Acute Impairment of Mitochondrial Trafficking by β-Amyloid Peptides in Hippocampal Neurons (original) (raw)
Abstract
Defects in axonal transport are often associated with a wide variety of neurological diseases including Alzheimer's disease (AD). β-Amyloid (Aβ) is a major component of neuritic plaques associated with pathological conditions of AD brains. Here, we report that a brief exposure of cultured hippocampal neurons to Aβ molecules resulted in rapid and severe impairment of mitochondrial transport without inducing apparent cell death and significant morphological changes. Such acute inhibition of mitochondrial transport was not associated with a disruption of mitochondria potential nor involved aberrant cytoskeletal changes. Aβ also did not elicit significant Ca2+ signaling to affect mitochondrial trafficking. However, stimulation of protein kinase A (PKA) by forskolin, cAMP analogs, or neuropeptides effectively alleviated the impairment. We also show that Aβ inhibited mitochondrial transport by acting through glycogen synthase kinase 3β (GSK3β). Given that mitochondria are crucial organelles for many cellular functions and survival, our findings thus identify an important acute action of Aβ molecules on nerve cells that could potentially contribute to various abnormalities of neuronal functions under AD conditions. Manipulation of GSK3β and PKA activities may represent a key approach for preventing and alleviating Aβ cytotoxicity and AD pathological conditions.
Keywords: Aβpeptide, Alzheimer's disease, signal transduction, actin, calcium, cAMP, transport
Introduction
Alzheimer's disease (AD) involves progressive loss of memory and cognitive abilities and is highlighted by two pathological hallmarks in the brain: neuritic plaques containing β-amyloid (Aβ) aggregates and intracellular neurofilbrillar tangles consisting of hyperphosphorylated microtubule-associated tau proteins (Selkoe, 2003; Bossy-Wetzel et al., 2004). Aβ, generated by proteolytic cleavage of the transmembrane Aβ precursor protein (APP) through β-secretase and γ-secretase, is believed to be the culprit behind neurodegeneration and associated cognitive and behavioral abnormalities (Mattson, 2004). Although there is ample evidence that Aβ fibrils exert a variety of toxicity to neurons, recent studies indicate that intermediate Aβ aggregates can profoundly inhibit synaptic functions (Walsh et al., 2002; Wang et al., 2002; Lacor et al., 2004). The cellular mechanisms underlying Aβ inhibition on synaptic and other neuronal functions, however, remain elusive. Defects in axonal transport are associated with a wide variety of neurological diseases including AD (Terwel et al., 2002). It has been shown recently that transport deficits may represent an early step in AD pathogenesis because axonal swelling and reduced axonal transport were observed before apparent AD hallmarks (Stokin et al., 2005). Moreover, the axonal swellings and varicosities were found to contain abnormal accumulations of transport cargos, APPs, and Aβ, and such transport deficits appear to enhance APP processing and local Aβ production. Although mutations in kinesin motors could mimic the transport deficits (Stokin et al., 2005), whether and how other factors are involved in causing transport deficits remain unknown.
Among many organelles being transported along cytoskeletal tracks, mitochondria represent a crucial organelle for cell viability and functions. Mitochondria are not only the main source of energy for cellular reactions but also a crucial component in the regulation of intracellular Ca2+ concentrations ([Ca2+]i). Mitochondrial dysfunction and/or damage have been linked to a number of neurodegenerative diseases (Eckert et al., 2003; Schapira, 2006). Recent studies also indicate that mitochondria appear to accumulate at the synaptic sites and mitochondrial trafficking to the synapse is essential for normal synaptic functions (Li et al., 2004; Chang et al., 2006). In this study, we report that relatively low concentrations of Aβ could rapidly impair fast transport of mitochondria (within 10 min). We also show that the impairment of transport is not a result of nonspecific Aβ toxicity, but instead involves specific signaling in which elevation of cAMP or inhibition of glycogen synthase kinase 3β (GSK3β) attenuates Aβ effects. Considering that fast vesicular transport is essential for many cellular functions and mitochondria are a crucial organelle for neuronal survival and functions (e.g., synaptic transmission), our findings have thus elucidated an acute inhibitory action of Aβ molecules that may play an important role in Aβ impairment of neuronal functions and could potentially contribute to adverse events associated with AD pathogenesis.
Materials and Methods
Cell culture.
Hippocampal neurons from embryonic day 18 rats were obtained according to the method described previously (Banker and Cowan, 1977). Dissociated cells were plated in glass-bottom 35 mm culture dishes (MatTek, Ashland, MA) for culture and microscopy. The glass surface was pretreated with poly-d-lysine (Sigma, St. Louis, MO) for 2 h (500 μg/ml), and ∼75,000 cells were plated in each dish in DMEM (Invitrogen, Carlsbad, CA) containing 5% FBS and 5% horse serum (HyClone, Logan, UT). On the second day after plating, the culture medium was replaced by Neurobasal medium containing B27 and Glutamax (Invitrogen). Cells were maintained in a CO2 incubator at 37°C, and 0.5 vol of the culture medium was replaced with fresh Neurobasal every 3 d. Before each imaging experiment, the medium was replaced by Krebs'–Ringer's saline (in mm: 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4) (Bacci et al., 1999).
Aβ peptides and treatments.
Aβ25–35 and its reverse control Aβ35–25 (Sigma) were dissolved at 2 mm in water and kept frozen until use. Aβ1–40 and Aβ1–42 (American Peptide Company, Sunnyvale, CA) were first dissolved in a small volume of 10 mm NaOH and then diluted to 0.5 and 2 mm in double-distilled H2O (as the stock), respectively, with pH adjusted to 7.4. Aβ1–42 was prepared in two different ways so that different forms of Aβ1–42 aggregation could be obtained for cell application. To obtain mostly monomers of Aβ1–42, the 2 mm stock solution of Aβ1–42 in water was freshly prepared before the experiments and used immediately (hereafter referred to as “fresh”). To prepare aggregated Aβ1–42 fibrils, the 2 mm Aβ1–42 in water was incubated at 37°C for 1 week for aggregation (hereafter referred to as “aged”), aliquoted, and frozen until use. Similarly, Aβ1–40 (American Peptide Company) was dissolved at 0.5 mm in water and incubated at 37°C for 1 week, aliquoted, and frozen until use.
Bath application of different Aβ fragments was achieved through a two-step dilution procedure. At first, the Aβ stock solution was diluted in the Krebs'–Ringer's saline to a concentration of twice the designated concentration (2× working stock). The 2× working stock solution was then gently added to and mixed with the bath saline of the cells in an equal volume to achieve the desired final concentration. In a typical experiment, 1 ml of the 2× stock solution was added to 1 ml of the bath solution in the imaging chamber on the microscope stage. Different inhibitors and activators for signal transduction were normally added to the cells 20 min before the imaging.
Mitochondria imaging and quantification.
Mitochondria were stained by 20 nm MitoTracker Green or Red (Invitrogen, Eugene, OR) for 20 min, followed by three washes and 30 min incubation at 37°C for additional clearing of excessive dye. Two imaging systems were used for fluorescent time-lapse recording; each consisted of an inverted Nikon (Tokyo, Japan) microscope (Diaphot 300 or TE2000), a cooled CCD camera [CoolSnapFX (Roper Scientific, Duluth, GA) or SensiCam QE (Cooke, Romulus, MI)], an excitation shutter (DG-4 or Lambda 10–2; Sutter Instruments, Novato, CA), and the imaging software [MetaFluor (Universal Imaging Corporation, West Chester, PA) or IPLab (Scanalytics; BD Biosciences, Rockville, MD)]. Time-lapse recording of mitochondrial transport in hippocampal neurons was typically performed at a sampling rate of one frame every 5 s for 5 min, with the CCD exposure at 50 ms exposure and 2 × 2 binning. For each experiment, a 5 min control period before the application of Aβ fragments was performed, followed by 5 min time-lapse recording at various times afterward (typically at 30 or 45 min after Aβ application). All of the experiments were performed on the microscope stage with the 35 mm dish housed in a temperature-controlled chamber (Warner Instruments, New Haven, CT) with the temperature set at ∼35°C.
We counted the number of moving mitochondria in each 5 min time-lapse sequence (control and 30 min after treatment), as well as the number of total mitochondria, all within the same imaging field. Because no changes in the total mitochondrial number were observed, we used only the numbers of moving mitochondria to calculate the percentage of changes after Aβ treatment, of which 100% indicates that same numbers of moving mitochondria were present in both recording periods. To generate the movement traces of mitochondria, ImageJ (National Institutes of Health, Bethesda, MD) was used to first process the image sequence using the Zprojection function (maximum intensity), followed by division against the first frame to produce the final image of the moving traces.
To determine mitochondrial potentials, cells were labeled by JC-1 (Invitrogen) at 600 nm for 5 min at 37°C, followed by three washes and a 20–30 min incubation in Krebs'–Ringer's saline. Cells were then excited at 480 nm wavelength and imaged at both green (JC-1 monomers, low potential) and red (JC-1 aggregates, high potential) channels (Reers et al., 1995). We used a dual-view module (Optical Insights, Tucson, AZ) to simultaneously acquire both green and red fluorescence, which were then merged using ImageJ.
Ca2+ imaging.
Cells were loaded with 6 μm Fluo-4-AM (Invitrogen) for 30 min at 37°C, followed by washes and a 30 min incubation in Krebs'–Ringer's saline. Ca2+ imaging was performed for a 2 min control period before and a 16 min period after saline or Aβ25–35 application. Quantitative measurements of changes in [Ca2+]i were obtained by measuring the average fluo-4 fluorescence intensity of the cell body, which were subtracted from the average background intensity from cell-free regions. Changes in [Ca2+]i were represented by changes in relative fluo-4 fluorescence compared with baseline intensity, obtained during the 2 min control period (Δ_F_/_F_0).
Fluorescent immunostaining and quantitative analysis.
Hippocampal cells were fixed for 15 min at 37°C with freshly prepared 4% paraformaldehyde in PBS and permeabilized by 0.1% Triton X-100 for 10 min at room temperature. The cells were then incubated with blocking buffer (2% normal goat serum in PBS) for 30 min. βIII-Tubulin was detected using the polyclonal anti-Tuj1 antibody (Covance, Berkeley, CA) at a concentration of 0.5 μg/ml in PBS containing 1% normal goat serum. After an overnight incubation at 4°C, the cells were washed and incubated with 4 μg/ml Alexafluor 488-conjugated anti-rabbit antibody (Invitrogen), for 1 h at room temperature. After additional washing, the cells were then incubated for 30 min with rhodamine–phalloidin (1:200 in PBS; Invitrogen). Fluorescent images were acquired as described above and quantitatively analyzed using IPLab (Scanalytics). Briefly, the primary neurites and their growth cones were traced, and the average fluorescence intensities were measured. For comparison between the control and the Aβ-treated groups, all of the imaging settings were the same, and the measured intensities were normalized against the average of the corresponding control group. As a result, each control group produced a normalized value of 100%.
Cell viability assays.
Three different assays were used to examine changes in cell viability after different Aβ treatments. The first method used the fluorescent DNA dye Hoechst 33342 (Sigma) to quantify apoptotic cell death (Jordan et al., 1997). Here, hippocampal neurons with and without Aβ treatments were fixed with 4% paraformaldehyde (30 min) and incubated with 10 μg/ml Hoechst 33342 for 30 min. Fluorescent microcopy was performed to identify the apoptotic cells (fragmented or condensed nuclei) and the viable cells (oval-shaped nuclei with diffuse/uniform Hoechst staining). The percentage of apoptotic cells (of the total cells) was calculated and used for assessing Aβ effects on cell apoptosis. Approximately 2000 cells were counted for each group and repeated two to three times.
The second method involved double-fluorescent staining using a LIVE/DEAD Viability/Cytotoxicity kit (Invitrogen) that contains calcein AM and ethidium homodimer-1 (EthD-1) (Vaughan et al., 1995). Briefly, cells were incubated with 1 μm calcein AM and 2 μm EthD-1 for 30 min at 37°C, washed with Krebs'–Ringer's saline, and imaged by fluorescence microscopy. Similarly, four fields near the center of each dish were imaged, and the numbers of live (calcein labeled, green fluorescence) and dead (EthD-1 labeled, red fluorescence) cells were counted. Typically 200–500 cells from three independent experiments were examined for each condition.
The third method is the trypan blue exclusion assay (Ying et al., 2000), in which the hippocampal neurons were exposed to 0.05% (v/v) trypan blue (Sigma) for 5 min at different time points before and after Aβ treatment, followed by three rinses. The cells were immediately examined under a phase-contrast microscope, imaged at four random fields near the center of the dish, and returned to incubator. The microscopic images were used to quantify the numbers of cells with (dead) and without (live) the trypan blue staining. In this assay, the same dish of cells was monitored and imaged at several time points after Aβ treatment.
It should be noted that only the third assay involving trypan blue allows the continuous monitoring of the same population of cells over different time points, because trypan blue is a vital dye and can be washed out. The first and second methods can be used on cells for one time point, so different dishes are required for assays at different time points after Aβ treatments.
Results
Aβ acutely impairs fast transport of mitochondria in cultured CNS neurons
We first examined the effects of Aβ25–35 on mitochondrial transport, because this short Aβ fragment is known for its neurotoxicity (Pike et al., 1995). Incubation of hippocampal neurons 3 d in vitro (3 DIV) with 20 μm Aβ25–35 did not appear to affect their morphology (Fig. 1a, left panels) or mitochondrial morphology and distribution (Fig. 1a, middle panels). However, when time-lapse fluorescent sequences were reviewed, a significant reduction in moving mitochondria was observed after 30 min of Aβ25–35 exposure (see supplemental movie, available at www.jneurosci.org as supplemental material). To better depict the Aβ inhibition of mitochondrial movement, we digitally processed the time-lapse sequences to generate traces of moving mitochondria (see Materials and Methods). Although the control sequence produced many traces of moving mitochondria, very few traces were observed after 30 min Aβ25–35 (Fig. 1a, right panels). Quantitative analysis shows that 30 min exposure to 20 μm Aβ25–35 caused a significant reduction in moving mitochondria to ∼40% of that in the control period (Fig. 1b), whereas bath application of the control saline had no effects. We also tested the effects of Aβ25–35 on hippocampal neurons cultured for different days and found the similar inhibition on mitochondrial transport (Fig. 1b). All of the experiments hereafter were performed on 2–4 DIV cells. The Aβ25–35 impairment of mitochondrial transport was found to be dose dependent, and significant inhibition of mitochondrial transport became evident at 2 μm Aβ25–35 (Fig. 1c). Importantly, we found that the reverse peptide, Aβ35–25, did not exert any influence on the transport of mitochondria (Fig. 1c), thus demonstrating the specificity of observed Aβ25–35 impairment on mitochondrial movement.
Figure 1.
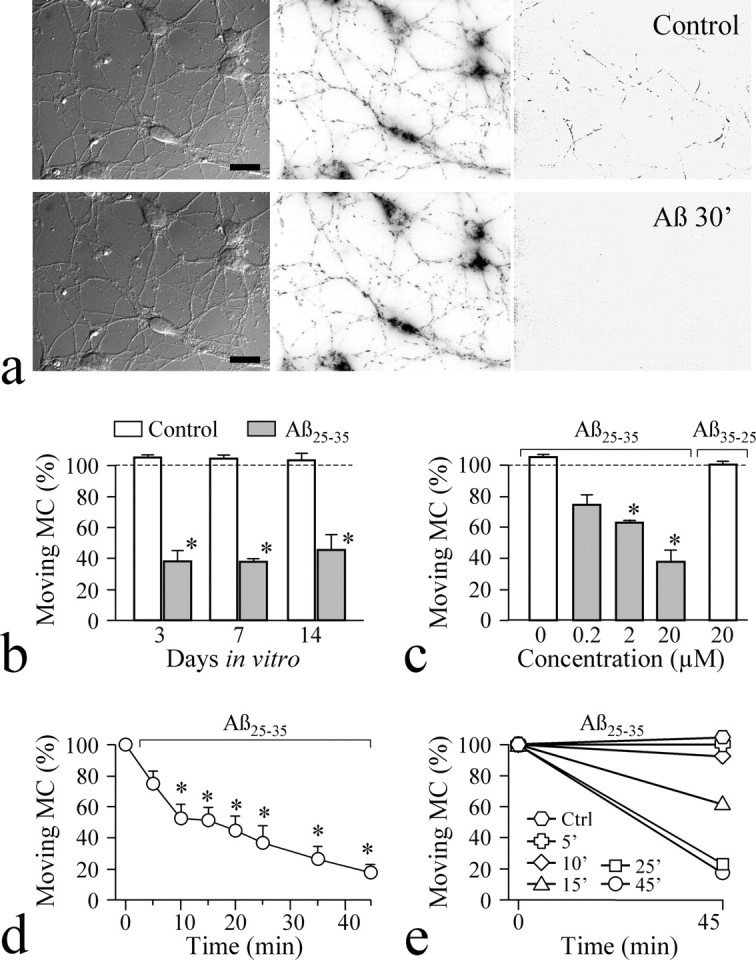
Acute Aβ impairment of mitochondrial transport in cultured neurons. a, Representative images in differential interference contrast of hippocampal neurons (left panels), their mitochondria (middle panels), and the movement traces (right panels) before (top row) and after (bottom row) 30 min Aβ25–35 treatment. For clarity, the fluorescent images were displayed in reversed grayscales. Scale bars, 10 μm. b, Percentages of moving mitochondria remaining after 30 min treatment of 3, 7, and 14 DIV cells with control saline (Control) and Aβ25–35. c, Dose dependence of Aβ25–35 inhibition of mitochondrial transport. The reserve peptide Aβ35–25 (20 μm) was the control. d, Time course of Aβ25–35 inhibition of mitochondrial movement in 3 DIV hippocampal cells. e, Exposure time of Aβ25–35 required for inhibition of mitochondrial transport. Cells (3 DIV) were incubated with 20 μm Aβ25–35 for various periods of time, followed by washout and imaging before and 45 min after the beginning of Aβ application. Ctrl, Control; MC, mitochondria. Error bars indicate SEM. *p < 0.01, significant difference from corresponding control (Student's t test).
We next examined the time dependence of Aβ25–35 inhibition of mitochondrial trafficking by performing time-lapse imaging at different time points after Aβ25–35 addition. It is apparent that mitochondrial transport was quickly affected after Aβ25–35 application, and at 10 min, significant impairment of mitochondrial transport was evident (Fig. 1d). To determine the time duration of Aβ25–35 exposure required for significant inhibition of mitochondrial transport, we exposed the neurons to Aβ25–35 for defined periods of time, washed out Aβ25–35, and performed imaging at 45 min after the onset of the experiments. We found that Aβ25–35 exposure over 15 min caused marked inhibition of mitochondrial transport (Fig. 1e). These data indicate that a brief exposure to Aβ25–35 (∼15 min) was sufficient to generate a long-lasting inhibitory effect on mitochondrial transport.
Although Aβ25–35 has been shown to mimic neurotoxicity of amyloid fragments, Aβ1–40 and Aβ1–42 are the two most relevant amyloid fragments found in AD brains. We therefore investigated whether these two amyloid peptides also exhibit acute inhibition of mitochondrial transport. Previous studies have shown that Aβ1–42 in solution undergoes a slow aggregation process that changes from monomers to fibrils; the latter is believed to contribute to AD pathological conditions (Dobeli et al., 1995; Parbhu et al., 2002; Tamagno et al., 2006). We thus prepared Aβ1–42 in two different forms, one freshly prepared and likely consisting of monomers and one aged over 1 week and likely containing mostly fibrils (Bhatia et al., 2000; Hiruma et al., 2003). We found that both Aβ1–42 preparations exhibited marked impairment on mitochondrial transport in hippocampal neurons, although aged Aβ1–42 appeared to be more potent (Fig. 2a). Similar acute impairment of mitochondrial transport was also observed with Aβ1–40 (Fig. 2b). Finally, we found that cultured cerebellar granule neurons also exhibited similar dose-dependent inhibition of mitochondrial transport in response to Aβ25–35 (Fig. 2c). Together, our results indicate that Aβ fragments exert acute and rapid inhibition of mitochondrial transport in cultured CNS neurons.
Figure 2.

Acute impairment of mitochondrial trafficking by other Aβ fragments. a, Inhibition of mitochondrial transport by different concentrations of either freshly prepared Aβ1–42 (mostly monomers) or aged Aβ1–42 (fibrils). The percentages of moving mitochondria were scored from time-lapse sequences. b, A significant decrease in mitochondrial transport was observed when hippocampal cells were incubated for 30 min with 5 μm Aβ1–40 (aged). c, Inhibition of mitochondrial transport in cerebellar granule neurons by Aβ25–35. *p < 0.01, significant difference from the corresponding control (Student's t test). Error bars indicate SEM. MC, Mitochondria; Conc., concentration.
Aβ impairment of mitochondrial transport does not involve changes in the actin cytoskeleton, mitochondrial potential, and Ca2+ signals
What are the mechanisms underlying the acute inhibition of mitochondrial transport by Aβ? Because different Aβ fragments appear to exhibit similar levels of impairment on the transport of mitochondria (see above), we used Aβ25–35 for the rest of the study. We first examined the possibility that Aβ peptides might inhibit transport by inducing excessive polymerization of the actin cytoskeleton (Hiruma et al., 2003). We used double immunostaining to examine the changes in the actin cytoskeleton and microtubules in hippocampal neurons. We found that there was no apparent difference in both cytoskeletal structures between the control hippocampal neurons and those exposed to 30 min 20 μm Aβ25–35 (Fig. 3a). Importantly, our quantitative analyses of the fluorescence intensities of the actin cytoskeleton at both the growth cone and the neurite processes, as well as of the microtubules, showed no difference between the control and Aβ-treated groups (Fig. 3b). To better examine the potential changes of the actin cytoskeleton in the same cells over different times after Aβ exposure, we next used high-resolution live imaging to examine the dynamics of green fluorescent protein (GFP)–actin expressed in hippocampal neurons. Our live imaging data show that Aβ25–35 exposure did not induce significant changes in the actin cytoskeleton in cultured hippocampal neurons of different ages (Fig. 3b,c). The neuronal morphology, branching, and dendritic spines highlighted by GFP–actin exhibited no changes in response to 20 μm Aβ25–35 for up to 50 min (Fig. 3b,c). Furthermore, cytochalasin B (CB), a drug that inhibits actin polymerization, did not prevent Aβ inhibition of mitochondrial transport. In the presence of 5 μm CB, Aβ25–35 still reduced moving mitochondria from the control 107.2 ± 6.3% (mean ± SEM; n = 4) to 31.9 ± 3.7% (n = 4; p < 0.001, Student's _t_ test), whereas CB alone slightly affected the percentage of moving mitochondria (87.0 ± 3.1%; _p_ > 0.1 compared with the control). Based on these imaging data (both immunofluorescence and live imaging) and pharmacological inhibition of actin polymerization, we conclude that Aβ impairment of mitochondrial transport was not a direct result of aberrant changes in the actin cytoskeleton.
Figure 3.

Aβ effects did not involve aberrant changes in the cytoskeleton. a, Double-immunofluorescent labeling of cultured hippocampal neurons with and without exposure to Aβ25–35 indicates no change in the cytoskeleton. The representative fluorescent images of a control and an Aβ-exposed hippocampal 3 DIV neuron are shown in the left two panels (green, microtubules; red, F-actin). Results of quantitative measurements of the fluorescent intensities of F-actin (Actin) and microtubules (MTs) along the neurite or at the growth cone (GC) are shown in the bar graph on the right. No difference was observed between the control (Ctrl) and the Aβ25–35-treated groups (p > 0.1, Student's _t_test). b, c, Live fluorescence images of GFP–actin in hippocampal neurons before and after Aβ treatment. Hippocampal neurons were transfected with a GFP–γ-actin construct (in pCS2+ vector) using the calcium phosphate method (Kohrmann et al., 1999) 24 h before imaging. The time-lapse sequences of a 3 DIV hippocampal neuron (b) and a 14 DIV neuron (c) expressing GFP–actin before and after 20 μm Aβ25–35 show no changes in the morphology and branching (b) and dendritic spines (c). In c, the first image was acquired at low magnification using a 40× lens. The boxed region was then imaged at a higher magnification using a 100× lens. The digits indicate minutes before and after Aβ application. Scale bars, 10 μm. Error bars indicate SEM.
Because the transport of mitochondria and their potentials are correlated (Miller and Sheetz, 2004) and Aβ induces mitochondrial dysfunction over the long term (Smith et al., 2002; Eckert et al., 2003), we examined the mitochondrial potential after Aβ exposure using dual-wavelength imaging of JC-1 (Reers et al., 1995). We found that a 30 min exposure to Aβ did not produce significant effects on the numbers of mitochondria exhibiting high and low potentials in hippocampal neurons (Fig. 4a). The percentage of mitochondria bearing high potential (indicated by red aggregates of JC-1) remained the same after Aβ exposure (Fig. 4b). Therefore, Aβ inhibition of mitochondrial transport is unlikely to be caused by changes in mitochondrial potential, but instead may involve specific signaling pathways.
Figure 4.

Aβ peptides did not induce significant changes in the mitochondrial potential. a, JC-1 imaging of mitochondrial potentials in hippocampal neurons before and after 30 min Aβ treatment. The differential interference contrast and merged fluorescence from both the green and red fluorescence of the same neurons are shown. Mitochondria with low membrane potential were stained green, whereas mitochondria with high potential displayed red aggregates of JC-1. Scale bar, 20 μm. b, Quantitative analysis of the percentage of mitochondria (MC) with red aggregates before and after 30 min Aβ treatment. The data presented in the bar graph came from five different cultures and contained ≥20 image fields (over 30 cells). Error bars indicate SEM. HP, High potential.
Aβ toxicity has been linked to Ca2+, and Aβ peptides could disrupt the Ca2+ homeostasis to induce cell death and malfunction (Abramov et al., 2003; Mattson and Chan, 2003). We therefore examined the role of Ca2+ in Aβ effects. Time-lapse fluo-4 imaging showed that 3 DIV hippocampal neurons maintained a stable level of intracellular Ca2+ concentrations ([Ca2+]i) before and after bath application of the control saline (Fig. 5a). When 20 μm Aβ25–35 was bath applied, the majority of the neurons maintained stable [Ca2+]i, whereas 5–7% of the cells (of >100 in total) displayed an elevation (Fig. 5b), consistent with previous observations that only [Ca2+]i in astrocytes was affected by Aβ (Abramov et al., 2004b). We next used a Ca2+-free saline (containing 1 mm EGTA) to prevent Ca2+ influx, and Ca2+ imaging revealed that the Ca2+-free saline effectively eliminated changes in [Ca2+]i associated with Aβ application (Fig. 5c), However, Aβ25–35 was still able to effectively inhibit the mitochondrial transport of hippocampal neurons in the Ca2+-free saline (Fig. 5d). To further test that changes in [Ca2+]i were not involved in Aβ impairment of mitochondrial transport, we loaded the hippocampal neurons with BAPTA-AM, a strong Ca2+ buffer that can effectively eliminate fluctuations in [Ca2+]i. We found that BAPTA buffering of [Ca2+]i did not prevent acute Aβ impairment of transport (Fig. 5e). These results, especially the inability to block Aβ impairment of transport by eliminating Ca2+ influx and buffering [Ca2+]i, strongly indicate that Ca2+ signaling is not involved in Aβ inhibition of mitochondrial transport.
Figure 5.

Ca2+ responses to Aβ application. a–c, Time-lapse measurements of the changes in [Ca2+]i by fluo-4 imaging from 10 individual cells undergoing three different treatments: control saline (Ctrl; a), 20 μm Aβ25–35 (b), and 20 μm Aβ25–35 in Ca2+-free Krebs'–Ringer's solution (c). Each trace depicts the relative change of the fluo-4 intensity (Δ_F_/_F_0) in one neuron before and after bath application of control or Aβ solution (indicated by the arrows). d, Impairment of mitochondrial transport of hippocampal neurons by different concentrations of Aβ25–35 in Ca2+-containing and Ca2+-free Krebs'–Ringer's solutions. e, Effects of BAPTA loading on mitochondrial transport with and without Aβ25–35. *p < 0.01 significant difference from the corresponding control (Student's t test). MC, Mitochondria. Error bars indicate SEM.
Involvement of protein kinase A and GSK3β in acute actions of Aβ peptides on mitochondrial transport
We next tested the involvement of the cAMP pathway in Aβ effects on mitochondrial transport. We first used specific antagonists to inhibit protein kinase A (PKA) and then examined the Aβ impairment of mitochondrial transport. We found that PKA inhibition by KT5720 or Rp-cAMP had no effects on mitochondrial transport and Aβ impairment (Fig. 6). This piece of data indicates that the Aβ impairment is not directly mediated by PKA. We next examined the effects of PKA activation on Aβ impairment of mitochondrial transport. When forskolin (30 μm) was used to stimulate adenylate cyclase to elevate intracellular cAMP levels, the impairment of mitochondrial transport by Aβ25–35 was completely eliminated (Fig. 6). Consistently, the membrane-permeable cAMP analog dibutyl-cAMP (db-cAMP) also effectively attenuated the acute inhibition of mitochondrial transport by Aβ25–35, whereas neither forskolin nor db-cAMP itself generated any effects on the transport of mitochondria (Fig. 6). Moreover, pituitary adenylate cyclase activating peptide, a neuropeptide widely expressed in the brain (Waschek, 2002), also abolished Aβ impairment of mitochondrial transport (Fig. 6). Together, these data indicate that Aβ25–35 did not impair mitochondrial transport by directly inhibiting PKA, but rather that PKA activity could modulate the Aβ acute actions.
Figure 6.

Involvement of PKA and GSK3β in acute Aβ impairment of mitochondrial transport. The bar graph depicts the percentages of moving mitochondria (MC) remaining after a 30 min treatment with different compounds with and without Aβ25–35. For clarity, results from cells exposed to Aβ25–35 are depicted in gray. Plus and minus signs indicate the presence and absence of the particular compounds in bath, respectively. Aβ25–35, 20 μm; Rp-cAMP, 50 μm; KT5720, 200 nm; Db-cAMP, 500 μm; forskolin, 30 μm; pituitary adenylate cyclase activating peptide (PACAP), 10 nm; lithium chloride (LiCl), 1 mm; valproic acid (VPA), 0.6 mm; SB415286, 10 μm. Each condition was repeated at least three times from different rounds of cultures and was averaged from 20–30 cells. *p < 0.01 (Student's t test). Error bars indicate SEM.
GSK3β has been implicated in AD pathogenesis and neurodegeneration (Eldar-Finkelman, 2002). Previous biochemical and functional assays have shown that Aβ peptides could activate GSK3β (Takashima et al., 1998; Cedazo-Minguez et al., 2003). Given that GSK3β could phosphorylate the motor protein kinesin-1 to affect fast axonal transport (Morfini et al., 2002), we investigated whether or not GSK3β could be a target of Aβ effects on mitochondrial transport. When lithium chloride, a known inhibitor of GSK3β, was applied (Williams et al., 2004), Aβ impairment of mitochondrial transport was essentially abolished (Fig. 6). Similarly, two other GSK3β inhibitors, valproic acid (Chen et al., 1999) and SB415286 (Coghlan et al., 2000), also effectively attenuated Aβ impairment of mitochondrial transport (Fig. 6). Considering that PKA can phosphorylate and inactivate GSK3β (Grimes and Jope, 2001), our data indicate an important role for GSK3β in the acute actions of Aβ on transport and suggest that PKA may alleviate Aβ inhibition by modulating GSK3β activity.
Cell death was only induced by long-term Aβ treatment
Long-term exposure to Aβ fragments is known to result in cell death in a variety of cells (Jordan et al., 1997; Troy et al., 2001; Eckert et al., 2003; Abramov et al., 2004a). However, the acute impairment of mitochondrial trafficking here was observed as early as 10 min after Aβ application without apparent morphological change related to cell death, indicating that cell death is unlikely to be a cause of mitochondrial transport impairment. To further evaluate this issue, we performed cell viability assays to examine Aβ-induced cell death and its time course in hippocampal neurons (2–4 DIV). We first used the fluorescent DNA dye Hoechst 33342 to identify apoptotic cells exhibiting condensed and fragmented nuclei (Fig. 7a, arrows in inset). Two types of Aβ treatments were performed: one involved 30 min exposure to 20 μm Aβ25–35, followed by three washes and incubation in the culture medium at 37°C, whereas the other had 20 μm Aβ25–35 continuously present in the bath solution. The cells were fixed, labeled by Hoechst 33342, and imaged at various time points before and after Aβ25–35 application. We found that Aβ25–35 did not induce significant cell apoptosis until 6 h after Aβ application (Fig. 7a). A significant number of apoptotic cells were observed at 24 h after Aβ25–35 application (Fig. 7a). Because the Hoechst labeling method depends on the morphological changes of nuclei for identification of apoptotic cells, it may take a longer time than the actual cell death. We therefore used a cell live/death assay in which calcein AM was used to label live cells and EthD-1 was used to label dead cells. Consistently, we found that 30 min exposure of hippocampal neurons to Aβ25–35 only induced marked cell death after 6 h.
Figure 7.
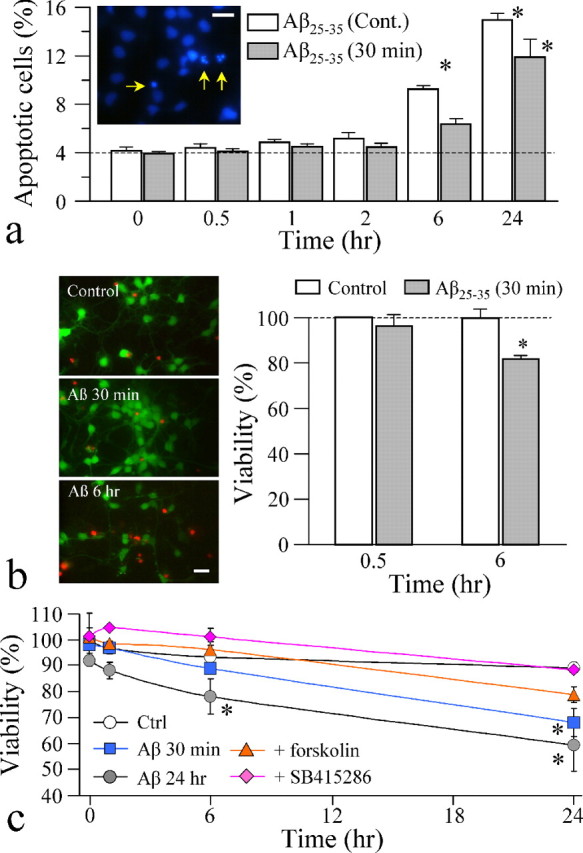
Cell death in hippocampal cultures induced by long-term exposure to Aβ peptides. a, Quantitative analysis of apoptotic cells by Hoechst 33342 labeling of nuclei. A representative fluorescent image of Hoechst-labeled hippocampal neurons is shown in the inset, in which arrows indicate the condensed/fragmented nuclei (apoptotic cells). Quantified results are presented in the bar graph showing the percentage of apoptotic cells at various times after Aβ25–35 application. The white bars show the results from cells incubated with 20 μm Aβ25–35 continuously, and the gray bars are from the cells treated with 20 μm Aβ25–35 for only 30 min and then washed out. Asterisks indicate a significant difference from the control (zero time point). Cont., Continuous present. b, Results from the viability assay using the Live/Dead kit. Representative fluorescent images of hippocampal neurons exposed to the control and Aβ25–35 peptides. The live cells were labeled by calcein AM (green), and the dead cells were labeled by EthD-1 (red). Quantified percentages of live cells are shown in the bar graph. The asterisk indicates a significant difference from the corresponding control. c, Quantitative analysis of the cell viability of the same population of hippocampal neurons over times using trypan blue dye. In most of the experiments, 20 μm Aβ25–35 was applied to the cells for 30 min and washed, except the group labeled by Aβ 24 h, in which 20 μm Aβ25–35 was applied for 24 h. Asterisks indicate a statistical significance when comparing with the control (Ctrl) at the corresponding time point. A total of 175–500 cells (from 2–3 experiments) were examined for each condition. Scale bars, 20 μm. Error bars indicate SEM.
To better examine the time course of cell death induced by Aβ peptides in the same population of cells in culture, we used trypan blue dye to perform the viability assay at various times after Aβ application (see Materials and Methods). Consistent with the results from the other two assays, we found that exposure of hippocampal neurons to Aβ25–35 (either 30 min or continuous exposure) only induced significant cell death after 6 h (Fig. 7c). At 24 h after Aβ application, the cell viability dropped to ∼70% (Fig. 7c). Importantly, we found that application of forskolin to elevate intracellular cAMP levels was able to mostly attenuate cell death induced by Aβ peptides (Fig. 7c). Similarly, inhibition of GSK3β by SB415286 completely prevented the cell death induced by Aβ25–35 (Fig. 7c). Together, these results show that Aβ-induced cell death is a long-term result and requires several hours, whereas Aβ impairment of mitochondrial trafficking occurs in minutes. As a result, the acute impairment of mitochondrial trafficking is unlikely to be a consequence of Aβ-induced cell death. Our data also suggest that both PKA and GSK3β are involved in Aβ actions on mitochondrial trafficking and cell viability.
Discussion
Aβ is a key molecule in AD pathogenesis. Many in vitro and in vivo studies have shown a variety of Aβ neurotoxicity including cell death and axonal transport deficits (Mattson, 2004; Zhu et al., 2005), but much of it appears to associate with Aβ aggregates and involve long-term actions (Ross and Poirier, 2004). Recently, soluble Aβ oligomers were found to inhibit synaptic connectivity (Walsh et al., 2002; Wang et al., 2002; Lacor et al., 2004), but the signaling transduction mechanisms remain elusive. In our study, we observed acute effects of Aβ molecules on the fast transport of mitochondria. The rapid onset of inhibition of mitochondrial transport (∼10 min) indicates a very early action of Aβ that is distinct from other long-term Aβ toxicity involved in cell death and degenerative sequences. Furthermore, we have excluded several possible nonspecific effects of Aβ on mitochondrial transport, including excessive actin polymerization and loss of mitochondria membrane polarization. Finally, we have presented evidence that Aβ likely acted through GSK3β to impair mitochondrial transport, and the impairment can be alleviated by PKA activation. Interestingly, both GSK3β and PKA appear to also be involved in Aβ-induced cell death, and PKA activation or GSK3β inhibition was able to attenuate Aβ-induced cell death. These findings support an important role of PKA and GSK3β in AD-related conditions (Bhat and Budd, 2002; Kaytor and Orr, 2002). It remains to be elucidated how Aβ activates GSK3β (directly or indirectly) and which downstream targets act on mitochondrial trafficking. Our preliminary experiments show that 30 min Aβ25–35 exposure did not induce significant elevation of tau phosphorylation, thus indicating that tau phosphorylation might not be the cause of transport impairment of mitochondria. It is also unclear whether the trafficking of other organelles was also affected by Aβ peptides. Finally, the presence of both axonal and dendritic processes (which contain microtubules with opposite polarities) in hippocampal cultures prevented us from determining whether anterograde and/or retrograde transport were selectively impaired. Nonetheless, our findings have identified an important acute effect of Aβ molecules on an extremely important organelle. Moreover, our results have not only provided significant insights into the signal transduction of Aβ effects but also indicated a potential strategy for alleviating Aβ toxicity to prevent or delay the onset of pathogenesis.
Mitochondria are responsible for local energy production, Ca2+ homeostasis, and clearance of reactive oxygen species, therefore localization and delivery of mitochondria to subcellular locations through transport are likely to be essential for many neuronal functions and for neuronal survival. Aβ impairment of mitochondrial transport could compromise many of these neuronal functions, leading to neuronal abnormalities that may underlie some AD pathological conditions. For example, mitochondrial trafficking is known to be essential for synaptic functions of CNS neurons, and the impairment of mitochondrial transport by Aβ could undermine the important contribution of mitochondria in synaptic development and functions (Li et al., 2004; Chang et al., 2006). Whether Aβ impairment of mitochondrial trafficking contributes directly or partially to Aβ effects on synaptic functions (Walsh et al., 2002; Wang et al., 2002; Lacor et al., 2004; Rowan et al., 2004) remains to be investigated. At this moment, it is unclear whether Aβ can also acutely impair the fast transport of other organelles. If true, it would be plausible that the acute Aβ inhibition of transport could also contribute to the cycle of transport deficits and Aβ production reported previously (Stokin et al., 2005). In this case, an imbalance of Aβ production and clearance in vivo might result in the accumulation of Aβ molecules, which could rapidly act on transport of important organelles to trigger a cascade of events leading to pathological conditions of AD. Clearly, future studies combining in vitro and in vivo models could test and validate these possibilities and lead to additional elucidation of the complex cellular steps involved in AD pathogenesis.
Footnotes
This work was supported by National Basic Research Program of China Grant 973 #2005CB522503 to Z.X. and by research grants from the National Institutes of Health (NS36241) and the National Science Foundation (0109776) to J.Q.Z.
References
- Abramov AY, Canevari L, Duchen MR. Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci. 2003;23:5088–5095. doi: 10.1523/JNEUROSCI.23-12-05088.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov AY, Canevari L, Duchen MR. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci. 2004a;24:565–575. doi: 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov AY, Canevari L, Duchen MR. Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta. 2004b;1742:81–87. doi: 10.1016/j.bbamcr.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Bacci A, Verderio C, Pravettoni E, Matteoli M. Synaptic and intrinsic mechanisms shape synchronous oscillations in hippocampal neurons in culture. Eur J Neurosci. 1999;11:389–397. doi: 10.1046/j.1460-9568.1999.00440.x. [DOI] [PubMed] [Google Scholar]
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–342. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Budd SL. GSK3beta signalling: casting a wide net in Alzheimer's disease. Neurosignals. 2002;11:251–261. doi: 10.1159/000067423. [DOI] [PubMed] [Google Scholar]
- Bhatia R, Lin H, Lal R. Fresh and globular amyloid beta protein (1–42) induces rapid cellular degeneration: evidence for AbetaP channel-mediated cellular toxicity. FASEB J. 2000;14:1233–1243. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Schwarzenbacher R, Lipton SA. Molecular pathways to neurodegeneration. Nat Med. 2004;10(Suppl):S2–S9. doi: 10.1038/nm1067. [DOI] [PubMed] [Google Scholar]
- Cedazo-Minguez A, Popescu BO, Blanco-Millan JM, Akterin S, Pei JJ, Winblad B, Cowburn RF. Apolipoprotein E and beta-amyloid (1–42) regulation of glycogen synthase kinase-3beta. J Neurochem. 2003;87:1152–1164. doi: 10.1046/j.1471-4159.2003.02088.x. [DOI] [PubMed] [Google Scholar]
- Chang DT, Honick AS, Reynolds IJ. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci. 2006;26:7035–7045. doi: 10.1523/JNEUROSCI.1012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Huang LD, Jiang YM, Manji HK. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem. 1999;72:1327–1330. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000;7:793–803. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Dobeli H, Draeger N, Huber G, Jakob P, Schmidt D, Seilheimer B, Stuber D, Wipf B, Zulauf M. A biotechnological method provides access to aggregation competent monomeric Alzheimer's 1–42 residue amyloid peptide. Biotechnology (NY) 1995;13:988–993. doi: 10.1038/nbt0995-988. [DOI] [PubMed] [Google Scholar]
- Eckert A, Keil U, Marques CA, Bonert A, Frey C, Schussel K, Muller WE. Mitochondrial dysfunction, apoptotic cell death, and Alzheimer's disease. Biochem Pharmacol. 2003;66:1627–1634. doi: 10.1016/s0006-2952(03)00534-3. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H. Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med. 2002;8:126–132. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Hiruma H, Katakura T, Takahashi S, Ichikawa T, Kawakami T. Glutamate and amyloid β-protein rapidly inhibit fast axonal transport in cultured rat hippocampal neurons by different mechanisms. J Neurosci. 2003;23:8967–8977. doi: 10.1523/JNEUROSCI.23-26-08967.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J, Galindo MF, Miller RJ. Role of calpain- and interleukin-1 beta converting enzyme-like proteases in the beta-amyloid-induced death of rat hippocampal neurons in culture. J Neurochem. 1997;68:1612–1621. doi: 10.1046/j.1471-4159.1997.68041612.x. [DOI] [PubMed] [Google Scholar]
- Kaytor MD, Orr HT. The GSK3 beta signaling cascade and neurodegenerative disease. Curr Opin Neurobiol. 2002;12:275–278. doi: 10.1016/s0959-4388(02)00320-3. [DOI] [PubMed] [Google Scholar]
- Kohrmann M, Haubensak W, Hemraj I, Kaether C, Lessmann VJ, Kiebler MA. Fast, convenient, and effective method to transiently transfect primary hippocampal neurons. J Neurosci Res. 1999;58:831–835. [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer's disease. Cell Calcium. 2003;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parbhu A, Lin H, Thimm J, Lal R. Imaging real-time aggregation of amyloid beta protein (1–42) by atomic force microscopy. Peptides. 2002;23:1265–1270. doi: 10.1016/s0196-9781(02)00061-x. [DOI] [PubMed] [Google Scholar]
- Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, Cribbs DH, Glabe CG, Cotman CW. Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25-35 region to aggregation and neurotoxicity. J Neurochem. 1995;64:253–265. doi: 10.1046/j.1471-4159.1995.64010253.x. [DOI] [PubMed] [Google Scholar]
- Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995;260:406–417. doi: 10.1016/0076-6879(95)60154-6. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Wang Q, Anwyl R. Mechanisms of the inhibitory effects of amyloid beta-protein on synaptic plasticity. Exp Gerontol. 2004;39:1661–1667. doi: 10.1016/j.exger.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Schapira AH. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- Smith MA, Drew KL, Nunomura A, Takeda A, Hirai K, Zhu X, Atwood CS, Raina AK, Rottkamp CA, Sayre LM, Friedland RP, Perry G. Amyloid-beta, tau alterations and mitochondrial dysfunction in Alzheimer disease: the chickens or the eggs? Neurochem Int. 2002;40:527–531. doi: 10.1016/s0197-0186(01)00123-1. [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- Takashima A, Honda T, Yasutake K, Michel G, Murayama O, Murayama M, Ishiguro K, Yamaguchi H. Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25-35) enhances phosphorylation of tau in hippocampal neurons. Neurosci Res. 1998;31:317–323. doi: 10.1016/s0168-0102(98)00061-3. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Bardini P, Guglielmotto M, Danni O, Tabaton M. The various aggregation states of beta-amyloid 1–42 mediate different effects on oxidative stress, neurodegeneration, and BACE-1 expression. Free Radic Biol Med. 2006;41:202–212. doi: 10.1016/j.freeradbiomed.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Terwel D, Dewachter I, Van Leuven F. Axonal transport, tau protein, and neurodegeneration in Alzheimer's disease. Neuromol Med. 2002;2:151–165. doi: 10.1385/NMM:2:2:151. [DOI] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J Neurochem. 2001;77:157–164. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- Vaughan PJ, Pike CJ, Cotman CW, Cunningham DD. Thrombin receptor activation protects neurons and astrocytes from cell death produced by environmental insults. J Neurosci. 1995;15:5389–5401. doi: 10.1523/JNEUROSCI.15-07-05389.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL. Soluble oligomers of beta amyloid (1–42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Waschek JA. Multiple actions of pituitary adenylyl cyclase activating peptide in nervous system development and regeneration. Dev Neurosci. 2002;24:14–23. doi: 10.1159/000064942. [DOI] [PubMed] [Google Scholar]
- Williams R, Ryves WJ, Dalton EC, Eickholt B, Shaltiel G, Agam G, Harwood AJ. A molecular cell biology of lithium. Biochem Soc Trans. 2004;32:799–802. doi: 10.1042/BST0320799. [DOI] [PubMed] [Google Scholar]
- Ying HS, Gottron FJ, Choi DW. Crawley JN, Gerfen CR, Rogawski MA, Sibley DR, Skolnick P, Wray S. Current protocols in neuroscience. Hoboken, NJ: Wiley; 2000. Assessment of cell viability in primary neuronal cultures; pp. 7.18.1–7.18.17. [Google Scholar]
- Zhu X, Moreira PI, Smith MA, Perry G. Alzheimer's disease: an intracellular movement disorder? Trends Mol Med. 2005;11:391–393. doi: 10.1016/j.molmed.2005.07.002. [DOI] [PubMed] [Google Scholar]