Synaptic Vesicle Mobility in Mouse Motor Nerve Terminals with and without Synapsin (original) (raw)
Abstract
We measured synaptic vesicle mobility using fluorescence recovery after photobleaching of FM 1-43 [_N_-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] stained mouse motor nerve terminals obtained from wild-type (WT) and synapsin triple knock-out (TKO) mice at room temperature and physiological temperature. Vesicles were mobile in resting terminals at physiological temperature but virtually immobile at room temperature. Mobility was increased at both temperatures by blocking phosphatases with okadaic acid, decreased at physiological temperature by blocking kinases with staurosporine, and unaffected by disrupting actin filaments with latrunculin A or reducing intracellular calcium concentration with BAPTA-AM. Synapsin TKO mice showed reduced numbers of synaptic vesicles and reduced FM 1-43 staining intensity. Synaptic transmission, however, was indistinguishable from WT, as was synaptic vesicle mobility under all conditions tested. Thus, in TKO mice, and perhaps WT mice, a phospho-protein different from synapsin but otherwise of unknown identity is the primary regulator of synaptic vesicle mobility.
Keywords: synapsin, FRAP, temperature, synaptic vesicle mobility, motor nerve terminal, actin
Introduction
Synaptic vesicle recruitment to active zones in presynaptic nerve terminals is important for maintaining neurotransmitter release during periods of prolonged nerve activity. Recruitment comes from synaptic vesicles that can reside several vesicle diameters away from fusion sites (Ceccarelli et al., 1972). Directed transport via an actin-myosin mechanism has been proposed to facilitate movement, based primarily on evidence of myosin involvement in vesicle recruitment (Prekeris and Terrian, 1997; Ryan, 1999; Verstreken et al., 2005) (but see Tokuoka and Goda, 2006). Direct evidence for an actin role, however, is mostly lacking (Sankaranarayanan et al., 2003; Richards et al., 2004; Gaffield et al., 2006) (but see Nunes et al., 2006). In addition to directed transport of vesicles, evidence for diffusion has been presented. Models for caged diffusion (Jordan et al., 2005), stick and diffuse (Shtrahman et al., 2005; Yeung et al., 2007), free diffusion (Holt et al., 2004; Rea et al., 2004), and hindered diffusion (Gaffield et al., 2006) have all suggested that diffusion may account entirely for synaptic vesicle movement. Most of these observations can be combined into a simple mechanism involving a protein that reversibly glues synaptic vesicles together. Free diffusion of synaptic vesicles would occur in terminals lacking this protein, such as in retinal cells, whereas synaptic vesicles in conventional terminals, which possess the protein, would be at least partially immobilized. During activity, this glue protein would change in some significant manner, thus releasing the synaptic vesicles, allowing them to diffuse to fusion sites.
The best candidate for the glue that binds synaptic vesicles is synapsin (De Camilli et al., 1983a,b; Huttner et al., 1983; Llinas et al., 1985). Decades of research have provided abundant evidence that synapsin binds to synaptic vesicles (Huttner et al., 1983; Benfenati et al., 1989) and to the actin cytoskeleton (Petrucci and Morrow, 1987). Synapsin can also dimerize (Hosaka and Sudhof, 1999). Together, these interactions have been proposed to immobilize synaptic vesicles (Hilfiker et al., 1999). In photoreceptors, which lack synapsin (Mandell et al., 1990), synaptic vesicles diffuse freely (Holt et al., 2004; Rea et al., 2004). In conventional terminals expressing synapsin (De Camilli et al., 1983a), synaptic vesicle mobility is restricted in a phosphorylation-dependent manner (Betz and Henkel, 1994; Kraszewski et al., 1995). During periods of nerve activity, synapsin becomes phosphorylated, which reduces its affinity for vesicles (Huttner et al., 1983; Torri-Tarelli et al., 1992) and results in synapsin dispersion (Chi et al., 2001). Studies on terminals from animals lacking all synapsin genes [_Drosophila_ larval muscle (Godenschwege et al., 2004); cultures of mouse CNS neurons (Gitler et al., 2004b)] found reduced synaptic vesicle numbers and reduced release during intense stimulation [especially in inhibitory synapses (Gitler et al., 2004b)] and support a model in which synapsin is particularly involved in anchoring vesicles belonging to the reserve pool (Hilfiker et al., 1999). None of these studies, however, addressed directly the core tenet of the synapsin hypothesis, namely that synapsin is the glue that regulates synaptic vesicle mobility.
Here we report results from a direct test of the synapsin hypothesis, which predicts that vesicles should be freely mobile in mice terminals lacking synapsin. We monitored vesicle mobility with fluorescence recovery after photobleaching (FRAP) and found all aspects of mobility regulation unchanged in motor nerve terminals from the synapsin triple knock-out (TKO) mouse.
Materials and Methods
Acute muscle preparation.
C57BL/6 wild-type (WT) mice or synapsin TKO mice [kindly provided by Paul Greengard (Rockefeller University, New York, NY) and Hung-Teh Kao (Nathan Kline Institute, Orangeburg, NY) (Gitler et al., 2004a,b)] were housed and handled in accordance with the University of Colorado Institutional Animal Care and Use Committee. The levator auris longus muscle was dissected from adult mice as described previously (Angaut-Petit et al., 1987) and pinned in a Sylgard-lined chamber. For nerve stimulation, the nerve was drawn into a suction electrode. The perfusing solution contained the following (in mm): 137 NaCl, 5 KCl, 1.8 CaCl2, 1 MgSO4, 12 NaHCO3, 1 NaH2PO4, and 11 glucose, pH 7.4 and bubbled with 95% O2/5% CO2 unless noted otherwise. During experiments, gravity-fed perfusion was maintained at a rate of ∼0.9 ml/min into a volume of 4–5 ml unless noted otherwise.
Muscles were loaded with FM 1-43 [_N_-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] (Invitrogen, Carlsbad, CA) by stimulating the nerve in the presence of 3.2 μm FM 1-43. After stimulation, the preparation recovered in the FM 1-43 for 10 min to allow for completion of endocytosis. The muscles were then washed for at least 15 min in normal saline solution, all of this done at 23°C. During FM 1-43 loading and destaining, muscles were bathed in 3 μm curare to prevent muscle contraction.
Agents.
Latrunculin A, okadaic acid, staurosporine, BAPTA-AM, β-actin antibody, and curare were purchased from Sigma (St. Louis, MO). μ-Conotoxin GIIIA was purchased from Bachem Bioscience (King of Prussia, PA). Stock solutions for latrunculin A, okadaic acid, staurosporine, and BAPTA-AM were made by dissolving into DMSO (final working DMSO concentration <0.5% v/v) and were stored at −20°C. Stock solutions for μ-conotoxin were made by dissolving into distilled water and were stored at −20°C. Stock solutions were thawed and mixed in normal saline solution for use.
Electrophysiology.
Temperature was maintained using dish heater DH-35 and in-line heater SF-28 controlled and monitored by dual automatic temperature controller TC-344B (all from Warner Instruments, Hamden, CT). Nerve stimulation was achieved using pulses from A310 Accupulser to A365 Stimulus Isolator (both from World Precision Instruments, Sarasota, FL). Electrophysiology was performed as described previously (Richards et al., 2003) except recording and analysis were done with custom written Matlab (MathWorks, Natick, MA) programs. Electrodes were pulled using a model P.80/PC Flaming Brown Micropipette puller (Sutter Instruments, Novato, CA). Electrodes were filled with 3 m KAc and resistances were between 15 and 30 MΩ. End plate potentials (EPPs) and quantum content data were corrected for nonlinear summation (Martin, 1955). Resting muscle potentials were more negative than −50 mV for recordings in curare (see Fig. 1C) and more negative than −60 mV for recordings in μ-conotoxin (see Figs. 7A, 8D,E).
Figure 1.
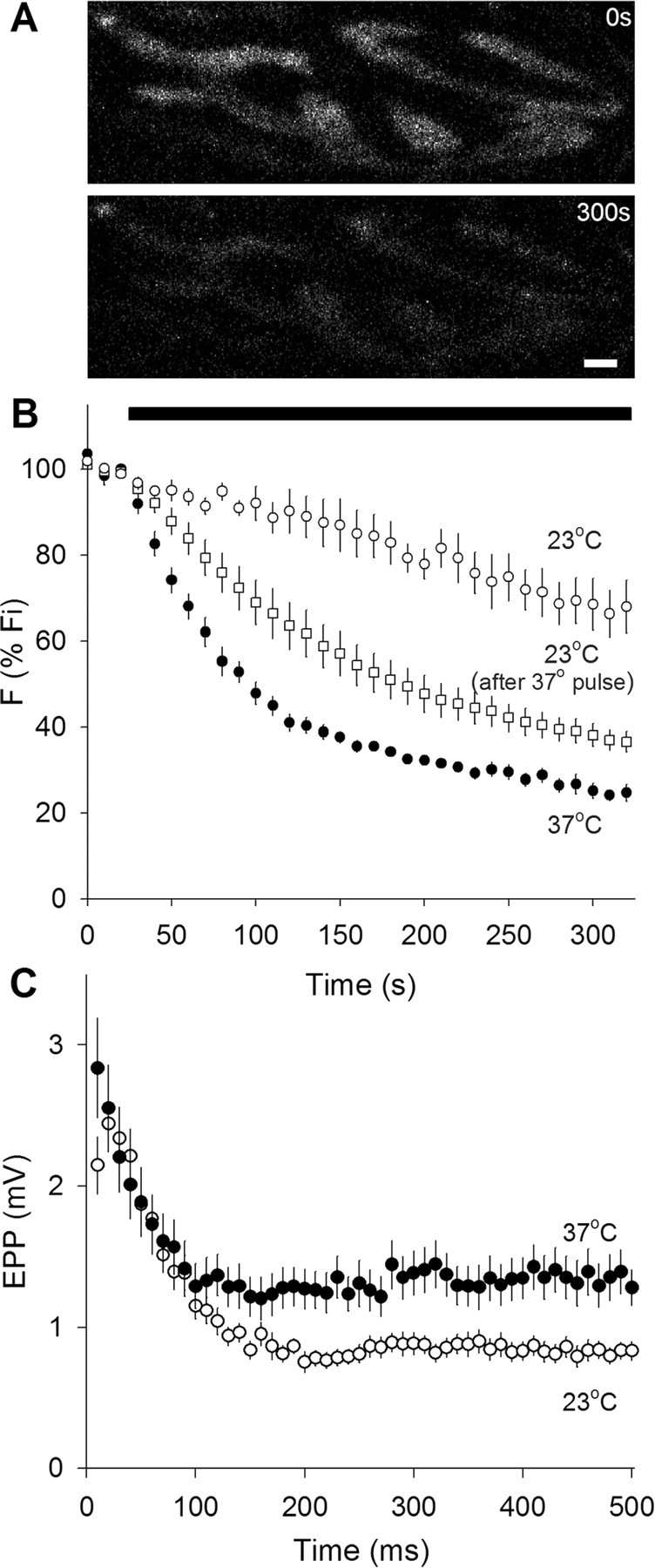
Synaptic vesicle fusion rate is higher at 37°C than at 23°C. A, Typical images of FM 1-43-labeled terminals before (top) and after (bottom) 30 Hz nerve stimulation for 300 s at 37°C. Scale bar, 2 μm. B, FM 1-43 destaining rates. FM 1-43 fluorescence intensity normalized to initial value. Nerve stimulation at 30 Hz during time marked by solid bar. Destaining was slow at 23°C (open circles; n = 4), and was much faster and more complete at 37°C (filled circles; n = 5). Destaining at 23°C could be accelerated if muscles were transiently warmed to 37°C and then cooled again to 23°C before stimulation (open squares; n = 4), suggesting that recycling of labeled vesicles was greatly slowed at 23°C. C, Electrophysiological recordings during nerve activity. End plate potential amplitudes (in 1.1 μm curare) for 50 shocks delivered at 100 Hz. Initial and steady-state amplitudes are higher at 37°C (filled circles; n = 17) than at 23°C (open circles; n = 18).
Figure 7.

Chelation of intracellular calcium with BAPTA-AM does not affect synaptic vesicle mobility at 37°C. A, BAPTA-AM reduces end plate potential amplitude. Typical end plate potential recordings taken from control (in 1.5 μm μ-conotoxin; black line) or BAPTA-AM-treated terminals (gray line). B, BAPTA-AM reduces temperature-dependent rise in miniature end plate potential frequency. Example recordings from miniature end plate potentials collected at 23°C, 37°C, or 37°C plus incubation with BAPTA-AM (100 μm for 1.5 h). C, Quantification of temperature-dependent rise in miniature end plate potential frequency and subsequent reduction in frequency after BAPTA-AM incubation. Each condition is significantly different from the other two (Student's t test; p < 0.01, n ≥ 9 for each condition). D, Fluorescence recovery plotted versus time for data collected before (open circles; n = 9; downward error bars) and after (filled circles; n = 22; upward error bars) BAPTA-AM (100 μm for 1.5 h) incubation. For clarity, error bars are shown in one direction only.
Figure 8.
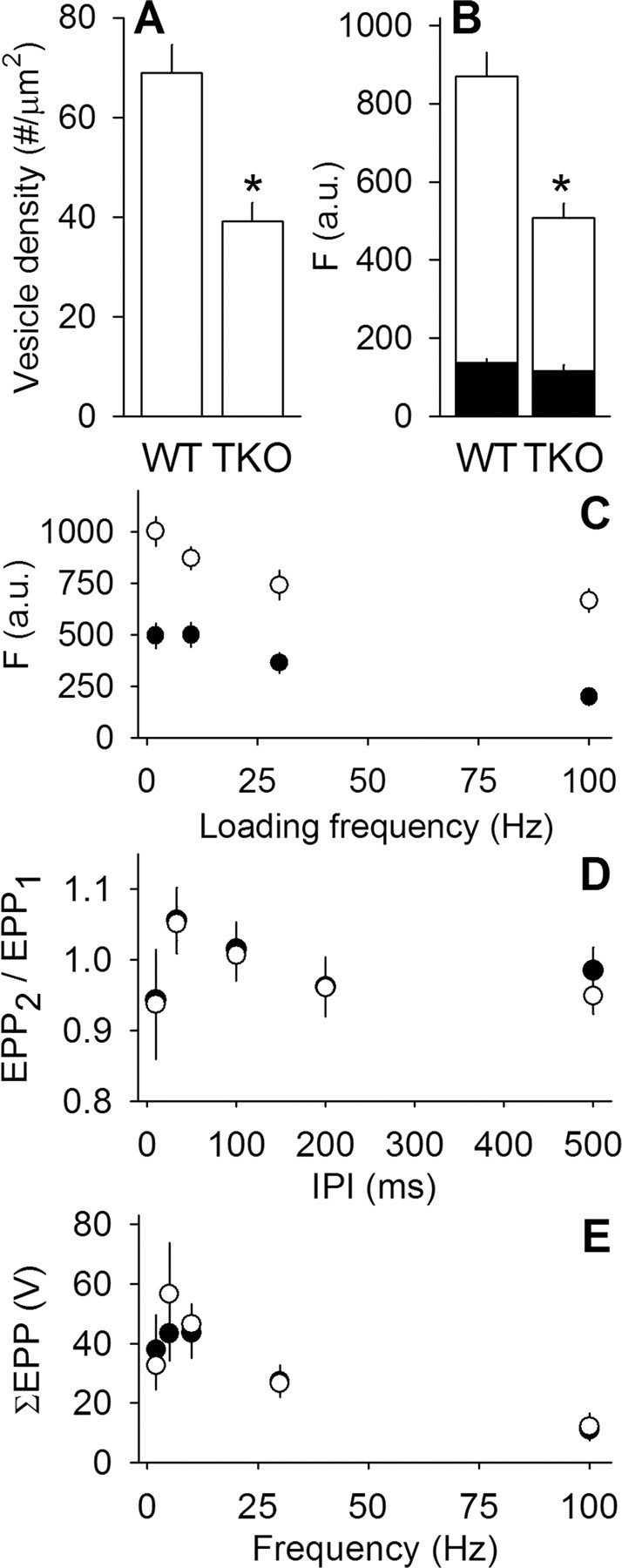
Motor nerve terminals lacking synapsin have fewer synaptic vesicles but normal electrophysiology. A, Quantification of synaptic vesicle numbers in synapsin TKO terminals. Synaptic vesicle density (plotted as number of vesicles per square micrometer of terminal area) is reduced by ∼50%. *p < 0.01, significant difference (Student's t test). n = 31 for WT and 36 for TKO. B, FM 1-43 fluorescence [arbitrary units (a.u.)] is reduced in synapsin TKO terminals. The standard loading protocol (30 Hz, 1 min nerve stimulation at 23°C) results in approximately half as much destainable fluorescence (open bars) in synapsin TKO compared with WT termi-nals. Background fluorescence is not significantly different (filled bars). *p < 0.01, significant difference in destainable fluorescence (Student's t test). n ≥ 15 terminals for each condition. C, FM 1-43 fluorescence (arbitrary units) after background subtraction for WT (open circles; n ≥ 12 for each data point) and synapsin TKO terminals (filled circles; n ≥ 15 for each data point) resulting from several different loading frequencies at 1800 shocks. D, Paired-pulse ratio, plotted as the ratio of the second end plate potential to the first end plate potential, is shown for several interpulse intervals (IPI) at 23°C. No significant difference between WT (open circles) and synapsin TKO terminals (filled circles) was found. n ≥ 5 for each data point. E, Summed end plate potential amplitudes are plotted for several different stimulus frequencies (each applied for 1800 shocks at 23°C). No significant difference between WT (open circles) and synapsin TKO terminals (filled circles) was found. n ≥ 5 for each data point.
Electron microscopy.
For electron micrographs, muscles were fixed using 2% paraformaldehyde, 2% glutaraldehyde in 0.1 m PBS, 0.12 m sucrose, and 0.1 mm CaCl2 for 45 min on ice. The muscles were then washed with PBS solution containing 0.12 m sucrose and 0.1 mm CaCl2 three times. The muscles were then embedded in Epon, sectioned (80–90 nm thickness), and imaged with a TECNAI G1 12 BioTwin microscope (FEI Company, Hillsboro, OR). Images were collected and analyzed digitally. For vesicle density in electron micrographs, the number of vesicles was counted by hand with image contrast adjusted in some cases to confirm vesicles. The average diameter of vesicles was calculated by looking at line profiles of several sample vesicles from each terminal and averaging. The terminal area was calculated by drawing a line around the terminal and calculating the area from the total number of pixels enclosed.
Imaging.
FRAP experiments were performed on resting nerve terminals as described previously (Gaffield et al., 2006). The bleaching protocol we used was the same as before when we found the bleach did not damage the nerve terminals (Gaffield et al., 2006). Temperature control was achieved as described for electrophysiology. For immunofluorescence, muscles were fixed for 10 min in −20°C methanol, washed in 1× PBS, blocked with 1% BSA dissolved in 1× PBS for 1 h, and then incubated with the β-actin antibody at 1:200 overnight. Terminals were imaged after washing in PBS. For temperature comparison of actin pattern, muscles were fixed in −20°C methanol from 23°C or after resting at 37°C for 15–30 min. For latrunculin-treated muscles, the contralateral muscle was used as a control. The treatment of each muscle was blind to the imager before imaging began. For analysis of fluorescence localization, terminals were outlined by hand, and the five pixels on either side perpendicular to the line were averaged to obtain a single average perimeter fluorescence value. The total fluorescence contained by and within the boundary of the line was also averaged. After background subtraction, the ratio of perimeter to total fluorescence was calculated.
Data analysis was performed using programs written in Matlab. Analysis of FRAP results including calculation of diffusion coefficients was as described previously (Gaffield et al., 2006).
Statistical analysis was performed using Microsoft (Redmond, WA) Excel, and plots and curve fitting were done using SigmaPlot. All experimental conditions were performed on at least two muscles from at least two different animals; terminal numbers are listed in the figure legends. All error bars indicate SEM. Significance was tested using the Student's t test, with p values <0.05 indicating a significant difference, although we report p values of <0.01 in most cases (see figure legends).
Results
Synaptic vesicle recycling is significantly impeded at room temperature
Several recent studies of murine synapses demonstrated reduced exocytic and recycling rates at room temperature compared with physiological temperature (Fernandez-Alfonso and Ryan, 2004; Micheva and Smith, 2005; Yang et al., 2005; Kushmerick et al., 2006). We set out to determine whether synaptic vesicle mobility was also affected by temperature using FRAP of FM 1-43-labeled vesicles.
As a prelude to the FRAP studies, we compared FM 1-43 destaining rates and evoked EPPs, as shown in Figure 1. We immediately noticed that mouse motor nerve terminals, which were readily loaded with FM 1-43 by nerve stimulation, were difficult and sometimes impossible to destain at room temperature. [We performed similar destaining experiments with FM 2-10 [_N_-(3-triethylammoniumpropyl)-4-(4-(diethylamino)styryl)pyridinium dibromide], a less hydrophobic version of FM 1-43, with similar results (data not shown)]. At 37°C, however, FM 1-43 destaining was fast, consistent, and reliable. Figure 1A shows raw images of a portion of a terminal before (top) and after (bottom) destaining at 37°C. Figure 1B shows quantified results. All preparations were loaded with FM 1-43 at room temperature, not 37°C, and then rested for 15–20 min (also room temperature) before destaining. In terminals destained at 37°C, warming required an additional 5–10 min. Nerve stimulation (30 Hz, black bar) caused rapid destaining at 37°C (filled circles); at room temperature, however, the initial destaining rate was approximately seven times slower (open circles) and more variable (note larger error bars). This large difference in destaining rate was unexpected. We wondered whether vesicle recycling might be greatly slowed at room temperature (requiring more than the 15–20 min rest period). To test this, we warmed stained preparations for ∼30 min and then cooled them back to 23°C, before destaining. As shown in Figure 1B (open squares), these terminals destained two to three times faster than those that were not given the 37°C pulse, although the destaining was still not as fast as at 37°C. Thus, it appears that the rate of vesicle recycling at room temperature is greatly slowed compared with physiological temperature. We did not measure in detail the time course of the warming effect, but it appeared that even a few minutes at 37°C was sufficient to cause rapid destaining during stimulation.
We compared FM 1-43 destaining with electrophysiological measurements of transmitter release (Fig. 1C). Amplitudes of evoked EPPs (curarized) were modestly increased at 37°C, and the plateau reached during repetitive stimulation was elevated by ∼50% compared with room temperature. This somewhat underestimates the true effect of temperature, because the blocking action of curare increases slightly as the temperature rises (Darveniza et al., 1979; Horrow and Bartkowski, 1983; Dickinson et al., 1995). Amplitudes of spontaneous miniature EPPs (MEPPs) were unchanged by temperature (1.04 ± 0.05 and 1.06 ± 0.18 mV for 23°C and 37°C, respectively). The changes in transmitter release were clearly much smaller than the sevenfold change in FM 1-43 destaining rate, suggesting that vesicles other than those labeled with FM 1-43 (whose recycling was retarded by the low temperature) were being released, reflecting an impressively adaptable exocytic regulatory mechanism. In summary, the overall exocytic rate was approximately doubled by increasing temperature from 23°C to 37°C, in good agreement with estimates using cultured hippocampal synapses (Micheva and Smith, 2005), hippocampal slices (Yang et al., 2005), and the calyx of Held (Kushmerick et al., 2006).
Synaptic vesicles in resting terminals are immobile at 23°C and mobile at 37°C
We next turned to direct measures of vesicle mobility. We performed FRAP experiments on motor nerve terminals loaded with FM 1-43 and maintained at either 23°C or 37°C. Figure 2A illustrates a typical FRAP experiment. After acquiring several control images of a terminal (left), we bleached a small region within the terminal (middle, arrow) and monitored subsequent recovery of fluorescence in the bleached region (right). Figure 2B shows the average time courses of recovery after photobleaching. At 23°C (gray circles), some recovery (∼20% over 60 s) is observed, but control experiments show that nearly all of the recovery is attributable to background fluorescence recovery (gray line). Background fluorescence accounts for 20–25% of the total fluorescence (Fig. 2B, inset) and is measured with FRAP after exhaustive stimulation to release all vesicular dye (Fig. 3) (cf. Gaffield et al., 2006). A very different result was obtained at 37°C (Fig. 2B, filled circles), with a significantly greater recovery than at room temperature, reflecting a large synaptic vesicle mobile fraction; background (black line) also was lower. From these data, we calculate a diffusion coefficient at 37°C of 0.005 μm2/s, which is similar to our estimate of 0.003 μm2/s for mobile synaptic vesicles in frog motor nerve terminals (Gaffield et al., 2006).
Figure 2.

Synaptic vesicle mobility is higher at 37°C than at 23°C. A, Images from a typical FRAP experiment. A portion of a terminal labeled with FM 1-43 and maintained at 37°C was imaged before the bleach (left), immediately after the bleach (middle), and 60 s later (right). The arrow indicates the bleached region. Scale bar, 1 μm. B, Quantification of FRAP experiments. Fluorescence recovery plotted versus time for terminals maintained at 23°C (gray circles; n = 12) or 37°C (black circles; n = 66). The gray line and black line show average background contribution to fluorescence recovery for 23°C and 37°C, respectively. Inset, Contribution of synaptic vesicles (open bars) and background (filled bars) to total fluorescence at the two temperatures. For details, see Figure 3. a.u., Arbitrary units. C, Mobile fraction is plotted versus temperature. Open circles indicate mobility measurements taken as terminals were warmed from 23°C to 37°C (n ≥ 12 for each data point). The filled circle indicates mobility measurements taken after preparations reached full mobility at 37°C and then were cooled back to room temperature. *p < 0.01, significant compared with 23°C; **p < 0.01, significant compared with 37°C after cooling (Student's t test).
Figure 3.
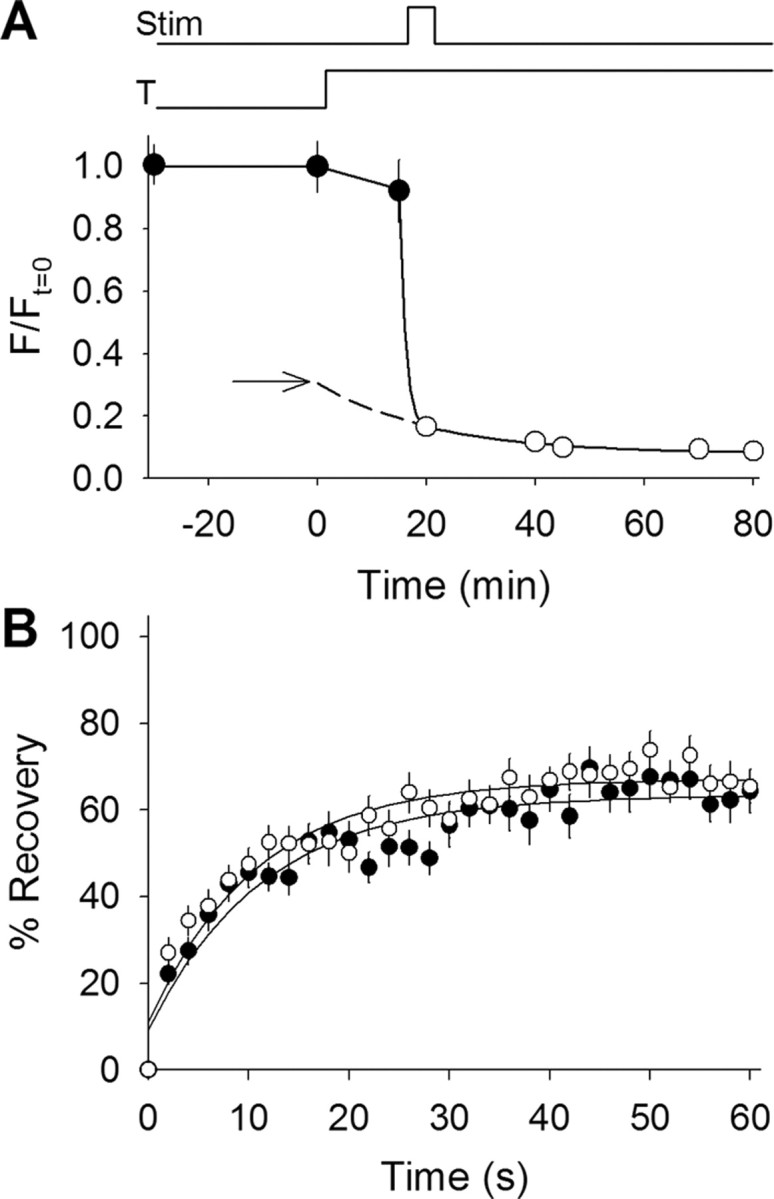
Background contributions to fluorescence recovery differ between 23°C and 37°C. A, Fluorescence measurements made before, during, and after a temperature increase from 23°C to 37°C and a destaining stimulus. Fluorescence remained constant at 23°C for 30 min (filled circles) and decreased slightly once the temperature was raised to 37°C (indicated by bottom line above plot; T). During stimulation (indicated by top line above plot; Stim) fluorescence fell to background levels. Background fluorescence (open circles) continued to decrease over time even after stimulation had ceased. The solid line through the open circles represents the best fit time constant (21 min) for background fluorescence loss. The dashed line extending from the best fit line indicates the time during which background fluorescence levels could not be measured. This includes the time required for stabilization of temperature and destaining of vesicular fluorescence. The dashed line was extended back to the zero time point (when heating began), thus providing an estimate of the initial background level at 23°C (arrow). Notice that most (if not all) of the fluorescence lost before stimulation was attributable to background and not synaptic vesicle fluorescence. Similar results were obtained for the synapsin TKO terminals used later (data not shown). B, Fluorescence recovery plotted versus time for the background fluorescence in WT (open circles; n = 18) and in the synapsin TKO terminals used later (filled circles; n = 25) measured at 37°C. The solid lines show single-exponential best fits with a time constant of ∼11 s for each trace. Little difference was observed between WT and synapsin TKO. The WT data were scaled to the background fluorescence fraction for plots shown in Figures 2B and 4A.
We performed additional FRAP experiments over a range of temperatures; the results are summarized in Figure 2C. Open circles show measurements of synaptic vesicle mobility as temperature was increased from 23°C to 37°C. A sharp increase in mobile fraction occurs between 32°C and 34°C. Why are vesicles immobile at 23°C? In Figure 1, we showed evidence that vesicles do not recycle effectively at 23°C. Perhaps they are immobilized as a result of incomplete rematuration, for example, failure to lose their clathrin coats (cf. Teng and Wilkinson, 2003). To test this, we pulsed the temperature to 37°C (Fig. 1B, open squares) and then repeated FRAP measurements at 23°C. Vesicle mobility was not increased by the pulse, thus showing that mature, releasable synaptic vesicles are immobilized at 23°C.
Glucose is required to maintain synaptic vesicle mobility at 37°C
We found that, to maintain vesicle mobility, it was necessary to perfuse the chamber with fresh saline solution continuously at 37°C. If perfusion stopped, vesicles became immobile with a time constant of ∼19 min (Fig. 4A,B). Vesicle immobilization also resulted if glucose was omitted from the perfusate (Fig. 4C). Raising glucose concentration (from 11 to 33 mm) preserved vesicle mobility for 1 h, even in the absence of perfusion (Fig. 4B). Omitting bubbling with 95% O2/5% CO2 had no significant effect, showing that atmospheric O2 was sufficient to maintain vesicle mobility (Fig. 4D) and that alkalinization did not affect mobility (data not shown). In summary, we could incubate preparations at 37°C without perfusion and maintain vesicle mobility if glucose concentration was increased to 33 mm.
Figure 4.

Glucose maintains high synaptic vesicle mobility at 37°C. A, Stopping perfusion greatly reduces vesicle mobility at 37°C. Fluorescence recovery is plotted versus time for terminals maintained at 37°C without any perfusion for >30 min (open circles; n = 9). Perfusion of the terminals with fresh, oxygenated saline solution containing 11 mm glucose (filled circles; n = 11) increased the mobility back to normal levels. The gray line indicates expected background contribution to recovery (as in Fig. 2B). B, Mobile fraction is plotted versus time. The line above the plot indicates when perfusion is on (up) or off (down). Data (filled circles; 10 min bins, n ≥ 3 for each data point) show a reduction in mobility with a time constant of ∼19 min after perfusion was stopped. Once perfusion began again, mobility recovered. The open circle (n = 12) shows mobile fraction when glucose was increased from normal (11 mm) to 33 mm. Mobility remained high in 33 mm glucose for at least 1 h after perfusion was stopped. C, Fluorescence recovery plotted versus time in terminals maintained at 37°C and perfused with oxygenated saline solution containing 0 mm glucose (filled circles; n = 14). For comparison, results collected from terminals perfused with solution containing 11 mm glucose are shown (gray line; data from Fig. 2B). D, Bubbling oxygen into the perfusing solution is not required to maintain synaptic vesicle mobility. Fluorescence recovery is plotted versus time comparing measurements taken from terminals maintained at 37°C with 11 mm glucose and bubbling oxygen (open circles) and 30 min after bubbling oxygen into the perfusing solution was stopped (filled circles; n = 21). Inset, Time course of gases leaving solution. Open squares show dissolved [CO2] (calculated by measuring pH) once perfusion stopped at time 0. The solid line shows exponential best fit with a time constant of ∼19 min. O2 is expected to leave at a slightly faster rate with an ∼16 min time constant attributable to faster diffusion rates (and similar effusion rates).
Depolymerization of actin filaments does not affect synaptic vesicle mobility
We wondered whether vesicle mobility at 37°C was actin dependent. We used a β-actin antibody to label actin filaments in the terminals. Typical actin staining patterns at 23°C and 37°C are shown in the top row of Figure 5A. Fluorescence tended to be localized around the periphery of the nerve terminal at 23°C, as reported for snake motor nerve terminals (Cole et al., 2000). However, perimeter staining was noticeably reduced at 37°C. We quantified this difference by comparing the ratios (average perimeter fluorescence)/(average total terminal fluorescence) for the two temperatures. Figure 5B summarizes the results, revealing a significant reduction in perimeter staining at 37°C (the total terminal fluorescence was reduced by ∼20%). We thought this difference might be attributable to poor initial fixation at 37°C, because proper protein fixation with methanol occurs at temperatures below −5°C (Kiernan, 1981). Therefore, we fixed 23°C terminals by adding 4°C methanol to the chamber and then moved it to −20°C (Fig. 5B, 23°C warm fix). The resulting peripheral staining value was not significantly different from normal cold fixation for 23°C, suggesting that the difference in staining pattern is real and might underlie the change in vesicle mobility with temperature.
Figure 5.
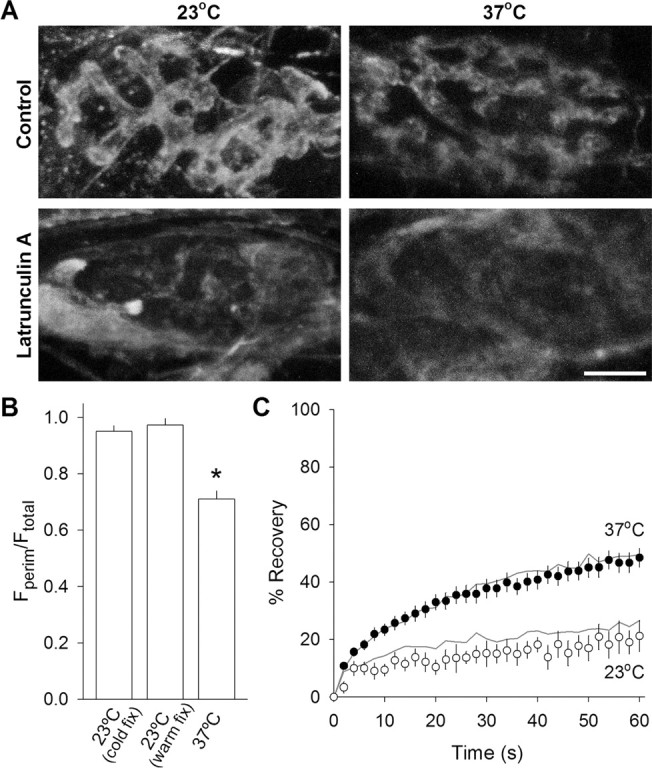
Latrunculin A disrupts actin but not synaptic vesicle mobility. A, Actin immunostaining. Typical images of β-actin staining pattern under four conditions: control (top row) and after latrunculin A treatment (15 μm for at least 45 min; bottom row) fixed after maintaining the terminals at either 23°C (left column) or 37°C (right column). The blurred staining seen in latrunculin A-treated terminals was outside of the terminal and was typical of all terminals surveyed. Scale bar, 5 μm. B, Quantification of fluorescence distribution of β-actin staining pattern. The ratio of the average perimeter fluorescence to average total fluorescence for each terminal is plotted for 23°C with cold fixative (left bar; n = 20), 23°C with a warm fixative (middle bar; n = 7), and 37°C (n = 16). A larger value indicates that fluorescence is distributed more peripherally. *p < 0.01, significantly different from other two conditions (Student's t test). C, Fluorescence recovery over time for latrunculin A-treated terminals (symbols) compared with controls (gray lines). Latrunculin A incubation had no significant effect at either 23°C (open circles; n = 5) or at 37°C (filled circles; n = 37).
We next examined the effect of actin filament disruption with latrunculin A. Treatment with this drug (15 μm) for 45 min (saline solution contained 33 mm glucose) abolished the actin staining pattern in nerve terminals at both temperatures. Total terminal fluorescence intensity was reduced by nearly 75% (Fig. 5A, bottom row; new staining sometimes appeared in structures located outside the terminal). Subsequent FRAP experiments on labeled synaptic vesicles (Fig. 5C) showed no significant effect on vesicle mobility at either temperature. In summary, actin filaments are likely not involved in regulating synaptic vesicle mobility under resting conditions.
Adjusting phosphorylation levels impacts synaptic vesicle mobility
Previous experiments on nerve terminals found that a phosphorylation event is involved in increasing synaptic vesicle mobility (Betz and Henkel, 1994; Kraszewski et al., 1995; Shtrahman et al., 2005; Gaffield et al., 2006). Could phosphorylation account for the increased synaptic vesicle mobility at 37°C? We used the nonspecific phosphatase inhibitor okadaic acid to increase phosphorylation in the nerve terminal. This drug (1 μm for 45 min, 33 mm glucose) increased synaptic vesicle mobility at both 23°C and 37°C (Fig. 6A). We next tried to block phosphorylation and thereby decrease synaptic vesicle mobility at 37°C. Previous experiments showed that the general kinase inhibitor staurosporine could block okadaic acid-dependent increases in synaptic vesicle mobility in frog motor nerve terminals (Henkel et al., 1996). Here we incubated our normal preparation with staurosporine (1 μm for 45 min, 33 mm glucose) and then performed FRAP experiments at 37°C (Fig. 6B). Staurosporine effectively abolished synaptic vesicle mobility at 37°C. In summary, phosphorylation increases vesicle mobility and can account for the mobility increase with increasing temperature.
Figure 6.
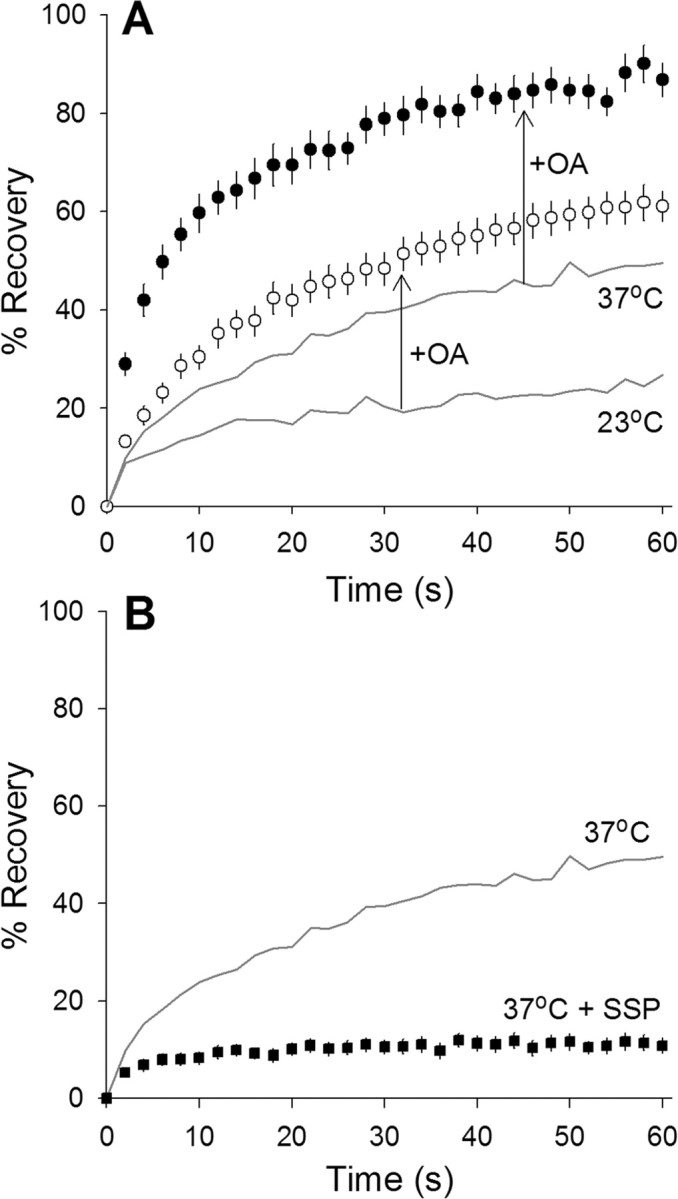
Phosphorylation regulates synaptic vesicle mobility. A, Okadaic acid (OA) mobilizes synaptic vesicles. Fluorescence recovery over time for terminals maintained at 23°C (open circles) or 37°C (filled circles) after application of okadaic acid (1 μm for at least 45 min). Okadaic acid increased mobility above controls at both 23°C (compare with bottom gray line) and 37°C (compare with top gray line). n ≥ 23 for each condition. B, Staurosporine (SSP) prevents synaptic vesicle mobilization. Fluorescence recovery over time for terminals maintained at 37°C (filled squares; n = 32) after application of staurosporine (1 μm for at least 45 min). Staurosporine blocks the expected temperature-dependent increase in synaptic vesicle mobility (gray line).
The vesicle mobility increase at 37°C is not blocked by BAPTA-AM
Mouse motor nerve terminals show increased spontaneous release rate linked to a possible rise in global calcium ion concentration on being warmed to 37°C (Katz et al., 1997). This rise in calcium might act to increase vesicle mobility, possibly by increasing the activity of calcium-dependent kinases. To test this, we incubated preparations with BAPTA-AM (100 μm) for 1.5 h at 37°C to reduce intraterminal free calcium levels. Although we did not measure calcium ion concentration directly, we showed that the BAPTA-AM treatment reduced the amplitude of evoked EPPs by ∼45% (Fig. 7A) and reversed the rise in MEPP frequency by approximately half (Fig. 7B,C). Vesicle mobility, however, was unaffected (Fig. 7D). Thus, if a temperature-dependent increase in calcium ion concentration does govern vesicle mobility at rest (by increasing phosphorylation levels), it must have a low threshold compared with evoked and spontaneous transmitter release.
Synaptic mobility is not different in presynaptic terminals from mice lacking synapsin
We next examined the most likely target for the phosphorylation action, synapsin. To test directly for the role of synapsin in synaptic vesicle mobility, we used a synapsin TKO mouse (Gitler et al., 2004a). Some results were similar to those shown previously for excitatory synapses in the CNS (Gitler et al., 2004b). For example, electron micrographs of WT and synapsin TKO terminals showed no major differences in overall structure, except a 50% reduction in the number and density of synaptic vesicles (measured as vesicle number per terminal cross-sectional area) (Fig. 8A). Terminal area and vesicle diameters were not significantly different, nor were vesicles restricted to any particular region of the terminal but were scattered like WT vesicles (quantified data not shown). Next, we measured uptake of FM 1-43 at 23°C and found a similar reduction in labeling in the TKO mice, with no change in background labeling (Fig. 8B). We labeled terminals at several different loading frequencies at 23°C and found similar TKO deficits at all loading frequencies (Fig. 8C). Contrary to results found by Gitler et al. (2004b) with electrophysiological experiments at 23°C, we found no transmission deficits. Quantum content (71.1 ± 6.9 and 79.0 ± 8.3 for WT and TKO, respectively), paired-pulse ratio (Fig. 8D), and summed EPP amplitudes during long trains (1800 stimuli) (Fig. 8E) were maintained normally with fewer vesicles.
We then measured FRAP in the TKO mice at 23°C, expecting to find highly mobile vesicles, perhaps approaching that seen in WT mice after okadaic acid treatment (Fig. 6A). The result, however, was entirely different, there being no detectable difference between WT and TKO vesicle mobile fractions with any loading frequency (Fig. 9A). We next looked to see whether temperature would still regulate synaptic vesicle mobility without synapsin. Figure 9, B and C, shows results from experiments similar to those in Figure 2, B and C (WT data are replotted here as gray lines). Not only did temperature increase synaptic vesicle mobility in synapsin TKO mice, but the mobile fraction increased with temperature in a manner similar to WT mice (Fig. 9C). Finally, as an additional test, we examined the effect of okadaic acid (Fig. 9D). Again, the WT and TKO results were nearly identical. Therefore, when synapsin is entirely absent, synaptic vesicle mobility is regulated by phosphorylation.
Figure 9.

Synapsin is not required for phosphorylation-dependent regulation of synaptic vesicle mobility. A, Mobile fraction is shown for terminals loaded with FM 1-43 at several frequencies (each applied for 1800 shocks at 23°C). None of the loading frequencies allowed access to a large mobile fraction in either WT (open squares; n ≥ 8 for each data point) or synapsin TKO terminals (filled squares; n ≥ 6 for each data point). B, Fluorescence recovery over time for terminals maintained at 23°C (open circles) and 37°C (filled circles). No major difference was observed between WT (gray lines; data from Fig. 2B) and synapsin TKO terminals (n = 10 for 23°C and 57 for 37°C). Background contribution to recovery (data not shown) was not significantly different between WT and synapsin TKO (see Figs. 3B, 8B). C, Mobile fraction is plotted versus bath temperature for synapsin TKO terminals (symbols; n ≥ 9 for each data point). Open circles indicate mobility measurements taken as nerve terminals were warmed. The filled circle shows data obtained after preparations reached full mobility at 37°C and then were cooled to room temperature. *p < 0.01, significant compared with 23°C; **p < 0.01, significant compared with 37°C (Student's t test). Gray line shows WT data from Figure 2C for comparison. D, Fluorescence recovery plotted versus time in okadaic-acid-treated terminals. Okadaic acid had nearly identical effects on both WT (gray lines; data from Fig. 6A) and synapsin TKO terminals at both 23°C (open circles; n = 23) and 37°C (filled circles; n = 23).
Discussion
To our knowledge, this work is the first direct test of the basic synapsin hypothesis, which holds that synapsin is the agent that binds and immobilizes vesicles in a phosphorylation-dependent manner (Llinas et al., 1985; Hilfiker et al., 1999; Evergren et al., 2007). We began by examining effects of temperature on vesicle mobility in WT terminals. Raising the temperature from ∼23°C (at which vesicles were virtually immobile in resting terminals) to the physiological range (36–37°C) caused a substantial increase in mobility, which could be blocked by kinase inhibition with staurosporine. The mobility increase could be mimicked at room temperature by blocking phosphatases with okadaic acid, together suggesting a phosphorylation-dependent step in mobility regulation. The temperature and phosphorylation dependence of synaptic vesicle mobility was indistinguishable in motor nerve terminals from synapsin TKO mice lacking all known synapsin genes. These findings show that synapsin is not uniquely required for phosphorylation-dependent regulation of synaptic vesicle mobility in mouse motor nerve terminals.
If synapsins were the only molecules to play this role, then vesicles would be freely mobile in the synapsin TKO mice. Instead, vesicle mobility was indistinguishable from that in WT mice. The identity of the vesicle glue that remains in the TKO mouse is unknown. It is possible that it arises as compensation for the missing synapsin and that it does not normally play such a role, which in the WT is reserved exclusively for synapsin. However, if this were the case, the glue that emerges in the TKO mouse would have to match very closely the properties of the WT glue in terms of its dependence on temperature and phosphorylation state. It seems more likely that the agent, whatever its identity, that binds synaptic vesicles in the TKO mouse motor nerve terminals is present normally and plays a role originally postulated to be that of synapsin (Llinas et al., 1985). Consistent with this, Siksou et al. (2007) have characterized filaments in rat CNS terminals using electron tomography and have detected similar filamentous structures in the synapsin TKO mouse.
The list of possible candidates can be narrowed to proteins absent or different in ribbon containing terminals [like synapsin is known to be (Mandell et al., 1990)]: first, because synaptic vesicles are freely mobile in these terminals (Holt et al., 2004; Rea et al., 2004; see discussion by Gaffield et al., 2006), and second, because phosphorylation resulting from okadaic acid application appears to hinder (rather than increase) synaptic vesicle mobility in photoreceptors (Guatimosim et al., 2002; Rea et al., 2004). One possible protein is rabphilin (Von Kriegstein et al., 1999). Okadaic acid application results in phosphorylation of synapsin and rabphilin (Lonart and Sudhof, 1998), and the synapsin I/II knock-out shows higher synapsin and rabphilin phosphorylation (Lonart and Simsek-Duran, 2006), reflecting similar pathways. Rabphilin may affect synaptic vesicle trafficking during endocytosis and exocytosis (Burns et al., 1998), but a knock-out of rabphilin initially was reported to have no synaptic transmission deficiencies (Schluter et al., 1999). More recently, a closer look at transmission found that loss of rabphilin resulted in improved recovery from depression (Deak et al., 2006), hinting at improved vesicle recruitment. However, a critical requirement for binding to SNAP-25 (synaptosome-associated protein of 25 kDa) led the authors of this study to conclude that rabphilin regulates vesicle priming (Deak et al., 2006). As for other possible glue proteins, okadaic acid does not change phosphorylation of dynamin 1, amphiphysin 1, or synaptojanin 1 (Bauerfeind et al., 1997), ruling out at least a few candidates.
Some studies have suggested a less unique role for synapsin, for example, one concerned with a subpopulation of synaptic vesicles [those belonging to the reserve pool (Hilfiker et al., 1999)] or one concerned especially with inhibitory synapses, which show a greater loss of transmission than do excitatory synapses in the synapsin TKO (Gitler et al., 2004b), paralleling the behavioral inclination toward seizures (Gitler et al., 2004a). The features of synapsin that led to early predictions of a glue, for example, the phosphorylation-dependent dissociation of synapsin from synaptic vesicles (for review, see Hilfiker et al., 1999) may actually play a role in regulating which vesicles are exocytosed during activity [observed as reduced docked vesicle number at inhibitory synapses of the synapsin TKO (Gitler et al., 2004b)] or in properly returning endocytosed vesicles into the vesicle cluster (for review, see Evergren et al., 2007). In this latter case, vesicles in resting terminals may act as a store for synapsin. If synapsin does act during exocytosis/endocytosis, then ribbon-type synapses (which lack synapsin; see above) must have different and possibly specialized recycling mechanisms that might promote the “streamlined” use of vesicles in these terminals (Rea et al., 2004).
Cultured neonatal CNS synapses from the TKO mouse have fewer synaptic vesicles than normal (Gitler et al., 2004b; Siksou et al., 2007), which we have confirmed at the neuromuscular junction. In fact, this characteristic, and its associated reduction in FM 1-43 fluorescence intensity, were the only two features by which we could reliably distinguish the synapsin TKO from WT neuromuscular junctions. Synaptic potentials, whether spontaneous or evoked by a variety of paradigms, as well as all aspects of synaptic vesicle mobility, were indistinguishable. Apparently, each vesicle in the synapsin TKO exocytosed more often than those in WT to maintain normal transmission. Neurotransmission deficits were reported in CNS terminals of the TKO after 50 shocks at 10 Hz, possibly attributable to rapid depletion of vesicles in the CNS terminals studied (Gitler et al., 2004b). Motor nerve terminals have considerably more vesicles (Rizzoli and Betz, 2005) and are probably less susceptible to the vesicle loss in the synapsin TKO. Thus, at the neuromuscular junction, it appears that synapsin, which is abundant (De Camilli et al., 1983a), plays no discernable role in short-term aspects of synaptic function. This of course raises the question of the function of those vesicles, comprising approximately half of the total, that disappear along with synapsin in the TKO mouse. They are evidently capable of exocytosis and recycling, because the WT mouse took up approximately twice as much FM 1-43 as the TKO mouse, although transmitter release was unchanged. The fact that WT terminals do maintain a large vesicle population would suggest that the role of those vesicles lost in the TKO is important and should be explored.
In a previous paper on the frog neuromuscular preparation (Gaffield et al., 2006), we suggested that phosphorylation of synapsin regulated vesicle diffusion by immobilizing reserve pool vesicles. In the mouse, the situation is similar. Vesicle mobility at physiological temperatures was unaffected by actin disruption, thus vesicles are likely diffusing. Additionally, vesicle mobility was regulated by phosphorylation. Therefore, phosphorylation regulates vesicle diffusion by immobilizing some vesicles. However, synapsin is not the phosphorylation target. Clearly, it will be of additional interest to identify the molecule (or molecules) that does (do) regulate vesicle mobility at the neuromuscular synapse.
Footnotes
This work was supported by research grants from the National Institutes of Health and the Muscular Dystrophy Association. We thank Steve Fadul for providing excellent technical assistance, Dot Dill for performing electron microscopy, Drs. Joe Johnson and Leah Sheridan for helpful discussions, Dr. John Caldwell for showing us the dissection, Dr. Rock Levinson for discussions on immunostaining, and Drs. Paul Greengard and Hung-Teh Kao for providing synapsin TKO mice.
References
- Angaut-Petit D, Molgo J, Connold AL, Faille L. The levator auris longus muscle of the mouse: a convenient preparation for studies of short- and long-term presynaptic effects of drugs or toxins. Neurosci Lett. 1987;82:83–88. doi: 10.1016/0304-3940(87)90175-3. [DOI] [PubMed] [Google Scholar]
- Bauerfeind R, Takei K, De Camilli P. Amphiphysin I is associated with coated endocytic intermediates and undergoes stimulation-dependent dephosphorylation in nerve terminals. J Biol Chem. 1997;272:30984–30992. doi: 10.1074/jbc.272.49.30984. [DOI] [PubMed] [Google Scholar]
- Benfenati F, Bahler M, Jahn R, Greengard P. Interactions of synapsin I with small synaptic vesicles: distinct sites in synapsin I bind to vesicle phospholipids and vesicle proteins. J Cell Biol. 1989;108:1863–1872. doi: 10.1083/jcb.108.5.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Henkel AW. Okadaic acid disrupts clusters of synaptic vesicles in frog motor nerve terminals. J Cell Biol. 1994;124:843–854. doi: 10.1083/jcb.124.5.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns ME, Sasaki T, Takai Y, Augustine GJ. Rabphilin-3A: a multifunctional regulator of synaptic vesicle traffic. J Gen Physiol. 1998;111:243–255. doi: 10.1085/jgp.111.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli B, Hurlbut WP, Mauro A. Depletion of vesicles from frog neuromuscular junctions by prolonged tetanic stimulation. J Cell Biol. 1972;54:30–38. doi: 10.1083/jcb.54.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Cole JC, Villa BR, Wilkinson RS. Disruption of actin impedes transmitter release in snake motor terminals. J Physiol (Lond) 2000;525:579–586. doi: 10.1111/j.1469-7793.2000.t01-2-00579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darveniza P, Morgan-Hughes JA, Thompson EJ. Interaction of di-iodinated 125I-labelled alpha-bungarotoxin and reversible cholinergic ligands with intact synaptic acetylcholine receptors on isolated skeletal-muscle fibres from the rat. Biochem J. 1979;181:545–557. doi: 10.1042/bj1810545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli P, Cameron R, Greengard P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. I. Its general distribution in synapses of the central and peripheral nervous system demonstrated by immunofluorescence in frozen and plastic sections. J Cell Biol. 1983a;96:1337–1354. doi: 10.1083/jcb.96.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Camilli P, Harris SM, Jr, Huttner WB, Greengard P. Synapsin I (Protein I), a nerve terminal-specific phosphoprotein. II. Its specific association with synaptic vesicles demonstrated by immunocytochemistry in agarose-embedded synaptosomes. J Cell Biol. 1983b;96:1355–1373. doi: 10.1083/jcb.96.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak F, Shin OH, Tang J, Hanson P, Ubach J, Jahn R, Rizo J, Kavalali ET, Sudhof TC. Rabphilin regulates SNARE-dependent re-priming of synaptic vesicles for fusion. EMBO J. 2006;25:2856–2866. doi: 10.1038/sj.emboj.7601165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson R, Lieb WR, Franks NP. The effects of temperature on the interactions between volatile general anaesthetics and a neuronal nicotinic acetylcholine receptor. Br J Pharmacol. 1995;116:2949–2956. doi: 10.1111/j.1476-5381.1995.tb15949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evergren E, Benfenati F, Shupliakov O. The synapsin cycle: a view from the synaptic endocytic zone. J Neurosci Res. 2007;85:2648–2656. doi: 10.1002/jnr.21176. [DOI] [PubMed] [Google Scholar]
- Fernandez-Alfonso T, Ryan TA. The kinetics of synaptic vesicle pool depletion at CNS synaptic terminals. Neuron. 2004;41:943–953. doi: 10.1016/s0896-6273(04)00113-8. [DOI] [PubMed] [Google Scholar]
- Gaffield MA, Rizzoli SO, Betz WJ. Mobility of synaptic vesicles in different pools in resting and stimulated frog motor nerve terminals. Neuron. 2006;51:317–325. doi: 10.1016/j.neuron.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Gitler D, Xu Y, Kao HT, Lin D, Lim S, Feng J, Greengard P, Augustine GJ. Molecular determinants of synapsin targeting to presynaptic terminals. J Neurosci. 2004a;24:3711–3720. doi: 10.1523/JNEUROSCI.5225-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, Augustine GJ. Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J Neurosci. 2004b;24:11368–11380. doi: 10.1523/JNEUROSCI.3795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godenschwege TA, Reisch D, Diegelmann S, Eberle K, Funk N, Heisenberg M, Hoppe V, Hoppe J, Klagges BR, Martin JR, Nikitina EA, Putz G, Reifegerste R, Reisch N, Rister J, Schaupp M, Scholz H, Schwarzel M, Werner U, Zars TD, Buchner S, Buchner E. Flies lacking all synapsins are unexpectedly healthy but are impaired in complex behaviour. Eur J Neurosci. 2004;20:611–622. doi: 10.1111/j.1460-9568.2004.03527.x. [DOI] [PubMed] [Google Scholar]
- Guatimosim C, Hull C, von Gersdorff H, Prado MA. Okadaic acid disrupts synaptic vesicle trafficking in a ribbon-type synapse. J Neurochem. 2002;82:1047–1057. doi: 10.1046/j.1471-4159.2002.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel AW, Simpson LL, Ridge RM, Betz WJ. Synaptic vesicle movements monitored by fluorescence recovery after photobleaching in nerve terminals stained with FM1-43. J Neurosci. 1996;16:3960–3967. doi: 10.1523/JNEUROSCI.16-12-03960.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M, Cooke A, Neef A, Lagnado L. High mobility of vesicles supports continuous exocytosis at a ribbon synapse. Curr Biol. 2004;14:173–183. doi: 10.1016/j.cub.2003.12.053. [DOI] [PubMed] [Google Scholar]
- Horrow JC, Bartkowski RR. Pancuronium, unlike other nondepolarizing relaxants, retains potency at hypothermia. Anesthesiology. 1983;58:357–361. doi: 10.1097/00000542-198304000-00010. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Sudhof TC. Homo- and heterodimerization of synapsins. J Biol Chem. 1999;274:16747–16753. doi: 10.1074/jbc.274.24.16747. [DOI] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan R, Lemke EA, Klingauf J. Visualization of synaptic vesicle movement in intact synaptic boutons using fluorescence fluctuation spectroscopy. Biophys J. 2005;89:2091–2102. doi: 10.1529/biophysj.105.061663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Protti DA, Ferro PA, Rosato S, Uchitel OD. Effects of Ca2+ channel blocker neurotoxins on transmitter release and presynaptic currents at the mouse neuromuscular junction. Br J Pharmacol. 1997;121:1531–1540. doi: 10.1038/sj.bjp.0701290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan JA. Histological and histochemical methods theory and practice. Oxford: Pergamon; 1981. [Google Scholar]
- Kraszewski K, Mundigl O, Daniell L, Verderio C, Matteoli M, De Camilli P. Synaptic vesicle dynamics in living cultured hippocampal neurons visualized with CY3-conjugated antibodies directed against the lumenal domain of synaptotagmin. J Neurosci. 1995;15:4328–4342. doi: 10.1523/JNEUROSCI.15-06-04328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushmerick C, Renden R, von Gersdorff H. Physiological temperatures reduce the rate of vesicle pool depletion and short-term depression via an acceleration of vesicle recruitment. J Neurosci. 2006;26:1366–1377. doi: 10.1523/JNEUROSCI.3889-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, McGuinness TL, Leonard CS, Sugimori M, Greengard P. Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proc Natl Acad Sci USA. 1985;82:3035–3039. doi: 10.1073/pnas.82.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonart G, Simsek-Duran F. Deletion of synapsins I and II genes alters the size of vesicular pools and rabphilin phosphorylation. Brain Res. 2006;1107:42–51. doi: 10.1016/j.brainres.2006.05.092. [DOI] [PubMed] [Google Scholar]
- Lonart G, Sudhof TC. Region-specific phosphorylation of rabphilin in mossy fiber nerve terminals of the hippocampus. J Neurosci. 1998;18:634–640. doi: 10.1523/JNEUROSCI.18-02-00634.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell JW, Townes-Anderson E, Czernik AJ, Cameron R, Greengard P, De Camilli P. Synapsins in the vertebrate retina: absence from ribbon synapses and heterogeneous distribution among conventional synapses. Neuron. 1990;5:19–33. doi: 10.1016/0896-6273(90)90030-j. [DOI] [PubMed] [Google Scholar]
- Martin AR. A further study of the statistical composition on the end-plate potential. J Physiol (Lond) 1955;130:114–122. doi: 10.1113/jphysiol.1955.sp005397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Smith SJ. Strong effects of subphysiological temperature on the function and plasticity of mammalian presynaptic terminals. J Neurosci. 2005;25:7481–7488. doi: 10.1523/JNEUROSCI.1801-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes P, Haines N, Kuppuswamy V, Fleet DJ, Stewart BA. Synaptic vesicle mobility and presynaptic F-actin are disrupted in a N-ethylmaleimide-sensitive factor allele of Drosophila. Mol Biol Cell. 2006;17:4709–4719. doi: 10.1091/mbc.E06-03-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucci TC, Morrow JS. Synapsin I: an actin-bundling protein under phosphorylation control. J Cell Biol. 1987;105:1355–1363. doi: 10.1083/jcb.105.3.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prekeris R, Terrian DM. Brain myosin V is a synaptic vesicle-associated motor protein: evidence for a Ca2+-dependent interaction with the synaptobrevin-synaptophysin complex. J Cell Biol. 1997;137:1589–1601. doi: 10.1083/jcb.137.7.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea R, Li J, Dharia A, Levitan ES, Sterling P, Kramer RH. Streamlined synaptic vesicle cycle in cone photoreceptor terminals. Neuron. 2004;41:755–766. doi: 10.1016/s0896-6273(04)00088-1. [DOI] [PubMed] [Google Scholar]
- Richards DA, Guatimosim C, Rizzoli SO, Betz WJ. Synaptic vesicle pools at the frog neuromuscular junction. Neuron. 2003;39:529–541. doi: 10.1016/s0896-6273(03)00405-7. [DOI] [PubMed] [Google Scholar]
- Richards DA, Rizzoli SO, Betz WJ. Effects of wortmannin and latrunculin A on slow endocytosis at the frog neuromuscular junction. J Physiol (Lond) 2004;557:77–91. doi: 10.1113/jphysiol.2004.062158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Ryan TA. Inhibitors of myosin light chain kinase block synaptic vesicle pool mobilization during action potential firing. J Neurosci. 1999;19:1317–1323. doi: 10.1523/JNEUROSCI.19-04-01317.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Atluri PP, Ryan TA. Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci. 2003;6:127–135. doi: 10.1038/nn1002. [DOI] [PubMed] [Google Scholar]
- Schluter OM, Schnell E, Verhage M, Tzonopoulos T, Nicoll RA, Janz R, Malenka RC, Geppert M, Sudhof TC. Rabphilin knock-out mice reveal that rabphilin is not required for rab3 function in regulating neurotransmitter release. J Neurosci. 1999;19:5834–5846. doi: 10.1523/JNEUROSCI.19-14-05834.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtrahman M, Yeung C, Nauen DW, Bi GQ, Wu XL. Probing vesicle dynamics in single hippocampal synapses. Biophys J. 2005;89:3615–3627. doi: 10.1529/biophysj.105.059295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siksou L, Rostaing P, Lechaire JP, Boudier T, Ohtsuka T, Fejtova A, Kao HT, Greengard P, Gundelfinger ED, Triller A, Marty S. Three-dimensional architecture of presynaptic terminal cytomatrix. J Neurosci. 2007;27:6868–6877. doi: 10.1523/JNEUROSCI.1773-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng H, Wilkinson RS. “Delayed” endocytosis is regulated by extracellular Ca2+ in snake motor boutons. J Physiol (Lond) 2003;551:103–114. doi: 10.1113/jphysiol.2003.041152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuoka H, Goda Y. Myosin light chain kinase is not a regulator of synaptic vesicle trafficking during repetitive exocytosis in cultured hippocampal neurons. J Neurosci. 2006;26:11606–11614. doi: 10.1523/JNEUROSCI.3400-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torri-Tarelli F, Bossi M, Fesce R, Greengard P, Valtorta F. Synapsin I partially dissociates from synaptic vesicles during exocytosis induced by electrical stimulation. Neuron. 1992;9:1143–1153. doi: 10.1016/0896-6273(92)90072-l. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJ, Koh TW, Zhou Y, Bellen HJ. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47:365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Von Kriegstein K, Schmitz F, Link E, Sudhof TC. Distribution of synaptic vesicle proteins in the mammalian retina identifies obligatory and facultative components of ribbon synapses. Eur J Neurosci. 1999;11:1335–1348. doi: 10.1046/j.1460-9568.1999.00542.x. [DOI] [PubMed] [Google Scholar]
- Yang XF, Ouyang Y, Kennedy BR, Rothman SM. Cooling blocks rat hippocampal neurotransmission by a presynaptic mechanism: observations using 2-photon microscopy. J Physiol (Lond) 2005;567:215–224. doi: 10.1113/jphysiol.2005.088948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung C, Shtrahman M, Wu XL. Stick-and-diffuse and caged diffusion: a comparison of two models of synaptic vesicle dynamics. Biophys J. 2007;92:2271–2280. doi: 10.1529/biophysj.106.081794. [DOI] [PMC free article] [PubMed] [Google Scholar]