Delayed Upregulation of ATP P2X3 Receptors of Trigeminal Sensory Neurons by Calcitonin Gene-Related Peptide (original) (raw)
Abstract
Recent evidence indicates a key role for the neuropeptide calcitonin gene-related peptide (CGRP) in migraine pain, as demonstrated by the strong analgesic action of CGRP receptor antagonists, although the mechanisms of this effect remain unclear. Most trigeminal nociceptive neurons releasing CGRP also express ATP-activated purinergic P2X3 receptors to transduce pain. To understand whether the CGRP action involves P2X3 receptor modulation, the model of trigeminal nociceptive neurons in culture was used to examine the long-term action of this peptide. Although 79% of CGRP-binding neurons expressed P2X3 receptors, acute application of CGRP did not change P2X3 receptor function. Nevertheless, after 1 h of CGRP treatment, strong enhancement of the amplitude of P2X3 receptor currents was observed together with accelerated recovery from desensitization. Receptor upregulation persisted up to 10 h (despite CGRP washout), was accompanied by increased P2X3 gene transcription, and was fully prevented by the CGRP antagonist CGRP8–37. Surface biotinylation showed CGRP augmented P2X3 receptor expression, consistent with confocal microscopy data indicating enhanced P2X3 immunoreactivity beneath the neuronal membrane. These results suggest that CGRP stimulated trafficking of P2X3 receptors to the cell-surface membrane. Using pharmacological tools, we demonstrated that this effect of CGRP was dependent on protein kinase A and PKC activation and was prevented by the trafficking inhibitor brefeldin A. Capsaicin-sensitive TRPV1 vanilloid receptors were not upregulated. The present data demonstrate a new form of selective, slow upregulation of nociceptive P2X3 receptors on trigeminal neurons by CGRP. This mechanism might contribute to pain sensitization and represents a model of neuronal plasticity in response to a migraine mediator.
Keywords: plasticity, pain, purinergic receptor, nociception, neuropeptide, receptor trafficking
Introduction
Calcitonin gene-related peptide (CGRP), a potent vasodilator and pro-inflammatory agent, is contained in vesicles of trigeminal nerve endings (Arulmani et al., 2004). Accumulating evidence suggests that CGRP has a key role in migraine, one of the commonest neurological disorders (Pietrobon and Striessnig, 2003; Goadsby, 2005). Although the CGRP plasma concentration closely correlates with the time course and severity of migraine (Sarchielli et al., 2000; Juhasz et al., 2005), the crucial role of this peptide is supported by the efficacy of CGRP antagonists for the treatment of migraine pain (Olesen et al., 2004; Edvinsson, 2005; Rudolf et al., 2005). Because CGRP does not have acute effects on the excitability of meningeal nociceptors (Levy et al., 2005), it is, however, difficult to understand how CGRP could induce persistent headache mediated by trigeminal ganglion (TG) neurons. This issue might be addressed by studying a model system that allows investigating structural and functional properties of TG neurons over time.
Because CGRP operates via G-protein-coupled receptors, it seems feasible to look for its downstream effectors among the pain-transducing receptors of TG neurons. On nociceptive sensory neurons, extracellular ATP is one of the main algogenic transmitters (Chizh and Illes, 2001; North, 2003) acting on ionotropic receptors containing the purinergic P2X3 subunit (Cockayne et al., 2000; Souslova et al., 2000). P2X3 receptors show rapid desensitization (North, 2003; Sokolova et al., 2004) controlled by intracellular messengers (Koshimizu et al., 1999). Therefore, P2X3 receptors and their desensitization properties appear to be one potential target for expressing the algogenic action of CGRP. In support of this hypothesis, other neuropeptides such as substance P and bradykinin facilitate P2X3 receptor signaling by speeding up recovery from desensitization, although with a faster time course (Paukert et al., 2001). Nevertheless, sensory neurons also use other pain-transducing receptors, such as TRPV1 (vanilloid receptor), that are activated by a wide range of stimuli [acidity, pressure, chemical irritants such as capsaicin, heat, etc. (Julius and Basbaum, 2001; Wang and Woolf, 2005)]. Using, as a model, cultures of mouse TG neurons, we demonstrated that sustained application of CGRP selectively potentiated P2X3 receptor function, while leaving TRPV1 receptors unaffected.
Materials and Methods
Cultured TG neurons.
Primary cultures of TG or thoraco-lumbar dorsal root ganglion (DRG) neurons were obtained from postnatal day 10–12 C57-Black/6Jico mice. Animals were anesthetized by diethyl ether and decapitated (in accordance with the Italian Animal Welfare Act and approved by the Local Authority Veterinary Service). Ganglia were isolated and dissociated for 10–20 min at 37°C in a solution containing 0.25 mg/ml trypsin, 1 mg/ml collagenase, and 0.2 mg/ml DNase (Sigma, St. Louis, MO) in F-12 medium (Invitrogen, San Diego, CA). Cells were used 24 h after plating.
Unless indicated otherwise, neurons were incubated with 1 μm CGRP for 1 h at 37°C. HPLC analysis demonstrated CGRP (dissolved in physiological solution) to be stable for at least 5 h at 37°C (Dr. O. Jahraus, personal communication). This observation is consistent with the reported stability (90%) of CGRP when incubated for 5 h at 37°C (Ichikawa et al., 2000). The following drugs were preapplied for 30 min and coapplied together with CGRP for 1 h at 37°C: the CGRP receptor antagonist CGRP8–37 (2 μm), actinomycin D (5 μg ml−1), the protein kinase A (PKA) inhibitor fragment 14-22 (3 μm), chelerythrine chloride (5 μm), brefeldin A (5 μg ml−1), forskolin (1 μm), and phorbol 12-myristate 13-acetate (PMA; 325 nm) (all from Sigma).
Patch-clamp recording.
After 1 d in culture, cells were superfused continuously (2 ml min−1) with physiological solution containing (in mm) 152 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES (pH adjusted to 7.4 with NaOH). Single cells were patch clamped in the whole-cell configuration by means of an L/M-EPC 7B patch-clamp amplifier (List Medical, Darmstadt, Germany) using pipettes with a resistance of 3–4 MΩ when filled with (in mm) the following: 140 KCl, 0.5 CaCl2, 2 MgCl2, 2 Mg2ATP3, 2 GTP, 10 HEPES, and 10 EGTA (pH adjusted to 7.2 with KOH). TG or DRG cells were voltage clamped at −60 mV (series resistance compensation, 70%). Currents were filtered at 1 kHz and acquired by means of a DigiData 1200 Interface and the pClamp 8.2 software (Molecular Devices, Sunnyvale, CA). To assess P2X3 receptor function, the potent synthetic agonist α,β-methylene-adenosine-5′-triphosphate (α,β-meATP) and the natural agonist ATP were applied with a fast superfusion system (Rapid Solution Changer RSC-200; BioLogic Science Instruments, Claix, France); the time for solution exchange was ∼30 ms. The rapid desensitization of the current during agonist application together with its block by the selective antagonist A-317491 (5-[[[(3-phenoxyphenyl)methyl][(1_S_)-1,2,3,4-tetrahydro-1-naphthalenyl]amino]carbonyl]-1,2,4-benzenetricarboxylic acid sodium salt) (1 μm; 95% inhibition; n = 13) (Jarvis, 2003) indicated its origin as a P2X3 receptor-mediated response. In a few experiments, α,β-meATP (200 μm; 10 ms pulse) was applied via a puffer pipette close to the recorded cell. Responses were measured in terms of peak amplitude and fitted with a logistic equation (Origin 6.0; Microcal, Northampton, MA) to express agonist potency in terms of EC50 values (concentration producing 50% of the maximum response). For standard tests of cell responsiveness, agonist applications (10 μm, 2 s; fast superfusion) were spaced at 5 min intervals to obtain full response recovery from desensitization (see Results). Paired-pulse experiments with α,β-meATP applications over shorter intervals were used to measure recovery from desensitization (Sokolova et al., 2004). The peak of α,β-meATP currents generated by the second pulse was expressed as the percentage of the peak amplitude of the control response so as to express recovery from desensitization as the time needed to regain 50% of the control peak amplitude (_t_½). To quantify the effect of a certain drug on the α,β-meATP-induced current amplitude for each drug-treated neuron, the peak response was expressed as a percentage of the mean peak current amplitude obtained from control neurons from sister dishes used in parallel. Capsaicin was applied at the standard test dose of 1 μm (2 s) to evoke reproducible inward currents. Recording of functional responses started just after the washout of the CGRP-containing medium and continued for 1–1.5 h (if not indicated otherwise).
Calcium imaging.
Cells were incubated for 40 min at 20–22°C in physiological solution containing Fluo3-AM (5 μm; Invitrogen, Eugene, OR), followed by a 30 min washout period. Fluorescence emission was acquired with a CCD camera (Coolsnap HQ; Roper Scientific, Duluth, GA) at 150 ms intervals. Data were collected from cells that produced a rapid response to a pulse of KCl (50 mm, 1 s), thus indicating they were neurons. Images were analyzed with the Metafluor software (Metafluor Imaging Series 6.0; Universal Imaging, Downingtown, PA). Intracellular Ca2+ transients were expressed as fractional amplitude increase (Δ_F_/_F_0, where F_0 is the baseline fluorescence level and Δ_F is the increment over baseline).
Real-time reverse transcription-PCR.
Total mRNA was extracted from TG cultures using Trizol reagent (Invitrogen). After DNase treatment (Ambion, Austin, TX), 1 μg of total RNA was retro-transcribed using SuperScript III (Invitrogen) with a mixture of oligo-dT and random primers (Invitrogen). cDNAs (30 ng) were amplified with JumpStart Taq ReadyMix (Invitrogen), specific oligonucleotide primers, and TaqMan fluorogenic probes (Applied Biosystems, Foster City, CA) in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems). Prevalidated assays, specific for amplification of mouse P2X3, β-tubulin III, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA (Applied Biosystems code numbers: Mm00523699_m1, Mm00727586_s1, and 4352339E FG m MGB) were used. The end-point PCR amplicons (analyzed on an agarose gel) had the expected length. Initial calibration of the samples gave similar amplification of the GAPDH housekeeping mRNA, and all assays were validated for linearity of amplification efficiency. Negative controls containing no template cDNA were run in each condition and gave no results. The relative mRNA expression of P2X3 in the different samples was normalized to the neuronal β-tubulin III mRNA content. Absolute calculations for relative mRNA transcript levels were performed using the comparative method between cycle thresholds of different reactions (Livak and Schmittgen, 2001).
Membrane biotinylation and Western immunoblot.
Total protein lysates of TG cultures were extracted 2 or 5 h after 1 h CGRP treatment. For the procedure of membrane protein biotinylation, intact TG neurons were incubated with 1 mg ml−1 Sulfo-NHS-Biotin (Pierce, Rockford, IL) for 30 min at 4°C. After quenching with 10 mm Tris-HCl, pH 7.5, cells were lysated for 30 min on ice in 40 μl of a buffer containing10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 20 mm EDTA, and 1% Triton X-100 plus protease inhibitors (Roche Products, Welwyn Garden City, UK). Pulldown of biotinylated proteins was obtained with ImmunoPure Immobilized Streptavidin beads (Pierce) for 2 h at 4°C. The beads were washed three times with radioimmunoprecipitation assay buffer and eluted with SDS-PAGE sample buffer (Invitrogen). Sample were separated on NuPAGE Novex 4–12% Bis-Tris gel (Invitrogen) and processed for Western immunoblot using antibodies against the P2X3 receptor (dilution 1:2000; Neuromics, Edina, MN), the TRPV1 receptor (dilution 1:1000; Alomone Laboratories, Jerusalem, Israel) or β-tubulin III (dilution 1:200; Chemicon, Temecula, CA). As secondary antibodies, specific HRP-conjugated antibodies were used and signals were detected with the enhanced chemiluminescence light system ECL (Amersham Biosciences, Piscataway, NJ). Biotinylation experiments resulted free of intracellular protein contaminants. For control of correct gel loading in biotinylation assays, we checked the β-tubulin III expression in the intracellular fraction. To quantify Western blot signals, band density was measured using CorelDraw Photopaint software (Corel, Ottawa, Ontario, Canada) and normalized with respect to the control.
Fluorescent markers.
For immunofluorescent staining, paraformaldehyde-fixed TG neurons were processed with antibodies against the P2X3 or the TRPV1 receptor (dilution 1:200; Alomone Laboratories) and the neuron-specific β-tubulin III (dilution 1:100; Chemicon). Immunofluorescence reactions were visualized using secondary antibodies AlexaFluor 488 or AlexaFluor 594 (dilution 1:500; Invitrogen). Confocal microscopy was performed by using a Zeiss (Thornwood, NY) LSM 510 microscope equipped with an argon–helium double laser, and images were analyzed and quantified with its dedicated software. Cells stained with the secondary antibody only showed no immunostaining. To visualize neurons sensitive to CGRP with standard fluorescence microscopy, we used live TG or DRG neurons incubated with 0.5 μm CGRP directly conjugated with rhodamine (CGRP-RITC; Phoenix, Belmont, CA) for 1 h at 4°C (Cottrell et al., 2005). Cells were then fixed and processed for indirect immunofluorescence for P2X3, TRPV1, or β-tubulin III proteins. An average of 500 cells were analyzed in each test, and data are the mean of three independent experiments.
Competition experiments (run in duplicate) with excess, unlabeled CGRP (10 μm; 15 min preincubation at 37°C) showed a minimal (<5% of control) fluorescence signal by subsequent application of CGRP-RITC (0.5 μm). These results were analyzed with the ImagePro Express software (Media Cybernetics, Silver Spring, MD).
Data analysis.
In each experiment, sister dishes of TG cultures were used to compare control neurons with neurons treated with CGRP or other drugs. Data are expressed as mean ± SEM. Statistical analysis was performed using the Student's t test, the Mann–Whitney rank sum test, or the ANOVA test (using the KyPlot software, version 2.0; Qualest). A p value of <0.05 was accepted as indicative of a statistically significant difference.
Results
Does CGRP induce rapid responses of trigeminal neurons?
Figure 1A shows an example of colocalization of CGRP binding and P2X3 immunoreactivity on TG cells in culture. The histograms of Figure 1B demonstrate the cell diameter distribution according to these two markers with overlap at the ∼20 μm value. On average, 79 ± 3% (n = 3 experiments) of CGRP-positive cells also expressed P2X3 receptors. Double fluorescence experiments with the neuronal marker β-tubulin III and CGRP-RITC confirmed that CGRP was bound by neurons (supplemental Fig. S1_A_,B, available at www.jneurosci.org as supplemental material) mainly of 15–25 μm somatic diameter.
Figure 1.
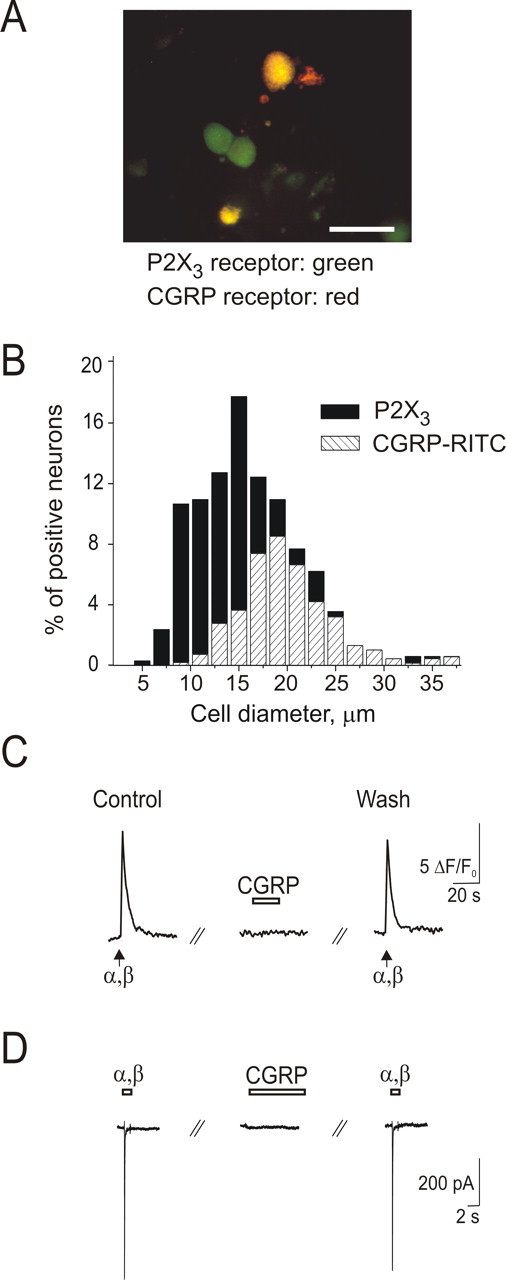
CGRP binds to P2X3-immunopositive TG neurons without acute changes in membrane current, intracellular Ca2+ level, or P2X3 receptor-mediated responses. A, Microphotograph depicts TG neurons labeled with rhodamine-conjugated CGRP (0.5 μm; red) and P2X3 receptor antibody (green). Scale bar, 50 μm. B, Somatic size distribution of TG cells immunostained with P2X3 receptor antibody (▪) and labeled with CGRP-RITC (▨). Data are from ∼1500 cells (3 independent experiments). C, Example of 20 s application of CGRP (1 μm; open bar) that has no effect on fast responses induced by α,β-meATP pulses (10 μm, 2 s; arrows) recorded as Ca2+ transients from a single neuron loaded with Fluo-3 AM. D, Example of lack of effect by CGRP (0.5 μm) applied for 6 min on the peak current induced by α,β-meATP (different cell from C). α,β, α,β-meATP.
To study the acute effects of CGRP on TG neurons in culture, we first investigated the action of this neuropeptide on the level of intracellular Ca2+. CGRP (0.5–1 μm) applied for 20–60 s did not induce Ca2+ signals in the majority of neurons (134 of 146 cells). In a subset of neurons (12 of 146), CGRP induced small Ca2+ transients (∼5% amplitude of those induced by 50 mm KCl). Figure 1C shows that the acute application of CGRP (1 μm, 20 s) neither induced Ca2+ response nor changed Ca2+ transients evoked by the P2X3 receptor agonist α,β-meATP (10 μm). Even longer (6 min) application of CGRP had no effect on α,β-meATP-induced Ca2+ responses (84 ± 8% of control; n = 9; p = 0.12).
Likewise, under patch-clamp conditions, α,β-meATP-induced membrane currents were not significantly changed (85 ± 4% of control; n = 4; p = 0.11) (Fig. 1D) by CGRP. Furthermore, there was no change in the rate of recovery of P2X3 receptors from desensitization tested at 30 s interval with paired pulses of α,β-meATP (11.3 ± 0.5% recovery in the control condition vs 9.3 ± 0.9% recovery after 6 min exposure to 0.5 μm CGRP; n = 4). CGRP (0.5–1 μm) per se produced no detectable membrane currents (n = 6) (Fig. 1D).
Delayed upregulation of P2X3 receptors by CGRP
The highest peak of CGRP in the effluent blood from the brain of migraine patients occurs 1 h from the start of the attack (Sarchielli et al., 2000). To find out whether TG neurons in culture might be suitable models to investigate the cellular action of CGRP, we explored whether this peptide could induce effects on P2X3 receptors over a longer timescale than the one of the previous acute experiments. Thus, sister culture dishes were used in parallel for control data and for experiments testing the consequences of 1 h CGRP application that was washed out before studying neurons with Ca2+ imaging or patch clamping.
After 1 h exposure to 1 μm CGRP, the amplitude of Ca2+ transients induced by α,β-meATP (expressed as a percentage of KCl responses on the same cell) was significantly potentiated (from 27 ± 2%, n = 86, to 45 ± 2%, n = 95; p < 0.0001) (Fig. 2A), whereas the amplitude of KCl-evoked responses remained unchanged. Consistent with this observation, the peak amplitude of membrane currents induced by α,β-meATP after 1 h CGRP exposure was significantly increased (184 ± 13% of control; n = 78; p < 0.00001) (Fig. 2B,C). This effect was fully blocked by the CGRP receptor antagonist CGRP8–37 (107 ± 20% of control; n = 11), which had no effect when applied alone (108 ± 24% of control; n = 15) (Fig. 2B,C). There was no change in cell input resistance after 1 h CGRP application (control: 874 ± 57 MΩ, n = 66; CGRP: 912 ± 60 MΩ, n = 65; p = 0.64), ruling out nonselective increases in cell responsiveness. CGRP application practically did not alter the number of cells immunoreactive for P2X3 receptors (from 67 ± 1 to 72 ± 1%; n = 3 experiments).
Figure 2.
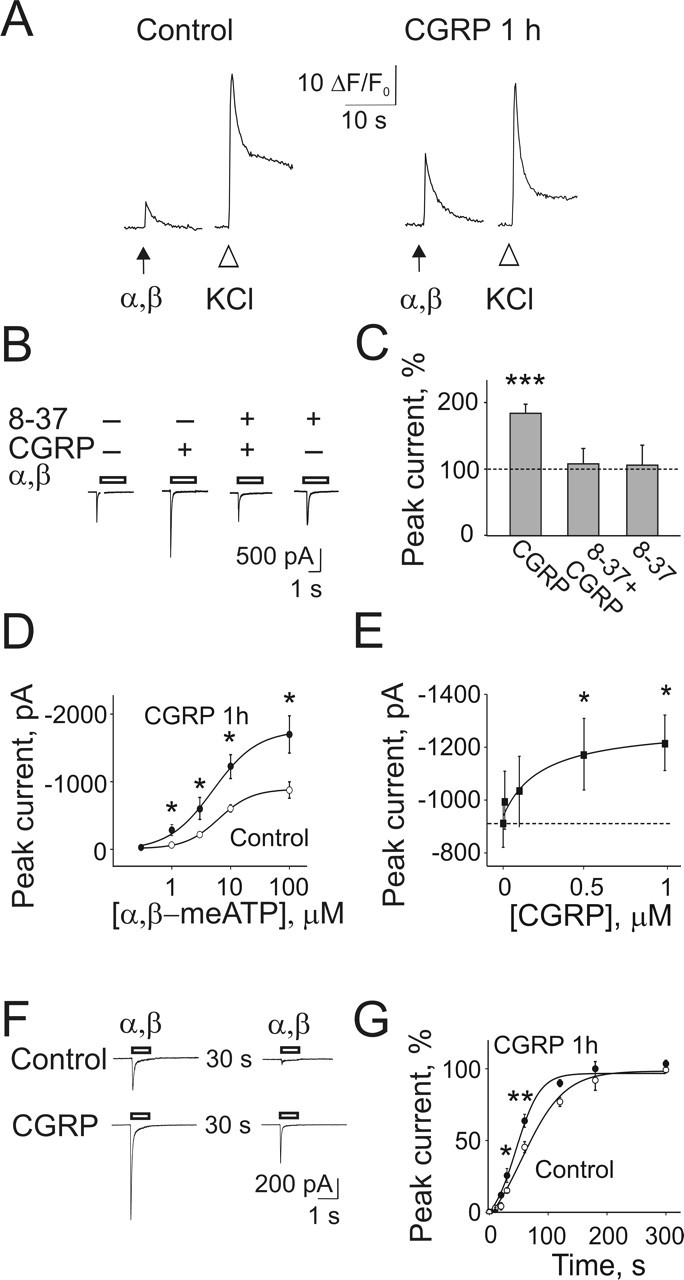
One-hour CGRP treatment upregulates P2X3 receptor-mediated responses in TG neurons. Sister culture dishes were used in parallel for control data and for experiments testing the consequences of a 1 h application of CGRP, washed out before studying neurons with Ca2+ imaging or patch clamping. A, Example of α,β-meATP (10 μm, 2 s; arrow) or KCl (50 mm, 1 s; open arrowheads) evoked Ca2+ transients in control condition. After 1 h CGRP (1 μm) treatment, the effect of α,β-meATP is comparatively larger, whereas the response to KCl is similar (different cell from control). B, Examples of currents evoked by α,β-meATP (10 μm, 2 s; open bars) in control or after a 1 h treatment with CGRP, in the presence or absence of the CGRP receptor antagonist CGRP8–37 (2 μm), which fully prevents the potentiation of current responses. C, CGRP significantly increases α,β-meATP-induced peak current (expressed as percentage of control; n = 78), an effect antagonized by CGRP8–37 (n = 11), which has no effect per se (n = 15). D, Dose–response curves for α,β-meATP in the control condition (○; n = 10) and after 1 h CGRP treatment (•; n = 12). The EC50 value is not altered by CGRP, whereas the peak current amplitude is increased. E, Concentration-dependent effect of CGRP on α,β-meATP-induced peak currents saturates near 1 μm. The dashed line indicates the mean value in the control condition (n = 16); n = 10–15 for CGRP treatments. F, G, CGRP accelerates P2X3 receptor recovery from desensitization evoked by α,β-meATP (open bar) tested in this example at a 30 s interval between pulses. CGRP treatment significantly decreases the half-time of recovery (measured as percentage of the first response amplitude and tested over an extended time interval; ○, n = 15 control; •, n = 11 CGRP). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.00001. α,β, α,β-meATP; 8-37, CGRP8–37.
Figure 2D shows that CGRP treatment enhanced α,β-meATP-induced currents without change in agonist potency (control: EC50 = 5.6 ± 0.7, n = 10; CGRP: 5.4 ± 0.8 μm, n = 12). The effect of CGRP was apparently saturated at 1 μm (Fig. 2E). If ATP was used instead of α,β-meATP as a P2X receptor agonist, there was similar potentiation of peak membrane currents after 1 h CGRP treatment (supplemental Fig. S2_A_, available at www.jneurosci.org as supplemental material).
On DRG neurons grown in identical culture conditions, CGRP (1 μm; 1 h exposure) did not significantly increase the peak amplitude of P2X3 receptor-mediated currents when compared with sister cultures (control: −521 ± 68 pA, n = 33; after CGRP: −611 ± 88 pA, n = 31). This difference between TG and DRG neurons prompted us to investigate the extent of colocalization of CGRP binding and P2X3 receptors in DRG neurons. Unlike TG neurons, only 17% of P2X3-immunoreactive DRG neurons (that represent the majority of neurons in this sensory ganglion) (Ruan et al., 2004) possessed CGRP binding, suggesting that topographic segregation of CGRP and P2X3 receptors accounted for the lack of peptide-evoked facilitation of P2X3 currents in DRG neurons.
Effects of CGRP on P2X3 receptor desensitization
Fast desensitization followed by slow recovery is a key property of P2X3 receptors (Cook et al., 1998; Sokolova et al., 2004). Using the patch-clamp technique and the paired-pulse protocol of agonist application, we tested whether desensitization of P2X3 receptors had been changed by sustained CGRP application. CGRP treatment for 1 h did not change the time constant (τfast) of current decay (control: 47 ± 2 ms; n = 52; after CGRP: 48 ± 2 ms, n = 49), indicating no effect of CGRP on the onset of P2X3 receptor desensitization (supplemental Fig. S2_B_, available at www.jneurosci.org as supplemental material). Nevertheless, 1 h CGRP treatment significantly accelerated recovery from desensitization (Fig. 2F,G) by decreasing the half-time of recovery (_t_½) from 75 ± 4 s (n = 15) to 51 ± 4 s (n = 11; p = 0.001). These data indicate that P2X3 receptors underwent two functional changes after CGRP treatment, namely enhanced responsiveness plus faster recovery from desensitization.
CGRP increased membrane expression of P2X3 receptors
To test whether the delayed changes produced by CGRP were mediated by translocation of P2X3 receptors to the membrane, we performed membrane biotinylation experiments. The amount of the P2X3 receptors on surface membranes was significantly increased 2 and 5 h after CGRP treatment, as demonstrated by Western blots of the purified receptor protein (Fig. 3A). This time-dependent enhancement was not observed when the antagonist CGRP8–37 was applied 30 min before CGRP and maintained throughout (Fig. 3A). CGRP also increased the ratio between the surface and the total protein content in control and 2 h CGRP-treated samples (2.5 ± 0.6-fold increment; n = 3 experiments).
Figure 3.
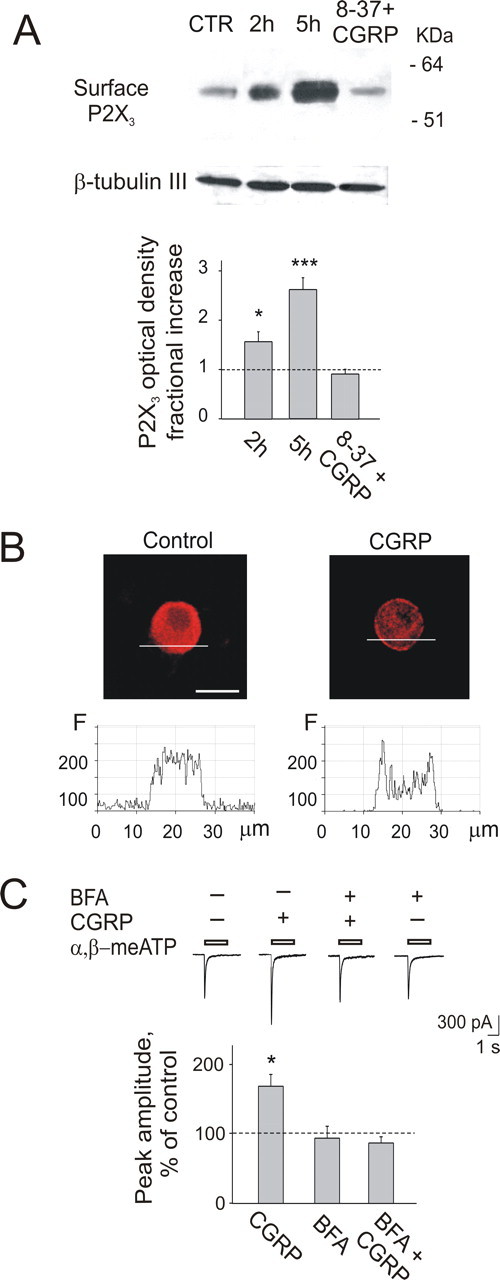
CGRP (1 μm, 1 h application) enhances expression of P2X3 receptors at membrane level. A, Example of Western immunoblots of the P2X3 receptor after membrane biotinylation, showing a single band for the membrane form of the receptor (57 kDa). Note the increase in P2X3 receptor expression after a 2 or 5 h washout of CGRP. When CGRP is coapplied with the receptor antagonist CGRP8–37 (8-37; 2 μm), there is no change in P2X3 receptor expression 2 h later. Control loading represents intracellular β-tubulin III. The plot shows protein expression increase as a fraction of control (histograms show P2X3 receptor expression measured with optical density in arbitrary units); n = 4 experiments, ∗p = 0.034 for 2 h; n = 3 experiments, ∗∗∗p = 0.0005 for 5 h. B, Example of confocal microscopy images of TG neurons treated (or untreated) with CGRP. P2X3 receptor immunoreactivity shows perimembrane location 1 h after CGRP treatment, whereas it has homogenous distribution in control conditions. Plots beneath confocal images are the fluorescence profiles (ordinate, fluorescence intensity in arbitrary units) obtained by scanning along the lines shown in the photographs. Scale bar, 20 μm. C, Top, The potentiating effect of CGRP on α,β-meATP-induced currents is prevented by brefeldin A (BFA; 5 μg/ml). Bottom, Quantification of blocking action of BFA on CGRP potentiation (n = 7, 12, or 11 for CGRP, BFA, or BFA+CGRP, respectively). ∗p < 0.05.
Confocal microscopy showed a ring-like structure enriched with P2X3 receptor immunoreactivity in the area immediately beneath the surface membrane in 51 ± 5% of the P2X3-positive neurons treated with CGRP (n = 118) (Fig. 3B). Conversely, only 3 ± 2% of neurons (n = 56) had a similar perimembrane signal in control (p < 0.0001) (Fig. 3B). To quantify these observations, we performed confocal line-profile analysis of neurons (Rathee et al., 2002) in control or after 1 h CGRP exposure by scanning along a line across the perimembrane region as indicated in Figure 3B. The plots corresponding to such scans are displayed below each cell image: note that, in control, P2X3 immunoreactivity was evenly distributed throughout the sampled region, whereas after CGRP treatment, the signal was concentrated near the membrane, giving rise to two peaks in correspondence with the membrane area. These data suggest that CGRP treatment stimulated trafficking of P2X3 receptors from cytoplasmic pools toward the membrane. In support for this notion, brefeldin A (5 μg ml−1), a selective inhibitor of the trafficking processes (Chardin and McCormick, 1999), prevented CGRP-induced potentiation of P2X3 receptor-mediated currents (86 ± 9%; n = 11) (Fig. 3C).
P2X3 potentiation is mediated by PKA and PKC
CGRP operates via G-protein-coupled receptors that, in many cell types, lead to activation of the cAMP/PKA cascade (Durham and Russo, 1999; Arulmani et al., 2004) and directly or indirectly to PKC activation (Drissi et al., 1998). Indeed, both PKA and PKC are involved in the enhancement of excitability of DRG neurons by CGRP (Natura et al., 2005). Thus, we first explored the role of PKA in the upregulation of P2X3 receptors. As shown in Figure 4, the PKA inhibitor 14-22 peptide (3 μm) completely blocked the CGRP-mediated P2X3 potentiation (76 ± 13% of control; n = 14; p = 0.18). Next, we tested the role of PKC in the action of CGRP on P2X3 receptors. The PKC inhibitor chelerythrine (5 μm) fully prevented the enhancement of P2X3 receptors by 1 h CGRP treatment (74 ± 15% of control; n = 8; p = 0.14) (Fig. 4). Both PKA and PKC inhibitors had no effect when applied alone (Fig. 4). Consistent with the involvement of PKA and PKC in the modulation of P2X3 receptors, we observed that forskolin (1 μm) or PMA (325 nm), activators of PKA or PKC, respectively, strongly enhanced currents mediated by P2X3 receptors (Fig. 4). In particular, α,β-meATP-induced currents were 211 ± 22% of control after 1 h forskolin treatment (n = 37; p < 0.001), whereas 1 h PMA treatment increased these currents to 249 ± 30% of control (n = 27; p < 0.001).
Figure 4.

Potentiation of P2X3-mediated currents by CGRP is PKA and PKC dependent. The plot summarizes the peak current amplitudes (expressed as percentage of the control value) of α,β-meATP-induced currents in TG neurons treated with CGRP and protein kinase inhibitors or activators. The PKA inhibitor 14-22 peptide (iPKA; 3 μm; n = 14) or the PKC inhibitor chelerythrine chloride (iPKC; 5 μm; n = 8) completely blocks the CGRP-mediated P2X3 receptor potentiation (n = 78), whereas there is no effect when they are applied alone (n = 36 and n = 11, respectively). Conversely, forskolin (1 μm; n = 37) or PMA (325 nm; n = 27) potentiates P2X3 receptor-mediated currents. ∗∗∗p < 0.001.
Persistence of CGRP effects
One point to be tested before considering the hypothesis that upregulated P2X3 receptor function triggered by CGRP might contribute to sustained pain is the persistence of receptor potentiation long after removal of CGRP. For this purpose, current responses to α,β-meATP were monitored at different times after CGRP washout after 1 h exposure to 1 μm CGRP. Figure 5A shows that the potentiation of the P2X3 receptor-mediated responses continued to grow after CGRP washout, peaking 5 h later and decreasing to control level after 24 h (n = 81 for control and n = 14–17 for CGRP data points). Likewise, improved recovery from P2X3 receptor desensitization was also a long-lasting feature persisting for several hours after CGRP washout (n = 6–15) (Fig. 5B).
Figure 5.
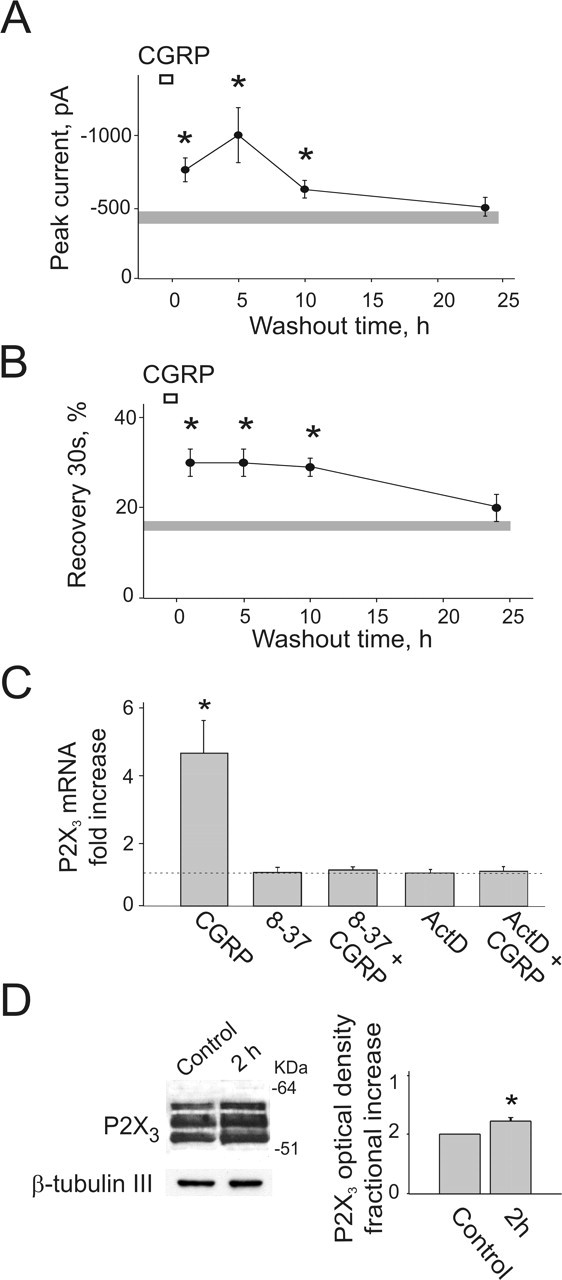
Long-lasting effects of CGRP on P2X3 receptors. A, Time course of potentiation of the P2X3 receptor-mediated responses after 1 h CGRP treatment (1 μm; open bar). Current responses (filled circles) to α,β-meATP application are monitored at different times after CGRP washout. Current amplitude is maximal after 5 h and gradually comes back to the control level at 24 h. The shaded horizontal bar indicates the SEM of values obtained from controls (n = 81), whereas data points for CGRP washout range from 14 to 17. ∗p < 0.05. B, Paired pulses of α,β-meATP (spaced by 30 s) are used to study P2X3 recovery from desensitization. The shaded horizontal bar indicates the SEM of values obtained from controls (n = 52). CGRP improves recovery for several hours after treatment with a gradual return to control levels; the number of neurons treated with CGRP ranges from 6 to 15; ∗p < 0.05. C, Long-lasting changes in P2X3 receptor potentiation after CGRP are supported by P2X3 receptor neosynthesis. A 1 h application of CGRP (1 μm) upregulates P2X 3 gene transcription (n = 4 experiments), an effect prevented by 2 μm CGRP8–37 (8-37; n = 3 experiments) or by 5 μg ml−1 actinomycin D (ActD; n = 3 experiments). ∗p = 0.01. D, Left, Example of Western blot analysis of P2X3 in total extracts from TG neurons showing increment of all glycosylated forms of the receptor after 1 h of CGRP treatment (bottom lanes show control loading with β-tubulin III). Right, Histograms for increase in P2X3 receptor expression measured with optical density (arbitrary units; n = 4 experiments; ∗p < 0.01).
Upregulation of P2X3 mRNA and protein synthesis could have been a mechanism responsible for such long-lasting facilitation of P2X3 receptor-mediated responses. To explore this issue, real-time PCR and Western blotting experiments were performed. Figure 5C shows that 1 h incubation with CGRP (1 μm) significantly upregulated P2X3 transcription (4.6 ± 1-fold increment in mRNA levels; n = 4 experiments; p = 0.01), an effect absent in cells incubated with CGRP plus the receptor antagonist CGRP8–37 (1.1 ± 0.1-fold increment; n = 3) (Fig. 5C). P2X3 mRNA neosynthesis was completely prevented by actinomycin D (1.1 ± 0.1-fold; n = 3 experiments) (Fig. 5C), thus demonstrating that CGRP activated gene transcription. Increased P2X3 mRNA synthesis was coupled to translation into new P2X3 protein as shown by Western blot experiments in which the differently glycosylated intracellular forms of the P2X3 receptor polypeptides (50–57 kDa) (Vulchanova et al., 1997) were significantly increased 1 h after CGRP treatment (1.2 ± 0.05-fold increment; n = 4 experiments; p = 0.01) (Fig. 5D).
We also investigated whether CGRP altered the number of P2X3-immunoreactive cells. P2X3 immunofluorescence analysis, however, revealed that the number of immunoreactive cells was not increased after 1 h CGRP application (67 ± 1% control vs 72 ± 1% after CGRP; n = 3 experiments).
CGRP has no effect on TRPV1 receptor function
Because TG neurons also express capsaicin-sensitive TRPV1 receptors as transducers of nociception (Julius and Basbaum, 2001; Wang and Woolf, 2005), we explored whether CGRP treatment (1 μm) preincubated for 1 h could modify them. Currents evoked by 1 μm capsaicin (−101 ± 21 pA; n = 40) were not changed by CGRP (−107 ± 19 pA; n = 37) (see example in supplemental Fig. 1S_C_, available at www.jneurosci.org as supplemental material). The number of cells sensitive to capsaicin (40 ± 5% in control condition; n = 17 experiments) was also unchanged by CGRP (41 ± 4%; n = 16), a result validated with TRPV1 immunofluorescence analysis (36 ± 1% of positive cells in control and 37 ± 1% after CGRP; n = 3 experiments for each condition). Finally, Western immunoblotting of extracts from TG neurons showed no difference in expression of TRPV1 protein by TG neurons treated with 1 μm CGRP for 1 h (n = 3 experiments; data not shown). We next examined whether TG neurons labeled with fluorescent CGRP were also immunoreactive for the TRPV1 receptors: colocalization of such signals was observed in 12 ± 3% neurons only (n = 3 experiments) (supplemental Fig. S1_D_, available at www.jneurosci.org as supplemental material).
Discussion
The principal finding of the present study is the demonstration that P2X3 receptors of TG neurons were selectively upregulated by CGRP. This slow modulation was mediated by neosynthesis and increased trafficking of P2X3 receptors to the plasma membrane. The present model thus outlines a mechanism for the persistent sensitization of a nociceptive system in chronic pain states such as migraine, associated with a high level of CGRP.
CGRP action on P2X3 receptors
Short-lasting application of CGRP had no direct effect on the membrane current or resistance of trigeminal neurons in culture, except for a few isolated cells showing a subtle rise in intracellular Ca2+. These observations accord with a recent in vivo study demonstrating lack of effect of acute administration of CGRP to meningeal nociceptors (Levy et al., 2005). Because we were interested in delayed effects of CGRP on an extended timescale that resembles the duration of pain in migraine, we took advantage of cultured TG neurons.
A role of ATP in migraine was first suspected in conjunction with the vascular theory of this disorder. Because the focus on migraine pathophysiology has shifted to neuronal dysfunction, we considered the possibility that ATP released during a pain attack (Burnstock, 2000) contributes to headache by activating ionotropic P2X3 receptors in trigeminal sensory neurons (Cook and McCleskey, 1997; Ruan et al., 2004). In keeping with this hypothesis, we detected frequent coexpression of P2X3 receptors with CGRP binding sites in mouse TG neurons in culture, thus providing the substrate for P2X3 receptor modulation by CGRP at single-cell level.
Although CGRP enhanced the maximum response of P2X3 receptors, it did not alter the agonist potency, suggesting an increase in the number of functional P2X3 receptors rather than in agonist sensitivity. The faster recovery of P2X3 receptors from desensitization would have significantly contributed to potentiation of P2X3 receptor function, because such receptors possess an unusually long recovery that curtails their ability to generate pain signals (Cook et al., 1998). This phenomenon is different from the modulation of desensitization by other algogenic peptides such as substance P or bradykinin occurring over a much shorter (seconds, minutes) time course (Paukert et al., 2001). Slow facilitation of P2X3 receptor function by CGRP also differs from the time course of the CGRP-evoked block of nicotinic receptors (Di Angelantonio et al., 2003).
Under the present experimental conditions, we did not observe CGRP-evoked modulation of TRPV1 receptor function, probably because the likelihood of detecting such an interaction was limited by the fact that few TRPV1-expressing neurons coexpressed CGRP binding sites. For analogous reasons, we did not detect potentiation by CGRP of P2X3 receptors on DRG neurons that showed infrequent colocalization of CGRP binding and P2X3 immunoreactivity in accordance with a low expression of CGRP receptors in rat DRG neurons (Natura et al., 2005). The overall outcome of CGRP action therefore appears to bias pain signaling toward purinergic-based rather than vanilloid-based mechanisms, and this phenomenon seems to be specific to TG.
Dynamics of CGRP action
The CGRP receptor consists of a G-protein-coupled complex that operates by increasing intracellular cAMP and activation of downstream effectors (Drissi et al., 1998; Durham and Russo, 1999; Poyner et al., 2002), leading to rapid changes in ion channel activity apparently unrelated to nociception and perhaps responsible for other physiological effects of the peptide (Zona et al., 1991; Di Angelantonio et al., 2003). The present study detected another feature of CGRP, namely delayed and persistent modulation of P2X3 receptors even after washout of the peptide. The reason for the gradual return of P2X3 receptor function to control level remains unclear and requires future studies to identify its mechanisms.
The potentiation of P2X3 receptors was accompanied by significantly larger expression, already after 2 h, of the mature form of these receptors at membrane level. The enhanced trafficking of pre-existing P2X3 receptors and their incorporation into the neuronal membrane was supported by biochemical and confocal microscopy evidence, suggestive of intense translocation of this receptor. Because selective inhibitors of PKA and PKC blocked P2X3 potentiation by CGRP, it is likely that activation of these kinases mediated the action of CGRP. This notion is corroborated by the facilitation of P2X3 receptor function by activators of PKA or PKC, indicating a key role of both enzymes in trafficking of P2X3 receptors. We suggest that PKA and PKC are just a part of a more complex scenario in which each step of P2X3 receptor potentiation is highly regulated and involves activation of several factors.
Real-time reverse transcription (RT)-PCR experiments showed that CGRP could induce P2X 3 gene transcription. Previous reports demonstrated that CGRP activated gene transcription via cAMP-dependent cAMP response element-binding protein (CREB) activation (Seybold et al., 2003; Anderson and Seybold, 2004). It is likely that CREB is involved in P2X 3 gene transcription, because analysis of the genomic P2X3 promoter region (GenBank accession number gi 84781757 ref NM_145526.2) indicates the presence of CRE and CRE-BP elements (nucleotide −92), both equally conserved in the two parallel strands of the DNA sequence (score 86.1% with TFSEARCH, www.cbrc.jp/research/db/TFSEARCHJ.html). The enhanced P2X3 mRNA synthesis after CGRP could account for a larger, delayed expression of P2X3 receptors at membrane level, providing a molecular mechanism for long-lasting sensitization of P2X3 receptors. Although both RT-PCR and Western blot yielded unidirectional results, we observed a degree of mismatch in the amplitude of RT-PCR and Western blot signals. A discrepancy between transcription and translation is not an uncommon phenomenon, as amply discussed in a recent review (Lewandowski and Small, 2005), because of the complex relationship between mRNA and protein, as transcription and translation are governed by independent mechanisms.
Pathophysiological implications
There is growing interest in the role of CGRP in migraine (Pietrobon and Striessnig, 2003; Goadsby, 2005). In situ experiments reveal that CGRP is expressed in human trigeminal neurons (Moreno et al., 1999; Tajti et al., 1999) from which it is released during migraine attacks (Edvinsson and Uddman, 2005). Novel CGRP antagonists are currently investigated for treating migraine pain (Olesen et al., 2004; Rudolf et al., 2005). Nevertheless, the mechanisms responsible for the algogenic action of CGRP remain little understood. The present model study suggests that the action of CGRP is linked to sensitization of P2X3 receptors of trigeminal nociceptors. Behavioral studies on P2X3−/− mice and the analgesic action of P2X3 receptor antagonists indicate P2X3 receptors to be involved in chronic pain (Cockayne et al., 2000; Jarvis, 2003; North, 2003). Our study showed that 1 h exposure to CGRP was sufficient for a large effect on P2X3 receptors, a timescale that should mimic the development of migraine pain. Hence, trigeminal P2X3 receptors could be potential targets for future analgesics designed to treat chronic pain syndromes such as migraine. Furthermore, our data predict that a CGRP receptor antagonist should be most efficient for analgesia during the early phase of a migraine attack.
Footnotes
This work was supported by grants from Telethon (GGP 04037) and FIRB (MIUR). We are most grateful to Dr. Serena Zacchigna and Prof. Mauro Giacca (International Centre for Genetic Engineering and Biotechnology, Trieste, Italy) for their help with real-time RT-PCR experiments and to Asya Giniatullina for her work in preliminary experiments of Ca2+ imaging. We are greatly indebted to Dr. Oliver Jahraus (Phoenix Europe, Karlsruhe, Germany) for performing HLPC measurements of CGRP stability.
References
- Anderson LE, Seybold VS (2004). Calcitonin gene-related peptide regulates gene transcription in primary afferent neurons. J Neurochem 91:1417–1429. [DOI] [PubMed] [Google Scholar]
- Arulmani U, Maassenvandenbrink A, Villalon CM, Saxena PR (2004). Calcitonin gene-related peptide and its role in migraine pathophysiology. Eur J Pharmacol 500:315–330. [DOI] [PubMed] [Google Scholar]
- Burnstock G (2000). P2X receptors in sensory neurones. Br J Anaesth 84:476–488. [DOI] [PubMed] [Google Scholar]
- Chardin P, McCormick F (1999). Brefeldin A: the advantage of being uncompetitive. Cell 97:153–155. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Illes P (2001). P2X receptors and nociception. Pharmacol Rev 53:553–568. [PubMed] [Google Scholar]
- Cockayne DA, Hamilton SG, Zhu QM, Dunn PM, Zhong Y, Novakovic S, Malmberg AB, Cain G, Berson A, Kassotakis L, Hedley L, Lachnit WG, Burnstock G, McMahon SB, Ford AP (2000). Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature 407:1011–1015. [DOI] [PubMed] [Google Scholar]
- Cook SP, McCleskey EW (1997). Desensitization, recovery and Ca2+-dependent modulation of ATP-gated P2X receptors in nociceptors. Neuropharmacology 36:1303–1308. [DOI] [PubMed] [Google Scholar]
- Cook SP, Rodland KD, McCleskey EW (1998). A memory for extracellular Ca2+ by speeding recovery of P2X receptors from desensitization. J Neurosci 18:9238–9244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell GS, Roosterman D, Marvizon JC, Song B, Wick E, Pikios S, Wong H, Berthelier C, Tang Y, Sternini C, Bunnett NW, Grady EF (2005). Localization of calcitonin receptor-like receptor and receptor activity modifying protein 1 in enteric neurons, dorsal root ganglia, and the spinal cord of the rat. J Comp Neurol 490:239–255. [DOI] [PubMed] [Google Scholar]
- Di Angelantonio S, Giniatullin R, Costa V, Sokolova E, Nistri A (2003). Modulation of neuronal nicotinic receptor function by the neuropeptides CGRP and substance P on autonomic nerve cells. Br J Pharmacol 139:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drissi H, Lasmoles F, Le Mellay V, Marie PJ, Lieberherr M (1998). Activation of phospholipase C-beta1 via Galphaq/11 during calcium mobilization by calcitonin gene-related peptide. J Biol Chem 273:20168–20174. [DOI] [PubMed] [Google Scholar]
- Durham PL, Russo AF (1999). Regulation of calcitonin gene-related peptide secretion by a serotonergic antimigraine drug. J Neurosci 19:3423–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L (2005). Clinical data on the CGRP antagonist BIBN4096BS for treatment of migraine attacks. CNS Drug Rev 11:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L, Uddman R (2005). Neurobiology in primary headaches. Brain Res Brain Res Rev 48:438–456. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ (2005). Calcitonin gene-related peptide antagonists as treatments of migraine and other primary headaches. Drugs 65:2557–2567. [DOI] [PubMed] [Google Scholar]
- Ichikawa T, Ishihara K, Kusakabe T, Hiruma H, Kawakami T, Hotta K (2000). CGRP modulates mucin synthesis in surface mucus cells of rat gastric oxyntic mucosa. Am J Physiol Gastrointest Liver Physiol 279:G82–G89. [DOI] [PubMed] [Google Scholar]
- Jarvis MF (2003). Contributions of P2X3 homomeric and heteromeric channels to acute and chronic pain. Expert Opin Ther Targets 7:513–522. [DOI] [PubMed] [Google Scholar]
- Juhasz G, Zsombok T, Jakab B, Nemeth J, Szolcsanyi J, Bagdy G (2005). Sumatriptan causes parallel decrease in plasma calcitonin gene-related peptide (CGRP) concentration and migraine headache during nitroglycerin induced migraine attack. Cephalalgia 25:179–183. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI (2001). Molecular mechanisms of nociception. Nature 413:203–210. [DOI] [PubMed] [Google Scholar]
- Koshimizu T, Koshimizu M, Stojilkovic SS (1999). Contributions of the C-terminal domain to the control of P2X receptor desensitization. J Biol Chem 274:37651–37657. [DOI] [PubMed] [Google Scholar]
- Levy D, Burstein R, Strassman AM (2005). Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol 58:698–705. [DOI] [PubMed] [Google Scholar]
- Lewandowski NM, Small SA (2005). Brain microarray: finding needles in molecular haystacks. J Neurosci 25:10341–10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-DΔCT method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Moreno MJ, Cohen Z, Stanimirovic DB, Hamel E (1999). Functional calcitonin gene-related peptide type 1 and adrenomedullin receptors in human trigeminal ganglia, brain vessels, and cerebromicrovascular or astroglial cells in culture. J Cereb Blood Flow Metab 19:1270–1278. [DOI] [PubMed] [Google Scholar]
- Natura G, von Banchet GS, Schaible HG (2005). Calcitonin gene-related peptide enhances TTX-resistant sodium currents in cultured dorsal root ganglion neurons from adult rats. Pain 116:194–204. [DOI] [PubMed] [Google Scholar]
- North RA (2003). The P2X3 subunit: a molecular target in pain therapeutics. Curr Opin Investig Drugs 4:833–840. [PubMed] [Google Scholar]
- Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, Pollentier S, Lesko LMBIBN 4096 BS Clinical Proof of Concept Study Group. (2004). Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med 350:1104–1110. [DOI] [PubMed] [Google Scholar]
- Paukert M, Osteroth R, Geisler HS, Brandle U, Glowatzki E, Ruppersberg JP, Grunder S (2001). Inflammatory mediators potentiate ATP-gated channels through the P2X3 subunit. J Biol Chem 276:21077–21082. [DOI] [PubMed] [Google Scholar]
- Pietrobon D, Striessnig J (2003). Neurobiology of migraine. Nat Rev Neurosci 4:386–398. [DOI] [PubMed] [Google Scholar]
- Poyner DR, Sexton PM, Marshall I, Smith DM, Quirion R, Born W, Muff R, Fischer JA, Foord SM (2002). International Union of Pharmacology. XXXII. The mammalian calcitonin gene-related peptides, adrenomedullin, amylin, and calcitonin receptors. Pharmacol Rev 54:233–246. [DOI] [PubMed] [Google Scholar]
- Rathee PK, Distler C, Obreja O, Neuhuber W, Wang GK, Wang SY, Nau C, Kress M (2002). PKA/AKAP/VR-1 module: a common link of Gs-mediated signaling to thermal hyperalgesia. J Neurosci 22:4740–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan HZ, Moules E, Burnstock G (2004). Changes in P2X3 purinoceptors in sensory ganglia of the mouse during embryonic and postnatal development. Histochem Cell Biol 122:539–551. [DOI] [PubMed] [Google Scholar]
- Rudolf K, Eberlein W, Engel W, Pieper H, Entzeroth M, Hallermayer G, Doods H (2005). Development of human calcitonin gene-related peptide (CGRP) receptor antagonists. 1. Potent and selective small molecule CGRP antagonists. 1-[N2-[3,5-dibromo-N-[[4-(3,4-dihydro-2(1H)-oxoquinazolin-3-yl)-1-piperidinyl] carbonyl]-d-tyrosyl]-l-lysyl]-4-(4-pyridinyl)piperazine: the first CGRP antagonist for clinical trials in acute migraine. J Med Chem 48:5921–5931. [DOI] [PubMed] [Google Scholar]
- Sarchielli P, Alberti A, Codini M, Floridi A, Gallai V (2000). Nitric oxide metabolites, prostaglandins and trigeminal vasoactive peptides in internal jugular vein blood during spontaneous migraine attacks. Cephalalgia 20:907–918. [DOI] [PubMed] [Google Scholar]
- Seybold VS, McCarson KE, Mermelstein PG, Groth RD, Abrahams LG (2003). Calcitonin gene-related peptide regulates expression of neurokinin1 receptors by rat spinal neurons. J Neurosci 23:1816–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova E, Skorinkin A, Fabbretti E, Masten L, Nistri A, Giniatullin R (2004). Agonist-dependence of recovery from desensitization of P2X3 receptors provides a novel and sensitive approach for their rapid up or downregulation. Br J Pharmacol 141:1048–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souslova V, Cesare P, Ding Y, Akopian AN, Stanfa L, Suzuki R, Carpenter K, Dickenson A, Boyce S, Hill R, Nebenuis-Oosthuizen D, Smith AJ, Kidd EJ, Wood JN (2000). Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature 407:1015–1017. [DOI] [PubMed] [Google Scholar]
- Tajti J, Uddman R, Moller S, Sundler F, Edvinsson L (1999). Messenger molecules and receptor mRNA in the human trigeminal ganglion. J Auton Nerv Syst 76:176–183. [DOI] [PubMed] [Google Scholar]
- Vulchanova L, Riedl MS, Shuster SJ, Buell G, Surprenant A, North RA, Elde R (1997). Immunohistochemical study of the P2X2 and P2X3 receptor subunits in rat and monkey sensory neurons and their central terminals. Neuropharmacology 36:1229–1242. [DOI] [PubMed] [Google Scholar]
- Wang H, Woolf CJ (2005). Pain TRPs. Neuron 46:9–12. [DOI] [PubMed] [Google Scholar]
- Zona C, Farini D, Palma E, Eusebi F (1991). Modulation of voltage-activated channels by calcitonin gene-related peptide in cultured rat neurones. J Physiol (Lond) 433:631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]