Mitochondrial Trafficking to Synapses in Cultured Primary Cortical Neurons (original) (raw)
Abstract
Functional synapses require mitochondria to supply ATP and regulate local [Ca2+]i for neurotransmission. Mitochondria are thought to be transported to specific cellular regions of increased need such as synapses. However, little is known about how this occurs, including the spatiotemporal distribution of mitochondria relative to presynaptic and postsynaptic sites, whether mitochondria are dynamically recruited to synapses, and how synaptic activity affects these trafficking patterns. We used primary cortical neurons in culture that form synaptic connections and show spontaneous synaptic activity under normal conditions. Neurons were cotransfected with a mitochondrially targeted cyan fluorescent protein and an enhanced yellow fluorescent protein-tagged synaptophysin or postsynaptic density-95 plasmid to label presynaptic or postsynaptic structures, respectively. Fluorescence microscopy revealed longer dendritic mitochondria that occupied a greater fraction of neuronal process length than axonal mitochondria. Mitochondria were significantly more likely to be localized at synaptic sites. Although this localization was unchanged by inhibition of synaptic activity by tetrodotoxin, it increased in dendritic synapses and decreased in axonal synapses during overactivity by veratridine. Mitochondrial movement and recruitment to synapses also differed between axons and dendrites under basal conditions and when synaptic activity was altered. Additionally, we show that movement of dendritic mitochondria can be selectively impaired by glutamate and zinc. We conclude that mitochondrial trafficking to synapses is dynamic in neurons and is modulated by changes in synaptic activity. Furthermore, mitochondrial morphology and distribution may be optimized differentially to best serve the synaptic distributions in axons and dendrites. Last, selective cessation of mitochondrial movement in dendrites suggests early postsynaptic dysfunction in neuronal injury and degeneration.
Keywords: morphology, mitochondria, excitotoxicity, transport, synaptophysin, PSD-95
Introduction
Mitochondria are vital to the function of cells because they are the main source of energy and because they regulate [Ca2+]i homeostasis. Because mitochondria are thought to be synthesized perinuclearly, they must be trafficked appropriately to meet demands throughout the cell (Wong-Riley, 1989; Miller, 1992; Davis and Clayton, 1996). This is particularly important in neurons in which processes can extend great distances from the cell body. Mitochondrial movement is dynamic, with individual organelles exhibiting various velocities and motility patterns in axons and dendrites (Overly et al., 1996; Ligon and Steward, 2000a). Kinesins and dyneins transport the organelles on microtubules and actin filaments, likely through interaction with accessory proteins such as Milton and syntabulin (Hirokawa et al., 1990; Nangaku et al., 1994; Morris and Hollenbeck, 1995; Tanaka et al., 1998; Ligon and Steward, 2000b; Stowers et al., 2002; Cai et al., 2005). Mitochondrial morphology is also dynamic and can be regulated, for example, through fusion and fission (for review, see Rube and van der Bliek, 2004). It is thought that an elongated morphology may confer bioenergetic advantages to ATP generation and dispersal (for review, see Skulachev, 2001). However, there is very little evidence directly relating mitochondrial morphology to mitochondrial function.
Although mitochondria are thought to be trafficked to specific cellular regions with high metabolic demand or elevated [Ca2+]i, few specific targets for mitochondrial recruitment have been unambiguously identified (Morris and Hollenbeck, 1993; Yi et al., 2004). An obvious site that requires mitochondria on neuronal processes is the synapse. Electron microscopy demonstrated mitochondria closely associated with synapses and tethered to vesicle release sites (Kageyama and Wong-Riley, 1982; Rowland et al., 2000). Synaptic transmission requires mitochondrial ATP generation and control of local [Ca2+]i for neurotransmitter exocytosis, vesicle recruitment, activation of ion conductances, signaling at metabotropic receptors, potentiation of neurotransmitter release, and synaptic plasticity (Bindokas et al., 1998; Zucker, 1999; David and Barrett, 2000, 2003; Zenisek and Matthews, 2000; Calupca et al., 2001; Billups and Forsythe, 2002; Vanden Berghe et al., 2002; Kann et al., 2003; Yang et al., 2003). However, recent studies in mutant Drosophila neurons with perinuclearly clustered mitochondria also suggest that direct mitochondrial localization to presynaptic terminals may be less critical for neurotransmission processes under physiological conditions than for Ca2+ buffering and mobilization of reserve pool vesicles during tetanic stimulation (Guo et al., 2005; Verstreken et al., 2005). The question then arises whether and how dynamic mitochondrial trafficking patterns and regulation of morphology mediate appropriate synaptic support, both presynaptically and postsynaptically. Some evidence exists for mitochondrial recruitment to dendritic spines during morphogenesis in hippocampal neurons after repetitive depolarizing stimulation (Li et al., 2004). However, the normal trafficking patterns of mitochondria to synapses in spontaneously active neurons remains to be elucidated.
Because mitochondria are important for supporting synaptic transmission, it is likely that impairment of mitochondrial transport could result in synaptic dysfunction in addition to inadequate distribution of ATP and sequestration of [Ca2+]i. This is particularly critical in injured neurons because many insults are known to stop mitochondrial movement (Rintoul et al., 2003; Malaiyandi et al., 2005; Reynolds and Santos, 2005). The work of Li et al. (2004) and Guo et al. (2005) suggests that different mechanisms account for mitochondrial distribution in axons and dendrites. For example, formation of dendritic spines but not of synaptic boutons may require local recruitment of mitochondria. Therefore, if mitochondrial movement in axons and dendrites has different susceptibilities to neuronal insults, the result could be selective dysfunction of presynaptic or postsynaptic neurotransmission. In this study, we used cultured primary rat cortical neurons in which we previously demonstrated abundant expression of synaptic proteins and spontaneous [Ca2+]i transients that are synchronous between cells and mediated by glutamatergic, cholinergic, and GABAergic signaling (Chang and Reynolds, 2006) (K. E. Dineley, D. T. W. Chang, and I. J. Reynolds, unpublished observations). We show not only the differential trafficking patterns of mitochondria to presynaptic terminals and postsynaptic densities but also reveal dynamic modulation of mitochondrial trafficking, morphology, and distribution that accompany changes in synaptic activity. Additionally, we provide evidence for heightened vulnerability of dendritic mitochondria to movement cessation after neurotoxic glutamate and zinc exposure, suggestive of dysfunction in dendrites before axons.
Materials and Methods
Materials.
All reagents were purchased from Sigma (St. Louis, MO) unless otherwise noted.
Cell culture.
All animals used for this study were maintained in accordance with the University of Pittsburgh Guidelines for the Care and Use of Animals. Primary neurons were dissociated for culture from the cortices of embryonic day 17 Sprague Dawley rat pups and grown under serum-free conditions as described previously (Malaiyandi et al., 2005).
Transfection of cortical cultures.
The mitochondrially targeted cyan fluorescent protein (mito-CFP) construct was obtained from Clontech (Mountain View, CA) and targets cytochrome c oxidase subunit VIII. The postsynaptic density-95 (PSD-95)–enhanced yellow fluorescent protein (eYFP) and synaptophysin–eYFP plasmids were provided by Dr. Ann Marie Craig (University of British Columbia, Vancouver, British Columbia, Canada). The mito-eYFP construct obtained from Dr. Roger Tsien (University of California, San Diego, La Jolla, CA) uses a cytochrome c oxidase subunit IV mitochondrial localization sequence (Llopis et al., 1998). Plasmids were purified and amplified using the Qiagen (Valencia, CA) Plasmid Maxi kit according to the instructions of the manufacturer. Cotransfections were performed on neurons after 11 d in vitro, using 1 μg each of mito-CFP and PSD-95–eYFP or synaptophysin–eYFP and 2.5 μl of Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) in an added volume of 250 μl of DMEM. Transfections with mito-eYFP were performed with 1 μg of plasmid and otherwise identical conditions. Media was completely replaced with conditioned media 6–8 h after transfection, and neurons were imaged 3–4 d after transfection.
Imaging.
Cells were perfused with HBSS adjusted to pH 7.4 with NaOH and composed of the following (in mm): 137 NaCl, 5 KCl, 10 NaHCO3, 20 HEPES, 5.5 glucose, 0.6 KH2PO4, 0.6 Na2HPO4, 1.4 CaCl2, and 0.9 MgSO4. HBSS was warmed to 37°C and flowed at a rate of 5 ml/min throughout the duration of each experiment. For fluorescence recording, we used a BX61WI Olympus Optical (Tokyo, Japan) microscope, a CCD camera (Hamamatsu, Shizouka, Japan) and a Lambda-LS xenon arc lamp light source with a Lambda 10-2 optical filter changer (Sutter Instruments, Novato, CA). Imaging was performed at a rate of one frame every 10 s at a magnification of 60× except when noted, using Compix (Cranberry, PA) imaging systems and SimplePCI software (Compix). Dual-wavelength fluorescence imaging was performed using the following excitation wavelengths: 440/495 nm for mito-CFP and eYFP-tagged synaptic components and 340/380 nm for fura-2 AM. Single-wavelength fluorescence imaging of mito-eYFP was performed at a 495 nm excitation wavelength.
Measurement and pharmacologic modulation of spontaneous [Ca2+]i fluxes.
Spontaneous [Ca2+]i transients were represented by fluxes in fura-2 AM ratios. Cells were loaded with 5 μm fura-2 AM/HBSS (Invitrogen) for 15–20 min at 37°C. Neurons were imaged at a rate of one frame every 3 s at 40× magnification. [Ca2+]i was represented as the fura-2 ratio of cell bodies after subtraction of background fluorescence using SimplePCI. Drug treatments were 1 h preincubation with 200 nm tetrodotoxin (TTX) prepared from a 1 mm stock dissolved in distilled water, 24 h preincubation with 1 μm TTX, or 30 min pretreatment with 250 nm veratridine prepared from a 1 mm stock dissolved in ethanol. Experiments were performed during the 30 min after each pretreatment with continuous perfusion of 200 nm TTX or 250 nm veratridine.
Analysis of mitochondrial length, occupancy, and number.
SimplePCI functions were used to determine average mitochondrial length, occupancy, and number in a subfield that excluded proximal processes clearly containing more than one mitochondrion in their cross-section. User-defined thresholds for pixel intensity and object size were used to identify the total number of measurable mitochondria in a frame. Length was calculated from skeletonized objects, which consisted of a one-pixel-wide line running the length of each mitochondrial object. All neuronal processes in the field were then converted into line objects, their total length was calculated, and mitochondrial occupancy was computed as total mitochondrial length/process length. A Student's t test was used for comparisons between axonal and dendritic controls. A one-way ANOVA with Dunnett's multiple comparison test to control was used to analyze data in axons or dendrites after pharmacologic treatments.
Analysis of synapse number, mitochondrial localization, and movement parameters relative to synaptic sites.
Synapses were designated using only the eYFP image so that the observer was blinded to the location of mitochondria. Clusters of eYFP-labeled PSD-95 and synaptophysin proteins were identified as stable synaptic components if they were ∼1 μm in diameter and did not display net lateral movement >1 μm during the course of each 30 min experiment. Therefore, small or mobile fluorescent clusters were excluded from our analysis. The total number of synaptophysin or PSD-95 clusters identified in this manner was divided by the total neuronal process length in a given subfield to yield the number of synapses per micrometer. A Student's t test was used for comparisons between axonal and dendritic controls. A one-way ANOVA with Dunnett's multiple comparison test to control was used to analyze data in axons or dendrites after pharmacologic treatments.
Using SimplePCI, rectangular regions of interest (ROIs) with a width of 1.1 μm were placed around each identified synaptic component cluster using the eYFP image. As controls, ROIs were placed along the same neuronal processes in locations that were void of eYFP-labeled clusters during the 30 min experiment. Mitochondrial parameters were then analyzed from imaging files containing the ROIs and only the CFP image so that the observer was blinded to the synaptic contents of each ROI. The mitochondrial localization and movement parameters that were measured are shown in Table 1. Mitochondria occupying at least half of the width of an ROI were considered to be localized in that ROI. A mitochondrion was considered to occupy an ROI for as long as the mitochondrial edge did not completely move out of the ROI. The number of mitochondria moving past each ROI was counted over the entire course of each movie, and those that localized in the ROI according to the definition above were considered to stop during their trajectory. The length of time that these moving mitochondria stopped in ROIs was also measured. A Student's paired t test was used for comparisons between synaptic and nonsynaptic sites in the same cells. A one-way ANOVA with Dunnett's multiple comparison test to control was used to analyze data in axons or dendrites after pharmacologic treatments.
Table 1.
Measured parameters of mitochondrial movement
| Parameter | Method of measurement |
|---|---|
| Mitochondrial localization frequency | Fraction of ROIs that contain mitochondria |
| Mitochondrial residence time | Fraction of total movie length (∼30 min) that the mitochondria initially found in an ROI remained stationary |
| Number of passing mitochondria per minute | Calculated from the number of mitochondria moving past each ROI during the entire movie |
| Fraction of passing mitochondria that stop | Fraction of mitochondria moving past an ROI that localized in that ROI for at least one imaging frame |
Measurement of drug-induced changes in mitochondrial movement and morphology.
For experiments with glutamate treatment, axons were identified in cortical neurons by selective expression of synaptophysin–eYFP, and dendrites were identified by selective expression of PSD-95–eYFP. Cells were cotransfected with mito-CFP. Proximal and distal axon segments were imaged 3–4 and 10–12 imaging fields away from the cell soma, which corresponded to 0.2–0.5 and 1–1.4 mm, respectively. Cells were perfused for 10 min with HBSS, followed by 10 min with 30 μm glutmate/1 μm glycine, followed by a 10 min HBSS wash. Mitochondrial length was measured as described above using individual frames corresponding to the start of HBSS perfusion, the end of glutamate treatment, and the end of HBSS wash. Mitochondrial roundness was computed from the same frames as length using a SimplePCI function as 4π × area/√perimeter. Overall mitochondrial movement was measured during the first 190 s of HBSS perfusion, the last 190 s of glutamate treatment, and the last 190 s of HBSS wash as described previously (Rintoul et al., 2003). Briefly, a custom Visual Basic macro was used to quantify movement as average event count/average number of mitochondrial pixels, in which event count is the number of corresponding pixels that vary by at least 20 fluorescence units between two consecutive frames, and the number of mitochondrial pixels is the number of pixels per frame that are 20 fluorescence units above background fluorescence. The value of 20 units was chosen because a masking function in SimplePCI software effectively identified mitochondria as pixels 20 units above background fluorescence.
For experiments involving FCCP (carbonyl cyanide _p_-trifluoromethoxyphenylhydrazone), oligomycin, zinc, rotenone, and 4-bromo-A23187 (4-Br-A23187) treatment, neurons were transfected with mito-eYFP, and axons and dendrites were imaged at a rate of one frame every 6 s. Movement was analyzed using the macro described above during the following 2 min time periods: before drug treatment, at the end of drug perfusion, and at the end of a 15–20 min HBSS wash. The treatment conditions were 5 min perfusion with 750 nm FCCP, 10 min perfusion with 10 μm oligomycin, 5 min perfusion with 2 μm rotenone, 5 min perfusion with 1 μm 4-Br-A23187, or 10 min perfusion with 3 μm ZnCl2/20 μm Na-pyrithione. ZnCl2 treatment was followed by a 5 min perfusion with 25 μm zinc chelator N,N,N′,_N′_-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN). CaCl2 was omitted from HBSS in experiments involving FCCP and ZnCl2. For each cell, mitochondrial movement during treatment and after washout was normalized to movement before treatment. A Student's paired t test was used for statistical analysis of movement recovery.
For all experiments, transfected neurons were selected for imaging if they exhibited moving mitochondria with the elongated morphology characteristic of healthy cells. Each independent experiment consisted of one cell per coverslip treated with a given drug, repeated on two to six coverslips each from two to five different cultures.
Results
Many studies examining synaptic mitochondria use electron microscopy and are limited to static images (Rowland et al., 2000; Sakata and Jones, 2003; Briones et al., 2005). More recently, synaptic function was investigated in mutant neurons that exhibited abnormal distribution and transport of mitochondria (Guo et al., 2005; Verstreken et al., 2005). However, it is difficult to account for the dynamics of normal mitochondrial movement and morphology in these models. Li et al. (2004) recently demonstrated recruitment of mitochondria to dendritic spines undergoing morphogenesis. We now provide new insight into the normal trafficking of mitochondria to both presynaptic and postsynaptic sites, thereby advancing our understanding of how the mitochondrial pool is allotted to meet dynamic demands throughout the cell.
Mitochondria are visualized concurrently with presynaptic and postsynaptic labels
Mitochondria were imaged simultaneously with presynaptic or postsynaptic sites by cotransfecting primary cortical neurons with a mito-CFP and either the presynaptic vesicle protein synaptophysin or the postsynaptic density protein PSD-95 tagged to an eYFP. A single neuron transfected with mito-CFP, synaptophysin-eYFP, and PSD-95–monomeric red fluorescent protein (mRFP) is shown in Figure 1A. Figure 1, B and C, shows labeled mitochondria with PSD-95 clusters in a dendrite and with synaptophysin clusters in axon branches, respectively. Axonal and dendritic differences in synaptic density and mitochondrial morphology and distribution were readily observed. Localization of mitochondria to a subset of presynaptic and postsynaptic sites was also seen. These mitochondrial morphology and distribution patterns were quantified and are discussed below.
Figure 1.

Mitochondrial localization and trafficking were visualized relative to presynaptic and postsynaptic sites by cotransfecting neurons with fluorescent proteins. A, Primary cortical neuron transfected with mito-CFP (red), presynaptic label synaptophysin–eYFP (green), and postsynaptic label PSD-95–mRFP (blue). B, Dendritic segment from a transfected neuron showing mito-CFP-labeled mitochondria (green) and PSD-95–eYFP clusters (red). C, Axonal branches from a transfected neuron showing mito-CFP-labeled mitochondria (green) and synaptophysin–eYFP clusters (red). Images are representative of three to five cultures. Scale bars, 10 μm.
Time-lapse fluorescence imaging permitted us to measure multiple parameters of mitochondrial movement patterns. Imaging movies demonstrated the variability and complexity of mitochondrial trafficking to presynaptic and postsynaptic sites (supplemental data 1, available at www.jneurosci.org as supplemental material). Specifically, we observed mitochondria that (1) localized to synapses for long or short time periods, (2) paused at synaptic sites as they traveled unidirectionally along a neuronal process, (3) shuttled bidirectionally between multiple neighboring synapses, (4) moved to and briefly resided at a synapse before seeming to divide, leaving one fragment at the synapse while the remaining fragment moved elsewhere, and (5) elongated to bridge nearby synapses. To study the more accessible characteristics of mitochondrial trafficking to synapses, we measured the parameters listed in Table 1. Three populations of mitochondria with specific movement patterns emerged from our data, as diagramed in Figure 2A. These included relatively immobile mitochondria that remained stationary for long time periods (generally >15 min) and mobile mitochondria that paused at specific locations for shorter (<20 s) or longer (mean of 1.5–2 min; data not shown) time periods. We distinguished sites at which mitochondria halted by whether they contained a synaptic component. The trafficking patterns of these mitochondrial populations are discussed in detail below.
Figure 2.

Populations of mitochondria with different movement patterns were observed, and their trafficking patterns were analyzed in a blinded manner. A, Mitochondria that were relatively immobile tended to remain stationary at synaptic and nonsynaptic sites for >15 min. Relatively mobile mitochondria exhibited saltatory movement, making stops at synaptic and nonsynaptic sites for shorter or longer time periods. B, Representative axonal image prepared for blinded data analysis shows ROIs placed around synaptophysin clusters (red boxes) and ROIs placed at nonsynaptic locations (blue boxes). C, Corresponding mitochondrial image with ROIs, shown here before box colors were made uniform, was used to measure the localization and trafficking parameters listed in Table 1. Scale bar, 10 μm.
Because mitochondria localized to synapses are thought to be functional in providing ATP or regulating local [Ca2+]i, we predicted that synaptic overactivity would induce mitochondrial recruitment to synapses and synaptic quiescence would release localized mitochondria to perform other duties. The spontaneous synaptic firing of neurons in culture was overactivated by veratridine, an inhibitor of Na+ channel inactivation. As a counterpart, we silenced synaptic activity with TTX, a blocker of voltage-sensitive Na+ channels in excitable membranes. Synaptic activity was monitored by the fluorescent [Ca2+]i indicator fura-2 AM, as the spontaneous and synchronous [Ca2+]i transients that cultured neurons exhibit when firing. As shown in Figure 3, veratridine caused intense, high-magnitude [Ca2+]i spiking activity, whereas TTX treatment completely suppressed [Ca2+]i transients. We confirmed that these activity patterns were maintained throughout the duration of our drug treatments, and drug was continuously perfused on cells during each imaging experiment.
Figure 3.

Spontaneous synaptic activity of cultured neurons was pharmacologically modulated to determine the effects on mitochondrial trafficking. A, Spontaneous [Ca2+]i fluxes in the cell somas of all neurons in an imaging field measured by fura-2 AM were indicative of basal synaptic activity and were amplified in magnitude by 250 nm veratridine. B, [Ca2+]i spiking was inhibited by 200 nm TTX. Traces represent 10–13 cells from a single coverslip. Experiments were repeated two to three times each on three separate cultures.
Mitochondrial morphology and distribution differ in axons and dendrites and are differentially modulated by changes in synaptic activity
The transfected presynaptic and postsynaptic markers were used as guides for differentiating axons from dendrites. PSD-95 was clearly expressed in all but one process, and synaptophysin was expressed in only one process. Additionally, axons were morphologically distinguished from dendrites by their extensive length, small diameter, lack of attenuation, and right-angle branch points. The morphology of mitochondria and their distribution relative to synaptic sites was clearly variable between axons and dendrites (Fig. 1). First, mitochondria were more fully distributed through dendrites, occupying a large fraction of neuronal process length. In contrast, mitochondria were visibly shorter and more sparsely distributed throughout axons. The distribution of presynaptic sites and postsynaptic densities also differed; axons were far less densely populated by synaptophysin clusters than were dendrites populated by PSD-95 clusters.
In accordance with these observations, we found that mitochondria occupied 26.3 ± 0.02% of any given dendritic segment, which was significantly greater than the 17.4 ± 0.02% mitochondrial occupation of any given axonal segment (p < 0.01) (Fig. 4A). This increase in mitochondrial occupancy of dendritic process length was attributed to a significant increase in mitochondrial length, 2.2 ± 0.1 μm in dendrites compared with 1.4 ± 0.1 μm in axons (p < 0.0001) (Fig. 4B). However, the number of discrete mitochondria per micrometer of neuronal process was equivalent in axons and dendrites (Fig. 4C). Interestingly, the increased mitochondrial length and occupancy in dendrites correlated with a twofold increase in density of PSD-95 clusters on dendrites compared with synaptophysin clusters on axons (p < 0.01) (Fig. 4D). Therefore, mitochondria seem to be more fully distributed through processes that have high synaptic density.
Figure 4.

Mitochondrial distribution and morphology differed between axons and dendrites and was differentially modulated by changes in synaptic activity. A, Mitochondrial occupancy of axons was significantly lower than that of dendrites. This was measured as the total length of all mitochondria divided by the length of all neuronal processes in a given field. Treatment with 1 μm TTX for 24 h or 250 nm veratridine for 30 min reduced mitochondrial occupancy of axons but increased occupancy of dendrites. B, Mitochondrial length was significantly shorter in axons than in dendrites. Treatment with TTX for 24 h increased mitochondrial length in dendrites. Veratridine also reduced mitochondrial length in axons. C, Number of mitochondria per micrometer of neuronal process was similar between axons and dendrites and was not affected by changes in synaptic activity. D, Significantly more stationary clusters of PSD-95 populated dendrites compared with synaptophysin clusters on axons. TTX and veratridine treatments reduced the number of stationary PSD-95 clusters. Values are shown as mean ± SE from two to three coverslips each from four to five separate cultures. p < 0.05 was considered significant, in which + represents comparisons between axons and dendrites of untreated cells, and ∗ represents comparisons between untreated cells and pharmacologically treated cells.
When synaptic activity was modulated, mitochondrial morphology and distribution surprisingly changed in opposite directions on axons compared with dendrites. Figure 4A demonstrates that 24 h TTX treatment reduced mitochondrial occupancy of axons from 17.4 ± 0.02 to 11.2 ± 0.01% (p < 0.05) but increased mitochondrial occupancy of dendrites from 26.2 ± 0.02 to 38.8 ± 0.03% (_p_ < 0.01). The change in mitochondrial occupancy in dendrites was coincident with a significant increase in mitochondrial length but no significant change in the number of mitochondria in neuronal processes (Fig. 4_B_,_C_). Veratridine treatment resulted in a significantly shorter mitochondrial length and a trend toward reduced mitochondrial occupancy in axons but increased mitochondrial occupancy in dendrites. It is important to note that all pharmacologic manipulations of synaptic activity reduced the number of stationary PSD-95 clusters per micrometer of dendritic processes (_p_ < 0.05) (Fig. 4_D_). However, treatments never altered the total number of labeled clusters (data not shown); thus, the effects seen in Figure 4_D_ are a result of increased cluster mobility. For our measurements, mobility was considered to be net movement >1 μm over the course of 30 min. Our criterion for cluster motility was necessarily strict to permit data to be analyzed blindly. In summary, these results demonstrate that mitochondrial morphology was dynamically modulated by global changes in synaptic activity. Furthermore, axonal and dendritic mitochondria responded differently to single pharmacologic manipulations, suggesting that distinct mechanisms operate within these neuronal compartments to alter mitochondrial morphology and distribution.
Mitochondria localize to synaptic sites on axons and dendrites for extended time periods
We observed mitochondria localized to subsets of presynaptic and postsynaptic sites for varying time periods. To measure mitochondrial localization frequency at synaptic sites, we placed ROIs of width 1.1 μm around synaptophysin and PSD-95 clusters as well as throughout neuronal processes in which such clusters were absent (Fig. 2B). The presence of mitochondria in ROIs and the length of time those mitochondria remained stationary were then blindly scored (Fig. 2C). During a 30 min span of imaging, we found that the mitochondria that were localized in ROIs tended to reside there for at least 15 min and therefore likely represented a generally stationary pool of mitochondria (Fig. 2A).
Our results demonstrate that mitochondria localized significantly to presynaptic and postsynaptic sites. The percentage of synaptophysin clusters that contained mitochondria was 36.1 ± 0.06% compared with the 14.3 ± 0.03% chance of finding mitochondria at locations on axons that did not contain synaptophysin clusters (p < 0.05) (Fig. 5A). Furthermore, mitochondria that localized to labeled presynaptic terminals remained stationary for 77.0 ± 0.09% of the movie duration, significantly more than the 47.4 ± 0.07% that mitochondria resided at nonsynaptic sites (p < 0.05) (Fig. 5B). Interestingly, veratridine treatment caused mitochondria in axons to (1) localize to synaptic and nonsynaptic sites at similar frequencies and (2) remain stationary at synaptic sites for similar time periods as at nonsynaptic sites (Fig. 5A,B).
Figure 5.

Mitochondria localized significantly to presynaptic and postsynaptic sites, and their distribution relative to synapses changed in response to altered synaptic activity. A, Mitochondrial localization frequency to presynaptic sites was measured as the fraction of synaptophysin clusters that contained mitochondria (white bars) compared with the fraction of randomly selected sites void of synaptophysin clusters that contained mitochondria (black bars). Mitochondria localized preferentially to synaptophysin clusters in untreated and TTX-treated cells but not in veratridine-treated cells. B, Residence times of the mitochondria localized to synaptic and nonsynaptic sites in A were compared. Residence time was calculated as the length of time that the colocalized mitochondria remained stationary divided by the duration time of the imaging movie. Mitochondria resided significantly longer at synaptophysin clusters than nonsynaptic sites in untreated and TTX-treated cells but not veratridine-treated cells. C, Mitochondrial localization frequency at PSD-95 clusters was measured as in A. Mitochondria localized preferentially to PSD-95 clusters, and veratridine treatment increased this localization frequency relative to untreated cells. D, Residence time of mitochondria at PSD-95 clusters was measured as described in B. Treatment with 200 nm TTX for 1 h reduced mitochondrial residence times in dendrites, yet mitochondria resided at PSD-95 clusters longer than at nonsynaptic sites. Values are shown as mean ± SE from two to three coverslips each from three to five separate cultures. p < 0.05 was considered significant, in which ∗ represents comparisons between synaptic sites and nonsynaptic sites in the same cells, and + represents comparisons between untreated cells and pharmacologically treated cells.
Mitochondria also localized to postsynaptic sites on dendrites but exhibited different patterns of distribution than on axons. Mitochondria colocalized to 39.3 ± 0.05% of PSD-95 clusters compared with 27.4 ± 0.05% at sites that did not contain postsynaptic densities (p < 0.05) (Fig. 5C). The modest colocalization of mitochondria at postsynaptic sites can be attributed to the high fractional occupancy of dendrites by mitochondria and thus increased frequency of finding mitochondria at nonsynaptic sites on dendrites compared with axons (Fig. 4A). We found that mitochondria were relatively stationary in dendrites, and, as a result, they resided at both synaptic and nonsynaptic sites for nearly the entire duration of our 30 min imaging experiments (Fig. 5D). However, results from TTX treatment show that, after 1 h, mitochondria remained stationary for shorter time periods and actually resided at sites containing PSD-95 clusters significantly longer than at sites void of clusters (p < 0.05) (Fig. 5D). Conversely, veratridine treatment caused mitochondria to associate with 59.2 ± 0.06% of PSD-95 clusters compared with 39.3 ± 0.05% of clusters in control (p < 0.05) (Fig. 5C). These results indicate that mitochondria significantly colocalize with a subset of presynaptic and postsynaptic sites and selectively reside at presynaptic terminals for extended time periods. Furthermore, changes in synaptic activity can modulate mitochondrial distribution to synaptic sites differently in axons and dendrites.
Mitochondrial movement is greater in axons than dendrites and increases during synaptic inactivity
We measured mitochondrial movement as the number of mitochondria that move past ROIs placed along neuronal processes over the course of 30 min. It is important to note that our measurement of mitochondrial movement, or flux, can be influenced by both the number of moving mitochondria and the velocity with which they move. Mitochondrial movement past synaptic sites was identical to nonsynaptic sites (data not shown). This suggests that we were measuring a population of highly mobile mitochondria that made brief stops lasting well under 30 min. We therefore distinguish this population of mitochondria from the generally immobile population described above and in Figure 5.
Remarkably, more mitochondrial movement was observed in axons compared with dendrites: 0.13 ± 0.02 versus 0.03 ± 0.008 mitochondria moved past an ROI per minute in axons and dendrites, respectively (p < 0.001) (Fig. 6A). Synaptic silence by TTX for 1 h caused mitochondrial movement to increase dramatically in all neuronal processes, with flux rates of 0.21 ± 0.02 mitochondria/ROI/min (p < 0.05) in axons and 0.09 ± 0.01 mitochondria/ROI/min (p < 0.01) in dendrites. Movement remained somewhat elevated in dendrites after 24 h of TTX treatment but completely returned to control values in axons. Conversely, veratridine treatment had no significant effects on movement in either axons or dendrites.
Figure 6.
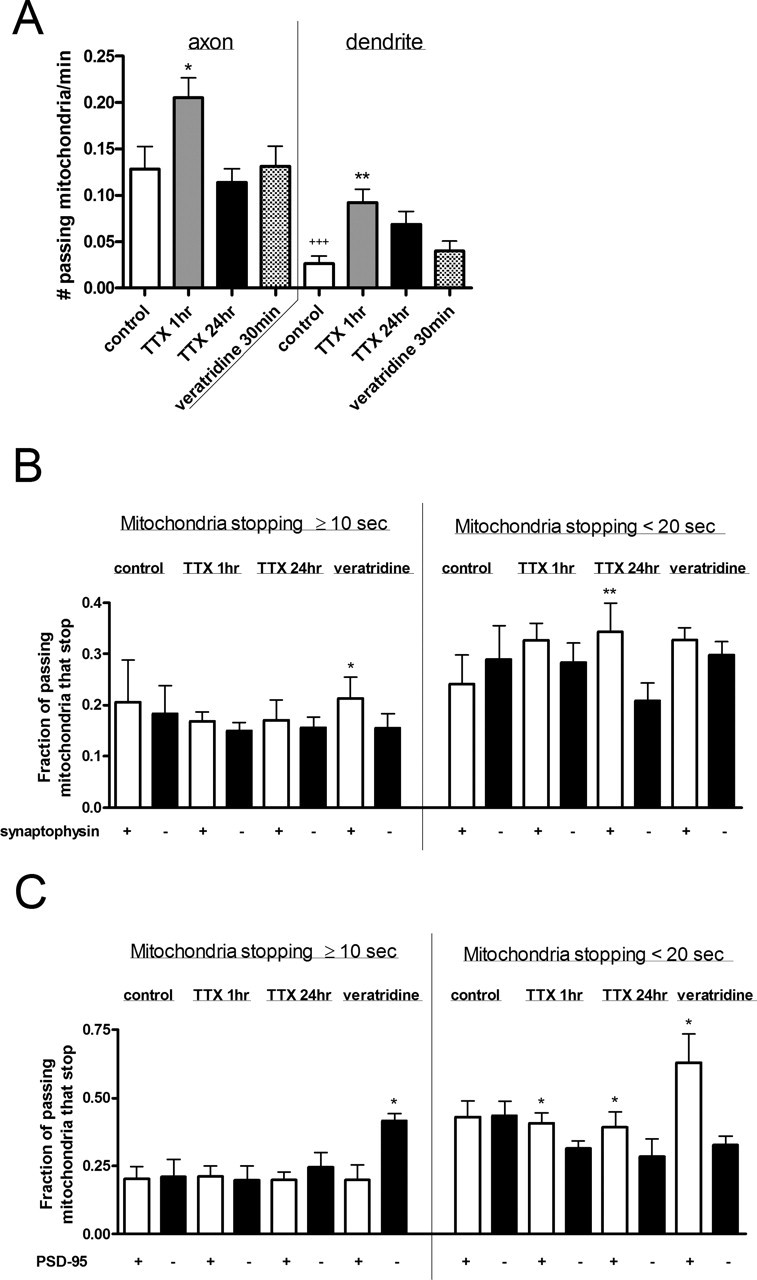
Mitochondrial movement patterns differed between axons and dendrites and was altered in response to changes in synaptic activity. A, Mitochondrial movement was greater in axons than dendrites and was increased in all processes by 1 h treatment with 200 nm TTX. Movement was measured as the number of mitochondria passing a given point on a neuronal process over time. B, The fraction of passing mitochondria measured in A that paused at synaptophysin clusters (white bars) on axons was compared with the fraction that paused at nonsynaptic sites (black bars). Mitochondria that stopped for at least two imaging frames, equivalent to ≥10 s (mean 1.5 min), showed a preference toward synaptophysin clusters after 30 min treatment with 250 nm veratridine. Mitochondria that stopped for one imaging frame, equivalent to <20 s, showed a preference toward synaptophysin clusters after 24 h treatment with 1 μm TTX. C, The fraction of passing mitochondria measured in A that paused at PSD-95 clusters (white bars) on dendrites was compared with the fraction that paused at nonsynaptic sites (black bars). Mitochondria that stopped for at least two imaging frames, equivalent to ≥10 s (mean of 2 min), showed a preference toward PSD-95 clusters after veratridine treatment. Mitochondria that stopped for one imaging frame, equivalent to <20 s, showed a preference toward nonsynaptic sites after TTX and veratridine treatments. Values are shown as mean ± SE from two to three coverslips each from three to five separate cultures. p < 0.05 was considered significant. In A, + represents comparisons between axons and dendrites of untreated cells, and ∗ represents comparisons between untreated cells and pharmacologically treated cells. In B and C, ∗ represents comparisons between synaptic sites and nonsynaptic sites in the same cells.
The dramatic increase in mitochondrial movement in both axons and dendrites after 1 h TTX treatment suggests a transition of mitochondria from the stationary state to the mobile state, as well as an increase in mobility of already moving mitochondria (Fig. 6A). In support of this, we observed that dendritic but not axonal mitochondria remained stationary for significantly shorter time periods after 1 h TTX (Fig. 5B,D). Therefore, moving mitochondria likely have increased velocity in axons and dendrites; additionally, more mitochondria probably become mobile in dendrites. Our results are in agreement with the effects of TTX in hippocampal dendrites (Li et al., 2004).
Saltatory movement of mitochondria is modulated by changes in synaptic activity
Overall mitochondrial movement in neuronal processes can be primarily attributed to a mobile subset of mitochondria that is distinct from a stationary subset of mitochondria. Mobile mitochondria have been reported to account for 5–20 or 35–45% of the total mitochondrial pool in cultured hippocampal neurons (Overly et al., 1996; Ligon and Steward, 2000a). These mitochondria exhibit saltatory movement, pausing briefly along their trajectory and also changing direction midcourse. As diagramed in Figure 2A, some mitochondria, most often those that travel with high velocity, paused for increments under 20 s; others paused for longer time periods, on average 1.5 min in axons and 2 min in dendrites (data not shown). Therefore, we separated these two groups of mitochondria as those that stopped for <20 s (one frame) and those that stopped for ≥10 s (at least two frames), as shown in supplemental data 2 (available at www.jneurosci.org as supplemental material). Some obligatory overlap in these groups exists because of limitations of time-lapse image acquisition, but no mitochondria were counted in both groups.
Our measurements of mitochondria that stopped at synaptic sites and at sites that did not contain synaptophysin or PSD-95 clusters indicate that such movement patterns change in response to altered synaptic activity. In the basal state, moving mitochondria were equally likely to stop at a given location regardless of whether a synapse was present (Fig. 6B,C). However, among mitochondria that paused for <20 s, a greater fraction stopped at labeled presynaptic terminals after 24 h TTX treatment (p < 0.01), and a greater fraction stopped at PSD-95 clusters after 1 and 24 h TTX compared with sites that did not contain synaptic components (p < 0.05). The same pattern of selectivity toward pausing at synaptic sites was seen after veratridine treatment among mitochondria that stopped for ≥10 s on axons and <20 s on dendrites (p < 0.05). There was also a notable increase in the fraction of mitochondria that stopped for ≥10 s at nonsynaptic sites relative to postsynaptic sites on dendrites after veratridine treatment (p < 0.05). Therefore, saltatory movement to both synaptic and nonsynaptic sites was altered by changes in synaptic activity.
Mitochondrial movement and morphology are differentially susceptible to neurotoxins in axons and dendrites
Our data demonstrate that axonal and dendritic mitochondria have different patterns of movement, morphology, and distribution under control conditions and when synaptic activity is altered (Figs. 4–6). We next sought to determine whether drugs that trigger mitochondrial or neuronal injury impair mitochondrial movement or cause morphologic remodeling of mitochondria selectively in one compartment over the other. Mitochondrial movement is thought to be important for neuronal health, and mitochondrial morphology may impart some functional property to the organelles. Therefore, if mitochondria in certain neuronal processes are more vulnerable to cessation of movement or morphological remodeling during injury, then those processes may also be more likely to degenerate. This could provide a mechanism for selective axonal or dendritic dysfunction and degeneration in neuronal injury and disease.
We and others previously showed that mitochondria in injured and apoptotic neurons and other cell types stop moving and adopt a punctate, fragmented morphology rather than the traditional thin, elongated shape (Bossy-Wetzel et al., 2003; Rintoul et al., 2003; Vanden Berghe et al., 2004). The remodeling of mitochondrial morphology can be directly visualized during an acute excitotoxic glutamate exposure and is thought to be mediated by Ca2+ influx into the cell (Rintoul et al., 2003). To further that investigation, we examined the ability of glutamate to stop movement and cause rounding of mitochondria in dendrites, proximal axon segments, and distal axon segments. Remodeling of mitochondrial morphology was observed in dendrites (Fig. 7A,B) but not in axonal segments at least 200 μm from the cell body (Fig. 7C,D). Quantification revealed that glutamate reduced the length of dendritic mitochondria to approximately one-fourth the initial length and well below the length of axonal mitochondria (Fig. 7E). Additionally, only dendritic mitochondria rounded after glutamate treatment (Fig. 7F).
Figure 7.
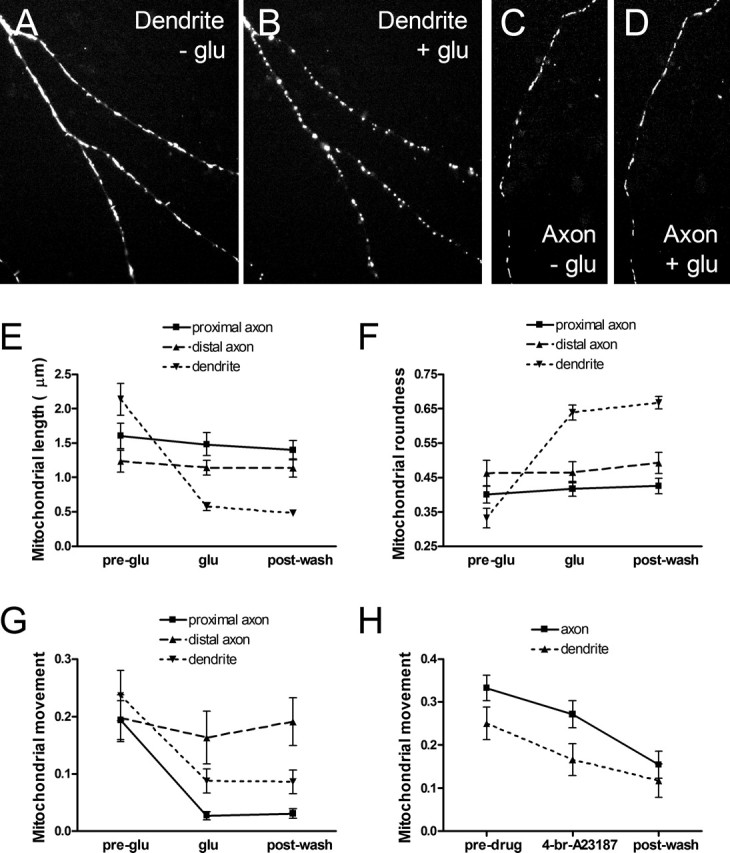
Acute treatment with excitotoxic glutamate concentrations caused remodeling of mitochondrial morphology solely in dendrites and cessation of mitochondrial movement in dendrites and proximal axon segments. A, B, Dendritic mitochondria displayed morphological remodeling and are shown before and after a 10 min treatment with 30 μm glutamate (glu)/1 μm glycine. C, D, Axonal mitochondria did not demonstrate morphological remodeling and are shown before and after glutamate treatment. E, Mitochondrial length shortened in dendrites but not axons after glutamate treatment. No recovery was observed after a 10 min wash. F, Mitochondria rounded selectively in dendrites after glutamate treatment with no recovery after a 10 min wash. G, Mitochondrial movement (measured as average event count/average number of mitochondrial pixels) in dendrites and proximal axon segments decreased significantly after glutamate treatment but was unaffected in distal axon segments. H, Mitochondrial movement was reduced in all neuronal processes when [Ca2+]i was uniformly increased by a 5 min treatment with 1 μm 4-Br-A23187, a Ca2+ ionophore. Note that the difference in mitochondrial morphology precludes the absolute values for axonal and dendritic movement to be compared by our measurement technique in G and H. Also, movement values should not be compared between G and H because images were acquired at different rates. Values are shown as mean ± SE from two to three coverslips each from three to four separate cultures. p < 0.05 was considered significant.
Analysis of mitochondrial movement after glutamate treatment revealed an interesting susceptibility of mitochondria in dendrites and proximal axon segments to cessation, whereas mitochondria in distal axons were unaffected (Fig. 7G). We tested the hypothesis that the spatial differences in cessation of movement were attributable to [Ca2+]i gradients after glutamate treatment, which were expected to be elevated near glutamate receptors on dendrites and the cell body, and decline progressively from the cell body into the axonal compartment as [Ca2+]i is buffered. Perfusion of cells with the Ca2+ ionophore 4-Br-A23187 reduced mitochondrial movement in all neuronal processes (Fig. 7H). At higher concentrations of ambient Ca2+, 4-Br-A23187 also caused rounding of mitochondria in all cellular compartments (data not shown). Therefore, short-term excitotoxic glutamate treatment reduces mitochondrial movement before it causes rounding, and these effects are most likely attributable to the distribution of elevated [Ca2+]i, which is particularly elevated in the dendritic compartment in which glutamate receptors are located.
We next tested whether other drugs that impair mitochondrial function or are related to neurotoxicity also stopped mitochondrial movement selectively in dendrites. Unlike glutamate treatment, which causes elevated [Ca2+]i primarily in the dendritic compartment, the drugs we tested here presumably gain equivalent entry into axons and dendrites. We examined the effects of acute (1) depolarization of mitochondrial membrane potential (ψm) by FCCP, (2) inhibition of mitochondrial ATP synthase by oligomycin, (3) elevation of intracellular zinc to neurotoxic concentrations followed by chelation with TPEN, and (4) inhibition of electron transport chain (ETC) complex I by rotenone. In contrast to glutamate, these four drug treatments reduced mitochondrial movement to a similar degree between axons and dendrites.
However, we found an interesting difference in acute recovery of mitochondrial movement after washout of drug (Fig. 8). Only axonal mitochondria regained movement after FCCP (p < 0.01). This recovery could be attributable to a faster rate of mitochondrial repolarization in axons, perhaps because the electrochemical gradient can be better reestablished in mitochondria of smaller volume. However, we observed mitochondria in the cell body fully repolarize 8–10 min after FCCP washout, well before dendritic mitochondria recover movement (data not shown). In contrast to the partially reversible effects of FCCP, mitochondrial movement in neither axons nor dendrites recovered from nearly irreversible inhibition of ATP synthase or ETC complex I by oligomycin and rotenone, respectively. These data confirm that mitochondrial ATP production is important for mitochondrial movement throughout all neuronal processes. Last, only axonal mitochondria regained movement after zinc/TPEN treatment (p < 0.05). It is important to note that mitochondrial movement in axons did not recover after zinc treatment unless TPEN was applied (data not shown). In summary, mitochondrial movement in dendrites was shown to be selectively impaired or unable to acutely recover from glutamate, FCCP, and zinc/TPEN treatment. This has important implications for abnormal dispersion of ATP and sequestration of Ca2+ by mobile mitochondria in dendrites after neuronal insults.
Figure 8.

Axonal and dendritic mitochondrial movement were equally susceptible to cessation after FCCP, oligomycin, rotenone, and zinc exposures, but only axonal mitochondria acutely recovered movement after washout of FCCP and chelation of zinc. A, Mitochondrial movement (measured as average event count/average number of mitochondrial pixels) in axons significantly recovered 15 min after depolarization with 5 min 750 nm FCCP treatment and 20 min after exposure to 10 min 3 μm ZnCl2/20 μm Na-pyrithione (pyr) treatment followed by chelation with 5 min 25 μm TPEN. Movement did not recover after 10 min treatment with 10 μm oligomycin or 5 min treatment with 2 μm rotenone. B, Mitochondrial movement in dendrites did not acutely recover after treatment with FCCP, oligomycin, or zinc/pyrithione followed by TPEN. Movement continued to decrease significantly in dendrites 20 min after rotenone was washed out. Values were normalized to mitochondrial movement before treatment and are shown as mean ± SE from two to six coverslips each from two to four separate cultures. p < 0.05 was considered significant.
Discussion
Our results demonstrate that mitochondrial morphology, distribution, and movement differ in axons and dendrites, yet mitochondria in both compartments localize to and remain stationary at a subset of synaptic sites. Furthermore, we reveal that these dynamic properties of mitochondria change under conditions of synaptic silence and overactivity. Last, we provide the first evidence for selective dendritic alterations in mitochondrial morphology and movement after neurotoxic glutamate and zinc treatments that may prove crucial to our understanding of excitotoxic and ischemic cell death.
Axonal and dendritic mitochondria differ in morphology, distribution, and movement
We show that the number of mitochondria per micrometer of neuronal process is similar in axons and dendrites, but mitochondria occupy a smaller fraction of axonal process length because of a shorter morphology (Fig. 4A–C). Furthermore, we correlate this with a lower density of synaptophysin clusters than postsynaptic densities (Fig. 4D). It is possible that longer mitochondria can more efficiently distribute energy to multiple, closely apposed sites of high metabolic demand on dendrites (for review, see Skulachev, 2001). This explanation coincides with evidence that dendritic mitochondria are more metabolically active than axonal mitochondria in hippocampal neurons (Overly et al., 1996). Three-dimensional ultrastructural reconstructions demonstrated long filamentous networks of mitochondria in dendrites and discrete mitochondrial bodies in axons of hippocampal neurons from ground squirrels and rats (Popov et al., 2005). Although we did not observe such extensive mitochondrial networks in rat cortical neurons, we certainly found similar patterns of mitochondrial distribution and morphology.
Presynaptic terminals require mitochondria for sequestering Ca2+, powering the plasma membrane Ca2+-ATPase and releasing Ca2+ for posttetanic potentiation (Tang and Zucker, 1997; Zenisek and Matthews, 2000; Calupca et al., 2001; Medler and Gleason, 2002). Mitochondrial ATP generation may also be important for the dynamic actin rearrangements associated with vesicle cycling and synaptic plasticity (for review, see Dillon and Goda, 2005). Therefore, increased mitochondrial transport in axons is probably necessary for delivery of organelles to synaptophysin clusters that are spaced farther apart and distributed along much greater distances than postsynaptic densities on dendrites (Figs. 4D, 6A). Mitochondrial movement in our experiments could be elevated by both a larger number of motile mitochondria and an increased velocity in axons, as was concluded in hippocampal neurons by Overly et al. (1996). Furthermore, it is possible that a shorter mitochondrial morphology is maintained in axons so that molecular motors can more efficiently transport the organelles.
Manipulation of spontaneous synaptic activity of cultured neurons demonstrated that mitochondrial morphology and distribution can be differentially altered in axons and dendrites by a single drug treatment. Changes in mitochondrial length are likely attributable to elongation or shortening of mitochondria so that mitochondrial surface area rather than volume is mainly affected. Although this type of morphology change might not affect mitochondrial functional capacity, it might have an important impact on the surface area over which ATP can diffuse or Ca2+ can be buffered. Mitochondrial fission and fusion are also known to occur rapidly and could cause a change in volume and functional capacity by loss or gain of mitochondrial proteins. However, this is the less likely scenario in our experiments because the number of mitochondria per micrometer remains stable during all drug treatments. Yet we cannot exclude the possibility that the large mitochondrial pool near the cell soma is trafficked into processes for fusion, and achievement of a homeostatic change in mitochondrial dynamics could explain the increased length but unchanged number of dendritic mitochondria observed after 24 h TTX treatment. Dendritic mitochondria may lengthen after prolonged TTX treatment in an attempt to bridge stationary postsynaptic densities that became fewer and farther apart (Fig. 4B,D). The increase in PSD-95 cluster movements we observed may represent changes in the motility, restructuring, and turnover of spines, all of which are known to be modulated by synaptic activity (Kirov and Harris, 1999; Okabe et al., 1999; Konur and Yuste, 2004). Additionally, PSD-95 proteins themselves exhibit a degree of dynamics (Rasse et al., 2005). In summary, we provide the first evidence for the distinct compartmental regulation of mitochondrial morphology and distribution in axons and dendrites, both in the basal state and during altered synaptic activity.
Mitochondrial localization and trafficking to presynaptic and postsynaptic sites
Presynaptic terminals have been described as containing abundant mitochondria (Kageyama and Wong-Riley, 1982; Nguyen et al., 1997; Popov et al., 2005). It was therefore surprising that we found mitochondria at only ∼36% of presynaptic labels in resting neurons (Fig. 5A). This is similar to an ultrastructural study finding mitochondria at ∼41% of synaptic boutons in axons of rat hippocampal neurons (Shepherd and Harris, 1998). Given the ability of mitochondria to rapidly move through processes, it is likely that transient metabolic demands are met by moving mitochondria or by nearby but not specifically localized mitochondria. Conversely, synaptic sites requiring longer-term support, perhaps for posttetanic potentiation and mobilization of reserve pool vesicles, may act as stable “docking sites” for mitochondria (Tang and Zucker, 1997; Verstreken et al., 2005). Indeed, we found that mitochondria that localized to presynaptic sites remained stationary for the majority of our 30 min imaging experiments (Fig. 5B). Furthermore, our examination of developing neurons that have not yet formed functional synapses revealed significantly more mitochondrial movement, presumably attributable to the absence of synaptic docking sites (Chang and Reynolds, 2006).
In general, the spontaneous synaptic activity exhibited by our cortical cultures despite mitochondrial localization at only a subset of presynaptic terminals agrees with reports of normal neurotransmission during physiological levels of stimulation in mutant dMiro and drp1 Drosophila axons with abnormally distributed mitochondria (Guo et al., 2005; Verstreken et al., 2005). Those studies also concluded that presynaptic mitochondrial functions are especially important during overstimulation. However, we did not observe recruitment of mitochondria to synaptophysin clusters during veratridine treatment other than a modest increase in the fraction of moving mitochondria that briefly paused at presynaptic sites (Figs. 5A,B, 6B). In fact, the redistribution trend of relatively stationary mitochondria was more toward nonsynaptic sites on axons, possibly to power ion channels for membrane repolarization (Fig. 5A,B). It is possible that the normal mitochondrial distribution still allowed adequate Ca2+ buffering, because [Ca2+]i transients continued to show full recovery (Fig. 3A).
The distribution of mitochondria relative to postsynaptic densities and dendritic spines is not clearly defined. Unlike presynaptic terminals, mitochondria in hippocampal neurons are not usually found in dendritic spines, although they have been seen penetrating the bases of thorny excrescences (Popov et al., 2005). Our results show PSD-95 clusters colocalized with mitochondria more frequently than did nonsynaptic sites (Fig. 5C). Stimulation with veratridine caused significantly more PSD-95 clusters to be associated with mitochondria and more moving mitochondria to briefly pause at postsynaptic densities (Figs. 5C, 6C). This suggests that mitochondrial proximity is important for supporting processes at excitatory postsynaptic sites, such as local [Ca2+]i regulation in conjunction with smooth endoplasmic reticulum (Pivovarova et al., 2002). Additionally, local ATP supply may power synapse-associated polyribosome complexes and clathrin-dependent endocytic machinery at postsynaptic sites (Steward and Levy, 1982; Racz et al., 2004). Unfortunately, we could not precisely resolve spine morphology at PSD-95 clusters; thus, it is possible that subtypes of spines, such as those with shorter necks or fewer Ca2+ pumps, are more dependent on mitochondrial uptake of Ca2+ that diffuses into the dendrite (Pivovarova et al., 1999, 2002; Majewska et al., 2000). Some PSD-95 clusters that recruited mitochondria after stimulation could also represent spines undergoing morphogenesis (Li et al., 2004). Alternatively, mitochondria may be recruited to provide ATP for ubiquitination and proteasome-dependent degradation of PSD-95, which has been shown to occur after NMDA treatment of hippocampal neurons in the time frame of our experiments (Colledge et al., 2003).
Interestingly, we found that, despite the remarkable increase in mitochondrial movement after 1 h TTX (Fig. 6A), there were negligible or modest changes in the fraction of passing mitochondria that stopped at synaptic and nonsynaptic sites (Fig. 6B,C). In other words, mitochondria increased overall mobility but without targeted destinations. Furthermore, the fraction of synaptic sites associated with mitochondria remained unaffected after 24 h TTX (Fig. 5A,C). Therefore, we conclude that inactive synapses still serve as targets for mitochondrial localization. This implies that (1) structural rather than activity-dependent mechanisms may cause mitochondria to stop at synaptic sites and (2) presynaptic and postsynaptic compensation mechanisms may require mitochondrial support, as evidenced by a moderate preference for moving mitochondria to stop at synaptic sites during TTX treatment (Fig. 6B,C).
Dendrite-specific impairments of mitochondrial movement in injury
Our results suggest that inadequate mitochondrial movement may be a more important pathologic process in dendrites than axons during glutamate- and zinc-mediated injury. Dendrites may consequently experience abnormal ATP distribution, [Ca2+]i dysregulation, and production of harmful reactive oxygen species by damaged mitochondria that are improperly removed. In light of our findings that mitochondrial morphology, localization, and trafficking to synapses are dynamic in the basal state and when synaptic activity is altered, we predict that impaired mitochondrial movement will also lead to aberrant neurotransmission.
We showed previously that acute exposure to excitotoxic glutamate concentrations caused mitochondria to stop moving and round in neuronal processes attributable to elevated Ca2+ influx through NMDA receptors that disrupts cytosolic integrity (Rintoul et al., 2003). We now confirm that this is a regional effect related to the distribution of glutamate receptors on dendrites because axonal mitochondria exhibited the same response only when [Ca2+]i was uniformly elevated with a Ca2+ ionophore (Fig. 7). Furthermore, our observation that mitochondria in proximal but not distal axons also stopped moving after glutamate treatment suggests that movement cessation precedes morphological remodeling and [Ca2+]i is relatively well buffered spatially. However, despite preserved mitochondrial movement in the majority of the axon, impaired trafficking to and from the cell body could have consequences on the distribution of mitochondria and clearance of damaged organelles by retrograde transport (Hollenbeck, 1993).
We also demonstrate that chelation of neurotoxic but non-ψm-depolarizing zinc concentrations caused a selective recovery of movement by axonal mitochondria (Fig. 8) (Malaiyandi et al., 2005). In these experiments, zinc was applied concurrently with the zinc ionophore pyrithione, so axons and dendrites received the same zinc exposure. We reported previously that the Zn2+/TPEN treatment conditions used here cause irreversible, phosphatidylinositol 3 (PI3)-kinase-dependent cessation of mitochondrial movement in undistinguished neuronal processes and >75% cell death in cultured cortical neurons (Malaiyandi et al., 2005). We now advance those findings by showing that zinc chelation actually leads to significant acute recovery of mitochondrial movement selectively in axons, which does not recover without chelation. Therefore, the TPEN-independent PI3-kinase signaling cascade might be activated by acute zinc exposure only in dendrites or at a much slower rate in axons. These data strongly suggest that our previous finding of significant neurotoxicity after acute Zn2+/TPEN treatment is related to a dendritic pathology in which impaired mitochondrial movement is an early event.
Footnotes
This work was supported by National Institutes of Health Grants NS049560 (I.J.R.) and T32 GM008424 (D.T.W.C.). We thank Dr. Ann Marie Craig for generous provision of the eYFP-tagged PSD-95 and synaptophysin plasmids. We are also grateful to Dr. Kirk Dineley for his work on spontaneous activity and to Dr. Olga Vergun, Dr. Donald DeFranco, and Reynolds laboratory members for helpful discussions and critical reading of this manuscript.
References
- Billups B, Forsythe ID (2002). Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci 22:5840–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindokas VP, Lee CC, Colmers WF, Miller RJ (1998). Changes in mitochondrial function resulting from synaptic activity in the rat hippocampal slice. J Neurosci 18:4570–4587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA (2003). Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol 15:706–716. [DOI] [PubMed] [Google Scholar]
- Briones TL, Suh E, Jozsa L, Rogozinska M, Woods J, Wadowska M (2005). Changes in number of synapses and mitochondria in presynaptic terminals in the dentate gyrus following cerebral ischemia and rehabilitation training. Brain Res 1033:51–57. [DOI] [PubMed] [Google Scholar]
- Cai Q, Gerwin C, Sheng Z-H (2005). Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J Cell Biol 170:959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calupca MA, Prior C, Merriam LA, Hendricks GM, Parsons RL (2001). Presynaptic function is altered in snake K+-depolarized motor nerve terminals containing compromised mitochondria. J Physiol (Lond) 532:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DTW, Reynolds IJ (2006). Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience in press. [DOI] [PubMed]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD (2003). Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40:595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett EF (2000). Stimulation-evoked increases in cytosolic [Ca2+] in mouse motor nerve terminals are limited by mitochondrial uptake and are temperature-dependent. J Neurosci 20:7290–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett EF (2003). Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol (Lond) 548:425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AF, Clayton DA (1996). In situ localization of mitochondrial DNA replication in intact mammalian cells. J Cell Biol 135:883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon C, Goda Y (2005). The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci 28:25–55. [DOI] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE (2005). The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47:379–393. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Yoshida T, Kawashima T (1990). Brain dynein (MAP1C) localizes on both anterogradely and retrogradely transported membranous organelles in vivo. J Cell Biol 111:1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ (1993). Products of endocytosis and autophagy are retrieved from axons by regulated retrograde organelle transport. J Cell Biol 121:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama GH, Wong-Riley MT (1982). Histochemical localization of cytochrome oxidase in the hippocampus: correlation with specific neuronal types and afferent pathways. Neuroscience 7:2337–2361. [DOI] [PubMed] [Google Scholar]
- Kann O, Kovacs R, Heinemann U (2003). Metabotropic receptor-mediated Ca2+ signaling elevates mitochondrial Ca2+ and stimulates oxidative metabolism in hippocampal slice cultures. J Neurophysiol 90:613–621. [DOI] [PubMed] [Google Scholar]
- Kirov SA, Harris KM (1999). Dendrites are more spiny on mature hippocampal neurons when synapses are inactivated. Nat Neurosci 2:878–883. [DOI] [PubMed] [Google Scholar]
- Konur S, Yuste R (2004). Imaging the motility of dendritic protrusions and axon terminals: roles in axon sampling and synaptic competition. Mol Cell Neurosci 27:427–440. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K-I, Hayashi Y, Sheng M (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119:873–887. [DOI] [PubMed] [Google Scholar]
- Ligon LA, Steward O (2000a). Movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J Comp Neurol 427:340–350. [DOI] [PubMed] [Google Scholar]
- Ligon LA, Steward O (2000b). Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J Comp Neurol 427:351–361. [DOI] [PubMed] [Google Scholar]
- Llopis J, McCaffery JM, Miyawaki A, Farquhar MG, Tsien RY (1998). Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc Natl Acad Sci USA 95:6803–6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewska A, Brown E, Ross J, Yuste R (2000). Mechanisms of calcium decay kinetics in hippocampal spines: role of spine calcium pumps and calcium diffusion through the spine neck in biochemical compartmentalization. J Neurosci 20:1722–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaiyandi LM, Honick AS, Rintoul GL, Wang QJ, Reynolds IJ (2005). Zn2+ inhibits mitochondrial movement in neurons by phosphatidylinositol 3-kinase activation. J Neurosci 25:9507–9514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medler K, Gleason EL (2002). Mitochondrial Ca2+ buffering regulates synaptic transmission between retinal amacrine cells. J Neurophysiol 87:1426–1439. [DOI] [PubMed] [Google Scholar]
- Miller RJ (1992). Neuronal Ca2+: getting it up and keeping it up. Trends Neurosci 15:317–319. [DOI] [PubMed] [Google Scholar]
- Morris R, Hollenbeck P (1995). Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol 131:1315–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ (1993). The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci 104:917–927. [DOI] [PubMed] [Google Scholar]
- Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N (1994). KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell 79:1209–1220. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Marin L, Atwood HL (1997). Synaptic physiology and mitochondrial function in crayfish tonic and phasic motor neurons. J Neurophysiol 78:281–294. [DOI] [PubMed] [Google Scholar]
- Okabe S, Kim HD, Miwa A, Kuriu T, Okado H (1999). Continual remodeling of postsynaptic density and its regulation by synaptic activity. Nat Neurosci 2:804–811. [DOI] [PubMed] [Google Scholar]
- Overly CC, Rieff HI, Hollenbeck PJ (1996). Organelle motility and metabolism in axons vs dendrites of cultured hippocampal neurons. J Cell Sci 109:971–980. [DOI] [PubMed] [Google Scholar]
- Pivovarova NB, Hongpaisan J, Andrews SB, Friel DD (1999). Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: spatial and temporal characteristics. J Neurosci 19:6372–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivovarova NB, Pozzo-Miller LD, Hongpaisan J, Andrews SB (2002). Correlated calcium uptake and release by mitochondria and endoplasmic reticulum of CA3 hippocampal dendrites after afferent synaptic stimulation. J Neurosci 22:10653–10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov V, Medvedev NI, Davies HA, Stewart MG (2005). Mitochondria form a filamentous reticular network in hippocampal dendrites but are present as discrete bodies in axons: a three-dimensional ultrastructural study. J Comp Neurol 492:50–65. [DOI] [PubMed] [Google Scholar]
- Racz B, Blanpied TA, Ehlers MD, Weinberg RJ (2004). Lateral organization of endocytic machinery in dendritic spines. Nat Neurosci 7:917–918. [DOI] [PubMed] [Google Scholar]
- Rasse TM, Fouquet W, Schmid A, Kittel RJ, Mertel S, Sigrist CB, Schmidt M, Guzman A, Merino C, Qin G, Quentin C, Madeo FF, Heckmann M, Sigrist SJ (2005). Glutamate receptor dynamics organizing synapse formation in vivo. Nat Neurosci 8:898–905. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ, Santos S (2005). Rotenone inhibits movement and alters morphology of mitochondria in cultured forebrain neurons. Soc Neurosc Abstr 31:1017.19. [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ (2003). Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci 23:7881–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland KC, Irby NK, Spirou GA (2000). Specialized synapse-associated structures within the calyx of Held. J Neurosci 20:9135–9144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rube DA, van der Bliek AM (2004). Mitochondrial morphology is dynamic and varied. Mol Cel Biochem 256–257:331–339. [DOI] [PubMed] [Google Scholar]
- Sakata JT, Jones TA (2003). Synaptic mitochondrial changes in the motor cortex following unilateral cortical lesions and motor skills training in adult male rats. Neurosci Lett 337:159–162. [DOI] [PubMed] [Google Scholar]
- Shepherd GMG, Harris KM (1998). Three-dimensional structure and composition of CA3→CA1 axons in rat hippocampal slices: implications for presynaptic connectivity and compartmentalization. J Neurosci 18:8300–8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skulachev VP (2001). Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci 26:23–29. [DOI] [PubMed] [Google Scholar]
- Steward O, Levy WB (1982). Preferential localization of polyribosomes under the base of dendritic spines in granule cells of the dentate gyrus. J Neurosci 2:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL (2002). Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36:1063–1077. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N (1998). Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 93:1147–1158. [DOI] [PubMed] [Google Scholar]
- Tang Y-G, Zucker RS (1997). Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron 18:483–491. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe P, Kenyon JL, Smith TK (2002). Mitochondrial Ca2+ uptake regulates the excitability of myenteric neurons. J Neurosci 22:6962–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe P, Hennig GW, Smith TK (2004). Characteristics of intermittent mitochondrial transport in guinea pig enteric nerve fibers. Am J Physiol Gastrointest Liver Physiol 286:G671–G682. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJT, Koh T-W, Zhou Y, Bellen HJ (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47:365–378. [DOI] [PubMed] [Google Scholar]
- Wong-Riley MTT (1989). Cytochrome oxidase: an endogenous metabolic marker for neuronal activity. Trends Neurosci 12:94–101. [DOI] [PubMed] [Google Scholar]
- Yang F, He X-P, Russell J, Lu B (2003). Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na+-Ca2+ exchanger and protein kinase C. J Cell Biol 163:511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Weaver D, Hajnoczky G (2004). Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol 167:661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenisek D, Matthews G (2000). The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron 25:229–237. [DOI] [PubMed] [Google Scholar]
- Zucker RS (1999). Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol 9:305–313. [DOI] [PubMed] [Google Scholar]