca2+]i levels (from the HCSP) to a greater extent than exocytosis elicited by higher [Ca2+]i levels (from the RRP) in both chromaffin cells (Yang et al., 2007) and insulin-secreting cells (Fig. 1B). This mirrors the higher sensitivity of the HCSP to phorbol ester application (Yang et al., 2002; Wan et al., 2004; Yang and Gillis, 2004). This effect of expressing S187E does not depend on whether or not the endogenous SNAP-25 is disabled by BoNT/E (compare Figs. 1B, 5C). Neither S187E expression (Fig. 5D) nor phorbol ester application (Gillis et al., 1996; Wan et al., 2004; Yang and Gillis, 2004) change the [Ca2+]i-dependent rate constants of exocytosis. It has previously been reported that S187E expression in chromaffin cells leads to an increase in the rate of the sustained component of exocytosis after photoelevation of [Ca2+]i, but not an increase in RRP size (Nagy et al., 2002). The increase in the “sustained” component by S187E has been interpreted as an increase in the rate of vesicle recruitment. Phorbol ester treatment has also been reported to increase the rate of vesicle resupply in chromaffin cells (Smith, 1999). In Aplysia sensory-to-motor synapses, expression of phospho-mimetic SNAP-25 mutants has been reported to slow the rate of synaptic depression in a manner similar to phorbol ester application (Houeland et al., 2007). This is also consistent with PKC phosphorylation of SNAP-25 leading to vesicle mobilization.
Expression of phosphomimetic Ser187 mutants does not reproduce all effects of phorbol esters on exocytosis. For example, expressing the S187E mutation does not lead to the 1.5- to 2-fold increase in the size of the RRP that is observed with phorbol ester application in endocrine cells. In addition, the HCSP is increased approximately sixfold with phorbol ester application, but only approximately twofold during expression of S187E. Nevertheless, Ser187 mutations can block these effects of phorbol esters. A possible reason for this is that phosphorylation of Ser187 is necessary, but not sufficient for these phorbol ester effects attributable to involvement of other proteins such munc-13 or munc-18. Another possibility, indirectly suggested because of the relatively weak binding to the phospho-specific antibody (Fig. 4B), is that the S187E mutation imperfectly mimics phosphorylation of native SNAP-25. Experiments using the potentially phospho-mimetic Asp substitution for Ser187 have not been particularly informative in this regard because expression of this SNAP-25 mutant in chromaffin cells leads to an inhibition of release from the RRP (Nagy et al., 2002; Yang et al., 2007).
The molecular mechanisms whereby PKC phosphorylation of SNAP-25 enhance vesicle mobilization and highly Ca2+-sensitive exocytosis remain incompletely understood, although it has recently been shown that the S187E mutation increases binding of purified, bacterially expressed SNAP-25 to syntaxin (Yang et al., 2007). This contrasts with an early report demonstrating that treatment of PC12 cell lysates with PKC and ATPγS reduces the amount of syntaxin coimmunoprecipitating with SNAP-25 (Shimazaki et al., 1996), but is consistent with a report demonstrating an increase in the ternary SNARE complex in PC12 cells expressing the S187E SNAP-25 mutant (Xu et al., 2004). Because formation of the syntaxin-SNAP-25 dimer is believed to be the initial, rate-limiting step in SNARE complex assembly, enhancement of heterodimer formation might be expected to enhance early steps of priming vesicles for exocytosis.
Footnotes
This work was supported by National Institutes of Health Grant RO1 NS40453 (K.D.G.). We thank Dr. Philippe Halban (University of Geneva, Geneva, Switzerland) for providing constructs for BoNT/E and toxin-resistant SNAP-25a.
References
- Andres-Mateos E, Renart J, Cruces J, Solis-Garrido LM, Serantes R, de Lucas-Cerrillo AM, Aldea M, Garcia AG, Montiel C. Dynamic association of the Ca2+ channel alpha1A subunit and SNAP-25 in round or neurite-emitting chromaffin cells. Eur J Neurosci. 2005;22:2187–2198. doi: 10.1111/j.1460-9568.2005.04385.x. [DOI] [PubMed] [Google Scholar]
- Asfari M, Janjic D, Meda P, Li G, Halban PA, Wollheim CB. Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology. 1992;130:167–178. doi: 10.1210/endo.130.1.1370150. [DOI] [PubMed] [Google Scholar]
- Ashery U, Betz A, Xu T, Brose N, Rettig J. An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol. 1999;78:525–532. doi: 10.1016/s0171-9335(99)80017-x. [DOI] [PubMed] [Google Scholar]
- Blasi J, Chapman ER, Link E, Binz T, Yamasaki S, De Camilli P, Sudhof TC, Niemann H, Jahn R. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 1993;365:160–163. doi: 10.1038/365160a0. [DOI] [PubMed] [Google Scholar]
- Braha O, Dale N, Hochner B, Klein M, Abrams TW, Kandel ER. Second messengers involved in the two processes of presynaptic facilitation that contribute to sensitization and dishabituation in Aplysia sensory neurons. Proc Natl Acad Sci USA. 1990;87:2040–2044. doi: 10.1073/pnas.87.5.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Rosenmund C. Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci. 2002;115:4399–4411. doi: 10.1242/jcs.00122. [DOI] [PubMed] [Google Scholar]
- DiCiommo DP, Bremner R. Rapid, high level protein production using DNA-based Semliki Forest virus vectors. J Biol Chem. 1998;273:18060–18066. doi: 10.1074/jbc.273.29.18060. [DOI] [PubMed] [Google Scholar]
- Duncan RR, Don-Wauchope AC, Tapechum S, Shipston MJ, Chow RH, Estibeiro P. High-efficiency Semliki Forest virus-mediated transduction in bovine adrenal chromaffin cells. Biochem J. 1999;342:497–501. [PMC free article] [PubMed] [Google Scholar]
- Ellis-Davies GC, Kaplan JH. Nitrophenyl-EGTA, a photolabile chelator that selectively binds Ca2+ with high affinity and releases it rapidly upon photolysis. Proc Natl Acad Sci USA. 1994;91:187–191. doi: 10.1073/pnas.91.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley MF, Scheller RH, Madison DV. SNAP-25 Ser187 does not mediate phorbol ester enhancement of hippocampal synaptic transmission. Neuropharmacology. 2003;45:857–862. doi: 10.1016/s0028-3908(03)00283-1. [DOI] [PubMed] [Google Scholar]
- Genoud S, Pralong W, Riederer BM, Eder L, Catsicas S, Muller D. Activity-dependent phosphorylation of SNAP-25 in hippocampal organotypic cultures. J Neurochem. 1999;72:1699–1706. doi: 10.1046/j.1471-4159.1999.721699.x. [DOI] [PubMed] [Google Scholar]
- Gillis KD. Admittance-based measurement of membrane capacitance using the EPC-9 patch-clamp amplifier. Pflügers Arch. 2000;439:655–664. doi: 10.1007/s004249900173. [DOI] [PubMed] [Google Scholar]
- Gillis KD, Möbner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- Gonelle-Gispert C, Halban PA, Niemann H, Palmer M, Catsicas S, Sadoul K. SNAP-25a and -25b isoforms are both expressed in insulin-secreting cells and can function in insulin secretion. Biochem J. 1999;339:159–165. [PMC free article] [PubMed] [Google Scholar]
- Gonelle-Gispert C, Molinete M, Halban PA, Sadoul K. Membrane localization and biological activity of SNAP-25 cysteine mutants in insulin-secreting cells. J Cell Sci. 2000;113:3197–3205. doi: 10.1242/jcs.113.18.3197. [DOI] [PubMed] [Google Scholar]
- Gonelle-Gispert C, Costa M, Takahashi M, Sadoul K, Halban P. Phosphorylation of SNAP-25 on serine-187 is induced by secretagogues in insulin-secreting cells, but is not correlated with insulin secretion. Biochem J. 2002;368:223–232. doi: 10.1042/BJ20020896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houeland G, Nakhost A, Sossin WS, Castellucci VF. PKC modulation of transmitter release by SNAP-25 at sensory-to-motor synapses in Aplysia. J Neurophysiol. 2007;97:134–143. doi: 10.1152/jn.00122.2006. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Kataoka M, Sekiguchi M, Shimazaki Y, Sato K, Takahashi M. Two distinct mechanisms underlie the stimulation of neurotransmitter release by phorbol esters in clonal rat pheochromocytoma PC12 cells. J Biochem (Tokyo) 2000;128:407–414. doi: 10.1093/oxfordjournals.jbchem.a022768. [DOI] [PubMed] [Google Scholar]
- Kataoka M, Kuwahara R, Iwasaki S, Shoji-Kasai Y, Takahashi M. Nerve growth factor-induced phosphorylation of SNAP-25 in PC12 cells: a possible involvement in the regulation of SNAP-25 localization. J Neurochem. 2000;74:2058–2066. doi: 10.1046/j.1471-4159.2000.0742058.x. [DOI] [PubMed] [Google Scholar]
- Martell AE, Smith RH. Critical stability constants. New York: Plenum; 1974–1989. [Google Scholar]
- Nagy G, Matti U, Nehring RB, Binz T, Rettig J, Neher E, Sorensen JB. Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J Neurosci. 2002;22:9278–9286. doi: 10.1523/JNEUROSCI.22-21-09278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Neher E. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflügers Arch. 1988;411:204–211. doi: 10.1007/BF00582316. [DOI] [PubMed] [Google Scholar]
- Rettig J, Heinemann C, Ashery U, Sheng ZH, Yokoyama CT, Catterall WA, Neher E. Alteration of Ca2+ dependence of neurotransmitter release by disruption of Ca2+ channel/syntaxin interaction. J Neurosci. 1997;17:6647–6656. doi: 10.1523/JNEUROSCI.17-17-06647.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Sudhof TC, Takahashi M, Rosenmund C, Brose N. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- Sadoul K, Berger A, Niemann H, Weller U, Roche PA, Klip A, Trimble WS, Regazzi R, Catsicas S, Halban PA. SNAP-23 is not cleaved by botulinum neurotoxin E and can replace SNAP-25 in the process of insulin secretion. J Biol Chem. 1997;272:33023–33027. doi: 10.1074/jbc.272.52.33023. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Rossetto O, Catsicas S, Polverino de Laureto P, DasGupta BR, Benfenati F, Montecucco C. Identification of the nerve terminal targets of botulinum neurotoxin serotypes A, D, and E. J Biol Chem. 1993;268:23784–23787. [PubMed] [Google Scholar]
- Shimazaki Y, Nishiki T, Omori A, Sekiguchi M, Kamata Y, Kozaki S, Takahashi M. Phosphorylation of 25-kDa synaptosome-associated protein. Possible involvement in protein kinase C-mediated regulation of neurotransmitter release. J Biol Chem. 1996;271:14548–14553. doi: 10.1074/jbc.271.24.14548. [DOI] [PubMed] [Google Scholar]
- Shoji-Kasai Y, Itakura M, Kataoka M, Yamamori S, Takahashi M. Protein kinase C-mediated translocation of secretory vesicles to plasma membrane and enhancement of neurotransmitter release from PC12 cells. Eur J Neurosci. 2002;15:1390–1394. doi: 10.1046/j.1460-9568.2002.01972.x. [DOI] [PubMed] [Google Scholar]
- Smith C. A persistent activity-dependent facilitation in chromaffin cells is caused by Ca2+ activation of protein kinase C. J Neurosci. 1999;19:589–598. doi: 10.1523/JNEUROSCI.19-02-00589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Terbush DR, Holz RW. Activation of protein kinase C is not required for exocytosis from bovine adrenal chromaffin cells. The effects of protein kinase C(19–31), Ca/CaM kinase II(291–317), and staurosporine. J Biol Chem. 1990;265:21179–21184. [PubMed] [Google Scholar]
- Voets T. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 2000;28:537–545. doi: 10.1016/s0896-6273(00)00131-8. [DOI] [PubMed] [Google Scholar]
- Wan QF, Dong Y, Yang H, Lou X, Ding J, Xu T. Protein kinase activation increases insulin secretion by sensitizing the secretory machinery to Ca2+ J Gen Physiol. 2004;124:653–662. doi: 10.1085/jgp.200409082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Xu T, Ashery U, Kollewe A, Matti U, Antonin W, Rettig J, Neher E. Exocytotic mechanism studied by truncated and zero layer mutants of the C-terminus of SNAP-25. EMBO J. 2000;19:1279–1289. doi: 10.1093/emboj/19.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M. Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron. 2007;54:275–290. doi: 10.1016/j.neuron.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Wiser O, Bennett MK, Atlas D. Functional interaction of syntaxin and SNAP-25 with voltage-sensitive L- and N-type Ca2+ channels. EMBO J. 1996;15:4100–4110. [PMC free article] [PubMed] [Google Scholar]
- Wu XS, Wu LG. Protein kinase C increases the apparent affinity of the release machinery to Ca2+ by enhancing the release machinery downstream of the Ca2+ sensor. J Neurosci. 2001;21:7928–7936. doi: 10.1523/JNEUROSCI.21-20-07928.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu NJ, Yu YX, Zhu JM, Liu H, Shen L, Zeng R, Zhang X, Pei G. Inhibition of SNAP-25 phosphorylation at Ser187 is involved in chronic morphine-induced down-regulation of SNARE complex formation. J Biol Chem. 2004;279:40601–40608. doi: 10.1074/jbc.M406896200. [DOI] [PubMed] [Google Scholar]
- Xu T, Binz T, Niemann H, Neher E. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci. 1998;1:192–200. doi: 10.1038/642. [DOI] [PubMed] [Google Scholar]
- Yang Y, Gillis KD. A highly Ca2+-sensitive pool of granules is regulated by glucose and protein kinases in insulin-secreting INS-1 cells. J Gen Physiol. 2004;124:641–651. doi: 10.1085/jgp.200409081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Udayasankar S, Dunning J, Chen P, Gillis KD. A highly Ca2+-sensitive pool of vesicles is regulated by protein kinase C in adrenal chromaffin cells. Proc Natl Acad Sci USA. 2002;99:17060–17065. doi: 10.1073/pnas.242624699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Craig TJ, Chen X, Ciufo LF, Takahashi M, Morgan A, Gillis KD. Phosphomimetic mutation of Ser-187 of SNAP-25 increases both syntaxin binding and highly Ca2+-sensitive exocytosis. J Gen Physiol. 2007;129:233–244. doi: 10.1085/jgp.200609685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H. Protein kinase C potentiates transmitter release from the chick ciliary presynaptic terminal by increasing the exocytotic fusion probability. J Physiol (Lond) 1999;515:169–180. doi: 10.1111/j.1469-7793.1999.169ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Hille B, Xu T. Sensitization of regulated exocytosis by protein kinase C. Proc Natl Acad Sci USA. 2002;99:17055–17059. doi: 10.1073/pnas.232588899. [DOI] [PMC free article] [PubMed] [Google Scholar]