Neural Stem/Progenitor Cells Participate in the Regenerative Response to Perinatal Hypoxia/Ischemia (original) (raw)
Abstract
Perinatal hypoxia/ischemia (H/I) is the leading cause of neurologic injury resulting from birth complications. Recent advances in critical care have dramatically improved the survival rate of infants suffering this insult, but ∼50% of survivors will develop neurologic sequelae such as cerebral palsy, epilepsy or cognitive deficits. Here we demonstrate that tripotential neural stem/progenitor cells (NSPs) participate in the regenerative response to perinatal H/I as their numbers increase 100% by 3 d and that they alter their intrinsic properties to divide using expansive symmetrical cell divisions. We further show that production of new striatal neurons follows the expansion of NSPs. Increased proliferation within the NSP niche occurs at 2 d after perinatal H/I, and the proliferating cells express nestin. Of those stem-cell related genes that change, the membrane receptors Notch1, gp-130, and the epidermal growth factor receptor, as well as the downstream transcription factor Hes5, which stimulate NSP proliferation and regulate stem cellness are induced before NSP expansion. The mechanisms for the reactive expansion of the NSPs reported here reveal potential therapeutic targets that could be exploited to amplify this response, thus enabling endogenous precursors to restore a normal pattern of brain development after perinatal H/I.
Keywords: regeneration, cerebral palsy, Notch, self-renewal, neurogenesis, subventricular zone
Introduction
Perinatal hypoxia/ischemia (H/I) is a disruption of blood and oxygen delivery to the brain and represents a primary cause of neurologic injury in the newborn. Perinatal H/I occurs in 1–2 of 1000 live term births, and ∼50% of surviving preterm infants sustain permanent brain damage from this type of injury (Volpe, 2000). Neurologic sequelae include cerebral palsy, epilepsy, and cognitive deficits. Current therapeutic strategies are aimed at preventing brain damage, but, at present, there are no effective means to repair the brain once damage has occurred. Moreover, because accumulating evidence suggests that most of these insults occur in utero, prevention may prove difficult, demanding that regenerative strategies be pursued to reduce the morbidity associated with these events.
The brain possesses a limited capacity for endogenous regeneration after various insults, including ischemia, traumatic injury, and focal apoptosis (Magavi et al., 2000; Arvidsson et al., 2002; Parent et al., 2002; Goings et al., 2004). Most of these investigations focused on neuronal production after injury to the adult brain, but perinatal H/I occurs during a critical period of brain growth and development. Neurogenesis alone would, therefore, be insufficient to properly restore development. White matter and its resident oligodendrocyte progenitors are especially vulnerable to perinatal brain injuries (Ness et al., 2001; Back et al., 2002). It is well established that neural stem cells (NSCs) and multiple classes of transit-amplifying progenitors, some of which express qualities of a true stem cell under certain conditions, reside in the subventricular zone (SVZ) of the mammalian brain throughout life (Reynolds et al., 1992; Palmer et al., 1995; Sanai et al., 2004). Because it is still not possible to discern NSCs from multipotential progenitors that possess limited self-renewal, we adopted the term neural stem/progenitor cells (NSPs) to encompass both populations. NSPs provide the greatest potential to regenerate not only neurons but also the various macroglia required to reconstitute a fully functional brain.
The demonstration that neurogenesis occurs after ischemic injury presents two possible alternatives: (1) ischemic injury directly affects committed neuroblasts, which then expand and mature into neurons, or (2) ischemic injury affects unrestricted precursors that subsequently produce new neuroblasts. We hypothesized that ischemic injury first stimulates tripotential precursors. If true, this would provide additional hope that a broader range of cells could be replaced, a necessity for achieving functional recovery after an ischemic insult.
To test our hypothesis, we analyzed NSPs at early recovery time points using a rat model of perinatal H/I. Here we demonstrate that perinatal H/I increases the proliferation of cells that are positionally and phenotypically NSPs. We show that twice as many NSP-derived neurospheres can be generated from the affected SVZ, that precursors within these neurospheres exhibit more frequent symmetric divisions, and that a greater proportion of the spheres generated from the damaged hemisphere are multipotential. Finally, we find that perinatal H/I induces the expression of multiple genes within SVZ known to influence stem cellness. These results encourage the pursuit of therapies to amplify the compensatory response initiated by the endogenous tripotential, self-renewing precursors of the SVZ.
Parts of this paper have been published previously (Snyder, 2001; Felling et al., 2003).
Materials and Methods
Perinatal hypoxia/ischemia model.
Timed pregnant Wistar rats (Charles River Laboratories, Wilmington, DE) were maintained at the Pennsylvania State College of Medicine by the Department of Comparative Medicine, an Association for Assessment and Accreditation of Laboratory Animal Care accredited facility. Animal experimentation was in accordance with research guidelines set forth by Pennsylvania State University and the Society for Neuroscience policy on the use of animals in neuroscience research. All animals were fed high-fat laboratory chow (Harlan Teklad, Madison, WI). After normal delivery, the litter size was adjusted to 10 pups per litter. Cerebral H/I was produced in 6-d-old rats [day of birth being postnatal day 0 (P0)] by a permanent unilateral common carotid ligation, followed by systemic hypoxia (Rice et al., 1981; Vannucci and Vannucci, 1997). Briefly, pups were lightly anesthetized with isofluorane (4% induction, 2% maintenance). Once fully anesthetized, a midline neck incision was made and the right common carotid artery (CCA) was identified. The CCA was separated from the vagus nerve and then ligated using 3-0 silk. The incision was then sutured, and animals were returned to the dam for 3 h. The pups were prewarmed for 20 min in jars submerged in a 37°C water bath. They were then exposed to 1.5 h of 8% O2/92% N2. After this hypoxic interval, the pups were returned to their dam for recovery periods of 1, 2, and 3 d, at which time, they were either decapitated for neurosphere assays or anesthetized and killed by intracardiac perfusion for immunohistochemistry. Sham-operated animals were anesthetized, and the carotid was isolated from the vagus but not ligated. They were not subjected to a hypoxic interval. Control animals were separated from the dam for the same amount of time as experimental animals but were otherwise not manipulated. In all cases, the contralateral and ipsilateral hemispheres from experimental animals were examined separately.
Tissue fixation and bromodeoxyuridine labeling.
Animals received intraperitoneal injections of 50 mg/kg bromodeoxyuridine (BrdU) either once 1 h before being killed at 48 h (see Fig. 2) or twice daily on post-H/I days 3, 4, and 5, followed by perfusion at 3 (n = 6) and 5 (n = 6) weeks of recovery (see Fig. 4). Animals were anesthetized with a mixture of ketamine (75 mg/kg) and xylazine (5 mg/kg) before intracardiac perfusion with 3% paraformaldehyde in PBS. The brain was then extracted and blocked, with cuts made at ∼2 mm from the anterior pole of the brain and 3 mm from the posterior pole of the cerebrum. The sections were then incubated in 3% paraformaldehyde for 2 h and cryoprotected using 20% sucrose with NaN3 overnight. The 20% sucrose solution was replaced with 30% sucrose for 4–6 h. The tissue was removed and frozen in embedding medium (O.C.T.; Miles, Elkart, IN) on a dry ice/ethanol slush. Frozen coronal sections (12 μm) were cut on a cryostat and mounted on Superfrost Plus microscope slides. Sections at 48 recovery were stained with anti-BrdU (1:10; Cappel, West Chester, PA) as described previously (Levison et al., 1996).
Figure 2.

Hypoxia/ischemia increases the number of BrdU+ cells adjacent to or among the ependymal lining in the ipsilateral SVZ. Control and experimental animals received a single intraperitoneal injection of BrdU 1 h before being killed at 48 h recovery from hypoxia/ischemia. Cryostat tissue sections were immunostained for BrdU incorporation (brown) and counterstained with hematoxylin (blue). A, BrdU labeling in a control brain. B, C, BrdU labeling in the ipsilateral hemisphere. Cells with BrdU-labeled nuclei occasionally appeared to have a cytoplasmic extension contacting the lateral ventricle in the ipsilateral SVZ (arrow in C). D, Quantitative results for the number of BrdU-positive cells per section from ipsilateral and contralateral hemispheres. *p < 0.05. Scale bar: A, B, 30 μm; C, 10 μm.
Figure 4.

A subset of newly generated cells differentiate along the neuronal lineage after perinatal H/I. Sections from the contralateral (A) and ipsilateral (B) hemisphere of animals killed at 3 weeks of recovery were processed for Dcx immunostaining. Whereas Dcx staining was primarily restricted to the SVZ in the contralateral hemisphere (as well as in controls), there were numerous cells with migratory profiles streaming out of the SVZ into the striatum of the ipsilateral hemisphere. When BrdU was injected between 3 and 5 d recovery after H/I and animals killed at 3 weeks after H/I, numerous BrdU+/Dcx+ cells could be found in the ipsilateral striatum (C). Another cohort of animals was perfused 5 weeks after H/I, and BrdU+/NeuN+ cells could be found in the ipsilateral striatum. All images were captured using a Zeiss LSM 410 confocal microscope. Scale bars: A, B, 100 μm; C, 50 μm; D, 10 μm.
Antibodies and immunohistochemistry.
Immunohistochemistry was performed on cryostat brain sections collected at 48 h of recovery. Primary antibodies were mouse monoclonal IgG2a anti-proliferating cell nuclear antigen (PCNA) (1:50; Santa Cruz Biotechnology, Santa Cruz, CA) and mouse monoclonal IgG1 anti-nestin (1:5; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). Secondary antibodies were FITC-conjugated goat anti-mouse (GAM) IgG2a (1:100; Southern Biotech, Birmingham, AL) and tetramethylrhodamine isothiocyanate-conjugated goat anti-mouse IgG1 (1:100; Southern Biotechnology). Sections were thawed in cold PBS and incubated for 10 min in 1% H2O2 in PBS, followed by incubation in 0.5% NP-40 in PBS for 10 min. The slides were washed in PBS and blocked with PGB super block (10% BSA, 0.05% NaN3, and 10% normal goat serum in PBS) for 1 h at room temperature. The super block was aspirated, and primary antibodies were applied in a 1:5 dilution of PGB super block with 0.05% Triton X-100 and incubated overnight at 4°C. The slides were rinsed extensively with PBS and then incubated in secondary antibodies for 1 h at 37°C. The sections were washed, counterstained with 4′,6′-diamidino-2-phenylindole (DAPI), and coverslipped with Gel/Mount (Biomeda, Foster City, CA). For Notch1 immunohistochemistry, cryosections were washed in PBS and treated with 5% BSA/0.2% Triton X-100 for 1 h. Polyclonal antibodies against the intracellular domain of the Notch receptor were diluted (1:50) in the same blocking solution and incubated overnight at 4°C. Sections were treated with biotin-labeled secondary antibodies and finally in chromogenic solution (DAB/oxygen peroxide). Sections were dehydrated, cleared, and mounted in Vectamount (Vector Laboratories, Burlingame, CA).
For BrdU/Doublecortin (Dcx) immunofluorescence, staining was performed on 40 μm free-floating sections. Sections for BrdU staining were pretreated with 2N HCl for 1 h at room temperature to denature DNA. Sections were blocked for 1 h in TBS with 10% donkey serum and 5% BSA. Primary antibodies were incubated for 24 h at 4°C. The following antibodies were used: anti-Dcx (goat polyclonal, Dcx-COOH terminus, 1:100; Santa Cruz Biotechnology); anti-neuronal-specific nuclear protein (NeuN) (mouse monoclonal, 1:100; Chemicon, Temecula, CA); anti-BrdU (rat monoclonal, 1:30; Accurate Chemical and Scientific Corporation, Westbury, NY). Secondary antibodies against the appropriate species were incubated for 2 h at room temperature (1:200; all from Jackson ImmunoResearch, West Grove, PA). Sections were analyzed using a Zeiss (Oberkochen, Germany) LSM410 confocal laser-scanning microscope. The following filter sets, with the indicated wavelengths (in nanometers) for the excitation laser line and emission filters were used: cyanine, 488/(510/540); rhodamine, 568/(590–610). Confocal Z sections were acquired and three dimensionally reconstructed using the Zeiss LSM software, cropped, adjusted, and optimized in Photoshop 9.0 (Adobe Systems, San Jose, CA).
The dorsolateral SVZ (SVZDL) was assessed at the level of the anterior commissure (−0.6 mm from bregma). Single-positive PCNA cells and double-positive PCNA/nestin cells were counted in the ependyma, subependyma, and total medial region of the SVZDL. The total medial region of the SVZDL represented a 80 × 100 μm field. The ependyma was demarcated as the ciliated pseudostratified layer of cells lining the intact ventricles and the most medial SVZ (SVZm) as those cells within 4 nuclear diameters from the ependymal layer. The Abercrombie correction was applied to all cell counts (Abercrombie, 1946). The data are presented as the number of PCNA+ or PCNA+/nestin+ cells per field in the ependyma, SVZm, and the total medial SVZDL (Fig. 1). All counts were conducted on three nonadjacent sections per animal from four experimental animals.
Figure 1.

Diagram of the brain regions analyzed. A, View from the top of the brain. Dashed lines represent the region that was blocked for neurosphere assays and RNA isolation. Dotted line indicates approximate level at which sections were taken for Figures 2–4. B, Coronal section at the level of the dotted line in A. The region bounded by dashed lines delimits the region dissected for neurosphere assays. The smaller region bounded by the dotted line was removed for RNA isolation. C, Representation of the medial SVZDL as shown in Figures 2–4. LV, Lateral ventricle; Ep, ependymal layer; SVZm, 3–4 cell diameters subjacent to Ep; Total, entire medial SVZ, including Ep and SVZm.
In situ hybridization.
Cryosections were thawed and postfixed for 15 min in 4% paraformaldehyde. After several rinses in phosphate buffer with 0.1% Tween 20 (PBT), they were treated with proteinase K (1 μg/ml in phosphate buffer) and then rinsed thoroughly in PBT, followed by another postfix in 4% paraformaldehyde, additional rinses, and treatment in 0.25% acetic anhydride in 100 mm triethanolamine. Sections were then rinsed and placed in humidified slide chambers and hybridized overnight at 65°C in hybridization solution (10 mm Tris, 100 mm EDTA, 600 mm NaCl, 0.25% SDS, 10% dextran sulfate, 1× Denhardt’s solution, 200 μg/ml yeast tRNA, and 50% formamide) with digoxigenin-labeled riboprobe against Hes5 generated according to the instructions of the manufacturer (Roche Products, Indianapolis, IN). After hybridization, slides were rinsed in 5× SSC and then 1× SSC/50% formamide at 65°C for 30 min and were then treated with RNase (20 μg/ml) in TNE (10 mm Tris, 1 mm EDTA, and 500 mm NaCl) for 30 min at 37°C. Slides were then washed in 2× SSC and 0.2× SSC for 20 min each at 65°C. After two rinses in MABT (100 mm maleic acid, 150 mm NaCl, and 0.1% Tween 20), sections were blocked with 1:1 mixture of 10% Blocking Reagent (Roche Products) and Tris-buffered saline for 15 min. Sections were incubated overnight at 4°C in alkaline phosphatase-conjugated anti-digoxigenin (1:500; Promega, Madison, WI) in blocking buffer. The following day, after thorough rinsing in MABT and preincubation with levamisole, sections were developed with nitroblue-tetrazolium-chloride/5-bromo-4-chlor-indolyl-phosphate.
Ventricular area measurements.
Images were taken at 10× magnification of the contralateral and ipsilateral ventricles of six animals 48 h after perinatal H/I using a BX-41 Olympus Optical (Thornwood, NY) microscope and a Jenoptik (Jena, Germany) ProgRes-14 camera. The borders of the ventricles were outlined using a drawing tool, and the areas were calculated using IPLab software (Scanalytics, Fairfax, VA). Means and averages were calculated, and statistical significance was determined by Student’s t test.
Primary neurosphere propagation.
Wistar pups (P7, P8, or P9) were killed by decapitation, and their brains were removed using aseptic techniques. Coronal sections were taken 2 mm from the anterior pole of the brain, excluding the optic tracts, and 3 mm posterior to the previous cut. The sections were then placed in dishes containing PGM solution (PBS with 1 mm MgCl2 and 0.6% dextrose), in which the SVZ was dissected out under a microscope. The tissue was mechanically minced and enzymatically dissociated for 5 min at 37°C using a solution of 0.01% trypsin/EDTA (Invitrogen, Carlsbad, CA) in PGM with 250 μg of DNaseI (Sigma, St. Louis, MO), after which an equal volume of 0.02% trypsin inhibitor (Sigma) in Pro-N media (DMEM/F-12 1:1 media containing 10 ng/ml d-biotin, 25 μg/ml insulin, 20 nm progesterone, 100 μm putrescine, 5 ng/ml selenium, 50 μg/ml apo-transferrin, and 50 μg/ml gentamycin) was added. The tissue was triturated in Pro-N media to obtain an even cell suspension. Triturations were done with progressively less media and smaller Eppendorf (Boulder, CO) filter tips with time allowing for the tissue to settle, and the supernatant was removed and placed in separate tubes before adding new media for the next round of trituration. The cell suspension was then passed through a 40 μm Nitex screen. The number of viable cells was determined with a hemocytometer by exclusion of 0.1% trypan blue dye. The cells were plated into plastic 12-well tissue culture plates at a density of 1 × 105 cells/ml in Pro-N media supplemented with 20 ng/ml epidermal growth factor (EGF) and 10 ng/ml FGF-2. Cell cultures were fed every 2 d by removing approximately half of the media and replacing it with an equal volume of fresh media.
Secondary neurosphere propagation.
Primary neurospheres were collected from 12-well plates after 6 d in vitro and pelleted by centrifugation at 1200 rpm for 5 min. The neurospheres were enzymatically dissociated for 5 min at 37°C in a 0.01% trypsin/EDTA solution in GHCKS buffer (11 mm glucose, 20 mm HEPES, 10 mm citrate, 4 mm KCl, 110 mm NaCl, and 0.002 g/L phenol red). Trypsin was inhibited by adding an equal volume of 0.02% trypsin inhibitor in Pro-N. The spheres were dissociated by trituration in Pro-N media using progressively less media and smaller Eppendorf filter tips. The number of viable cells was determined with a hemocytometer via exclusion of 0.1% trypan blue dye. The cells were plated into a plastic 12-well tissue culture plate at a density of 5 × 104 cells/ml in Pro-N media supplemented with 20 ng/ml EGF and 10 ng/ml FGF-2. Cell cultures were fed every 2 d by removing approximately half of the media and replacing it with an equal volume of fresh media.
Neurosphere quantitation.
A neurosphere was defined as a free-floating, cohesive cluster of at least eight cells, although the vast majority of neurospheres (>98%) were substantially larger than this. Plates were gently shaken before counting each well to ensure an even distribution of spheres. Ten random 10× fields were counted per well. The frequency of sphere-forming cells (i.e., NSPs) was calculated from the average number of spheres per field, the area of the field, and the area of the well. The number of NSPs per hemisphere was then extrapolated by applying the frequency of sphere-forming cells to the total number of cells obtained in the initial dissociation of the tissue.
Neurosphere immunohistochemistry staining.
Spheres were collected and resuspended in 5% horse serum in CNM-2 media (DMEM/F-12 1:1 containing 10 ng/ml d-biotin, 5 ng/ml insulin, 20 nm progesterone, 100 μm putrescine, 5 ng/ml selenium, 50 μg/ml apo-transferrin, 50 μg/ml gentamycin, and 150 μl of 0.5 m kynurenic acid) at an approximate density of 100–200 spheres/ml. A total of 100 μl of the neurosphere suspension was then plated in plastic 24-well tissue culture plates onto flame-sterilized coverslips precoated with 1% w/v poly-d-lysine and 10 μg/ml laminin. The spheres were allowed to attach in a 37°C incubator for a minimum of 1.5 h, after which 400 μl of CNM-2 supplemented to 5% horse serum was added to the well. After 16–20 h, this media was replaced with CNM-2 with 0.03% DMSO. Cultures were allowed to differentiate for 5 d, with media replenished on day 3. After the differentiation period, the cells were fixed using 2% paraformaldehyde for 15 min and washed twice with BCH (10% bovine calf serum in Earle’s basal medium with 4.8 mg/ml HEPES). Sections were stained at room temperature for 45 min with O4 culture supernatant diluted 1:3 in BCH supplemented with 10% lamb serum. After thoroughly rinsing, the cells were incubated for 45 min at room temperature in GAM IgM lissamine rhodamine sulfonyl chloride (1:200; The Jackson Laboratory, Bar Harbor, ME). The cells were then permeabilized with BCH-S (BCH with 0.5 mg/ml saponin) and stained for 45 min at room temperature with anti-TuJ1 (1:250; Promega) and anti-GFAP (1:200; Roche Products) in saponin diluent (BCH-S supplemented with 10% lamb serum). Cells were then incubated for 45 min at room temperature in GAM IgG FITC (1:400; The Jackson Laboratory) and goat anti-rabbit amino-4-methylcoumarin-3-acetic acid (1:200; The Jackson Laboratory) in saponin diluent. The coverslips were rinsed and mounted onto microscope slides with Gel/Mount (Biomeda) and allowed to dry overnight. Colonies were scored according to the types of cells present. Images of stained cells were collected using a SenSys cooled-coupled device camera (CRI, Woburn, MA) interfaced with IPLab scientific imaging software (Scanalytics) on an Olympus Optical BX-40 microscope.
Isolation of total RNA.
The entire SVZ was dissected out of control, ipsilateral, and contralateral hemispheres, snap frozen on a dry ice/ethanol slush, and immediately stored at −80°C. Dissections were directed toward the angles of the ventricles to avoid any confounding effects of ventricular hypertrophy subsequent to striatal degeneration (Fig. 1). Tissue samples were then thawed directly into 0.5 ml Trizol reagent (Molecular Research Center, Cincinnati, OH) and homogenized using a tissue homogenizer. A total of 100 μl of chloroform was added, and the samples were centrifuged at 13,000 rpm for 15 min at 4°C. The aqueous phase was then transferred to a new tube. After adding 250 μl of 70% EtOH, the aqueous phase was applied to an RNeasy Mini-spin column (Qiagen, Valencia, CA) to remove contaminants from the RNA, according to the instructions of the manufacturer. The concentration of total RNA was determined by measuring optical density on a spectrophotometer (BD, Franklin Lakes, NJ). RNA samples were stored at −80°C until needed.
Microarray analysis.
Distinct samples of pooled total mRNA were generated from five injured hemispheres or uninjured control hemispheres in each of two biological replications. Probes were generated for chemiluminescent detection using the TrueLabeling-RT kit according to the instructions of the manufacturer (Superarray, Frederick, MD). Probes were then hybridized to a nylon membrane Stem Cell Gene Array (Superarray) in a rotisserie hybridization oven (Hybaid, Franklin, MA) and developed according to the instructions of the manufacturer. Images were obtained using a UVP imaging system with LabWorks software (UVP, Upland, CA). A grid was overlaid onto the image of the array, and local background corrections were used. The intensity of each spot was normalized to the average expression of glyceraldehyde-3-phosphate dehydrogenase and compared between injured and uninjured SVZ samples. Only those genes that were induced or repressed at least twofold in each replication are listed in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Table 1.
Primer sets used in RT-PCR
| Transcript | Primers |
|---|---|
| EGFR (TaqMan) | Applied Biosystems TaqMan Assay ID Rn00580398_m1 |
| Notch1 (Lux) | 5′-CAC GTA CTG CGA GCT GCC CTA CG[FAM]G-3′ |
| 5′-GGC AGG TGC CTC CGT TCT-3′ | |
| 18S (TaqMan) | Applied Biosystems TaqMan Assay ID Hs99999901_s1 |
| 18S (Lux) | Invitrogen catalog #115HM-01 |
Real-time PCR.
Total RNA was isolated from at least five individual SVZs per group using TriReagent (Molecular Research Center, Cincinnati, OH) and RNeasy kits (Qiagen) according to the protocol of the manufacturers. A total of 2 μg of RNA was reverse transcribed to cDNA using the Qiagen Omniscript RT kit supplemented with random nonamer primer (Sigma) and RNaseIN (Promega). Primer pairs specific for the genes of interest were designed using Lux Primer Design software (Invitrogen) or obtained from cataloged TaqMan gene expression assays (Applied Biosystems, Foster, City, CA) (Table 1). Duplicates for each sample were amplified in 96-well plates using Platinum-UDP Supermix kit according to the instructions of the manufacturer (Invitrogen) and analyzed on an ABI Prism 7700 Sequence Detection System (Applied Biosystems). Fold changes in gene expression relative to a housekeeping gene were obtained using the Relative Expression Software Tool for groupwise comparison and statistical analysis of relative expression results in real-time PCR, and experiment variability is given as the coefficient of variation (CV) for the target gene in the sample groups (Pfaffl et al., 2002).
Statistics.
A power analysis was calculated at a 0.05 level of significance, which recommended a sample size of five or greater to obtain a sample size that would provide at least 85% power for finding mean differences that are 1.0 and 1.5 (respectively) times larger than the associated SDs. The normality of each dataset was verified using the Shapiro-Wilks numerical test for normality. Student’s t tests for independent samples were conducted to analyze differences between ipsilateral and untouched control hemispheres and between contralateral and untouched control hemispheres. Neurosphere differentiation data were analyzed by two-way ANOVA, followed by Bonferroni’s/Dunn’s post hoc tests. Statistical tests were conducted using SPSS software (SPSS, Chicago, IL) or SAS software (SAS Institute, Cary, NC).
Results
Markers of cell proliferation increase in the medial SVZ after perinatal H/I
The SVZ represents the postnatal remnant of the embryonic germinal zones and continues to produce several cell types after birth (Brazel et al., 2003). The cellular architecture of the SVZ has been highly characterized, revealing that NSCs reside in the most medial region of the SVZ, subjacent to the ependymal layer (Garcia-Verdugo et al., 1998; Chiasson et al., 1999; Capela and Temple, 2002). To determine how cell proliferation in the SVZ is affected by perinatal H/I, we injected BrdU into rat pups at 48 h after the insult and perfused them 1 h later. In the injured brain, many BrdU+ cells were found adjacent to or among the ependymal lining at 48 h, whereas almost all BrdU+ cells in control pups were ∼5–6 cell diameters removed from the ependymal lining (Fig. 2A–C). BrdU-labeled cells in sham-operated animals with hypoxia showed the same distribution as uninjured control animals after 48 h of recovery (Snyder, 2001). Restricting counts to the NSC niche, i.e., cells within 4 cell diameters of the ependymal layer, revealed an increase in the number of BrdU+ cells in the ipsilateral hemisphere compared with the uninjured contralateral hemisphere (Fig. 2D). Despite this very selective increase in BrdU labeling within the NSP niche, there was no difference in the total number of BrdU+ cells within the entire ipsilateral SVZ compared with controls at this time point (data not shown).
Other sections were processed for PCNA immunohistochemistry at 48 h of recovery (Fig. 3A–C). PCNA is a protein expressed during S phase of the cell cycle and is frequently used as another marker of cell proliferation (Celis et al., 1986). BrdU and PCNA staining yielded similar expression patterns. There was approximately a fourfold increase in the number of PCNA+ cells in the NSC niche in the ipsilateral hemisphere of injured pups compared with controls (p < 0.0003) (Fig. 3G, SVZm). A smaller increase also occurred in the contralateral hemisphere (p < 0.002) (Fig. 3G, SVZm). Again, there was no significant difference between control and injured pups in the number of PCNA+ cells found in the entire medial SVZ (Fig. 3G, Total).
Figure 3.
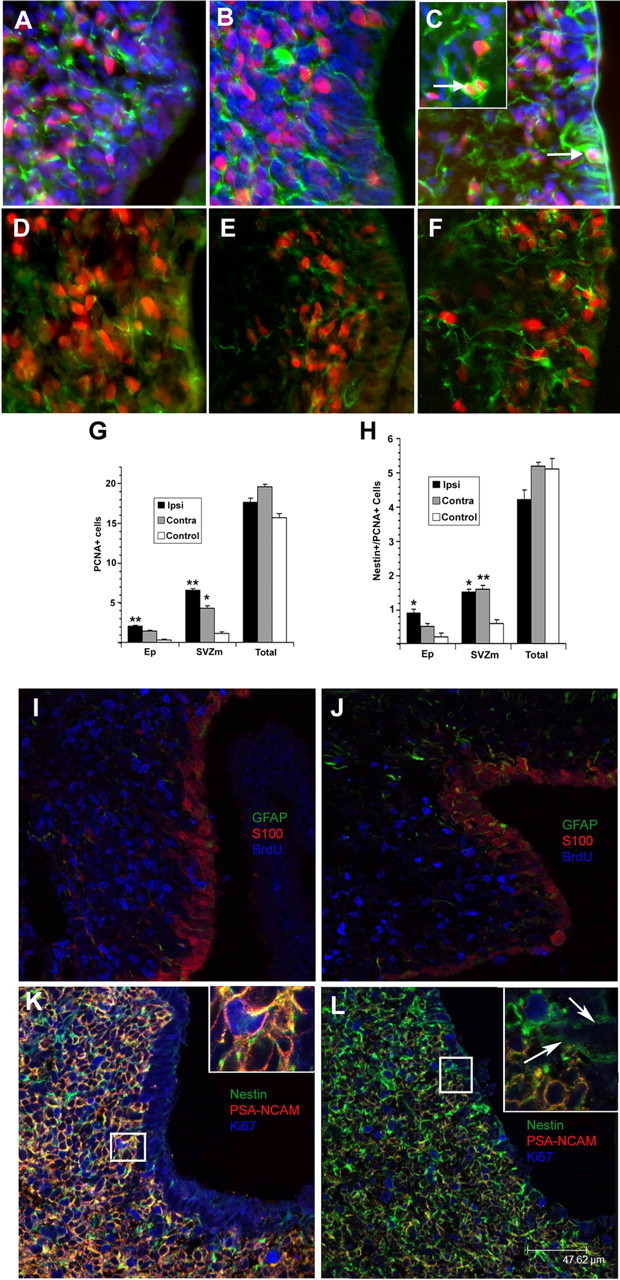
Hypoxia/ischemia increases the number of proliferating precursors within the NSP niche. A–H, Cryostat tissue sections from control and experimental animals were immunostained for PCNA (red) and nestin (green). DAPI (blue) is the nuclear stain in D–F. Panels represent control (A, D), H/I contralateral (B, E), and H/I ipsilateral (C, F) hemispheres. The number of PCNA+ cells (G) or PCNA/nestin double-positive cells (H) per field of view was quantified for the entire medial SVZ (Total), the ependymal layer (Ep), and those cells within 4 nuclear diameters of the ependymal layer (SVZm). Counts were conducted on three nonadjacent sections per animal. Data represent the mean ± SEM of four independent experiments. *p < 0.002 versus control; **p < 0.0003 versus control by two-tailed t test. I–L, Cryostat sections from ipsilateral (J, L) and contralateral (I, K) SVZs were stained for GFAP, S100, and BrdU (I, J) or nestin, PSA-NCAM, and Ki67 (K, L), and images were obtained on a laser-scanning confocal microscope. In I, J, BrdU administration was the same as in Figure 2.
To establish which cells were dividing in the SVZ after perinatal H/I, we combined immunohistochemistry for markers of dividing cells with markers for precursor class. Double immunohistochemistry for PCNA and nestin, an intermediate neurofilament frequently used to identify NSPs, revealed more than twice as many double-positive cells in the most medial region of both the contralateral and ipsilateral SVZs of H/I pups compared with controls (p < 0.002) (Fig. 3D–F,H, SVZm). This increase persisted into the ependymal layer in the ipsilateral hemisphere but not in the contralateral hemisphere of injured animals (Fig. 3H, EP). Once again, there was no significant difference throughout the entire dorsolateral SVZ in the number of PCNA/nestin double-positive cells (Fig. 3H, Total). BrdU staining was also combined with S100, an ependymal cell marker, and GFAP, an astrocytic marker. Confocal imaging of these sections revealed that, whereas GFAP expression was increased in the ipsilateral hemisphere, there was no noticeable change in GFAP staining in the most medial aspect of the SVZ, and there was no increase in the prevalence of BrdU+/GFAP+ or BrdU+/S100+ cells in the medial aspect (M. J. Romanko, unpublished data) (Fig. 3I,J). Confocal images were also obtained from sections stained for Ki67, nestin, and PSA-neural cell adhesion molecule (NCAM). In the contralateral hemisphere, PSA-NCAM was abundant throughout the hemisphere, and most dividing cells expressed both PSA-NCAM and nestin. In the ipsilateral hemisphere, however, PSA-NCAM+ cells were less abundant, and cycling cells positive for Ki67 were primarily nestin+ and PSA-NCAM− (Fig. 3K,L). Nestin+ processes could also frequently be seen extending from Ki67+ subependymal cells, penetrating the ependymal layer and contacting the ventricular surface (Fig. 3L, arrows).
To examine the fate of newborn cells after perinatal H/I, BrdU was injected intraperitoneally on post-H/I days 3–5, which is the time point used in published studies to document neurogenesis after ischemia. Animals were killed after 3 and 5 weeks of recovery. By 3 weeks after H/I, a large number of Dcx+ cells with morphologies of migrating neuroblasts were found in ipsilateral striatum, which appeared to be migrating from the SVZ (Fig. 4B). Dcx+ cells with migratory profiles were rare in the contralateral striatum (Fig. 4A). Immunostaining performed at 3 weeks of recovery after H/I revealed BrdU/Dcx double-positive cells in the striatum, in some cases located several millimeters from the SVZ (Fig. 4C). Furthermore, at 5 weeks of recovery, BrdU+/NeuN+ cells could be identified in the damaged striatum using confocal microscopy (Fig. 4D). These results corroborate observations of other investigators, but some using a similar injury model failed to see survival of NeuN+ cells through 5 weeks of recovery (Zhang et al., 2001; Arvidsson et al., 2002; Parent et al., 2002; Plane et al., 2004; Ong et al., 2005). The simplest explanation for the difference between the study by Ong et al. (2005) and ours is that they used Sprague Dawley rats whereas we used Wistar rats, and it is well established that there are significant specifies differences in the vulnerability and response to perinatal H/I (Sheldon et al., 1998).
More neurospheres can be isolated from the SVZ after perinatal H/I
The clonal neurosphere assay is a critical method for quantifying numbers of NSPs in the brain. To determine whether the dividing cells observed after perinatal H/I were NSPs, we microdissected the SVZ from control, contralateral, and ipsilateral hemispheres, dissociated the cells, and cultured them at clonal density in the presence of EGF and FGF-2 in which they formed primary neurospheres. Quantitative analysis of the number of neurospheres revealed no difference at 1 or 2 d after the injury; however, almost twice as many neurospheres were generated from the ipsilateral hemisphere than from uninjured controls by 3 d of recovery (p < 0.05) (Fig. 5A). In contrast, there was no significant difference in the number of neurospheres generated from the contralateral hemisphere (Fig. 5B). Repeated experiments using 10-fold lower cell-seeding densities and sham-operated animals rather than normal controls yielded similar results, and similar differences were seen between groups whether the number of neurospheres was determined as a total per hemisphere or a percentage of total cells plated (data not shown). A similar increase in the numbers of neurospheres has been reported 7 d after ischemic injury in adult rats (Zhang et al., 2004b). After 6 d in vitro, neurospheres from the injured hemisphere after 3 d of recovery were noticeably larger in diameter than those from contralateral or control hemispheres (Fig. 5C–F). This increase in diameter translates to more than a threefold increase in volume compared with sham neurospheres and more than a 2.3-fold increase compared with contralateral neurospheres.
Figure 5.
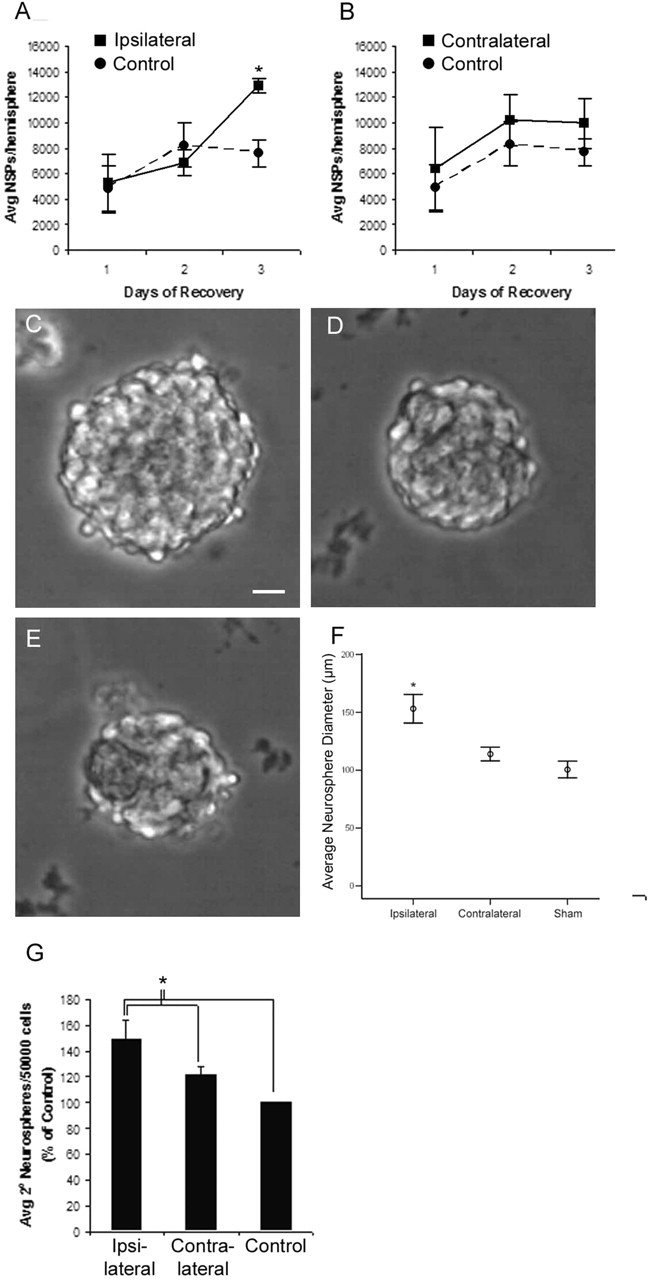
Hypoxia/ischemia increases the number of colony-forming NSPs and the self-renewal of NSPs in the ipsilateral SVZ. Control and experimental animals were sterilely decapitated at the designated time points, and the SVZs were dissociated into single-cell suspensions. These suspensions were cultured in the presence of FGF-2 (10 ng/ml) and EGF (20 ng/ml). Each replication consisted of pools of two to three animals per group. The number of NSPs per hemisphere for controls was compared with H/I ipsilateral (A) and H/I contralateral (B) hemispheres. Representative neurospheres from ipsilateral H/I (C), contralateral H/I (D), and control (E) SVZs demonstrate the larger size of spheres from the ipsilateral SVZ. Scale bar, 20 μm. The diameter of 10 neurospheres from each of four animals per condition was measured (F; error bars represent a 95% confidence interval). Primary neurospheres generated at 3 d of recovery were dissociated and subcultured to determine the relative self-renewal rates of each group (G). Values represent the mean ± SEM of four independent experiments. *p < 0.05 versus control by two-tailed t test.
Unilateral injury to the brain results in an expansion of the ventricle ipsilateral to the injury, a phenomenon that we observed previously at later time points of recovery (Levison et al., 2001). Because ventricular distortion could alter cell counts as well as the physical isolation of the SVZ, we performed measurements of ventricular area at 48 h of recovery. An analysis of six animals per group revealed a slight increase in the size of the ipsilateral ventricle; however, this change was not statistically significant at the time point at which these studies were conducted (Table 2). It is, therefore, unlikely that changes in ventricular size contribute to these results.
Table 2.
Ventricular area measurements at 48 h recovery
| Area (μm2) | ||
|---|---|---|
| Contralateral | Ipsilateral | |
| Mean | 11,609.3 | 15,031.2 |
| SD | 4096.9 | 8433.4 |
NSPs divide symmetrically more often after perinatal H/I
One of the defining characteristics of a stem cell is the ability to self-renew through asymmetric cell divisions. Under normal circumstances, asymmetric divisions maintain the population of NSPs at the same size. Alternatively, symmetric cell divisions can occur in which an NSP can generate two NSP progeny, thereby expanding the population of NSPs (Potten and Loeffler, 1990). Changes in the mode of cell division can be assessed by passaging primary neurospheres. If all NSPs undergo asymmetric cell divisions, every primary neurosphere would generate a single secondary neurosphere; however, if some symmetric divisions occur, a single primary neurosphere will give multiple secondary neurospheres because of the presence of multiple NSPs within that primary neurosphere. Secondary neurosphere assays, therefore, can be indicative of the relative frequency of symmetric cell divisions occurring within a population of cells (Reynolds and Weiss, 1996).
Because we observed an increase in the number of neurospheres generated from the damaged hemisphere, we postulated that this was attributable to an increase in symmetrical divisions rather than recruitment of quiescent stem cells. To test this hypothesis, we performed secondary neurosphere assays to determine the frequency of symmetric divisions after perinatal H/I. We dissociated primary neurospheres, seeded them at low density, and established the number of secondary spheres formed after 6 d in vitro. Primary spheres from the ipsilateral H/I hemisphere generated ∼30% more secondary neurospheres than contralateral or uninjured controls (p < 0.05) (Fig. 5G).
A greater proportion of neurospheres are multipotent after perinatal H/I
To verify that the neurospheres obtained after perinatal H/I were derived from multipotential precursors, we examined the progeny generated after differentiating single neurospheres for 5 d. We assessed differentiation by immunostaining for lineage-specific neural cell markers and quantifying the percentage of neurospheres that were unipotential, bipotential, or multipotential (Fig. 6). Neurospheres generated from both control brains and the contralateral hemisphere of injured brains demonstrated a low frequency of tripotency, although ∼50% produced at least two types of cells (Fig. 6A,B). Previous experiments have also shown no difference in tripotency between sham-operated and untouched controls (6.6 and 3.8%, respectively) (Snyder, 2001). In contrast, >75% of neurospheres from the ipsilateral hemisphere of the injured pups produced at least two cell types, and nearly 50% exhibited tripotency (p < 0.005 for ipsilateral tripotent vs control tripotent) (Fig. 6C,D).
Figure 6.

Hypoxia/ischemia increases the proportion of multipotential SVZ-derived neurospheres. NSPs were harvested from animals at 48 h of recovery and cultured in vitro as neurospheres in the presence of both FGF-2 and EGF. Spheres were plated onto coverslips and differentiated in growth factor free medium for 5 d. Neurospheres from control (A), contralateral (B), and ipsilateral (C) SVZs were immunostained for neuronal, astrocytic, and oligodendrocytic markers, and the percentage of glial or neuron/glial producing spheres was quantified. D depicts a representative multipotential clone from the ipsilateral SVZ stained for a neuronal marker (Tuj1, green), an astrocytic marker (GFAP, blue), and an oligodendrocyte marker (O4, red). Values embedded in the chart are the mean numbers of colonies counted from six independent experiments with three to four animals per group per experiment. *p < 0.005 for the percentage of ipsilateral multipotential spheres versus control multipotential spheres using ANOVA, followed by Bonferroni’s/Dunn’s post hoc test.
Perinatal H/I induces the expression of Notch1, gp130, and EGF receptor mRNA
Investigations into the properties of NSPs are beginning to reveal which molecules regulate neural stem cellness (Gaiano and Fishell, 2002; Cai et al., 2004). Because the increased frequency of symmetric divisions after perinatal H/I suggests an alteration of the intrinsic properties of the NSPs, we investigated molecular changes induced by the injury. We isolated total RNA from the SVZ at 48 h of recovery from the insult and performed a microarray analysis with 288 genes related to neural precursors (SuperArray Stem Cell Array). This analysis revealed induction of 35 transcripts, including a number of growth factors known to support the proliferation of NSCs, as well as many markers that identify stem cellness. In contrast, only four stem cell-related transcripts from the array were downregulated in the SVZ after perinatal H/I (supplemental Table S1, available at www.jneurosci.org as supplemental material). We isolated RNA from the entire SVZ and validated the expression of three receptors and one nonreceptor regulator of NSP proliferation and maintenance using real-time PCR. These genes were Notch1, EGFR, gp130, and Numb.
Signaling through the epidermal growth factor receptor (EGFR) is a critical mitogenic pathway for NSPs during development and postnatally (Reynolds et al., 1992; Tropepe et al., 1999; Kornblum et al., 2000; Gritti et al., 2002). Several ligands act through the EGFR to activate multiple signaling cascades, including the Janus kinase–signal transducer and activator of transcription and mitogen-activated protein kinase pathways. Previous studies have shown that perinatal H/I induces the EGFR ligand heparin-binding–EGF (Tanaka et al., 1999); therefore, we asked whether perinatal H/I also affects the capacity of SVZ cells to respond to these growth factors. Using quantitative reverse transcription (qRT)-PCR analysis, we observed nearly a threefold increase in the expression of EGFR mRNA relative to 18S compared with sham-operated controls and an approximate twofold increase compared with the contralateral hemisphere 48 h after the injury (p < 0.005) (Fig. 7A). The mild increase in EGFR expression in the contralateral hemisphere compared with sham controls was not statistically significant.
Figure 7.
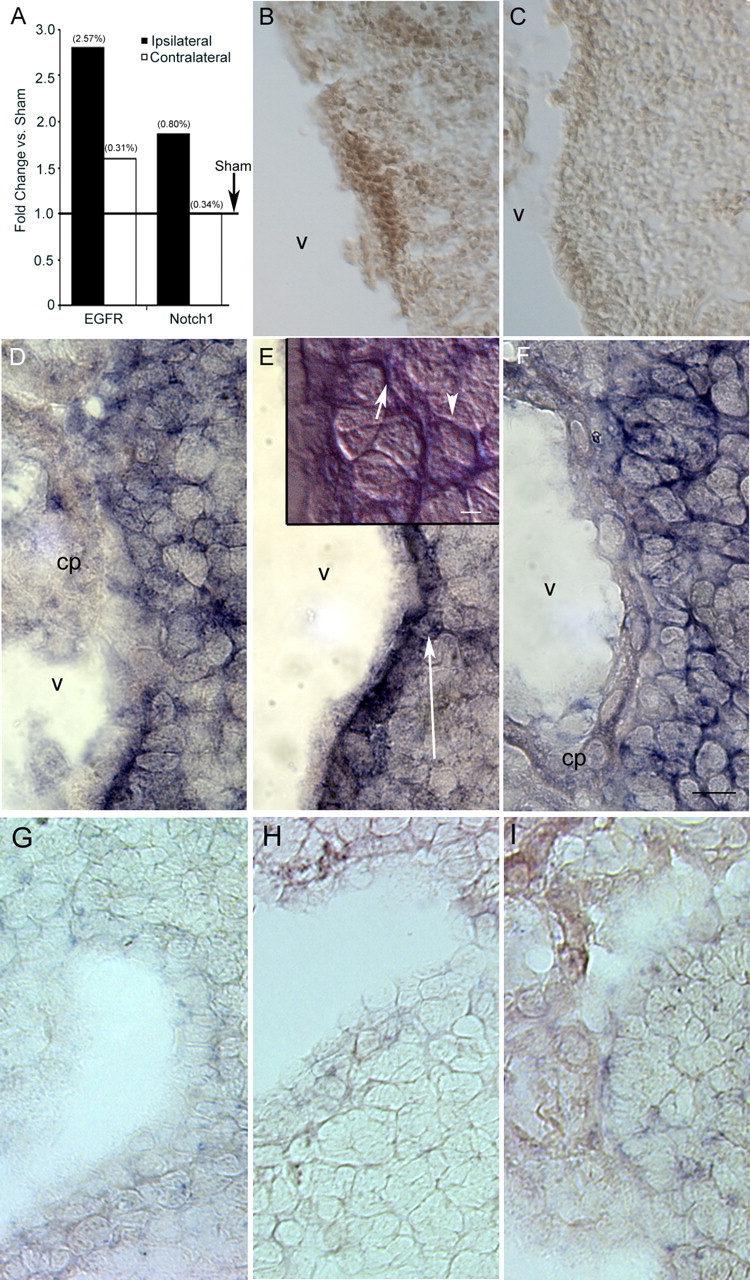
Signaling pathways involved in control of NSP fate are induced by perinatal H/I. Ipsilateral H/I and control hemispheres were dissected out after 48 h of recovery. A, Total RNA was isolated and amplified by qRT-PCR using primers specific for EGFR and Notch1 and normalized to expression of 18S. Values in parentheses indicate CV for the target gene in the sample groups; n = 10 for ipsilateral and contralateral conditions, and n = 4 for sham condition. *p < 0.05 versus sham; †p < 0.05 versus contralateral by pairwise fixed reallocation randomization. Immunostaining for Notch1 was performed on cryostat sections from H/I (B) and control (C) animals. In situ hybridization was performed on cryostat sections of contralateral (D, G), ipsilateral (E, H), and control (F, I) hemispheres using a digoxigenin-labeled RNA probe for Hes5 (D–F) and Hes1 (G–I). Arrows in E delineate the region of increased Hes5 expression. Inset in E shows high-magnification 100× Nomarski image of the Hes5+ region showing a subependymal Hes5+ cell body (arrowhead) with processes (arrow) projecting through the ependyma. There was no evident change in Hes1 expression. Scale bars: inset in E, 4 μm; F, 10 μm. cp, Choroid plexus; V, ventricle.
Notch1 is a cell-surface receptor that regulates cell fate in both the developing and adult brain (Gaiano and Fishell, 2002). It has been demonstrated in vivo and in vitro that Notch1 is essential for the maintenance of neural precursors (Hitoshi et al., 2002). Using qRT-PCR, we confirmed a twofold induction of Notch1 mRNA (p < 0.05) (Fig. 7A) compared with sham-operated and contralateral SVZs at 48 h of recovery from perinatal H/I. Immunostaining for the Notch1 receptor also revealed intensely stained pockets of cells in the ipsilateral hemisphere 48 h after perinatal H/I (Fig. 7B,C). We further confirmed upregulation of this signaling pathway with in situ hybridization, demonstrating strong expression of Hes5, an effector of Notch1 signaling, within the NSP niche of the SVZ (Fig. 7D–F). Higher-magnification Nomarski imaging of this region revealed that Hes5-expressing cell bodies were located subependymally, with some processes projecting through the ependymal layer and apparently contacting the ventricle (Fig. 7E, inset). The lack of any appreciable change in the expression of Hes1 indicates that these results are not a manifestation of nonspecific binding in the ipsilateral hemisphere. In addition to these results, we also confirmed by semiquantitative RT-PCR a twofold increase in the expression of gp130 (data not shown), a member of the component of the interleukin-6 family of cytokine receptors that may influence Notch1 expression (Chojnacki et al., 2003) and an increase in the expression of leukemia inhibitory factor (M. Covey, personal communication), a molecule implicated in NSP self-renewal that signals through gp130. Furthermore, we confirmed maintenance of normal expression levels of Numb, another molecule that has been implicated in precursor cell division (Shen et al., 2002) (data not shown).
Discussion
Here we provide evidence of an NSP response that temporally precedes the increase in neurogenesis after a perinatal H/I insult. We demonstrate the following: more proliferating cells in the medial SVZ after perinatal H/I that express markers of immature multipotent precursors (nestin) but not markers of more restricted progenitors (PSA-NCAM); a subset of cells actively dividing during the early recovery period differentiate along the neuronal lineage; nearly twice as many neurospheres can be isolated from the ipsilateral hemisphere of the damaged brain by 3 d after perinatal H/I, and neurospheres from the ipsilateral hemisphere are larger than those from the contralateral hemisphere or sham-operated animals; a greater proportion of neurospheres cultured after perinatal H/I are tripotent, and expansive symmetric divisions are maintained in vitro after perinatal H/I to produce more secondary neurospheres; finally the NSP-related genes EGFR, Notch1, and Hes5 are induced within the injured NSP niche. These data represent the first evidence that self-renewing, tripotential NSPs initiate a regenerative response to perinatal H/I.
Neurogenesis occurs after ischemic injury in both adult and immature rodents (Felling and Levison, 2003; Plane et al., 2004; Ong et al., 2005). Proliferation peaks 1 week after an ischemic insult, but increases have been observed as early as 2 d after H/I (Jin et al., 2001; Zhang et al., 2001; Li et al., 2002; Iwai et al., 2003). Some cells proliferating at 1 week appear to migrate from the SVZ into the damaged striatum to differentiate into phenotypically mature striatal neurons (Arvidsson et al., 2002; Parent et al., 2002). We confirmed these results in our animal model. Moreover, we established recently that robust compensatory neurogenesis also occurs in the rat neocortex subsequent to perinatal H/I (Yang et al., 2006). Furthermore, we demonstrated a doubling in the number of NSPs in the SVZ between 2 and 3 d after perinatal H/I, a time point well situated to supply an increased precursor pool for these restorative events.
We documented increased proliferation in the medial SVZ after 2 d of recovery using three different markers for dividing cells. Importantly, these studies provide only a snapshot of cells dividing at the time point of analysis and are likely an underestimate of the total number of cells proliferating after the injury. In the ipsilateral hemisphere, these cycling cells coexpressed nestin, a marker of immature neural precursors. Cells expressing PSA-NCAM, a marker for restricted progenitors, were primarily negative for proliferation markers in the ipsilateral hemisphere. In contrast, in the contralateral hemisphere, many of the proliferating cells did express PSA-NCAM. Similarly, proliferating cells were negative for GFAP and S100β, indicating that the proliferating cells were neither reactive astrocytes nor ependymal cells. That there was no change in the proliferating GFAP+ cells in the medial aspect of the SVZ does not contradict our conclusion that the NSPs are proliferating, because it has not been established that the neural stem cells in the perinatal brain are GFAP+. These data suggest that proliferation increases selectively in the immature multipotential NSP population.
The observed increase in proliferating cells was restricted to the most medial cell layers of the SVZ 48 h after the injury (Figs. 2D, 3G,H), a critical point because the architecture of the SVZ is such that the most immature cells lie in close proximity to the ventricle, and lineage restriction increases with progression toward the more lateral part of the region (Garcia-Verdugo et al., 1998). Thus, the increased proliferation is occurring preferentially in the NSP compartment of the SVZ. Moreover, using nestin as a marker of NSPs, we find a significant increase in the number of PCNA/nestin double-positive cells, again unique to the most medial region, providing more conclusive evidence that these dividing cells are NSPs. One consideration for the differences in cell proliferation between the medial and lateral portions of the SVZ is the cell death that occurs after injury. Our laboratory has demonstrated recently that cell death is uncommon in the medial region compared with the lateral SVZ (Romanko et al., 2004). Thus, proliferation may be increased within the lateral compartment as well but offset by extensive cell death.
The neurosphere assay has been the gold standard for quantifying numbers of NSPs. Using this assay, we find that there are nearly twice as many NSPs present in the brain by 3 d after perinatal H/I (Fig. 5E). This is substantiated by the observation that more nestin+ cells in the NSP niche are proliferating 1 d earlier. We show, in addition to increased numbers of neurospheres after cerebral injury, that a significantly larger percentage of these neurospheres are tripotential. Although much effort has been directed at detecting new neurons after brain injury, our data demonstrating an increase in tripotent cells is critical because it suggests an increased capacity for producing oligodendrocytes as well, a population known to be highly vulnerable to this insult in the immature brain (Ness et al., 2001; Back et al., 2002). Our in vitro results are corroborated by the reports of others of increased oligodendrogenesis in vivo after brain injury (Liu et al., 2002; Tanaka et al., 2003; Zaidi et al., 2004).
We used secondary neurosphere assays to demonstrate that the NSPs continue to undergo expansive symmetrical divisions in vitro. Although other mechanisms could explain the increase in secondary neurosphere formation, such as alterations in NSP survival, the difference in secondary neurosphere formation among the groups certainly suggests an intrinsic change in the behavior of these cells after perinatal H/I. Assuming that this expansion reflects the continuation of a mode of cell division that the NSPs adopted in vivo, these data provide a mechanism for the increased number of NSPs. The observations of more frequent symmetric expanding cell divisions favor a mechanism of increased proliferation of active NSPs rather than the recruitment of dormant NSPs into the cell cycle. Our results are similar to those reported by Zhang et al. (2004a) who also analyzed the plane of cell division of SVZ cells in vivo during recovery from ischemia and concluded that the plane of division was altered, supporting a change in the mode of cell division of precursors in the SVZ during recovery from ischemia.
An important point to consider is that the contralateral hemisphere also exhibits mildly increased cell proliferation within the medial SVZ according to our markers of cell proliferation. However, although there was a slight increase in the number of neurospheres obtained from the contralateral hemisphere compared with controls, this difference did not reach significance. Furthermore, there was no change in the differentiation potential of the contralateral neurospheres and no increase in the expression of EGFR and Notch1 within the contralateral SVZ (Fig. 7A). These data suggest that some of the dividing cells observed in the contralateral hemisphere may represent more restricted transit-amplifying cells. In the perinatal rat model we use, the contralateral hemisphere becomes hypoxic, although it does not become ischemic. Although it is possible that hypoxia alone induced these changes, it is more likely that diffusible factors from the injured hemisphere, communicating via the lateral ventricles, contributed to changes in the contralateral hemisphere, because there is a noticeable effect on the contralateral hemisphere that is not observed as a consequence of a sham operation, followed by a similar hypoxic episode (Snyder, 2001).
Initial molecular analyses demonstrating induction of receptors critical to NSP proliferation and fate determination support an increase in tripotential neurospheres after perinatal H/I (Fig. 7A). EGF receptor signaling is critical to the proliferation of NSPs, and Notch1 and gp130 are receptors that maintain the NSP population (Reynolds and Weiss, 1996; Tropepe et al., 1997; Gaiano et al., 2000; Doetsch et al., 2002; Chojnacki et al., 2003). Importantly, there is no induction of EGFR or Notch1 in the contralateral SVZ, decreasing the likelihood that the mild increases in proliferation on the contralateral side are a result of true NSP proliferation. Induction of EGF and Notch signaling pathways within the injured SVZ could promote NSP proliferation and prevent cells from differentiating by increasing the rate of symmetric cell division after brain injury. The cumulative effects of these receptors could support an increase in NSP numbers, and a rigorous analysis of these mechanisms in the context of this injury is the focus of our ongoing studies.
The past decade has seen tremendous advances in our understanding of the brain; however, translation of this knowledge into clinical settings has been comparatively slow. Many neurological diseases remain incurable and severely burden our healthcare system. Perinatal H/I is the leading cause of brain damage resulting from birth complications, and our current capabilities offer no means of restoring normal development after injuries have occurred. Our data indicate that this injury mobilizes endogenous NSPs and that these cells may be participating in regeneration. Future efforts at identifying barriers to more complete regenerative responses will be essential to understand how to more effectively harness the potential of NSPs, and understanding the mechanisms underlying this response will present targets for the therapeutic use of these vital cells.
Footnotes
This work was supported by National Institutes of Health Grants MH 59950 and HD 30705 (S.W.L.) and NS0469903-01 (R.J.F.) and by the Italian Institutes of Health. The nestin monoclonal antibody developed by S. Hockfield (Yale University, New Haven, CT) was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa (Department of Biological Sciences, Iowa City, IA). We thank Dr. Gord Fishell for his generous donation of the plasmid used for Hes5 in situ hybridization.
References
- Abercrombie M (1946). Estimation of nuclear population from microtome sections. Anat Rec 94:239–247. [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002). Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 8:963–970. [DOI] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM (2002). Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci 22:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazel CY, Romanko MJ, Rothstein RP, Levison SW (2003). Roles of the mammalian subventricular zone in brain development. Prog Neurobiol 69:49–69. [DOI] [PubMed] [Google Scholar]
- Cai J, Cheng A, Luo Y, Lu C, Mattson MP, Rao MS, Furukawa K (2004). Membrane properties of rat embryonic multipotent neural stem cells. J Neurochem 88:212–226. [DOI] [PubMed] [Google Scholar]
- Capela A, Temple S (2002). LeX/ssea-1 is expressed by adult mouse CNS stem cells, identifying them as nonependymal. Neuron 35:865–875. [DOI] [PubMed] [Google Scholar]
- Celis JE, Madsen P, Nielsen S, Celis A (1986). Nuclear patterns of cyclin (PCNA) antigen distribution subdivide S-phase in cultured cells—some applications of PCNA antibodies. Leuk Res 10:237–249. [DOI] [PubMed] [Google Scholar]
- Chiasson BJ, Tropepe V, Morshead CM, van der Kooy D (1999). Adult mammalian forebrain ependymal and subependymal cells demonstrate proliferative potential, but only subependymal cells have neural stem cell characteristics. J Neurosci 19:4462–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnacki A, Shimazaki T, Gregg C, Weinmaster G, Weiss S (2003). Glycoprotein 130 signaling regulates Notch1 expression and activation in the self-renewal of mammalian forebrain neural stem cells. J Neurosci 23:1730–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Petreanu L, Caille I, Garcia-Verdugo JM, Alvarez-Buylla A (2002). EGF converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron 36:1021–1034. [DOI] [PubMed] [Google Scholar]
- Felling RJ, Levison SW (2003). Enhanced neurogenesis following stroke. J Neurosci Res 73:277–283. [DOI] [PubMed] [Google Scholar]
- Felling RJ, Snyder MJ, Romanko MJ, Levison SW (2003). Neural stem cells react to perinatal hypoxic/ischemic brain insults. Soc Neurosci Abstr 29:42.6. [Google Scholar]
- Gaiano N, Fishell G (2002). The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci 25:471–490. [DOI] [PubMed] [Google Scholar]
- Gaiano N, Nye JS, Fishell G (2000). Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron 26:395–404. [DOI] [PubMed] [Google Scholar]
- Garcia-Verdugo JM, Doetsch F, Wichterle H, Lim DA, Alvarez-Buylla A (1998). Architecture and cell types of the adult subventricular zone: in search of the stem cells. J Neurobiol 36:234–248. [DOI] [PubMed] [Google Scholar]
- Goings GE, Sahni V, Szele FG (2004). Migration patterns of subventricular zone cells in adult mice change after cerebral cortex injury. Brain Res 996:213–226. [DOI] [PubMed] [Google Scholar]
- Gritti A, Bonfanti L, Doetsch F, Caille I, Alvarez-Buylla A, Lim DA, Galli R, Verdugo JM, Herrera DG, Vescovi AL (2002). Multipotent neural stem cells reside into the rostral extension and olfactory bulb of adult rodents. J Neurosci 22:437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitoshi S, Alexson T, Tropepe V, Donoviel D, Elia AJ, Nye JS, Conlon RA, Mak TW, Bernstein A, van der Kooy D (2002). Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev 16:846–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai M, Sato K, Kamada H, Omori N, Nagano I, Shoji M, Abe K (2003). Temporal profile of stem cell division, migration, and differentiation from subventricular zone to olfactory bulb after transient forebrain ischemia in gerbils. J Cereb Blood Flow Metab 23:331–341. [DOI] [PubMed] [Google Scholar]
- Jin K, Minami M, Lan JQ, Mao XO, Batteur S, Simon RP, Greenberg DA (2001). Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci USA 98:4710–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblum HI, Yanni DS, Easterday MC, Seroogy KB (2000). Expression of the EGF receptor family members ErbB2, ErbB3, and ErbB4 in germinal zones of the developing brain and in neurosphere cultures containing CNS stem cells. Dev Neurosci 22:16–24. [DOI] [PubMed] [Google Scholar]
- Levison SW, Ducceschi MH, Young GM, Wood TL (1996). Acute exposure to CNTF in vivo induces multiple components of reactive gliosis. Exp Neurol 141:256–268. [DOI] [PubMed] [Google Scholar]
- Levison SW, Rothstein RP, Romanko MJ, Snyder MJ, Meyers RL, Vannucci SJ (2001). Hypoxia/ischemia depletes the rat perinatal subventricular zone of oligodendrocyte progenitors and neural stem cells. Dev Neurosci 23:234–247. [DOI] [PubMed] [Google Scholar]
- Li Y, Chen J, Chopp M (2002). Cell proliferation and differentiation from ependymal, subependymal and choroid plexus cells in response to stroke in rats. J Neurol Sci 193:137–146. [DOI] [PubMed] [Google Scholar]
- Liu Y, Silverstein FS, Skoff R, Barks JD (2002). Hypoxic-ischemic oligodendroglial injury in neonatal rat brain. Pediatr Res 51:25–33. [DOI] [PubMed] [Google Scholar]
- Magavi SS, Leavitt BR, Macklis JD (2000). Induction of neurogenesis in the neocortex of adult mice. Nature 405:951–955. [DOI] [PubMed] [Google Scholar]
- Ness JK, Romanko MJ, Rothstein RP, Wood TL, Levison SW (2001). Perinatal hypoxia-ischemia induces apoptotic and excitotoxic death of periventricular white matter oligodendrocyte progenitors. Dev Neurosci 23:203–208. [DOI] [PubMed] [Google Scholar]
- Ong J, Plane JM, Parent JM, Silverstein FS (2005). Hypoxic-ischemic injury stimulates subventricular zone proliferation and neurogenesis in the neonatal rat. Pediatr Res 58:600–606. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Ray J, Gage FH (1995). FGF-2-responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Mol Cell Neurosci 6:474–486. [DOI] [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM (2002). Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol 52:802–813. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L (2002). Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plane JM, Liu R, Wang TW, Silverstein FS, Parent JM (2004). Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis 16:585–595. [DOI] [PubMed] [Google Scholar]
- Potten CS, Loeffler M (1990). Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development 110:1001–1020. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S (1996). Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol 175:1–13. [DOI] [PubMed] [Google Scholar]
- Reynolds BA, Tetzlaff W, Weiss S (1992). A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J Neurosci 12:4565–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JE III, Vannucci RC, Brierley JB (1981). The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9:131–141. [DOI] [PubMed] [Google Scholar]
- Romanko MJ, Rothstein RP, Levison SW (2004). Neural stem cells in the subventricular zone are resilient to hypoxia/ischemia whereas progenitors are vulnerable. J Cereb Blood Flow Metab 24:814–825. [DOI] [PubMed] [Google Scholar]
- Sanai N, Tramontin AD, Quinones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, Lawton MT, McDermott MW, Parsa AT, Manuel-Garcia Verdugo J, Berger MS, Alvarez-Buylla A (2004). Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 427:740–744. [DOI] [PubMed] [Google Scholar]
- Sheldon RA, Sedik C, Ferriero DM (1998). Strain-related brain injury in neonatal mice subjected to hypoxia-ischemia. Brain Res 810:114–122. [DOI] [PubMed] [Google Scholar]
- Shen Q, Zhong W, Jan YN, Temple S (2002). Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development 129:4843–4853. [DOI] [PubMed] [Google Scholar]
- Snyder MJ (2001). Hypoxia/ischemia depletes neural stem cells from the subventricular zone. In: Master’s thesis The Pennsylvania State University.
- Tanaka K, Nogawa S, Suzuki S, Dembo T, Kosakai A (2003). Upregulation of oligodendrocyte progenitor cells associated with restoration of mature oligodendrocytes and myelination in peri-infarct area in the rat brain. Brain Res 989:172–179. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Sasahara M, Ohno M, Higashiyama S, Hayase Y, Shimada M (1999). Heparin-binding epidermal growth factor-like growth factor mRNA expression in neonatal rat brain with hypoxic/ischemic injury. Brain Res 827:130–138. [DOI] [PubMed] [Google Scholar]
- Tropepe V, Craig CG, Morshead CM, van der Kooy D (1997). Transforming growth factor-α null and senescent mice show decreased neural progenitor cell proliferation in the forebrain subependyma. J Neurosci 17:7850–7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropepe V, Sibilia M, Ciruna BG, Rossant J, Wagner EF, van der Kooy D (1999). Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol 208:166–188. [DOI] [PubMed] [Google Scholar]
- Vannucci RC, Vannucci SJ (1997). A model of perinatal hypoxic-ischemic brain damage. Ann NY Acad Sci 835:234–249. [DOI] [PubMed] [Google Scholar]
- Volpe JJ (2000). In: Neurology of the neonate Ed 4 Philadelphia: Saunders.
- Yang Z, Covey MV, Bitel CL, Levison SW (2006). Neonatal hypoxic/ischemic brain injury initiates and sustains neocortical neuronal replacement from the subventricular zone. J Neurosci in press.
- Zaidi AU, Bessert DA, Ong JE, Xu H, Barks JD, Silverstein FS, Skoff RP (2004). New oligodendrocytes are generated after neonatal hypoxic-ischemic brain injury in rodents. Glia 46:380–390. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhang Z, Zhang C, Zhang L, Robin A, Wang Y, Lu M, Chopp M (2004a). Stroke transiently increases subventricular zone cell division from asymmetric to symmetric and increases neuronal differentiation in the adult rat. J Neurosci 24:5810–5815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Zhang Z, Wang L, Wang Y, Gousev A, Zhang L, Ho KL, Morshead C, Chopp M (2004b). Activated neural stem cells contribute to stroke-induced neurogenesis and neuroblast migration toward the infarct boundary in adult rats. J Cereb Blood Flow Metab 24:441–448. [DOI] [PubMed] [Google Scholar]
- Zhang RL, Zhang ZG, Zhang L, Chopp M (2001). Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience 105:33–41. [DOI] [PubMed] [Google Scholar]