NMDA Receptor Activation Potentiates Inhibitory Transmission through GABA Receptor-Associated Protein-Dependent Exocytosis of GABAA Receptors (original) (raw)
Abstract
The trafficking of postsynaptic AMPA receptors (AMPARs) is a powerful mechanism for regulating the strength of excitatory synapses. It has become clear that the surface levels of inhibitory GABAA receptors (GABAARs) are also subject to regulation and that GABAAR trafficking may contribute to inhibitory plasticity, although the underlying mechanisms are not fully understood. Here, we report that NMDA receptor activation, which has been shown to drive excitatory long-term depression through AMPAR endocytosis, simultaneously increases expression of GABAARs at the dendritic surface of hippocampal neurons. This NMDA stimulus increases miniature IPSC amplitudes and requires the activity of Ca2+ calmodulin-dependent kinase II and the trafficking proteins _N_-ethylmaleimide-sensitive factor, GABA receptor-associated protein (GABARAP), and glutamate receptor interacting protein (GRIP). These data demonstrate for the first time that endogenous GABARAP and GRIP contribute to the regulated trafficking of GABAARs. In addition, they reveal that the bidirectional trafficking of AMPA and GABAA receptors can be driven by a single glutamatergic stimulus, providing a potent postsynaptic mechanism for modulating neuronal excitability.
Keywords: synaptic plasticity, GABAA receptor trafficking, CaMKII, NSF, GABARAP, GRIP
Introduction
Synaptic plasticity has been implicated in learning and memory (for review, see Martin et al., 2000), synaptogenesis (Chen et al., 2007), schizophrenia (Spencer and McCarley, 2005), autism (Rubenstein and Merzenich, 2003), and addiction (Ungless et al., 2001). Many forms of plasticity at excitatory synapses have been characterized, including NMDA receptor (NMDAR)-dependent long-term potentiation (LTP) and long-term depression (LTD), which are mediated by insertion or removal of AMPA-type glutamate receptors (AMPARs) from the postsynaptic membrane (Malenka and Bear, 2004). Numerous types of plasticity at inhibitory synapses have also been identified (for review, see Gaiarsa et al., 2002), including several thought to be expressed postsynaptically (Kano et al., 1992; Morishita and Sastry, 1996; Ouardouz and Sastry, 2000).
Increasing evidence suggests that GABAA receptor (GABAAR) redistribution may be involved in some forms of inhibitory plasticity. GABAARs exhibit multiple forms of trafficking (for review, see Luscher and Keller, 2004; Michels and Moss, 2007): they are constitutively cycled at synapses (Kittler et al., 2000) and are trafficked to the surface after kindling (a model of epileptogenesis) (Nusser et al., 1998), insulin application (Wan et al., 1997), and Ca2+ calmodulin-dependent kinase II (CaMKII) activation (Wei et al., 2004). GABAARs can be removed from the neuronal surface in the hippocampus after status epilepticus (Naylor et al., 2005), in the amygdala during fear conditioning (Chhatwal et al., 2005), and by treatment with phorbol esters (Connolly et al., 1999), the GABAAR agonist muscimol (Barnes, 1996), or tumor necrosis factor α (Stellwagen et al., 2005). In the hippocampus, NMDARs couple to changes in GABAergic transmission in a manner consistent with membrane removal of GABAARs (Wang et al., 2003), whereas in the deep cerebellar nuclei (DCN) NMDARs increase GABAergic transmission, possibly through insertion of GABAARs (Ouardouz and Sastry, 2000). However, there has been no direct evidence that glutamatergic signaling regulates GABAARs trafficking despite reports of cross talk between excitatory and inhibitory synapses.
Several proteins are known to regulate GABAAR trafficking (Chen and Olsen, 2007), including GABAAR-associated protein (GABARAP). Expression of GABARAP in heterologous cells increases GABAAR channel conductance (Everitt et al., 2004), surface expression, and clustering (Chen et al., 2000, 2005). Overexpression of GABARAP in cultured hippocampal neurons increases surface GABAAR levels (Leil et al., 2004). These data along with the fact that GABARAP interacts with microtubules (Wang et al., 1999), trafficking proteins including _N_-ethylmaleimide-sensitive factor (NSF) (Kittler et al., 2001), and synaptic scaffolding proteins such as glutamate receptor interacting protein (GRIP) (Kittler et al., 2004) and gephyrin (Kneussel et al., 2000) suggest that GABARAP could play a role in receptor delivery to synapses.
Here, we show that NMDAR activation, a stimulus that induces LTD at excitatory synapses, increases surface GABAAR expression in hippocampal neurons and thereby potentiates inhibitory transmission. In investigating the mechanism underlying this change, we found that the NSF-dependent trafficking is triggered by calcium-dependent activation of CaMKII. In addition, we provide novel evidence that GABARAP is a central component of the machinery driving the activity-regulated delivery of GABAARs to the membrane, and that GRIP, previously recognized for its role in AMPAR trafficking, is also essential to this process.
Materials and Methods
Primary hippocampal cultures
Hippocampi were isolated from postnatal day 0 (P0) rat pups, and the dentate gyri were removed. After dissociation with papain (Worthington Biochemical, Lakewood, NJ) and mechanical trituration, the cells were plated at ∼75,000 cells per 12 mm poly-l-lysine-coated coverslip. Cultures were incubated in MEM (Invitrogen, Grand Island, NY) with fetal bovine serum (Invitrogen) for 24–36 h and subsequently maintained in Neurobasal media (Invitrogen) supplemented with B27 (Invitrogen) and Glutamax (Invitrogen). Experiments were performed on cultures from 14 to 21 d in vitro (DIV).
Antibodies and reagents
Antibodies (Abs) used in the study included the following: GABAA β2/3 (mouse monoclonal clone 62–3G1; Upstate Biotechnology, Lake Placid, NY), GAD-65 (rabbit polyclonal; Chemicon, Temecula, CA), glutamate receptor 1 (GluR1) (rabbit polyclonal; Calbiochem, La Jolla, CA), GluR2 (mouse monoclonal; Chemicon), GRIP (mouse monoclonal; BD Transduction Laboratories, San Jose, CA; rabbit polyclonal, Upstate Biotechnology), β-actin (mouse monoclonal; Chemicon), NSF (mouse monoclonal; BD Biosciences, San Jose, CA), NMDA receptor 1 (NR1) (Chemicon), GABARAP (for immunoprecipitation, rabbit polyclonal directed to full-length human GABARAP; Santa Cruz Biotechnology, Santa Cruz, CA; for immunocytochemistry, rabbit polyclonal directed against an N-terminal fragment of the human GABARAP protein, Abcam, Cambridge, MA). Cy3, FITC, and HRP-conjugated Abs raised in donkey were obtained from Jackson ImmunoResearch (West Grove, PA). NMDA, CNQX, DNQX, KN-93 (_N_-[2-[[[3-(4-chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-_N_-(2-hydroxyethyl)-4-methoxybenzenesulfonamide), okadaic acid, picrotoxin (PTx), and CaMKII autoinhibitory peptide (AIP) were purchased from Tocris Cookson (Ellisville, MO). BAPTA, BAPTA-AM, EDTA, HEPES, neocuproine, and ATPγS were obtained from Sigma (St. Louis, MO), and cypermethrin was obtained from Calbiochem. NSF-SNAP (_S_-nitroso penicillamine) inhibitory peptides were purchased from AnaSpec (San Jose, CA). GABARAP-GABAAR inhibitory peptides were obtained from Invitrogen (Carlsbad, CA). All drugs were diluted in H20 except CNQX and DNQX (DMSO). Botulinum neurotoxin light chain type B (BoNT/B) was purchased from List Biological Laboratories (Campbell, CA).
Immunocytochemical methods
Assay for surface GABAARs.
Hippocampal cells were treated with NMDA (20 μm; CNQX, 10 μm) for 2 min at 37°C followed by a 13 min recovery in conditioned media. Cells were then fixed under nonpermeablizing conditions with 4% paraformaldehyde. After blocking with Tris-buffered saline (TBS) containing 4% bovine serum albumin (BSA), cells were labeled with an antibody recognizing an extracellular epitope of the GABAA β2/3 subunits. The integrity of the cell membrane under these blocking conditions was confirmed by applying antibody against the cytosolic protein GRIP, which exhibited little staining (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). For double labeling with GAD or GRIP, the cells were then permeabilized for 15 min with TBS/4%BSA/0.1% Triton X-100 and labeled with either a GAD-65 or GRIP rabbit polyclonal antibody. A donkey anti-mouse secondary Ab conjugated with Cy3 was used to label surface GABAA β2/3 receptors, and an FITC-conjugated donkey anti-rabbit secondary antibody was used to visualize GAD-65/GRIP.
Double labeling for surface GABAARs and surface GluR1.
Hippocampal cells were live labeled with an antibody to the extracellular region of GluR1 for 30 min before NMDA. NMDA stimulation, fixation, blocking, and surface GABAA β2/3 labeling was then done as described above. An FITC-conjugated donkey anti-rabbit secondary antibody was used to visualize surface GluR1, and a Cy3-conjugated donkey anti-mouse secondary was used for surface GABAA β2/3.
Image acquisition and analysis.
Neurons were imaged using a Hamamatsu Orca ER camera attached to an inverted Nikon fluorescent microscope with a 60× Plan Apo lens. Exposure times were adjusted to ensure signals throughout the neuron fell within the linear range of the camera. Images were analyzed using MetaMorph software (Universal Imaging, Downingtown, PA). For analysis of synaptic GABAAR intensity, images of individual cells were taken for both GAD and GABAAR labeling. Images of GABAAR staining were background-subtracted and thresholded to include only signals approximately twofold greater than the diffuse labeling in dendritic shafts. Regions were automatically generated around GAD puncta using MetaMorph and transferred to images of GABAAR staining. Intensities of these synaptically localized regions were measured and compared in both control and drug-treated cells. In parallel analysis, GABAAR puncta in background-subtracted and thresholded images of GABAAR staining were counted and normalized to the length of dendrite analyzed. Additionally, integrated signal intensity values of fluorescence were determined for the punctate GABAAR labeling and normalized to the area of dendrite. This analysis gave similar results to those measuring changes in intensity at GAD puncta [GABAARs at GAD, 62.7 ± 9.1% increase with NMDA treatment (n = 6); GABAAR puncta alone, 67.2 ± 12.1% increase with NMDA treatment (n = 6)] (Fig. 1b). Consequently, this latter technique was used to measure GABAAR surface intensity in subsequent experiments (Fig. 2, see Figs. 4–7). Additional experiments in which the GABAAR antibody was applied to live, intact neurons at 4°C after NMDA treatment showed a similar increase (50.9 ± 15.1% increase), confirming that changes represent differences in surface receptor levels (see Fig. 4a, 0 min). Data are graphed as a percentage change from labeling in untreated control cells. For all experiments, n values represent individual experiments in which 7–20 cells are imaged in each condition.
Figure 1.
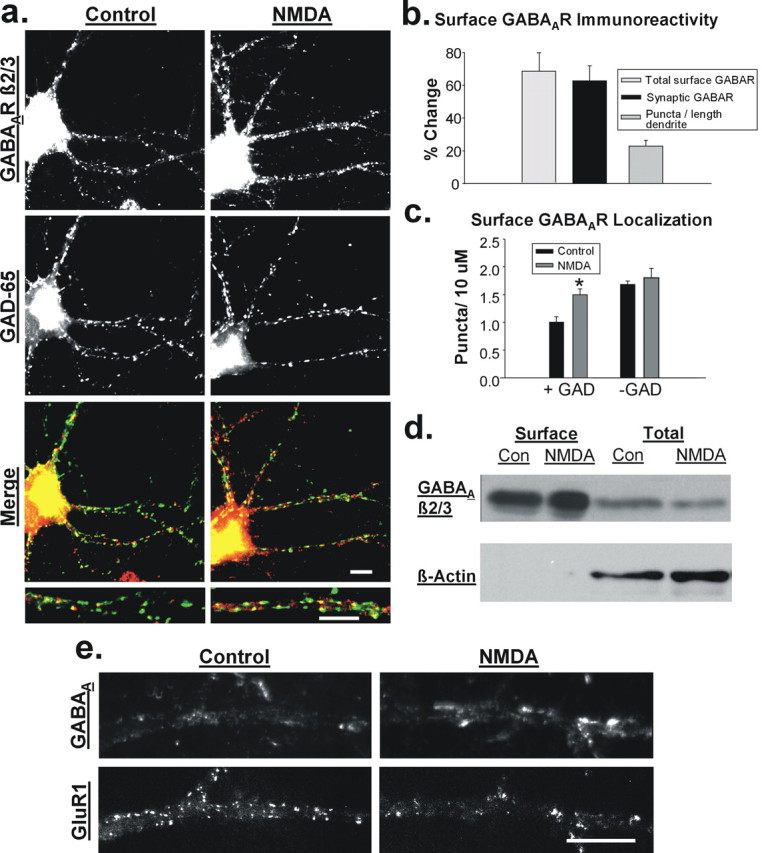
NMDAR activation increases surface expression of GABAA receptors. a, Immunolabeling for β2/3-containing GABAARs at the surface membrane (top) and the inhibitory presynaptic marker GAD-65 (middle) in untreated control (left) and NMDA-treated (20 μm, 2 min; right) hippocampal neurons. Merged images (bottom) show increased labeling of GABAARs (red) and enhanced colocalization with GAD-65 (green) after NMDA treatment. Scale bars, 10 μm. b, Quantitation of immunocytochemical data showing that NMDA increases the intensity of surface GABAAR labeling (light gray), intensity of synaptic, GAD-65-overlapping GABAAR puncta (black bar), as well as the overall number of GABAAR puncta per 10 μm length of dendrite (dark gray bar) (n = 5). c, NMDA increases the number of GAD-65-positive GABAAR puncta (n = 5; *p < 0.001) but not GAD-65-negative GABAAR puncta per length of dendrite (_n_ = 5; _p_ > 0.1). d, Representative blot of biotinylated proteins from control (Con) and NMDA-treated (20 μm, 3 min) hippocampal slices shows an increase in the surface-to-total GABAAR ratio using a β2/3-subunit antibody. Reprobe of blot for β-actin verifies the purity of the surface protein fraction. e, Representative images of control and NMDA-treated cells labeled for both surface β2/3-containing GABAARs and surface GluR1-containing AMPARs. Scale bar, 10 μm.
Figure 2.

NMDA-induced increase in surface GABAA receptors requires Ca2+ and CaMKII. a, Surface expression of β2/3-containing GABAARs in representative cultured hippocampal neurons after control, NMDA, and NMDA treatment in the presence of inhibitors of GABAARs (picrotoxin), CaMKII (KN-93), and calcineurin (cypermethrin). Scale bar, 10 μm. b, Blocking GABAARs with picrotoxin during the NMDA stimulus did not alter the increase in GABAAR expression at the dendritic surface (n = 4), but chelating Ca2+ with BAPTA-AM (n = 4) or inhibiting CaMKII (n = 6) prevents the NMDA-induced elevation in GABAAR surface expression. c, Summary of data showing that calcineurin and PP1 (blocked with okadaic acid) activity are not required for NMDA to increase GABAAR surface levels (n = 5).
Figure 4.

NMDAR activation does not reduce basal GABAAR endocytosis. The rate of GABAAR endocytosis was determined by live-labeling control and NMDA-treated cells with GABAAR antibody, incubating for various time points to allow for endocytosis, and analyzing the reduction in surface receptor labeling over time. a, Representative images of surface GABAAR expression at 0, 15, and 30 min in control (left) and NMDA-treated (right) cells. Scale bar, 10 μm. b, Graph of immunocytochemical data in a demonstrating that the rate of GABAAR endocytosis is not altered by NMDAR activation.
Figure 5.
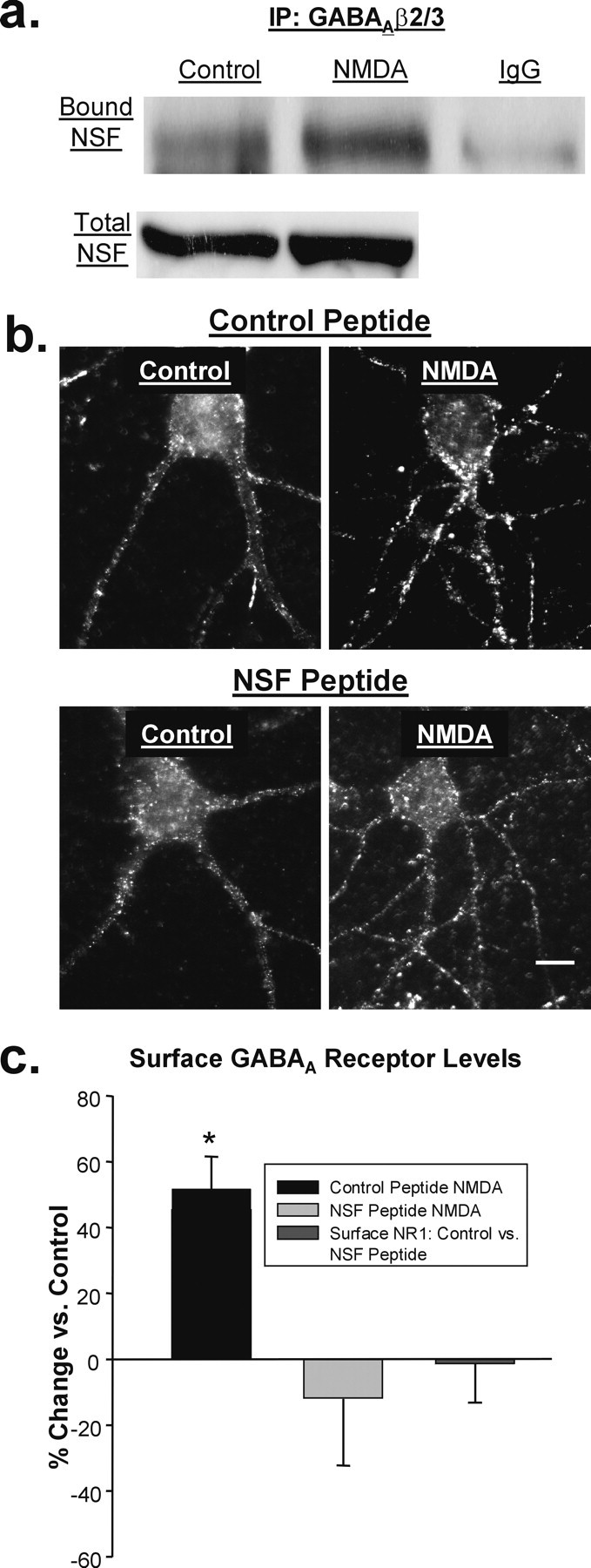
Delivery of GABAA receptors after NMDA is NSF dependent. a, Representative blot of β2/3-GABAAR-immunoprecipitated (top, left two lanes) or IgG-immunoprecipitated (top, right lane) and total (bottom) NSF from control and NMDA-treated hippocampal slices. b, Images of GABAAR surface expression in hippocampal neurons treated with a cell-permeable, TAT-conjugated peptide designed to block the interaction of NSF with α- and β-SNAP (bottom) or a scrambled control version of the peptide (top). Scale bar, 10 μm. c, The NSF-inhibitory peptide, but not the control peptide, prevents the increase in GABAAR surface expression in NMDA-treated cells (n = 8; *p < 0.01). Surface NMDA receptor labeling with an NR1 antibody was unaffected by the NSF peptide (n = 3).
Figure 6.

GABARAP is central to NMDAR-mediated GABAA receptor insertion. a, GABARAP-immunoprecipitated (left panel, left two lanes) or IgG-immunoprecipitated (left panel, right lane) and total (right panel) β2/3-GABAARs from NMDA-treated hippocampal slices display increased binding of GABARAP to the GABAA receptor compared with control (Con) as shown in a representative blot. b, Immunolabeling of surface GABAARs in representative hippocampal neurons 72 h after transfection with control or GABARAP siRNA. Surface GABAAR expression was increased after NMDA application in control siRNA cells but not in GABARAP siRNA cells. Scale bar, 10 μm. c, Quantification of immunocytochemical data in b (n = 5; *p < 0.05). d, Images of surface GABAAR expression in cells treated with a TAT-linked peptide that blocks the interaction of GABARAP and GABAARs (bottom) or a scrambled control peptide (top). Scale bar, 10 μm. e, Graph of data in d illustrating that the interaction between GABARAP and GABAAR is not required to maintain basal surface GABAARs but is necessary for the NMDA-induced increase in surface GABAAR expression (n = 5; *p < 0.05).
Figure 7.

GABARAP may act through GRIP to increase surface GABAAR expression. a, Coimmunoprecipitation of GABARAP and GRIP is elevated in NMDA-treated hippocampal slices. A representative blot shows GABARAP-immunoprecipitated (top, left two lanes) or IgG-immunoprecipitated (top, right lane) and total (bottom) GRIP. Con, Control. b, Staining of cultured hippocampal neurons for surface GABAARs (top) and GRIP (middle) in control (left) and NMDA-treated (right) cells. Merged images (bottom) display increased colocalization of surface GABAARs (red) and GRIP (green) after NMDA. c, Immunolabeling of surface GABAARs in representative hippocampal neurons 72 h after transfection with control or GRIP1 and GRIP2 siRNA. Surface GABAAR expression was increased after NMDA application in control siRNA cells (top) but not in GRIP1 and GRIP2 siRNA cells (bottom). Scale bar, 10 μm. d, Quantification of immunocytochemical data in c (n = 4; *p < 0.05).
Drug treatments.
Inhibitors of CaMKII (KN-93), protein phosphatase 2B (PP2B) (cypermethrin), protein phosphatase 1 (PP1) (okadaic acid) were applied at 1 μm for 30 min before treatment with NMDA and were also present during the recovery period. BAPTA-AM (100 μm) was applied for 20 min before treatment. In experiments using picrotoxin (100 μm), the GABAAR antagonist was applied during the NMDA treatment only. We also used a cell-permeable, TAT-conjugated peptide designed to inhibit NSF-mediated exocytosis by interfering with NSF-SNAP association (Lledo et al., 1998). The NSF peptide mimics the SNAP binding site for NSF, with the sequence QSFFSGLFGGSSKIEEACE–GGG–YGRKKRRQRRR. This peptide or a scrambled control peptide (GFAESLFQSIEKESGFSCG–GGG–YGRKKRRQRRR) was applied to cells at a concentration of 10 μm for 30 min before NMDA. Cells were fixed 15 min after NMDA treatment and processed for immunocytochemical analysis as described previously. Similarly, a TAT-conjugated peptide designed to disrupt GABARAP-GABAAR binding was used, with the sequence RTGAWRHGRIHIRIAKMD–GGG–YGRKKRRQRRR. The scrambled control peptide had the sequence HARHRGWHRIKIDIGRAT–GGG–YGRKKRRQRRR. These peptides were applied for 30 min before NMDA treatment at 10 μm.
Small interfering RNA experiments.
Hippocampal cultures, 11–13 DIV, were transfected with negative control small interfering RNA (siRNA) or siRNAs against GABARAP or GRIP1/2 using lipofectamine 2000 (Invitrogen) according to a procedure specified by the manufacturer. Two sets of predesigned siRNA sequences targeted to each GRIP protein family coding sequence (GRIP1 exons 5 and 21; and GRIP2 exons 7 and 25) were purchased from Ambion (Austin, TX). GABARAP siRNA (targeting exons 1 and 2) and control siRNA also were purchased from Ambion. After incubation for 2 h, the cells were washed in conditioned media and kept at 37°C. Neurons were treated with drugs as specified and immunocytochemically processed for labeling of surface GABAA β2/3 receptors after 3 d. Determination of protein knock-down was done immunocytochemically by labeling for GABARAP (rabbit polyclonal; Abcam) or GRIP after permeablization with TBS containing 4% BSA and 0.1% Triton X-100 for 30 min.
Biochemical methods
Hippocampal slices and drug treatments.
Hippocampal slices (400 μm) were prepared from 14- to 21-d-old Sprague Dawley rats (Taconic, Germantown, NY). Slices were kept in artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 1 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, saturated with 95% O2/5% CO2. After equilibration to 37°C, slices were incubated in ACSF alone (control), NMDA (20 μm with 10 μm CNQX in ACSF) for 3 min, followed by a 12 min recovery in ACSF.
Surface protein biotinylation.
After drug treatments, hippocampal slices were washed twice with 4°C ACSF and incubated in HEPES-buffered ACSF containing 1 mg/ml EZ-link Sulfo-NHS-LC Biotin (Pierce Biotechnology, Rockford, IL) for 1 h at 4°C. After two washes with cold ACSF, slices were homogenized in lysis buffer (Tris HCl, pH 7.6, 0.1% Triton X-100 with protease inhibitor cocktail (Complete Mini, EDTA-free; Roche Diagnostics, Mannheim, Germany). Protein extracts were centrifuged for 5 min at 1000 rpm, subjected to a second round of homogenization, and spun for an additional 5 min at 5000 rpm. Protein concentration of the supernatants was measured using a BCA protein assay (Pierce). Biotinylated protein (100–200 μg) was incubated with UltraLink Immobilized Streptavidin (Pierce Biotechnology) for 2 h at 4°C. Streptavidin-protein complexes were washed four times with PBS and spun for 1 min at 4000 rpm. Bound proteins were separated from beads, denatured by boiling in SDS sample buffer, and separated on 10% SDS-PAGE gels together with the total protein lysates (20 μg). After transferring proteins to ECL nitrocellulose membranes (GE Healthcare, Piscataway, NJ), blots were probed with GABAA β2/3 receptor antibody. Blots were then reprobed for the intracellular protein β-actin to ensure the purity of the cell surface fraction.
Coimmunoprecipitation.
After drug treatments, hippocampal slices were chilled on ice and briefly washed with ice cold ACSF. Protein extracts were prepared by homogenization in HEPES-OH, pH 7.7, EDTA, neocuproine (HEN) buffer with ATPγS. Protein extract (500–1000 μg) was precleared on Protein G agarose (Sigma) for 2 h at 4°C. Supernatants were then incubated with 4 μg of GABAA β2/3 receptor antibody or 2 μg of GABARAP antibody overnight at 4°C with constant shaking. The antibody-bound complexes were incubated with Protein G agarose for 2 h at 4°C. The protein-bound beads were washed in HEN buffer and pelleted by centrifugation for 2 min at 2000 rpm. The beads were resuspended in SDS sample buffer, and the immune complexes were eluted by boiling. Total and immunoprecipitated proteins were separated by electrophoresis on 10% SDS-PAGE gels and transferred onto nitrocellulose membranes. The blots were probed with antibodies to NSF (1:2500), GABAA β2/3 (1:500), or GRIP (1:1000).
Western blot analysis.
In membranes probed with the appropriate HRP-conjugated secondary antibodies, the ECL Western Blotting Detection System (GE Healthcare) was used to visualize the bound antigens. The chemiluminescent signal was captured using Kodak BioMax light film (Fisher Scientific, Houston, TX). The films were imaged using Epson Perfection 1240U scanner, and the intensity of the bands was quantified with MetaMorph software. Bound/total ratios were normalized to the mean of the controls.
Electrophysiology
Whole-cell patch recordings were made from 2- to 3-week-old cultured hippocampal neurons or acute hippocampal slices prepared as described above. Miniature IPSCs (mIPSCs) were recorded at room temperature with a Multiclamp Amplifier (Molecular Devices, Sunnyvale, CA) using low-resistance electrodes (3–5 MΩ). The internal solution contained the following (in mm): 10 K gluconate, 1 EGTA, 10 HEPES, 125 KCl, 5 sucrose, 4 MgATP, pH 7.2. The extracellular solution contained the following (in mm): 119 NaCl, 26 NaHCO3, 10 Glucose, 2.5 KCl, 1 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, and adjusted to pH 7.4. The Cl− concentrations of these solutions are such that the Cl− reversal potential should equal 0 mV. Complete blockade of mIPSCs after application of 100 μm picrotoxin confirmed that currents were GABAA receptor mediated. Extracellular solution was infused with 95% O2/5% CO2 and contained 100 μm lidocaine and 25 μm DNQX. The stability of series and input resistances were confirmed throughout the experiment using Igor Pro software (Wavemetrics, Lake Oswego, OR). After a brief baseline recording period, during which cells were voltage clamped at −70 mV, NMDA (20 μm) was bath applied for 2 min (3 min for slice recordings) with cells maintained in current clamp. The agonist was then washed out, and after complete repolarization, cells were again voltage clamped at −70 mV, and mIPSC collection resumed. Miniature IPSCs were collected continuously for 7 min before and 20 min after NMDA application. Experiments with BoNT/B (5 nm), BAPTA (10 mm), or AIP (5 μm) in the recording pipette were performed in precisely the same manner. mIPSCs were analyzed using the Mini Analysis Program (Synaptosoft, Decatur, GA). Data points were obtained by binning mIPSC data in 2 min intervals and normalizing to the mean of the baseline amplitude/frequency time points.
Statistical analysis
Data were analyzed using two-tailed Student's t tests. All error bars represent the SEM.
Results
Chem-LTD NMDA receptor activation increases surface GABAA receptors
A chemical form of long-term depression (chem-LTD) of excitatory transmission in both cultured hippocampal neurons and CA1 pyramidal neurons in hippocampal slices can be induced by brief application of the specific agonist NMDA (2–3 min, 10–50 μm, with 10 μm CNQX) (Lee et al., 1998; Kamal et al., 1999; Snyder et al., 2005). The mechanism involves the internalization of postsynaptic AMPA receptors, similar to low-frequency-induced LTD (Carroll et al., 1999; Beattie et al., 2000). To examine what effect this stimulus may have on GABAAR expression, we monitored the surface levels of these receptors using an antibody directed against the extracellular region of the β2/3 subunits. The vast majority of GABAARs contain β-subunits (Sieghart, 1995), and β1 has negligible expression in the hippocampus (Sperk et al., 1997), indicating β2/3 subunits are expressed in the majority of synapses. We measured changes in surface dendritic GABAAR expression by imaging receptors immunolabeled in cultured hippocampal neurons under nonpermeablizing conditions. We also specifically determined changes in synaptic GABAAR levels by colabeling with an antibody to the inhibitory presynaptic terminal marker GAD-65 and then measuring the intensity of only those GABAAR puncta that colocalized with GAD-65. Our results showed that 15 min after NMDA treatment, the overall intensity of surface receptors was elevated (67.2 ± 12.1% increase with NMDA treatment; n = 6) (Fig. 1a,b, light gray bar) as was the intensity of synaptic, GAD-colocalized GABAAR puncta (62.7 ± 9.1% increase over control; n = 6; p < 0.05) (Fig. 1_a_,_b_, black bar). We also observed a more modest elevation in the number of surface GABAAR puncta per 10 μm of dendrite after NMDAR activation (control, 2.71 ± 0.04 puncta/10 μm; NMDA, 3.36 ± 0.13 puncta/10 μm; 22.9 ± 3.7% increase over control; _n_ = 10; _p_ < 0.001) (Fig. 1_a_,_b_, dark gray bar). By further analyzing the colocalization of GABAAR and GAD puncta, we found that the number of GABAAR puncta localized at GAD-positive synapses were selectively elevated after NMDA application compared with those nonsynaptic sites (Fig. 1_a_,_c_) (GAD+: control, 1.00 ± 0.1; NMDA, 1.50 ± 0.1 puncta/10 μm; _n_ = 5; _p_ < 0.001; GAD-: control, 1.68 ± 0.065; NMDA, 1.80 ± 0.17 puncta/10 μm; _n_ = 5; _p_ > 0.1). To establish whether NMDAR activation also induces the increased surface expression of GABAARs in a more intact preparation, we treated hippocampal slices with NMDA (3 min, 20 μm). Fifteen minutes after treatment, slices were incubated at 4°C with biotin to label surface proteins. After isolating these surface proteins on streptavidin beads and subjecting them to SDS-PAGE, we probed by Western blot for the GABAAR β2/3 subunits. After NMDA stimulation, the ratio of surface-to-total GABAAR was significantly increased compared with untreated control slices (Fig. 1d) [control, 1.000 ± 0.156; NMDA, 1.448 ± 0.244 (ratio normalized to control); n = 7; p < 0.05] providing further evidence for an increase in the surface levels of receptors.
NMDA simultaneously decreases AMPA receptors and increases GABAA receptors at the surface of hippocampal neurons
Because NMDA treatment is known to trigger AMPAR endocytosis in hippocampal neurons, we sought to test whether the trafficking of both AMPARs and GABAARs is simultaneously and oppositely regulated by the same signal within individual neurons. We performed double-labeling experiments in cultured neurons in which we analyzed the surface levels of GluR1-containing AMPA receptors and β2/3-containing GABAARs in the same cells after NMDA application. We found that the surface expression of GABAARs was increased in neurons in which surface GluR1 receptors were reduced (GABAA, 50.23 ± 13.89% change from control; p < 0.05; GluR1, −35.15 ± 4.72%; n = 7; p < 0.0005) (Fig. 1e). These experiments demonstrate that glutamatergic signaling can mediate both the loss of AMPARs and the addition of GABAARs at the surface of a single hippocampal neuron.
Increases in surface GABAA receptors require Ca2+ and CaMKII
GABAergic agonists have been shown to cause a redistribution of GABAARs (Barnes, 1996). Therefore, we initially tested whether possible activation of GABAARs resulting from NMDA treatment is critical to the mechanism underlying the trafficking of GABAARs to the surface. Incubation of cultured hippocampal neurons with the GABAAR antagonist picrotoxin (100 μm) during the NMDA application had no effect on the increase in surface GABAARs (NMDA, 79.90 ± 22.78% change from control; NMDA plus PTx, 64.20 ± 19.10%; n = 4) (Fig. 2a,b), indicating that GABAAR activation is not necessary for enhanced GABAAR surface expression after NMDAR stimulation.
An NMDAR-dependent rise in intracellular Ca2+ is likely to trigger the increased surface GABAAR levels, particularly because the same NMDA stimulation that initiates changes in GABAAR expression drives synaptic depression and AMPAR endocytosis through a Ca2+-dependent pathway (Beattie et al., 2000). Pretreatment of hippocampal neurons with BAPTA-AM (100 μm), a cell-permeable Ca2+ chelator, prevented the NMDA-induced increase in surface GABAARs (NMDA plus BAPTA-AM, −0.22 ± 8.58%; n = 4) (Fig. 2b), demonstrating the necessity of Ca2+ for the trafficking of GABAARs.
We next investigated the involvement of CaMKII as follows: (1) it is activated by Ca2+, (2) it is known to phosphorylate the GABAAR (Churn et al., 2002), (3) infusion of Ca2+-CaM has been shown to increase GABAAR currents in CA1 pyramidal neurons (Wei et al., 2004), and (4) Ca2+-CaM-dependent enhancement is blocked by addition of an autoinhibitory form of CaMKII (Wei et al., 2004). When we inhibited CaMKII in cultured neurons with KN-93 (1 μm), the NMDA-induced increase in GABAA receptor surface expression was blocked (NMDA plus KN-93, −31.01 ± 10.42%; n = 6) (Fig. 2a,b). We further investigated the possible roles of two phosphatases known to participate in AMPAR endocytosis during NMDA-induced LTD, calcineurin (PP2B) and PP1 (Mulkey and Malenka, 1992), using the inhibitors cypermethrin (1 μm) and okadaic acid (1 μm), respectively. Neither inhibitor significantly altered NMDA-mediated increases in surface GABAARs (NMDA, 89.98 ± 14.65% change from control; NMDA plus cypermethrin, 72.97 ± 26.41%; NMDA plus okadaic acid, 109.53 ± 33.16%; n = 5) (Fig. 2a,c). It therefore appears that NMDA stimulation activates two sets of signaling pathways, one involving phosphatases and leading to AMPAR endocytosis, and the other involving CaMKII and leading to the delivery of GABAARs to the membrane.
NMDA receptor activation potentiates inhibitory synaptic transmission
Our immunocytochemical data suggest that NMDA increases synaptic GABAAR expression in a Ca2+- and CaMKII-dependent manner. To establish whether this increase in surface receptors results in altered inhibitory synaptic transmission, we recorded picrotoxin-sensitive GABAAR-mediated mIPSCs in cultured hippocampal neurons before and after NMDA treatment (Fig. 3a,b, closed circles) (n = 11, *p < 0.05). Fifteen minutes after bath application of NMDA (20 μm), we observed a significant increase in the average mIPSC amplitude (baseline, 14.58 ± 0.66 pA; 15 min after NMDA, 18.63 ± 0.68 pA; p < 0.005) (Fig. 3a,b) as well as in the mIPSC frequency (baseline, 11.33 ± 1.34 Hz; 15 min after NMDA, 15.56 ± 1.03 Hz; p < 0.05) (Fig. 3a,b). NMDA did not significantly alter mIPSC decay kinetics, because decay half-times were similar before and after NMDA application (before NMDA, 8.40 ± 0.96 ms; after NMDA, 8.19 ± 0.88 ms). A role for postsynaptic exocytosis in mediating the potentiation was supported by the observation that the increase in mIPSC amplitude was blocked when the light chain of BoNT/B (5 nm), a potent inhibitor of SNARE (SNAP receptor)-dependent exocytosis, was present in the recording pipette (Fig. 3b, open circles) (n = 7).
Figure 3.

NMDA receptor activation potentiates inhibitory synaptic transmission. GABAAR-mediated miniature IPSCs were analyzed using whole-cell patch-clamp recording techniques in cultured hippocampal neurons (a, b) and CA1 pyramidal neurons in acute hippocampal slices (c, d). a, Averaged mIPSC traces (left) recorded from cultured hippocampal neurons before and after NMDA treatment; representative traces (right) demonstrate increased amplitude and frequency of mIPSCs after NMDA. b, Time course of averaged, normalized mIPSC amplitude (left) shows an increase above the baseline after NMDA treatment (closed circles; n = 11; *p < 0.05) but no increase when BnTX, BAPTA, or CaMKII AIP were included in the recording pipette. Time course of averaged, normalized mIPSC frequency (right) shows an increase after NMDA application both in the presence and absence of BnTX, BAPTA, or AIP. c, Averaged mIPSC traces (left) from CA1 pyramidal neurons before and after NMDA; representative traces (right) display increased mIPSC amplitude but not frequency after NMDA treatment. d, Time course of averaged, normalized mIPSC amplitude (left) and frequency (right) (n = 5; *p < 0.05). BnTX (open circles) prevented the NMDA-induced mIPSC amplitude potentiation.
To further establish that the inhibitory potentiation involves the same mechanism as the immunocytochemically detected increase in surface GABAARs, we monitored the effects of blocking elevations in calcium and CaMKII activity with BAPTA and CaMKII AIP. BAPTA and AIP applied through the recording pipette had no effect on mIPSC amplitude (BAPTA, 91.0 ± 19.4% of baseline at 20 min, n = 4, p > 0.1; AIP, 87.4 ± 15.4% of baseline at 20 min, n = 4, p > 0.1) or frequency (BAPTA, 89.2 ± 14.6% of baseline at 20 min, n = 4, p > 0.1; AIP, 78.6 ± 38.7% of baseline at 20 min, n = 4, p > 0.1) on their own. However, consistent with our immunocytochemical data, both BAPTA (10 mm) (Fig. 3b, closed triangles) (n = 4) and AIP (5 μm) (Fig. 3b, open triangles) (n = 5) prevented the mIPSC amplitude potentiation when applied through the recording pipette. The inhibition by BoNT/B, BAPTA, and AIP does not appear to result from effects on NMDAR function, because there was no significant difference in the extent of membrane depolarization resulting from NMDAR activation (control, 19.64 ± 2.92 mV; BoNT/B, 15.3 ± 1.62 mV, p > 0.1; BAPTA, 23.50 ± 2.60 mV, p > 0.1; AIP, 15.63 ± 2.25 mV, p > 0.1). Interestingly, the increase in mIPSC frequency was not blocked with postsynaptic BoNT/B, BAPTA, or AIP, suggesting that there is likely a change in presynaptic properties of inhibitory synapses after NMDA treatment as well (Fig. 3a,b).
To further establish whether the NMDA-mediated increase in GABAAR surface expression in hippocampal slices (Fig. 1d,e) also represents a change in synaptic receptor levels, we tested whether mIPSCs in slices were likewise enhanced. Miniature IPSCs were recorded in CA1 pyramidal neurons of acute hippocampal slices and, as in the cultured neurons, mIPSC amplitudes were potentiated by ∼25% after NMDA treatment (baseline, 22.22 ± 1.46 pA; 15 min after NMDA, 27.62 ± 1.75 pA) (Fig. 3c,d, left panels) (n = 5; p < 0.05). When BoNT/B was present in the recording pipette, however, there was no NMDA-dependent enhancement (Fig. 3d, open circles). In fact, there was a significant decrease in mIPSC amplitude in the presence of BoNT/B, suggesting that blockade of receptor insertion may reveal a depression of mIPSCs, perhaps consistent with a previous report of NMDA receptor-dependent inhibitory plasticity in the hippocampus after LTP-inducing stimuli (Wang et al., 2003). In contrast to the cultures, mIPSC frequency was not altered after NMDA treatment (baseline, 0.98 ± 0.17 Hz; 15 min after NMDA, 1.13 ± 0.32 Hz) (Fig. 3c,d, right panels). This suggests that this apparently presynaptic change in inhibition is either sensitive to the pattern of activity elicited by NMDA application in these two preparations or is absent in presynaptic terminals of CA3 pyramidal neurons of slices.
NMDAR activation does not affect the basal rate of GABAAR endocytosis
Our immunocytochemical, biochemical, and electrophysiological results are all consistent with the possibility that increases in surface GABAARs after NMDAR activation occur through the exocytosis of GABAARs. However, increased surface expression of receptors could also result from a decrease in the rate of removal of GABAARs, which are known to constitutively cycle into and out of the surface membrane (Kittler et al., 2000). We tested this possibility by determining whether NMDA treatment impacts the rate or extent of GABAAR endocytosis. Hippocampal neurons were treated with NMDA and, after 5 min, were placed at 4°C to stop receptor trafficking. An antibody to the extracellular N terminus of the GABAAR was applied to the live neurons to label surface receptors. The neurons were washed and returned to 37°C for varying lengths of time before fixation. Remaining surface GABAARs were immunocytochemically labeled, imaged, and analyzed. In untreated neurons, there was a significant decrease in surface labeling over time (15 min, 47.3 ± 15.0% of baseline; n = 4) indicative of a basal rate of receptor endocytosis (Fig. 4). NMDA-treated neurons showed a similar pattern of endocytosis (15 min, 49.1 ± 7.8% of baseline; n = 4) suggesting that NMDA treatment does not greatly affect the basal rate of GABAAR endocytosis. Thus, the increased surface expression of GABAARs after NMDA appears to be primarily attributable to receptor insertion into the dendritic membrane.
Delivery of GABAA receptors after NMDA exposure is NSF dependent
Ca2+ and CaMKII have many targets that could be important for the insertion of GABAARs. One of them, NSF (Hirling and Scheller, 1996), is activated by stimulation of NMDARs (Huang et al., 2005) and has been implicated in regulating GABAAR insertion (Goto et al., 2005) through its interaction with GABAAR β-subunits. If NSF were to mediate the insertion of GABAARs after NMDAR activation, we predicted that its binding to the GABAAR would at least transiently increase. To test this prediction, we performed coimmunoprecipitation experiments using protein derived from control and NMDA-treated hippocampal slices. We found that in slices treated with NMDA, the ratio of GABAAR-bound/total NSF was elevated almost threefold over controls (control, 1.02 ± 0.01; NMDA, 2.81 ± 0.59 ratio normalized to control; n = 3, p < 0.05) (Fig. 5a).
We tested the requirement for NSF activity in the delivery of GABAARs by using an NSF-inhibitory peptide that mimics the NSF binding site of α- and β-SNAP (Lledo et al., 1998). The peptide was made cell-permeable for use in our cultures by the addition of the 11 amino acid TAT sequence. When hippocampal neurons were preincubated for 30 min with the NSF peptide (10 μm), surface GABAAR staining was no longer elevated after NMDA treatment (NSF peptide plus NMDA, −10.69 ± 20.55% compared with peptide alone; n = 8) (Fig. 5b,c). However, neurons incubated with a scrambled version of the peptide (also linked to the TAT sequence) still displayed an increase (control peptide plus NMDA, 53.41 ± 10.34% increase compared with peptide alone; n = 7; p < 0.01) (Fig. 5b,c). To rule out a nonspecific effect of the NSF peptide on NMDAR expression, which could result in reduced responsiveness to NMDA and thereby prevent increases in GABAAR expression, we labeled surface NMDARs with an antibody to the NR1 subunit. Our results showed no difference in NR1 expression between cells treated with the control peptide and cells treated with the NSF-inhibitory peptide (−1.43 ± 11.77% change from control; n = 3) (Fig. 5c), confirming that exocytosis of GABAARs after NMDA exposure requires NSF.
GABARAP is central to GABAA receptor insertion after chem-LTD
The involvement of NSF in GABAAR exocytosis prompted us to look at one of its binding partners, GABARAP (Kittler et al., 2001), which for several years has been a primary focus of research on GABAAR trafficking. Although its overexpression increases surface GABAAR levels (Leil et al., 2004), understanding of the role of GABARAP is complicated by the fact that it is not enriched at synapses under basal conditions (Kittler et al., 2001) and that it is not necessary for maintaining basal surface levels of γ-subunit-containing GABAARs, as shown in a GABARAP knock-out mouse (O'Sullivan et al., 2005). These data do not exclude the possibility, however, that GABARAP is involved in the regulated delivery of GABAARs to the surface. We hypothesized that, like NSF, if GABARAP were involved in delivering GABAARs to the surface, more GABAARs should be bound to GABARAP after NMDAR stimulation. Using protein isolated from hippocampal slices, we found that more GABAARs coimmunoprecipitated with GABARAP in NMDA-treated slices than in controls (GABARAP-bound GABAAR/total GABAAR: control, 1.00 ± 0.08; NMDA, 1.97 ± 0.33 ratio normalized to control; n = 5; p < 0.05) (Fig. 6a).
To establish whether GABARAP is necessary for NMDA-mediated GABAAR exocytosis, we used RNAi to knock down the amount of GABARAP in our cultured neurons. Cells were transfected with control siRNA or siRNA targeting GABARAP for 72 h, resulting in an average reduction of 48.46 ± 7.57% in dendritic GABARAP fluorescence intensity (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Labeling for surface GABAARs showed no difference between control siRNA and GABARAP siRNA-treated cells (2.09 ± 12.4%; n = 5) (Fig. 6b,c), indicating either that, like the GABARAP knock-out mouse (O'Sullivan et al., 2005), GABARAP plays no role in maintaining basal surface levels of GABAARs, or that a ∼50% reduction in GABARAP is not sufficient to see an effect. When we applied NMDA to these cells, however, we found that this reduction was sufficient to prevent the increase in surface GABAARs seen in the control siRNA-treated cells (control siRNA plus NMDA: 59.15 ± 30.62% increase from control; GABARAP siRNA plus NMDA: −17.50 ± 11.55%; n = 5; p < 0.05) (Fig. 6b,c). To test for possible off-target effects of the siRNA, we determined that GABARAP siRNA did not alter AMPAR internalization after NMDA treatment (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), indicating intact NMDAR signaling and verifying that normal levels of endogenous GABARAP are necessary specifically for the NMDA-driven increase in surface GABAARs.
To confirm the role of GABARAP in NMDA-dependent GABAAR insertion, and to further test whether this increase specifically requires the interaction between GABARAP and the GABAAR, we disrupted this interaction with a cell-permeable, TAT-conjugated peptide mimicking the GABARAP-binding domain of the γ2-subunit of the GABAAR (Fig. 6d,e). Although a scrambled control peptide had no effect on the NMDA-mediated increase in GABAAR surface expression (48.9 ± 9.18% increase from control; n = 5; p < 0.05), when cells were pretreated with the GABARAP inhibitory peptide for 30 min, NMDA induced a significant decrease in GABAAR surface levels (−38.17 ± 5.15%; _n_ = 5; _p_ < 0.05). As reported previously (Kawaguchi and Hirano, 2007), disruption of the GABARAP-GABAAR interaction with the GABARAP inhibitory peptide had no effect on basal surface GABAAR expression (1.42 ± 13.03% change from control; _n_ = 5; _p_ > 0.1).
The GABARAP binding partner GRIP is necessary for increased surface GABAAR expression
GRIP has been reported to interact with GABARAP (Kittler et al., 2004) and is found at a population of inhibitory postsynapses (Dong et al., 1999; Charych et al., 2004; Li et al., 2005). Therefore, although it is known primarily for its role in the trafficking and/or stabilization of AMPARs (Kim et al., 2001; Braithwaite et al., 2002), GRIP could also be involved in mediating the GABARAP-dependent delivery of GABAARs to the surface. To test this, we used protein from untreated and NMDA-treated hippocampal slices and found that the amount of GRIP pulled down with GABARAP was approximately doubled in NMDA-treated slices (GABARAP-bound/total GRIP: control, 1.00 ± 0.16; NMDA, 1.96 ± 0.22; n = 6; p < 0.005) (Fig. 7a).
The increased association of GABARAP and GRIP after NMDA application suggests that GABAARs designated for surface insertion may, through GABARAP, be targeted to GRIP-containing inhibitory synapses. To test this possibility, cultured hippocampal neurons were labeled for both surface GABAARs and for GRIP, and we found that, after NMDA application, there was an increase in the proportion of surface GABAARs colocalized with GRIP (control, 37.3 ± 4.2%; NMDA, 50.3 ± 3.4%; n = 4; p < 0.01) (Fig. 7b).
Finally, we used siRNA to reduce the expression of both GRIP1 and GRIP2 to establish whether GRIP is a necessary mediator of NMDA-induced GABAAR delivery. GRIP1/2 siRNA reduced the immunolabeling of GRIP in cultured hippocampal neurons by 66.73 ± 10.17% on average compared with control siRNA cells (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). As reported previously (Hoogenraad et al., 2005), this knockdown did not result in a significant change in the average basal levels of surface GABAARs (29.50 ± 12.78%; n = 4; p > 0.1) (Fig. 7c,d). NMDA triggered a 72.05 ± 18.04% increase in surface GABAARs in cells transfected with control siRNA (n = 4; p < 0.05) but a 35.55 ± 18.98% decrease when applied to GRIP siRNA transfected cells (Fig. 7c,d). Ruling out a nonspecific effect, the GRIP siRNA does not interfere with NMDA-induced AMPAR internalization (our unpublished observation), indicating that NMDARs and the downstream signaling pathways remain intact. These data provide the first evidence of a role for GRIP in the trafficking of GABAARs and thus in the regulation of inhibitory synaptic transmission.
Discussion
It has become clear that plasticity at excitatory synapses is not always independent of that at inhibitory synapses. Previous reports have identified that LTP-inducing stimuli at glutamatergic synapses in the hippocampus can simultaneously potentiate excitatory and depress inhibitory transmission (Chevaleyre and Castillo, 2003; Ivenshitz and Segal, 2006). To our knowledge, however, there is no evidence of glutamatergic stimuli that concurrently depress excitatory transmission and enhance inhibition. Here, we show that NMDA receptor activation removes AMPA receptors from the membrane while simultaneously increasing expression of surface GABAARs. These newly inserted receptors are functional and synaptic, because NMDAR stimulation leads to potentiation of mIPSC amplitudes in neurons of both hippocampal cultures and acute slices. Furthermore, we identified key components of the machinery underlying this GABAAR trafficking. Activation of CaMKII is necessary to drive an NSF-mediated delivery of receptors that also requires GABARAP and its interacting partner GRIP. These findings provide evidence for a novel mechanism by which glutamatergic stimuli can enhance inhibition through postsynaptic GABAAR trafficking.
Studies in the DCN have provided intriguing evidence that NMDARs can drive changes in postsynaptic GABAAR number and/or function. High-frequency stimulation in the DCN has been shown to produce an NMDAR-dependent enhancement of mIPSC frequency but not amplitude (Ouardouz and Sastry, 2000). This phenomenon was blocked with postsynaptic infusions of BAPTA and tetanus toxin, suggesting that trafficking of GABAARs and/or release of postsynaptic agents that regulate GABAAR function could be triggered by NMDARs (Ouardouz and Sastry, 2000). Because frequency changes often indicate presynaptic modifications, it is also possible that alterations in presynaptic transmitter release could happen through retrograde signaling, although it is more likely that the postsynaptic unsilencing of GABAergic synapses mediates potentiation in the DCN (Ouardouz and Sastry, 2000). Our study directly demonstrates that NMDARs in hippocampal neurons can couple to the exocytosis of GABAARs, as evidenced by both immunocytochemical and biochemical analysis.
The mean amplitude of mIPSCs increased during NMDAR-dependent GABAergic potentiation, suggesting that GABAARs are inserted into previously functional synapses. Both our immunocytochemical and electrophysiological data argue against postsynaptic unsilencing as the primary mechanism of potentiation. We observed a large increase (∼45%) in the intensity of synaptic GABAAR puncta but a more modest increase (∼22%) in the number of such puncta (Fig. 1b). Although an increase in puncta number could indicate new, unsilenced synapses, this may also result from receptor levels increasing to above the immunocytochemical detection threshold at previously existing synapses. We did find increased mIPSC frequency after NMDA treatment in cultured neurons, but the fact that blocking exocytosis, Ca2+, or CaMKII with postsynaptic BoNT/B, BAPTA, or AIP, respectively, did not prevent the increase in mIPSC frequency suggests that NMDA also alters presynaptic properties of hippocampal inhibitory synapses. Furthermore, because we did not see changes in mIPSC frequency in CA1 neurons in hippocampal slices after NMDA application but did observe increased surface GABAAR levels (Fig. 1d), it appears that the increase in surface receptors happens at pre-existing synapses.
NMDAR-dependent LTD is known to be Ca2+ dependent (Mulkey and Malenka, 1992); therefore, it may not be surprising that NMDA-induced changes in GABAAR expression are similarly Ca2+ dependent. Our data further suggest that CaMKII plays an essential role in NMDAR-mediated insertion of GABAARs. Although our studies used KN-93 (Fig. 2a,b), which has been found to have non-CaMKII targets, the role of CaMKII was supported by prevention of mIPSC amplitude potentiation with the CaMKII-selective autoinhibitory peptide (Fig. 3b). It is possible that inhibition of CaMKII could alter NMDAR function and/or trafficking (Sessoms-Sikes et al., 2005; Mauceri et al., 2007) and in this way prevent NMDA-mediated mIPSC potentiation. However, because the extent of membrane depolarization recorded during NMDA treatment was unaffected by AIP, we find this explanation unlikely.
It is perhaps surprising that the same NMDA stimulus that results in internalization of AMPARs through activation of PP1 and PP2B (Kamal et al., 1999) depends instead on CaMKII for the increase in surface GABAARs. NMDAR activation of CaMKII is generally associated with high levels of Ca2+ influx through the NMDAR and the enhancement of glutamatergic transmission during LTP (Silva et al., 1992). However, the NMDA treatment used here has been found previously to cause translocation of CaMKII (Shen and Meyer, 1999). It is likely that a combination of factors, including the subcellular localization and balance of kinase and phosphatase activation under different signaling conditions, determines the direction of receptor trafficking at excitatory and inhibitory synapses. For example, a range of NMDAR stimulation levels may activate CaMKII to various extents, whereas some will also preferentially activate phosphatases (Zhabotinsky et al., 2006). Because these phosphatases can be localized by scaffolding proteins to spines (Yan et al., 1999) and may inactivate CaMKII themselves (Bradshaw et al., 2003), the level of CaMKII activity at excitatory synapses may be very low compared with that at inhibitory synapses on dendritic shafts. Although we have not yet identified downstream targets, CaMKII is already known to phosphorylate GABAARs (Churn et al., 2002) and can influence GABAAR properties (Churn et al., 2002; Wei et al., 2004), possibly including effects on GABAAR number (Wei et al., 2004).
We established that the activity of another potential CaMKII target, NSF (Hirling and Scheller, 1996), is essential for the targeting of receptors to the surface after NMDAR activation. NSF has been implicated in the exocytosis of many receptor types in the CNS, including GABAARs, AMPARs, β2-adrenergic receptors, and dopaminergic receptors (for review, see Zhao et al., 2007), so perhaps the specificity of NSF-mediated receptor delivery is conferred by associated proteins such as GABARAP. Several studies have shown that exogenous GABARAP can lead to increased surface GABAAR expression (Leil et al., 2004; Chen et al., 2005), but our data provide the first indication of a similar role for the endogenous protein. These results at first seem at odds with data from a GABARAP knock-out mouse that showed no alteration in GABAAR expression (O'Sullivan et al., 2005). However, using an acute knockdown of GABARAP with siRNA, we similarly demonstrate that normal levels of GABARAP are not necessary for maintaining basal surface GABAAR expression (Fig. 6c, black bar). Normal levels of GABARAP are required, however, for the activity-dependent delivery of new receptors such as occurs after NMDAR activation (Fig. 6c, dark gray bar).
A recent report suggests a more direct link between CaMKII activation and the activity of GABARAP. Kawaguchi and Hirano (2007) showed that the interaction of GABARAP with the γ2 subunit of the GABAAR is necessary for rebound potentiation (RP), a form of inhibitory synaptic potentiation that occurs in cerebellar Purkinje neurons after postsynaptic depolarization (Kano et al., 1992). RP was found to require a structural change in GABARAP mediated by CaMKII activity (Kawaguchi and Hirano, 2007). Interestingly, it is not clear that expression of RP, although similar to the NMDAR-dependent potentiation described here, is caused by trafficking of GABAARs to synapses. In fact, Kawaguchi and Hirano (2007) reported no change in surface GABAAR expression after RP induction. It remains possible that GABARAP has multiple functions in regulating GABAergic signaling: through altered GABAAR function as seen in Purkinje neurons and through enhanced GABAAR numbers as we found in the hippocampus.
That GABARAP binds microtubules (Wang et al., 1999), gephyrin (Kneussel et al., 2000), NSF (Kittler et al., 2001), and GRIP (Kittler et al., 2004) makes it a tantalizing candidate for regulating GABAAR trafficking. However, the functional consequences of these interactions are not well understood. We show that the binding of GABARAP to GABAARs and to GRIP can be modified by NMDAR activation, and thus the interactions of GABARAP are subject to activity-dependent regulation. Furthermore, peptide disruption demonstrates that the interaction between GABARAP and the GABAAR is essential for GABAAR targeting to the surface after NMDA. Together with our immunocytochemical colocalization studies, these data suggest that NMDAR activation drives GABARAP-bound GABAARs to a subset of GRIP-containing inhibitory synapses where they are inserted into the membrane through the activity of NSF.
What would be the physiological impact of potentiating inhibition while at the same time depressing excitation? The heterosynaptic nature of this regulation suggests that potentiated inhibitory synapses are likely to be near depressed excitatory synapses. This would act to locally and potently reduce the contribution of those excitatory synapses. It has also been reported that LTD induction results in an NMDAR-dependent depression of EPSP–spike (E-S) coupling (Daoudal et al., 2002). It is possible that the potentiation of inhibition we see could be partly responsible for this decrease in neuronal excitability, particularly because changes in inhibition can account for ∼60% of E-S uncoupling (Daoudal et al., 2002). In addition to the potential implications for synaptic plasticity, alterations in GABAAR number and/or function also have been linked to many neurological disorders, including anxiety, epilepsy, schizophrenia, and insomnia (Mohler, 2006). Therefore, a clear understanding of how activity affects GABAAR trafficking is an important goal requiring additional study.
Footnotes
This work was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke Grant NS 049661. We thank Dr. Diana Pettit for helpful comments on this manuscript.
References
- Barnes EM., Jr Use dependent regulation of GABAA receptors. Int Rev Neurobiol. 1996;39:53–76. doi: 10.1016/s0074-7742(08)60663-7. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- Bradshaw J, Kubota Y, Meyer T, Schulman H. An ultrasensitive Ca2+/calmodulin-dependent protein kinase II-protein phosphatase 1 switch facilitates specificity in postsynaptic calcium signaling. Proc Natl Acad Sci USA. 2003;100:10512–10517. doi: 10.1073/pnas.1932759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braithwaite S, Xia H, Malenka RC. Differential roles for NSF and GRIP/ABP in AMPA receptor cycling. Proc Natl Acad Sci USA. 2002;99:7096–7101. doi: 10.1073/pnas.102156099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, von Zastrow M, Nicoll RA, Malenka RC. Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci. 1999;2:454–460. doi: 10.1038/8123. [DOI] [PubMed] [Google Scholar]
- Charych E, Yu W, Li R, Serwanski DR, Miralles CP, Li X, Yang B, Pinal N, Walikonis R, de Blas AL. A four PDZ domain-containing splice variant form of GRIP1 is localized in GABAergic and glutamatergic synapses in the brain. J Biol Chem. 2004;279:38978–38990. doi: 10.1074/jbc.M405786200. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang H, Vicini S, Olsen RW. The g-aminobutyric acid type A (GABAA) receptor-associated protein (GABARAP) promotes GABAA receptor clustering and modulates the channel kinetics. Proc Natl Acad Sci USA. 2000;97:11557–11562. doi: 10.1073/pnas.190133497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Tracy T, Nam CI. Dynamics of postsynaptic glutamate receptor targeting. Curr Opin Neurobiol. 2007;17:1–6. doi: 10.1016/j.conb.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Chen ZW, Olsen RW. GABAA receptor associated proteins: a key factor regulating GABAA receptor function. J Neurochem. 2007;100:279–294. doi: 10.1111/j.1471-4159.2006.04206.x. [DOI] [PubMed] [Google Scholar]
- Chen ZW, Chang CS, Leil TA, Olcese R, Olsen RW. GABAA receptor-associated protein regulates GABAA receptor cell-surface number in Xenopus laevis oocytes. Mol Pharmacol. 2005;68:152–159. doi: 10.1124/mol.104.009878. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chhatwal J, Myers K, Ressler K, Davis M. Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J Neurosci. 2005;25:502–506. doi: 10.1523/JNEUROSCI.3301-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churn S, Rana A, Lee K, Parsons J, de Blas AL, Delorenzo R. Calcium/calmodulin-dependent kinase II phosphorylation of the GABAA receptor alpha1 subunit modulates benzodiazepine binding. J Neurochem. 2002;82:1065–1076. doi: 10.1046/j.1471-4159.2002.01032.x. [DOI] [PubMed] [Google Scholar]
- Connolly CN, Kittler JT, Thomas P, Uren JM, Brandon NJ, Smart TG, Moss SJ. Cell-surface stability of GABAA receptors: dependence on PKC activity and subunit composition. J Biol Chem. 1999;274:36565–36572. doi: 10.1074/jbc.274.51.36565. [DOI] [PubMed] [Google Scholar]
- Daoudal G, Hanada Y, Debanne D. Bidirectional plasticity of excitatory postsynaptic potential (EPSP)-spike coupling in CA1 hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 2002;99:14512–14517. doi: 10.1073/pnas.222546399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Zhang P, Song I, Petralia R, Liao D, Huganir RL. Characterization of the glutamate receptor-interacting proteins GRIP1 and GRIP2. J Neurosci. 1999;19:6930–6941. doi: 10.1523/JNEUROSCI.19-16-06930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt A, Luu T, Cromer B, Tierney M, Birnir B, Olsen RW, Gage P. Conductance of recombinant GABA channels is increased in cells co-expressing GABAA A receptor-associated protein. J Biol Chem. 2004;279:21701–21706. doi: 10.1074/jbc.M312806200. [DOI] [PubMed] [Google Scholar]
- Gaiarsa J, Caillard O, Ben-Ari Y. Long-term plasticity at GABAergic and glycinergic synapses: mechanisms and functional significance. Trends Neurosci. 2002;25:564–570. doi: 10.1016/s0166-2236(02)02269-5. [DOI] [PubMed] [Google Scholar]
- Goto H, Terunuma M, Kanematsu T, Misumi Y, Moss SJ, Hirata M. Direct interaction of N-ethylmaleimide-sensitive factor with GABAA receptor beta subunits. Mol Cell Neurosci. 2005;30:197–206. doi: 10.1016/j.mcn.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Hirling H, Scheller R. Phosphorylation of synaptic vesicle proteins: modulation of the alpha SNAP interaction with the core complex. Proc Natl Acad Sci USA. 1996;93:11945–11949. doi: 10.1073/pnas.93.21.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogenraad CC, Milstein AD, Ethell IM, Henkemeyer M, Sheng M. GRIP1 controls dendrite morphogenesis by regulating EphB receptor trafficking. Nat Neurosci. 2005;8:906–915. doi: 10.1038/nn1487. [DOI] [PubMed] [Google Scholar]
- Huang Y, Man H, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein CJ, Huganir RL, Snyder S. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Ivenshitz M, Segal M. Simultaneous NMDA-dependent long-term potentiation of EPSCs and long-term depression of IPSCs in cultured rat hippocampal neurons. J Neurosci. 2006;26:1199–1210. doi: 10.1523/JNEUROSCI.2964-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A, Ramakers GM, Urban IJ, De Graan PN, Gipsen WH. Chemical LTD in the CA1 field of the hippocampus from young and mature rats. Eur J Neurosci. 1999;11:3512–3516. doi: 10.1046/j.1460-9568.1999.00769.x. [DOI] [PubMed] [Google Scholar]
- Kano M, Rexhausen U, Dreessen J, Konnerth A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature. 1992;356:601–604. doi: 10.1038/356601a0. [DOI] [PubMed] [Google Scholar]
- Kawaguchi S-Y, Hirano T. Sustained structural change of GABAA receptor-associated protein underlies long-term potentiation at inhibitory synapses on a cerebellar Purkinje neuron. J Neurosci. 2007;27:6788–6799. doi: 10.1523/JNEUROSCI.1981-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Chung HJ, Lee H-K, Huganir RL. Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci USA. 2001;98:11725–11730. doi: 10.1073/pnas.211132798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Delmas P, Jovanovich JN, Brown DA, Smart TG, Moss SJ. Constitutive endocytosis of GABAA receptors by an association with the adaptic AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. J Neurosci. 2000;20:7972–7977. doi: 10.1523/JNEUROSCI.20-21-07972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Rostaing P, Schiavo G, Fritschy JM, Olsen RW, Triller A, Moss SJ. The subcellular distribution of GABARAP and its ability to interact with NSF suggest a role for this protein in the intracellular transport of GABA(A) receptors. Mol Cell Neurosci. 2001;18:13–25. doi: 10.1006/mcne.2001.1005. [DOI] [PubMed] [Google Scholar]
- Kittler JT, Arancibia-Carcamo IL, Moss SJ. Association of GRIP1 with a GABA(A) receptor associated protein suggests a role for GRIP1 at inhibitory synapses. Biochem Pharmacol. 2004;68:1649–1654. doi: 10.1016/j.bcp.2004.07.028. [DOI] [PubMed] [Google Scholar]
- Kneussel M, Havercamp S, Fuhrmann JC, Wang H, Wassle H, Olsen RW, Betz H. The gamma-aminobutyric acid type-A receptor (GABAAR)-associated protein GABARAP interacts with gephyrin but is not involved in receptor anchoring at the synapse. Proc Natl Acad Sci USA. 2000;97:8594–8599. doi: 10.1073/pnas.97.15.8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-K, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- Leil TA, Chen ZW, Chang CS, Olsen RW. GABAA receptor-associated protein traffics GABAA receptors to the plasma membrane in neurons. J Neurosci. 2004;24:11429–11438. doi: 10.1523/JNEUROSCI.3355-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li RW, Serwanski DR, Miralles CP, Li X, Charych E, Riquelme R, Huganir RL, de Blas AL. GRIP1 in GABAergic synapses. J Comp Neurol. 2005;488:11–27. doi: 10.1002/cne.20566. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Zhang X, Sudhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- Luscher B, Keller C. Regulation of GABAA receptor trafficking, channel activity, and functional plasticity of inhibitory synapses. Pharmacol Ther. 2004;102:195–221. doi: 10.1016/j.pharmthera.2004.04.003. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RGM. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Mauceri D, Gardoni F, Marcello E, Di Luca M. Dual role of CaMKII-dependent SAP97 phosphorylation in mediating trafficking and insertion of NMDA receptor subunit NR2A. J Neurochem. 2007;100:1032–1046. doi: 10.1111/j.1471-4159.2006.04267.x. [DOI] [PubMed] [Google Scholar]
- Michels G, Moss SJ. GABAA receptors: properties and trafficking. Crit Rev Biochem Mol Biol. 2007;42:3–14. doi: 10.1080/10409230601146219. [DOI] [PubMed] [Google Scholar]
- Mohler H. GABAA receptors in central nervous system disease: anxiety, epilepsy, and insomnia. J Recept Signal Transduct Res. 2006;26:731–740. doi: 10.1080/10799890600920035. [DOI] [PubMed] [Google Scholar]
- Morishita W, Sastry BR. Postsynaptic mechanisms underlying long-term depression of GABAergic transmission in neurons of the deep cerebellar nuclei. J Neurophysiol. 1996;76:59–68. doi: 10.1152/jn.1996.76.1.59. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Naylor D, Liu H, Wasterlain C. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci. 2005;25:7724–7733. doi: 10.1523/JNEUROSCI.4944-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Hajos N, Somogyi P, Mody I. Increased number of synaptic GABAA receptors underlies potentiation at hippocampal inhibitory synapses. Nature. 1998;395:172–177. doi: 10.1038/25999. [DOI] [PubMed] [Google Scholar]
- O'Sullivan GA, Kneussel M, Elazar Z, Betz H. GABARAP is not essential for GABA receptor targeting to the synapse. Eur J Neurosci. 2005;22:2644–2648. doi: 10.1111/j.1460-9568.2005.04448.x. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Sastry BR. Mechanisms underlying LTP of inhibitory synaptic depression in the deep cerebellar nuclei. J Neurophysiol. 2000;84:1414–1421. doi: 10.1152/jn.2000.84.3.1414. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessoms-Sikes S, Honse Y, Lovinger D, Colbran R. CaMKIIalpha enhances the desensitization of NR2B-containing NMDA receptors by an autophosphorylation-dependent mechanism. Mol Cell Neurosci. 2005;29:139–147. doi: 10.1016/j.mcn.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–167. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- Sieghart W. Structure and pharmacology of gamma-aminobutyric acidA receptor subtypes. Pharmacol Rev. 1995;47:181–234. [PubMed] [Google Scholar]
- Silva A, Stevens C, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Colledge M, Crozier RA, Chen WS, Scott JD, Bear MF. Role for A kinase-anchoring proteins (AKAPS) in glutamate receptor trafficking and long term synaptic depression. J Biol Chem. 2005;280:16962–16968. doi: 10.1074/jbc.M409693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer KM, McCarley RW. Visual hallucinations, attention, and neural circuitry: perspectives from schizophrenia research. Behav Brain Sci. 2005;28:774. [Google Scholar]
- Sperk G, Schwarzer C, Tsunashima K, Fuchs K, Sieghart W. GABAA receptor subunits in the rat hippocampus I: immunocytochemical distribution of 13 subunits. Neuroscience. 1997;80:987–1000. doi: 10.1016/s0306-4522(97)00146-2. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. Recruitment of functional GABAA receptors to postsynaptic domains by insulin. Nature. 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- Wang H, Bedford F, Brandon NJ, Moss SJ, Olsen RW. GABAA-receptor-associated protein links GABAA receptors and the cytoskeleton. Nature. 1999;397:69–72. doi: 10.1038/16264. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Haditsch U, Tu W, Cochrane K, Ahmadian G, Tran L, Paw J, Wang Y, Mansuy IM, Salter M, Lu YM. Interaction of calcineurin and type-A GABA receptor γ2 subunits produces long-term depression at CA1 inhibitory synapses. J Neurosci. 2003;23:826–836. doi: 10.1523/JNEUROSCI.23-03-00826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Zhang M, Zhu Y, Wang J. Ca(2+)-calmodulin signalling pathway up-regulates GABA synaptic transmission through cytoskeleton-mediated mechanisms. Neuroscience. 2004;127:637–647. doi: 10.1016/j.neuroscience.2004.05.056. [DOI] [PubMed] [Google Scholar]
- Yan Z, Hsieh-Wilson L, Feng J, Tomizawa K, Allen P, Fienberg A, Nairn A, Greengard P. Protein phosphatase 1 modulation of neostriatal AMPA channels: regulation by DARPP-32 and spinophilin. Nat Neurosci. 1999;2:13–17. doi: 10.1038/4516. [DOI] [PubMed] [Google Scholar]
- Zhabotinsky A, Camp R, Epstein I, Lisman J. Role of the neurogranin concentrated in spines in the induction of long-term potentiation. J Neurosci. 2006;26:7337–7347. doi: 10.1523/JNEUROSCI.0729-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Slevin J, Whiteheart S. Cellular functions of NSF: not just SNAPs and SNAREs. FEBS Lett. 2007;581:2140–2149. doi: 10.1016/j.febslet.2007.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]