Interaction of Cellular Prion and Stress-Inducible Protein 1 Promotes Neuritogenesis and Neuroprotection by Distinct Signaling Pathways (original) (raw)
Abstract
Understanding the physiological function of the cellular prion (PrPc) depends on the investigation of PrPc-interacting proteins. Stress-inducible protein 1 (STI1) is a specific PrPc ligand that promotes neuroprotection of retinal neurons through cAMP-dependent protein kinase A (PKA). Here, we examined the signaling pathways and functional consequences of the PrPc interaction with STI1 in hippocampal neurons. Both PrPc and STI1 are abundantly expressed and highly colocalized in the hippocampus in situ, indicating that they can interact in vivo. Recombinant STI1 (His6-STI1) added to hippocampal cultures interacts with PrPc at the neuronal surface and elicits neuritogenesis in wild-type neurons but not in PrPc-null cells. This effect was abolished by antibodies against either PrPc or STI1 and was dependent on the STI1 domain that binds PrPc. Binding of these proteins induced the phosphorylation/activation of the mitogen-activated protein kinase, which was essential for STI1-promoted neuritogenesis. His6-STI1, but not its counterpart lacking the PrPc binding site, prevented cell death via PKA activation. These results demonstrate that two parallel effects of the PrPc–STI1 interaction, neuritogenesis and neuroprotection, are mediated by distinct signaling pathways.
Keywords: cellular prion protein, MAPK, neuritogenesis, neuroprotection, PKA, STI1
Introduction
Cellular prion protein (PrPc) is a ubiquitous, glycosylphosphatidylinositol-anchored protein, the physiological functions of which are still under discussion. Several biological roles for PrPc have been proposed, such as protection against oxidative insults, neuronal adhesion, cell differentiation, and survival (Brown and Sassoon, 2002; Martins et al., 2002). Components of various signal transduction pathways, including the Src-related family member p59Fyn, phosphatidylinositol-3-kinase (PI3 kinase)/Akt, protein kinase A (PKA; cAMP-dependent protein kinase), and mitogen-activated protein kinases (MAPKs), have been suggested to mediate roles of PrPc in neuronal survival and neurite outgrowth (Chiarini et al., 2002; Chen et al., 2003; Santuccione et al., 2005).
PrPc is constitutively expressed in neurons and is abundant in regions such as the olfactory bulb, hippocampus, and synaptic neuropil in close spatiotemporal association with synapse formation. The localization of PrPc in elongating axons suggests a role in axon growth (Sales et al., 1998, 2002). Interestingly, interaction of PrPc with the extracellular matrix protein laminin has been shown to promote neuritogenesis and maintenance of neurites (Graner et al., 2000a,b).
Furthermore, strong evidence for a neuroprotective PrPc function derived from our description of a putative PrPc 66 kDa ligand (Martins et al., 1997), which was later identified as the stress-inducible protein 1 (STI1) (Chiarini et al., 2002; Zanata et al., 2002). STI1 was described in Saccharomyces cerevisiae, in which it was shown to mediate the heat shock response of heat shock protein 70 (Hsp70) genes (Nicolet and Craig, 1989). Murine STI1 has 97% amino acid identity with its human homolog designated Hop (Hsp70/Hsp90-organizing protein). STI1, as well as Hop, interacts with Hsp70 and Hsp90 at its N and C termini (van der Spuy et al., 2000; Carrigan et al., 2004). This interaction facilitates transfer of substrates from Hsp70 to Hsp90 and is important for proper protein folding and maturation (Hernandez et al., 2002).
Interaction of PrPc with either His6-STI1 or a peptide mimicking the PrPc binding domain of STI1 prevented programmed cell death of undifferentiated postmitotic retinal cells (Chiarini et al., 2002; Zanata et al., 2002). Engagement of PrPc increased intracellular cAMP and activated the MAPK pathway in retinal tissue. However, the neuroprotective effect required the activity of PKA rather than MAPK (Chiarini et al., 2002).
We have proposed that PrPc is part of a multiprotein complex that modulates various cellular functions, depending on both protein combination and cell type (Martins et al., 2002). Hence, it is important to dissect the cell signaling and biological significance of the association of PrPc with each one of its partners and in different cell types.
We have therefore examined the functional responses and signaling pathways induced by the interaction of STI1 with PrPc in hippocampal neurons, particularly the roles of the cAMP/PKA and MAPK pathways. Our data indicate that PrPc and STI1 colocalize in the hippocampus and that the interaction of STI1 with PrPc has pronounced effects on both neuritogenesis and survival in hippocampal neurons. The neuritogenesis was found to be dependent only on MAPK activity, whereas cAMP-dependent PKA mediates neuroprotection.
Materials and Methods
Materials
Mouse recombinant STI1 (His6-STI1) and PrPc (His6-PrPc) were purified as described previously (Zanata et al., 2002). Peptides corresponding to mouse STI1 amino acid sequences pepSTI1230–245 (230-ELGNDAYKKKDFDKAL-245) and pepSTI1422–437 (422-QLEPTFIKGYTRKAAA-437) were chemically synthesized by Neosystem (Strasbourg, France). Polyclonal antibodies anti-STI1 (Zanata et al., 2002) and anti-pepSTI1230–245 raised in rabbits were obtained from Bethyl Laboratories (Montgomery, TX), whereas anti-PrPc was produced in PrPc-null mice (Chiarini et al., 2002). The monoclonal antibody 6H4 against amino acids 144–152 of human PrPc was purchased from Prionics (Zurich, Switzerland), and anti-His-Tag-HRP and anti-HisTag antibodies were from Invitrogen (San Diego, CA) and Amersham Biosciences (Piscataway, NJ), respectively. The following chemical inhibitors for different signaling proteins were used: MAP/extracellular signal-regulated kinase (ERK) kinase (MEK) inhibitor 1,4-diamino-2,3-dicyano-1,4-bis (2-aminophenyltio)butadiene (U0126), PKA inhibitor KT5720, PKC inhibitors bisindolylmaleimide (Bim) and chelerethrin chloride (Chel), and phosphodiesterase inhibitor 3-isobutyl-1 methylxantine (IBMX; Sigma, St. Louis, MO). The inhibitors were from Calbiochem (La Jolla, CA), except for U0126, which was from Promega (Madison, WI). The PKA activator (forskolin) was purchased from LC Laboratories (Woburn, MA).
Immunohistochemistry
Embryonic day 17 (E17) mouse embryos were fixed with formalin and embedded in paraffin. Sections (3–4 μm) were deparaffinized, rehydrated, and subjected to epitope retrieval by microwave heating in 10 mm citrate buffer, pH 6.4. Additional treatment was made with 50 mm glycine, 0.02 g/L Triton X-100, 0.5 g/L nonfat dry milk, and 1.5 g/L nonimmune goat serum (Martins et al., 1999). Endogenous peroxidase was blocked with 0.3 g/L hydrogen peroxide. Sections were then incubated overnight at 4°C with the primary antibody anti-PrPc (1:1000) (Chiarini et al., 2002) or anti-STI1 (1:250) (Zanata et al., 2002) diluted in PBS plus 0.5 g/L nonfat dry milk and 1.5 g/L normal goat serum, followed by incubation for 60 min at 37°C with EnVision Labeled Polymer peroxidase (Dako, High Wycombe, UK). Color was developed using DAB (3,3′diaminobenzidine tetrahydrochloride; Sigma) and counterstained with hematoxylin. Sections were visualized in an Olympus (Melville, NY) IMT2-NIC microscope.
Immunofluorescence
Tissue. Brains from E17 wild-type and PrPc-null mice were removed and immediately frozen in liquid nitrogen. Cryostat sections (3 μm) were fixed in ice-cold acetone for 30 min, air dried, rehydrated, and blocked with TBS (20 mm Tris and 150 mm NaCl) containing 0.1% Triton X-100, 10% normal goat serum, and 50 μg/ml anti-mouse IgG at room temperature for 1 h. Brain sections were then incubated at room temperature for 16 h with anti-PrPc mouse serum (1:250) (Chiarini et al., 2002) and anti-STI1 rabbit serum (1:100) (Zanata et al., 2002) in TBS and 0.1% Triton X-100 with 1% normal goat serum. After washing, anti-mouse Alexa-568 (1:3000; Molecular Probes, Eugene, OR) and anti-rabbit FITC (1:1000; PharMingen, San Diego, CA) were added to the slices and incubated in the same buffer for 1 h at room temperature.
Cultured cells. Hippocampal neurons (1 × 105 cells) were plated on glass coverslips coated with poly-l-lysine, washed with PBS, and fixed for 20 min at room temperature with 4% paraformaldehyde and 0.12 m sucrose in PBS. For permeabilization, the cells were incubated with 0.2% Triton X-100 in PBS for 5 min at room temperature. After rinsing with PBS, cultured cells were treated for 1 h at room temperature with blocking solution containing 5% bovine serum albumin (BSA; Sigma) in PBS. For PrPc and STI1 staining, cells were incubated at room temperature for 1 h with anti-PrPc (1:100) and anti-STI1 (1:100) antibodies (Chiarini et al., 2002) diluted in 1% BSA in PBS. The reaction proceeded by incubation with secondary antibodies [anti-rabbit Cy3 (1:3000; Amersham Biosciences) or anti-mouse Alexa-488 (1:500; Molecular Probes)], followed by permeabilization for 4′,6-diamidino-2-phenylindole (DAPI) staining. After additional washes, the coverslips were mounted on slides using Fluoromount (Southern Biotechnology, Birmingham, AL). Immunolabeled cells were imaged with a Bio-Rad (Hercules, CA) Radiance 2100 laser scanning confocal system running the software Laser Sharp 3.0, coupled with a Nikon (Melville, NY) microscope (TE2000-U). Argon (488 nm) and green HeNe (543 nm) lasers were used to excite the fluorophores. Image processing was done with Photoshop (Adobe Systems, San Jose, CA).
Flow cytometry assay
A total of 106 hippocampal neurons from wild-type mice were preincubated in the absence or presence of (7.5 × 10–6 m) of recombinant His6-STI1 in blocking solution (0.5% BSA in PBS) for 1 h at 4°C. Cells were washed and incubated with anti-His-Tag antibody (1:300; Amersham Biosciences), followed by anti-mouse IgG conjugated to R-phycoerythrin (1:200; Dako), both for 1 h at 4°C. Analyses were performed using a Becton Dickinson (Mountain View, CA) FACScan cytometer, and data acquisition from 10,000 cells was done with the Consort 32 system Lysis II software (Becton Dickinson).
His tag pull down
A total of 8 × 106 neurons from wild-type embryos cultured on poly-l-lysine were incubated with His6-STI1 (1.2 × 10–6 m) for 20 min at 37°C, the medium was removed, and the cells were lysed with ice-cold (PBS) 0.5% NP-40 plus Complete Protease Inhibitor Cocktail (Roche, Basel, Switzerland) and centrifuged for 10 min at 6000 × g. The protein extract was incubated with 50 μl of Ni-NTA agarose beads (Qiagen, Hilden, Germany) for 3 h at room temperature. The beads were then washed with 1% NP-40 plus 20 mm imidazole. Bound material was eluted with Laemmli buffer at 100°C and analyzed by immunoblotting using anti-STI1 (1:10,000; Bethyl), anti-PrPc (1:1000), or anti-His-Tag-HRP (1:5000; Invitrogen) antibody.
Alexa Fluor 488 STI1 labeling
His6-STI1 (1 mg) labeling was performed using the Alexa Fluor 488 labeling kit (Molecular Probes), according to the manufacturer's instructions. Wild-type hippocampal neurons (4 × 105 cells) were incubated with His6-STI1 Alexa488 (1.2 × 10–6 m) for 1 h at 4°C, washed with PBS, and fixed for 20 min at room temperature with 4% paraformaldehyde and 0.12 m sucrose in PBS. Immunofluorescence reaction with anti-PrPc (1:100), followed by secondary antibody incubation (anti-mouse; 1:3000), was done as described above.
Immunoblotting analysis
Purified proteins (His6-STI1 and His6-STI1Δ230–245) or protein extracts prepared from wild-type (Prnp+/+) and PrPc-null (Prnp0/0) mice hippocampal cells (Bueler et al., 1992) in Laemmli buffer were resolved in 10% SDS-PAGE, followed by immunoblotting with polyclonal antibody anti-STI1 (1:10,000) or anti-pepSTI1230–245 (1:5000) (Zanata et al., 2002). Protein loading control was performed with a polyclonal antibody to actin (1:200; Sigma). Rabbit nonimmune purified IgG was used as the immunoblotting negative control.
Neuritogenesis assays
Primary hippocampal cultures were obtained from E17 brains of either wild-type (Prnp+/+; a strain generated by crossing F1 descendants from mating 129/SV and C57BL/6J) or PrPc-null (Prnp0/0) (Bueler et al., 1992) mice. The hippocampal structure was aseptically dissected in HBSS (Invitrogen) and treated with trypsin (0.06%) in HBSS for 20 min at 37°C. The protease was inactivated with 10% FCS in Neurobasal medium (Invitrogen) for 5 min. After three washes with HBSS, cells were mechanically dissociated in Neurobasal medium containing B-27 supplement (Invitrogen), glutamine (2 mm; Invitrogen), penicillin (100 IU), and streptomycin (100 μg/ml; Invitrogen). The cells (4 × 104 cells) were plated onto coverslips (13 mm) coated with 5 μg/ml poly-l-lysine (Sigma) and treated with recombinant His6-STI1 and His6-STI1Δ230–245; peptides pepSTI1230–245 and pepSTI1422–437; anti-PrPc (6H4; 6 μg/ml); or anti-pepSTI1230–245 (1:200) for 16 h at 37°C and in a 5% CO2 atmosphere. For analysis of signal transduction mechanisms, cells were preincubated for 1 h with specific inhibitors U0126, KT5720, Bim, and Chel dissolved in dimethylsulfoxide (DMSO) before stimulation of neuritogenesis by His6-STI1 for 16 h. Control cultures received the same concentration of DMSO (0.2%). The cells were fixed with 4% paraformaldehyde in 0.12 m sucrose in PBS for 20 min at room temperature, washed three times with PBS, and stained with hematoxylin.
Morphometric analyses were done using Image J software (National Institutes of Health, Bethesda, MD) and Neuron J plug in. The following parameters were measured: percentage of cells with neurites (cells with neurites of any size/total number of cells), number of neurites per cell (total number of neurites/number of cells with neurites), mean length of neurites per cell (total length of neurites/number of cells with neurites), and percentage of cells with neurites >30 μm, which represents three or more times the average cell body (number of cells with neurites >30 μm/total number of cells). Approximately, a total of 300 cells per sample were analyzed. Data are presented as mean ± SD from at least three independent experiments.
Construction of STI1Δ230–245
A recombinant PCR technique (Ausubel et al., 1993) was used to delete the PrPc binding site of STI1. We amplified cDNA fragments using pTRC-His A STI1 (Zanata et al., 2002) with internal primers 5′-AAAGAGAAGAAGCATTATGAC-3′ and 5′-GTCATAATGCTTCTTCTCTTT-3′ and external primers 5′-CCGCTCGAGGAGCAGGTGAATGAGCTAAAGGA-3′ and 5′-CGGGGTACCTCACCGAATTGCGATGAGACCC-3′. The PCR fragments were cloned between the _Xho_I and _Kpn_I restriction sites in the same vector. Sequencing analysis was done to check for the deletion. The expression and purification of this protein followed the same protocol as described previously (Zanata et al., 2002).
Overlay assay
The indicated amounts of recombinant His6-PrPc and BSA were adsorbed onto nitrocellulose membrane. The membrane was incubated with 5% nonfat milk in TBST (TBS and 0.05% Tween 20) for 1 h at room temperature and washed, followed by incubation with either wild-type His6-STI1 (4 μg/ml) or mutant His6-STI1Δ230–245 (4 μg/ml) for 16 h at 4°C. After extensive washing with TBST, membranes were incubated with polyclonal antibody anti-STI1 (1:10,000) (Zanata et al., 2002) for 2 h at room temperature, followed by a second incubation with peroxidase-coupled anti-rabbit IgG. The reaction was developed using enhanced chemiluminescence (Amersham Biosciences).
Kinase assays
p44/42 MAPK phosphorylation. Phosphorylation assays were done using the PhosphoPlus p44–42 MAPK (Thr202/Tyr204) antibody kit (Cell Signaling, Beverly, MA) according to the manufacturer's instructions. Briefly, primary hippocampal cell cultures (106 cells) from either Prnp+/+ or Prnp0/0 were plated on dishes pretreated with poly-l-lysine and stimulated with His6-STI1 (3.5 × 10–7 m) for different incubation periods, rinsed once with ice-cold PBS, and lysed in Laemmli buffer. For assaying MAPK phosphorylation, cell extracts were subject to SDS-PAGE, followed by immunoblotting with anti-phospho-MAPK and anti-total MAPK antibodies (Cell Signaling). The bands obtained after x-ray film exposure to the membranes were analyzed by densitometric scanning and quantified using the Scion (Frederick, MD) Image software. Values represent the ratio between phospho-MAPK (p42 plus p44) and total MAPK (p42 plus p44) for each sample. Untreated Prnp+/+ or Prnp0/0 values were set as 1.0, and the others are relative to it.
p44/42 MAPK activity. The p44/42 MAPK assay kit (Cell Signaling) was used for estimating the activity of MAPK in primary hippocampal cultures treated with His6-STI1 (3.5 × 10–7 m). Cells were disrupted in lysis buffer and centrifuged for 10 min at 4°C, and the supernatant was transferred to a fresh tube. Active MAPK was immunoprecipitated from 80 μg of total protein in each sample using an immobilized phospho-p44/42 MAPK monoclonal antibody (Cell Signaling). MAPK activity was evaluated by incubation with Elk-1 substrate, followed by electrophoresis and immunoblotting with anti-phospho Elk-1 (Cell Signaling).
PKA activity. Primary hippocampal neurons (1.5 × 106cells) cultured onto poly-l-lysine were preincubated with 100 μm IBMX (Sigma) for 1 h at 37°C and treated with or without PKA inhibitor (KT5720; 6 × 10–8 m) for 1 h at 37°C. His6-STI1 (1.2 × 10–6 m) or forskolin (10 μm) was incubated for 20 min at 37°C. The cells were washed with PBS and homogenized with ice-cold extraction buffer (150 mm NaCl, 20 mm MgCl2, 1% Triton X-100, and 25 mm Tris-HCl, pH 7.4) plus Complete Protease Inhibitor Cocktail (Roche). Cellular debris was removed by centrifugation at 6000 × g for 10 min. The PKA activity was determined by γ-ATP incorporation to a PKA-specific substrate provided by the PKA assay system kit (Upstate Biotechnology, Lake Placid, NY). The reaction was performed according to the manufacturer's instructions.
Cell death assay
Primary hippocampal cultures (4 × 104cells) from wild-type mice were grown onto poly-l-lysine-coated coverslips and preincubated with His6-STI1 (1.2 × 10–6 m) or control buffer (TBS) for 2 h, followed by staurosporine for 16 h. Cell cultures treated with signaling enzyme inhibitors received KT5720, U0126, Bim, or Chel 1 h before incubation with His6-STI1. The cell cultures were fixed with 4% paraformaldehyde plus 0.12 m sucrose in PBS, pH 7.4, for 20 min and stained with propidium iodide (Sigma) in PBS and 0.1% Triton X-100 for 20 min. Cell death induced by staurosporine (Favata et al., 1998) was detected as condensed and pyknotic profiles. At least three independent experiments were done, and three microscopic fields were counted in each group.
Statistical analysis
The results represent the mean ± SD of at least three independent experiments. Two-way ANOVA with a Tukey's HSD post hoc test was used for neuritogenesis, cell death assays, and experiments with inhibitors. The phospho-MAPK statistical analyses were done using single-mean Student's t test.
Results
Expression and colocalization of STI1 and PrPc in the hippocampus
STI1 is expressed in a variety of neurons and glia during neural development as well as in the adult nervous system, suggesting potential roles for this protein during development, plasticity, and regeneration (G.N.M. Hajj, R.M.P.S. Castro, T. Takiishi, M.H. Lopes, Z.S.P. Cook, and V.R. Martins, unpublished data). We compared the expression of STI1 and PrPc in the embryonic brain. Sections from Prnp+/+ (E17) mouse brain showed strong immunoreactivity for both STI1 (Fig. 1_A,C_) and PrPc (Fig. 1_B,D_) in the cerebral cortex and developing hippocampus (nonspecific staining with the respective irrelevant antibodies are shown in the insets). STI1 also showed high levels of expression in Prnp0/0 mouse brain (Fig. 1_F_), whereas PrPc was not detected (Fig. 1_G_), demonstrating the specificity of the PrPc staining. STI1 levels were examined in brain homogenates from Prnp+/+ and Prnp0/0 mice using SDS-PAGE, followed by immunoblotting. Figure 1_E_ shows that STI1 levels are similar in Prnp+/+ and Prnp0/0 mice brain, indicating that STI1 expression seems to be independent of PrPc.
Figure 1.
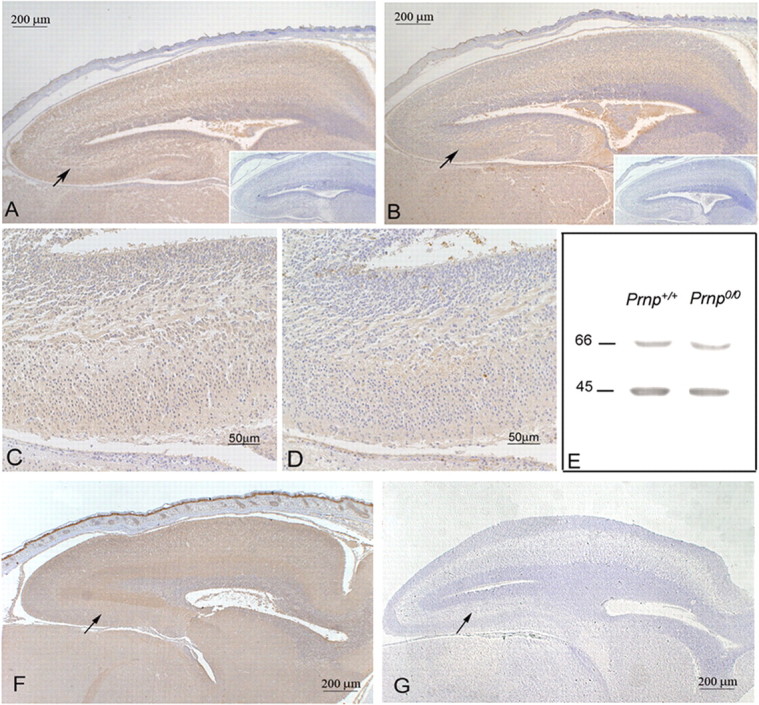
STI1 and PrPc are expressed in the hippocampus. A–D, F, G, Histological brain sections from wild-type (Prnp+/+; A–D) and PrPc -null (Prnp0/0; F, G) mouse embryos (E17) were immunostained for STI1 (A, C, F) or PrPc (B, D, G) and counterstained with hematoxylin. Higher magnifications of the indicated areas (arrows) in A and B are shown in C and D, respectively. Irrelevant antibody controls lack specific signals (A, B, insets). The hippocampus is indicated by arrows in F and G. E, Brain homogenates from wild-type (Prnp+/+) and PrPc-null (Prnp0/0) mice were immunoblotted using antibodies against STI1 (66 kDa) and actin (45 kDa).
When viewed through confocal fluorescence microscopy, brain sections from Prnp+/+ mice confirmed an abundant concentration of both STI1 (Fig. 2_D,G_) and PrPc (Fig. 2_E,H_) in the cortex and hippocampus. Figure 2_A–C_ depicts brain sections from Prnp0/0 mice, in which only STI1 (Fig. 2_A_) was detected. The distribution of both PrPc and STI1 in the hippocampus is relatively uniform, and immunolabeling is colocalized (Fig. 2_F,I_), suggesting that both proteins can interact in this region of the brain.
Figure 2.

Colocalization of STI1 and PrPc in the hippocampus. Histological brain sections of Prnp0/0 (A–C) and Prnp+/+ (D–I) mouse embryos (E17) were immunostained for STI1 (A, D, G) and PrPc (B, E, H). The hippocampal structure marked by the arrowheads in D and E is shown at a higher magnification in G and H. C, F, and I are merged images of immunostaining for STI1 (green) and PrPc (red). There is an extensive colocalization (yellow) of the two proteins in the developing hippocampus. Preimmune rabbit serum displayed no specific staining (A, inset).
In dissociated and permeabilized Prnp+/+ hippocampal neurons, PrPc and STI1 can be visualized in the cell body, perinuclear region, and along neurites, with a consistent colocalization (Fig. 3_Ah_). In unpermeabilized cells, PrPc was distributed along the plasma membrane of both the cell body and neurites (Fig. 3_Aa_). STI1, in contrast, was detected mainly on the surface of the cell soma (Fig. 3_Ab_), where it colocalizes with PrPc (Fig. 3_Ad_).
Figure 3.
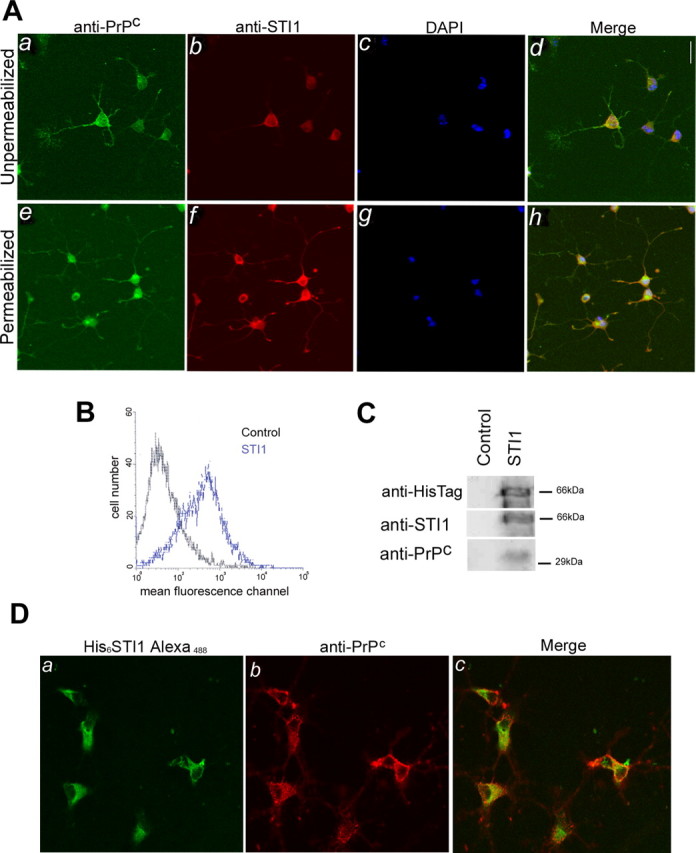
Endogenous and recombinant STI1 colocalize with PrPc on the surface of hippocampal neurons. A, Cultured, unpermeabilized, or permeabilized hippocampal neurons from Prnp+/+ mice were immunostained for PrPc (a, e) and STI1 (b, f). Cell nuclei were labeled with DAPI (c, g). In unpermeabilized cells, colocalization was observed at the surface of the cell soma (d). Permeabilized cells show extensive colocalization of both proteins in the cell body, perinuclear region, and neurites (h). B, Prnp+/+ primary neuronal cultures were either treated with (blue curve) or without (black curve) His6-STI1, followed by incubation with an anti-His-Tag antibody. Flow cytometry analysis detected His6-STI1 at the neuronal surface. C, Primary neuronal cultures were either treated with (STI1) or without (Control) His6-STI1. Cells were washed and lysed, and the extracts were incubated with Ni-NTA-agarose. The bound material was eluted off the beads and analyzed by immunoblotting using anti-HisTag, anti-STI1, and anti-PrPc antibodies. D, Hippocampal cultures were treated with His6-STI1-Alexa488 (a) and immunostained with anti-PrPc antibody (b). Extensive colocalization of both proteins was observed on the cell surface (c).
Previous studies have identified the involvement of PrPc in neuritogenesis. To evaluate whether PrPc-STI1 could play a role in hippocampal neurons, we added recombinant His6-STI1 to these cell cultures. Flow cytometry demonstrated that recombinant His6-STI1 binds to the surface of hippocampal neurons (Fig. 3_B_). Additionally, pull-down experiments were performed to confirm His6-STI1 binding to PrPc. Entire cells were treated with His6-STI1, followed by lysis and incubation of the extract with Ni-NTA agarose. Resin-bound His6-STI1- and STI1-associated proteins were assayed by immunoblotting with anti-STI1, anti-His-Tag, or anti-PrPc antibodies. Recombinant His6-STI1 binds to cellular PrPc (Fig. 3_C_), whereas PrPc did not associate with the resin in the absence of His6-STI1 (Fig. 3_C_, Control).
Cultured hippocampal neurons were incubated with His6-STI1Alexa488 and immunostained with anti-PrPc antibodies. Colocalization data confirmed that His6-STI1 binds to PrPc, particularly at the soma surface (Fig. 3_D_). Thus, these data indicate that recombinant His6-STI1 specifically binds resident PrPc at the cell surface.
STI1 promotes neuritogenesis
Based on the STI1 and PrPc colocalization in hippocampal neurons and in our previous description that interaction of PrPc with laminin induced neuritogenesis (Graner et al., 2000a), we investigated whether STI1 may promote neuritogenesis through the engagement of PrPc. Primary hippocampal cultures from Prnp+/+ or Prnp0/0 mouse embryos (E17) were treated either with recombinant wild-type His6-STI1 or with pepSTI1230–245 (the STI1 peptide that corresponds to the PrPc binding site at STI1). Treatments led to a substantial increase in the proportion of wild-type cells with neurites, as assessed through the counting either of processes of all sizes (Fig. 4_A_) or of processes >30 μm (Fig. 4_B_). In contrast, no differences were seen in either the number of neurites per cell (Fig. 4_C_) or the mean length of neurites per cell (Fig. 4_D_). These results indicate that STI1 enhanced only the neuritogenic response of hippocampal cells (percentage of responsive cells) and did not alter the final neurite characteristics (number or length). Conversely, Prnp0/0 neurons did not extend neurites in response to His6-STI1 or pepSTI1230–245 (Fig. 4_A,B_).
Figure 4.

STI1 and its PrPc binding domain induce PrPc-dependent neuritogenesis. Cultured hippocampal neurons from Prnp+/+ (filled bars) or Prnp0/0 (open bars) were grown on poly-l-lysine and incubated with His6-STI1 at 2 × 10–7 m (STI1 2) or 3.5 × 10–7 m (STI13.5), or with 1.6 × 10–5 m of STI1 peptide230–245 (pepSTI1). Neurons were fixed and stained, and the neurites were quantified. Values represent the means of at least three independent experiments. Error bars represent SD. *p < 0.005 versus poly-l-lysine (ANOVA and Tukey's _post hoc_ test). _**A**_, Percentage of cells with neurites of any size. _**B**_, Percentage of cells with neurites >30 μm. C, Number of neurites per cell. D, Mean length of neurites per cell. PL, Poly-l-lysine.
To characterize whether neuritogenesis promoted by STI1 depends on its specific interaction with PrPc, we constructed an STI1 deletion mutant lacking residues 230–245 (His6-STI1Δ230–245), therefore eliminating the domain previously characterized as containing the PrPc binding site (Zanata et al., 2002). Figure 5_A_ shows that His6-STI1Δ230–245 has the expected molecular weight and is recognized by the anti-STI1 serum (lane 1), but not by an antibody that recognizes the deleted residues (anti-pepSTI1230–245, lane 3). In contrast, wild-type His6-STI1 is recognized by both antibodies (lanes 2, 4). An overlay experiment was performed to test the binding capacities of the wild-type and mutant His6-STI1 to PrPc. His6-PrPc and BSA were immobilized onto a nitrocellulose membrane, incubated with wild-type His6-STI1 (lane 6) or His6-STI1Δ230–245 (lane 5), and immunoreacted with the anti-STI1 antibody. This experiment confirmed that wild-type His6-STI1 binds PrPc (lane 6), whereas mutant His6-STI1Δ230–245 does not (lane 5). The graph in Figure 5_B_ shows that His6-STI1Δ230–245 was unable to promote neuritogenesis in wild-type neurons, thus demonstrating that the PrPc binding domain on STI1 is necessary to induce the neuritogenic response.
Figure 5.

STI1-induced neuritogenesis depends on the PrPc binding site and PrPc expression. A, Wild-type His6-STI1 (lanes 2, 4) and His6-STI1Δ230–245 (lanes 1, 3) were immunoblotted with anti-STI1 (lanes 1, 2) and anti-pepSTI1230–245 antibodies (lanes 3, 4). Lanes 5 and 6 show the overlay assay, in which BSA (2μg) and His6-PrPc (1μg) were adsorbed onto a membrane that was incubated with His6-STI1 (lane 6) or His6-STI1Δ230–245 (lane 5), followed by immunoblotting with anti-STI1 antibody. B, The graph shows neuritogenesis of primary hippocampal neurons from Prnp+/+ embryos treated with either His6-STI1 or His6-STI1Δ230–245. *p<0.005, statistically significant compared with His6-STI1-treated neurons (ANOVA and Tukey's post hoc test). C, Hippocampal neurons were incubated with 3.5 × 10–7 m His6-STI1, followed by treatment with anti-pepSTI1230–245 serum or its preimmune control. Neurons in the presence of His6-STI1 were also treated with anti-PrPc monoclonal antibody (6H4) or control mouse IgG. Values represent the mean of at least three independent experiments. *p < 0.005 versus control (ANOVA and Tukey's post hoc test). Error bars represent SD. PL, Poly-l-lysine.
We further tested the effect of blocking the PrPc–STI1 interaction in wild-type cells. Hippocampal neurons treated with His6-STI1 in the presence of antibodies against PrPc or pep-STI1230–245 presented lower neuritogenic response compared with His6-STI1-treated cells (control). To irrelevant control mouse IgG or preimmune rabbit serum (Fig. 5_C_). Therefore, blocking neuronal PrPc with a specific antibody reproduced the results observed in Prnp0/0 neurons. This indicates that the data obtained with PrPc-null neurons were attributable to the absence of PrPc, rather than to any random events.
Together, the results demonstrate that a specific interaction of STI1 with PrPc promoted neuritogenesis and that the PrPc binding domain of STI1 is both necessary and sufficient for inducing this effect.
MAPK activity is required for neuritogenesis induced by STI1–PrPc interaction
We showed previously that engagement of PrPc with a peptide mimicking the PrPc binding site of the STI1 molecule produced the activation of both the cAMP/PKA and MAPK pathways (Chiarini et al., 2002). However, the protection against programmed cell death of retinal neurons produced by engagement of PrPc required the activity of PKA rather than MAPK. Interestingly, MAPKs have been implicated in neuritogenesis stimulated by cell adhesion molecules and neurotrophic factors (Doherty et al., 2000; Huang and Reichardt, 2001).
To evaluate whether MAPK activation is required for the STI1-PrPc neuritogenic response, we cultured hippocampal neurons in the presence of U0126, a specific inhibitor of the MAPK kinases MEK-1 and MEK-2, which blocks ERK1/2 phosphorylation (Favata et al., 1998). In addition, we tested a set of specific inhibitors of other signal transduction pathways.
The MAPK inhibitor significantly decreased the percentage of cells with neurites (Fig. 6_A_) and the percentage of cells with neurites >30 μm (Fig. 6_B_), whereas the mean number of neurites per cell (Fig. 6_C_) and the mean neurite length per cell (Fig. 6_D_) remained unaltered. Cresyl violet staining indicated no significant cell death among cells treated with U0126 (data not shown). Conversely, neither KT5720, a cell-permeable selective inhibitor of PKA, nor specific PKC inhibitors Chel and Bim (Audesirk et al., 1997) had any effect on neuritogenesis induced by His6-STI1 (Fig. 6_A,B_). Furthermore, these drugs did not alter the number of neurites or their length (Fig. 6_C,D_).
Figure 6.

The MAPK pathway is required for neuritogenesis induced by STI1. Prnp+/+ hippocampal neurons were plated on poly-l-lysine, pretreated for 1 h with inhibitors (2.5 × 10–5 m U0126, 5 × 10–7 m Bim, 10–7 m Chel, or 6 × 10–8 m KT5720), and incubated with His6-STI1 (3.5 × 10–7 m) for 16 h. Cells were fixed and stained with hematoxylin, and neurites were quantified. Values represent the mean and SD of at least three independent experiments. A, Percentage of cells with neurites of any size. B, Percentage of cells with neurites >30μm. C, Number of neurites per cell. D, Mean length of neurites per cell. *p < 0.005 versus control (ANOVA and Tukey's post hoc test). Inhi, Inhibitors; U0, U0126; Kt, KT5720.
To verify whether the STI1–PrPc interaction induces a MAPK cascade in hippocampal neurons, immunoblotting assays were performed to determine the phosphorylation/activation status of the MAPK proteins (Fig. 7_A_, pMAPK). Immunoblotting against total MAPK (Fig. 7_A_, MAPK) was performed as a protein loading control. The relative levels of MAPK phoshorylation were quantified in Prnp+/+ (Fig. 7_B_) and Prnp0/0 (Fig. 7_C_). Prnp+/+ neurons treated with His6-STI1 presented a rapid increase in the phosphorylation status of both the p42 and p44 forms of MAPK, whereas no effect was observed in Prnp0/0 cells.
Figure 7.

STI1 activates the MAPK pathway. A, Hippocampal neurons from Prnp+/+ and Prnp0/0 mice were grown on poly-l-lysine and treated with His6-STI1 (3.5 × 10–7 m) for 0.5, 1, or 5 min. Cell extracts were subjected to SDS-PAGE, followed by immunoblotting using anti-phospho-MAPK antibody. After stripping, the blots were reprobed with anti-total MAPK. MAPK activity was measured by phospho-MAPK immunoprecipitation, incubation with Elk-1 substrate, SDS-PAGE, and immunoblotting using anti-phospho-Elk-1. B, C, Prnp+/+ (B) and Prnp0/0 (C) immunoblots performed for MAPK phosphorylation and total MAPK were quantified by densitometry. The graphs represent the ratio between phospho-MAPK and total MAPK. *p < 0.05 versus respective untreated cells (single-mean Student's t test). D, Prnp+/+ and Prnp0/0 hippocampal neurons treated with His6-STI1Δ230–245 (3.5 × 10–7 m) or pep-STI1230–245 (1.6 × 10–5 m) for 1 min. Cell extracts were subjected to SDS-PAGE, followed by immunoblotting using anti-phospho-MAPK antibody. After stripping, the blots were reprobed with anti-total MAPK. pMAPK, Phospho-MAPK; pElk-1, phospho-Elk-1; CTR, control, untreated cells.
The functional activity of p42/p44 MAPK was determined by assessing its ability to phosphorylate a specific recombinant substrate, Elk1, which was then visualized by immunoblotting (Fig. 7_A_, pElk-1). His6-STI1 also induced p42/p44 MAPK activation in Prnp+/+ neurons but not in Prnp0/0 neurons (Fig. 7_A_). A higher basal phosphorylation and activity levels of MAPK were found in Prnp0/0 compared with wild-type cells (Fig. 7_A_), confirming our previous data in newborn mouse retina (Chiarini et al., 2002), as well as those of other authors in adult brain and cerebellum (Brown et al., 2002).
A substantial increase in MAPK phosphorylation was observed in Prnp+/+ neurons but not in Prnp0/0 cells treated with pepSTI1230–245. Conversely, His6-STI1Δ230–245, which has no effect in neuritogenesis, was also unable to induce MAPK phosphorylation either in Prnp+/+ or in Prnp0/0 neurons (Fig. 7_D_).
These results show that neuritogenesis induced by interaction of PrPc and STI1 depends on MAPK rather than the PKA or PKC pathways.
STI1 promotes neuroprotection in hippocampal neurons through cAMP-dependent protein kinase
We then tested whether STI1 induces neuroprotective responses in hippocampal neurons similar to retinal explants (Chiarini et al., 2002; Zanata et al., 2002). Primary hippocampal cultures from mouse embryos (E17) were treated with staurosporine (Streptomyces staurospores), a nonselective protein kinase inhibitor (Ruegg and Burgess, 1989) that is often used as a general inducer of apoptosis (Nicotera and Orrenius, 1998).
Hippocampal neurons from Prnp+/+ mice were sensitive to cell death induced by staurosporine in a dose-dependent manner (Fig. 8_A_). Treatment with His6-STI1 rescued Prnp+/+ neurons, depending on the presence of the PrPc binding site of STI1, because the mutated His6-STI1Δ230–245 had no effect on cell survival (Fig. 8_B_).
Figure 8.

STI1 rescues hippocampal neurons from staurosporine-induced cell death through the PKA signaling pathway. A, Hippocampal neurons from Prnp+/+ mice were plated on poly-l-lysine, either pretreated with or without His6-STI1 (1.2 × 10–6 m) for 1 h, and incubated with increasing concentrations of staurosporine (1, 2.5, and 5 × 10–8 m) for 16 h. B, hippocampal neurons from Prnp+/+ mice were plated on poly-l-lysine, either pretreated for 1 h with 1.2 × 10–6 m His6-STI1 or His6-STI1Δ230–245 or not treated. Cells were then treated with the inhibitors (5 × 10–5 m U0126, 5 × 10–7 m Bim, 10–7 m Chel, or 6 × 10–8 m KT5720) for 1 h. Cell death was induced by treatment with 2.5 × 10–8 m staurosporine for an additional 16 h. The cells were fixed and stained with propidium iodide, and the percentage of pyknotic profiles, indicating cell death, was estimated. C, Prnp+/+ neuronal cultures were treated with His6-STI1 and His6-STI1Δ230–245 for 20 min, and the PKA activity was analyzed. Forskolin was used as positive control, and the activity of the PKA inhibitor (KT5720) was also tested. Values represent the mean and SD of at least three independent experiments. *p < 0.05 versus control (ANOVA and Tukey's post hoc test). Stauro, Staurosporine; Inhi, inhibitors; U0, U0126; Kt, KT5720; Forsk, forskolin.
We have found previously that His6-STI1, pepSTI1230–245, and another PrPc-engaging peptide prevented programmed cell death of undifferentiated postmitotic retinal cells induced by anisomycin, through activation of PKA (Chiarini et al., 2002; Zanata et al., 2002). To address whether the PKA pathway is also involved in STI1-induced neuroprotection of hippocampal neurons, we tested the effects of specific signaling inhibitors. The neuroprotective effect of His6-STI1 was abrogated by the PKA inhibitor (KT5720), whereas PKC (Bim or Chel) and MAPK (U0126) inhibitors had no effect (Fig. 8_B_). In nonstimulated neurons (absence of STI1), inhibitors had no effect on staurosporine-induced cell death (supplemental Fig. 9, available at www.jneurosci.org as supplemental material).
The activity of PKA was also assessed to confirm its involvement in the neuroprotection stimulated by His6-STI1. Incubation of Prnp+/+ hippocampal cells with His6-STI1 was followed by an increase in PKA activity, comparable to that obtained with forskolin, a potent activator of PKA (Fig. 8_C_). In contrast, treatment with His6-STI1Δ230–245 was unable to induce PKA activation. The PKA inhibitor (KT5720) strongly inhibited the His6-STI1-stimulated PKA activity.
These results demonstrate the involvement of PKA in neuroprotection induced by the interaction of STI1 with PrPc in hippocampal neurons, similar to our previously described results in retinal tissue (Chiarini et al., 2002), whereas neither MAPK nor PKC are required for this effect.
Discussion
This investigation showed that (1) the cellular prion protein (PrPc) and its binding partner STI1 colocalize in hippocampal neurons, (2) interaction of STI1 with PrPc mediated by their cognate binding domains induces both neuritogenesis and neuroprotection of hippocampal neurons in culture, and (3) neuritogenesis induced by STI1 is mediated by the MAPK pathway, whereas neuroprotection induced by the same protein depends on a PKA pathway. These data add to the growing body of evidence that PrPc is involved in neurotrophic signaling within the CNS and helps clarify its mechanisms.
It is becoming increasingly clear that PrPc has a role in neuronal differentiation. Previous studies from our group showed that PrPc is the main cellular receptor for a peptide at the γ1-laminin chain. This specific interaction between laminin and PrPc induced neuritogenesis in mouse hippocampal neurons (Graner et al., 2000a).
We also showed the participation of PrPc in neurite adhesion and maintenance through its interaction with laminin (Graner et al., 2000b). PrPc has been proposed to be a cell-surface adhesion protein, and its early developmental distribution resembles that of adhesion molecules (Endo et al., 1989). Recent studies using a time-controlled transcardiac perfusion cross-linking procedure showed that proteins involved in cell adhesion and neurite out-growth can be found in the molecular microenvironment of PrPc in the living brain (Schmitt-Ulms et al., 2001, 2004). Furthermore, the laminin receptor, required for cell differentiation and growth, was identified as a cell-surface binding partner of PrPc (Gauczynski et al., 2001; Hundt et al., 2001). PrPc also interacts with glycosaminoglycans expressed on the cell surface (Shyng et al., 1995; Rieger et al., 1997; Pan et al., 2002). This is particularly relevant for brain development, because both laminin and glycosaminoglycans are developmentally regulated and contribute to axon growth and the formation of fiber tracts (Reichardt and Tomaselli, 1991). The abundance of PrPc in elongating axons suggests a role for the protein in axon growth (Sales et al., 2002), which is supported by data from Prnp0/0 mice, in which the hippocampal mossy fiber projection is disorganized (Colling et al., 1997).
PrPc activation by antibody cross-linking induces a signaling pathway that involves the tyrosine kinase p59fyn in a cell line capable of neuron-like differentiation (Mouillet-Richard et al., 2000). The p59fyn kinase has also been implicated in modulating axonal guidance of olfactory axons that express high levels of PrPc throughout life and mediate neuronal cell adhesion molecule (NCAM)-induced neurite outgrowth (Morse et al., 1998; Sales et al., 1998; Beggs et al., 1997; Kolkova et al., 2000). Accordingly, recent findings show that PrPc interacts directly with the NCAM, leading to stabilization of the latter in lipid rafts where fyn is activated, and induces NCAM-dependent neuritogenesis (Santuccione et al., 2005). Nevertheless, the PrPc binding site at the NCAM was not characterized.
Various studies provided evidence that PrPc is implicated in neuroprotection. PrPc has been reported to protect human primary neurons against Bax-induced cell death, either from its location at the cell surface through an unidentified signaling pathway or from the cytosol (Bounhar et al., 2001; Roucou et al., 2003). It was reported recently that several signal transduction pathways involved in survival are activated in mouse primary cerebellar granule neurons grown in PrP-coated tissue culture plates. In this preparation, homophilic PrPc interaction led to activation of PKA, Src-related tyrosine kinases, PI3 kinase/Akt, and MAPK/ERK kinases. Among downstream targets, increased Bcl-2 levels and decreased Bax levels were observed, consistent with PrPc-triggering survival signals (Chen et al., 2003).
Investigation of PrPc-interacting proteins is an important tool to understand not only cellular signaling triggered by, but also the biological function(s) related to, PrPc. This approach is more likely to reveal physiological functions than antibody-mediated cross-linking (Mouillet-Richard et al., 2000; Hugel et al., 2004; Monnet et al., 2004).
Accordingly, we have demonstrated that interaction of PrPc with its ligand STI1 protein activates a PKA-dependent signaling pathway to rescue retinal cells from induced apoptosis (Chiarini et al., 2002; Zanata et al., 2002). Recently, Onodera and colleagues (Sakudo et al., 2005) showed that PrPc cooperates with STI1 to regulate superoxide dismutase activity and consequently to modulate cell survival.
The current experiments provided clear evidence that neuritogenesis induced by STI1 depends on the expression of PrPc and that MAPK family members are required for this effect. In turn, neuroprotection induced by STI1–PrPc interaction in hippocampal neurons, similar to our previous studies in retinal cells, is mediated by a PKA signaling pathway.
It is interesting to note that the concentration of His6-STI1 required to promote neuroprotection is higher than the one necessary to induce neuritogenesis. This can be attributable to the activation threshold of the distinct MAPK and PKA signaling pathways involved in each phenomenon.
Therefore, we propose a model in which PrPc interacts with STI1 and transduces both a survival or protective signal through PKA and a neuritogenesis/differentiation signal through the MAPK pathway in neuronal cells (supplemental Fig. 10, available at www.jneurosci.org as supplemental material). The PrPc–STI1 complex may be formed by proteins either in the same cells (supplemental Fig. 10_A_, available at www.jneurosci.org as supplemental material) or in distinct cells (supplemental Fig. 10_B_, available at www.jneurosci.org as supplemental material). In our previous study, we speculated that STI1 could act as soluble neurotrophic factor (Chiarini et al., 2002) interacting with PrPc. STI1 has indeed been found released by certain tumor cell lines (Eustace and Jay, 2004), glial cells, and in lower amounts by neurons in culture (our unpublished results). Thus, there is a third possibility in which either astrocytes or neurons secrete STI1, which then may act as a paracrine or autocrine factor in neuronal differentiation (supplemental Fig. 10_C_, available at www.jneurosci.org as supplemental material).
The significance of the high levels of STI1 colocalized with PrPc at the surface of the cell soma rather than in neurites is not clear. The lack of detectable immunoreactivity to STI1 in neurites may be attributable either to low amounts of the protein at the surface or to a true differential distribution. This, as well as the relationships between STI1 and NCAM (Santuccione et al., 2005), both of which are involved in neuritogenesis mediated by PrPc, remain to be addressed.
The results presented here advance a new biological function for STI1, support a significant role for PrPc as a response mediator in both neuritogenesis and neuroprotection promoted by STI1, and provide additional insight into the molecular basis of STI1-induced intracellular signaling. The roles of PrPc in neuronal survival and neuritogenesis may be germane to loss-of-function components of the pathogenesis of prion diseases. Finally, the characterization of PrPc ligands with neurotrophic activity is relevant both for developmental neurobiology as well as for pathology. These molecules may represent new targets for therapeutic intervention in neurodegenerative conditions.
Footnotes
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (Grant 03/13189-2), Conselho Nacional de Desenvolvimento Cientifico e Tecnológico (CNPq), Fundacão de Amparo a Pesquisa do Estado do Rio de Janeiro, and Programa de Apoio a Núcleos de Excelência–Ministério da Ciência e Tecnologia. Fellowships from FAPESP (M.H.L. and G.N.M.H.), CNPq (G.L.M.), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (R.M.P.S.C.) are gratefully acknowledged. We are grateful to Camila Pinto Arantes and Tiago Góss dos Santos for excellent technical assistance and to Prof. Marco A. M. Prado for helpful discussions. We also thank Carlos F. Nascimento, Severino da Silva Ferreira, and Miyuki Fukuda da Silva for technical assistance.
Correspondence should be addressed to Dr. Vilma R. Martins, Laboratory of Cellular and Molecular Biology, Ludwig Institute for Cancer Research, Rua Prof Antonio Prudente 109/4A, 01509-010 São Paulo, São Paulo, Brazil. E-mail: vmartins@ludwig.org.br.
DOI:10.1523/JNEUROSCI.2313-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511330-10$15.00/0
*
M.H.L. and G.N.M.H. contributed equally to this work.
References
- Audesirk G, Cabell L, Kern M (1997) Modulation of neurite branching by protein phosphorylation in cultured rat hippocampal neurons. Brain Res Dev Brain Res 102: 247–260. [DOI] [PubMed] [Google Scholar]
- Beggs HE, Baragona SC, Hemperly JJ, Maness PF (1997) NCAM140 interacts with the focal adhesion kinase p125(fak) and the SRC-related tyrosine kinase p59(fyn). J Biol Chem 272: 8310–8319. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A (2001) Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem 276: 39145–39149. [DOI] [PubMed] [Google Scholar]
- Brown DR, Sassoon J (2002) Copper-dependent functions for the prion protein. Mol Biotechnol 22: 165–178. [DOI] [PubMed] [Google Scholar]
- Brown DR, Nicholas RS, Canevari L (2002) Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res 67: 211–224. [DOI] [PubMed] [Google Scholar]
- Bueler H, Fischer M, Lang Y, Bluethmann H, Lipp HP, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C (1992) Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356: 577–582. [DOI] [PubMed] [Google Scholar]
- Carrigan PE, Nelson GM, Roberts PJ, Stoffer J, Riggs DL, Smith DF (2004) Multiple domains of the co-chaperone Hop are important for Hsp70 binding. J Biol Chem 279: 16185–16193. [DOI] [PubMed] [Google Scholar]
- Chen S, Mange A, Dong L, Lehmann S, Schachner M (2003) Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol Cell Neurosci 22: 227–233. [DOI] [PubMed] [Google Scholar]
- Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R (2002) Cellular prion protein transduces neuroprotective signals. EMBO J 21: 3317–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colling SB, Khana M, Collinge J, Jefferys JG (1997) Mossy fibre reorganization in the hippocampus of prion protein null mice. Brain Res 755: 28–35. [DOI] [PubMed] [Google Scholar]
- Doherty P, Williams G, Williams EJ (2000) CAMs and axonal growth: a critical evaluation of the role of calcium and the MAPK cascade. Mol Cell Neurosci 16: 283–295. [DOI] [PubMed] [Google Scholar]
- Elion EA (1993) Constructing recombinant DNA molecules by the polymerase chain reaction. In: Current protocols in molecular biology, Chap 3 (Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JD, Smith JA, Struhl K, eds), p 3.17.1. New York: Wiley Interscience.
- Endo T, Groth D, Prusiner SB, Kobata A (1989) Diversity of oligosaccharide structures linked to asparagines of the scrapie prion protein. Biochemistry 28: 8380–8388. [DOI] [PubMed] [Google Scholar]
- Eustace BK, Jay DG (2004) Extracellular roles for the molecular chaperone, hsp90. Cell Cycle 3: 1098–1100. [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM (1998) Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 273: 18623–18632. [DOI] [PubMed] [Google Scholar]
- Gauczynski S, Peyrin JM, Haik S, Leucht C, Hundt C, Rieger R, Krasemann S, Deslys JP, Dormont D, Lasmezas CI, Weiss S (2001) The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J 20: 5863–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graner E, Mercadante AF, Zanata SM, Forlenza OV, Cabral AL, Veiga SS, Juliano MA, Roesler R, Walz R, Minetti A, Izquierdo I, Martins VR, Brentani RR (2000a) Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res Mol Brain Res 76: 85–92. [DOI] [PubMed] [Google Scholar]
- Graner E, Mercadante AF, Zanata SM, Martins VR, Jay DG, Brentani RR (2000b) Laminin-induced PC-12 cell differentiation is inhibited following laser inactivation of cellular prion protein. FEBS Lett 482: 257–260. [DOI] [PubMed] [Google Scholar]
- Hernandez MP, Sullivan WP, Toft DO (2002) The assembly and intermolecular properties of the hsp70-Hop-hsp90 molecular chaperone complex. J Biol Chem 277: 38294–38304. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugel B, Martinez MC, Kunzelmann C, Blattler T, Aguzzi A, Freyssinet JM (2004) Modulation of signal transduction through the cellular prion protein is linked to its incorporation in lipid rafts. Cell Mol Life Sci 61: 2998–3007. [DOI] [PubMed] [Google Scholar]
- Hundt C, Peyrin JM, Haik S, Gauczynski S, Leucht C, Rieger R, Riley ML, Deslys JP, Dormont D, Lasmezas CI, Weiss S (2001) Identification of interaction domains of the prion protein with its 37-kDa/67-kDa laminin receptor. EMBO J 20: 5876–5886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolkova K, Novitskaya V, Pedersen N, Berezin V, Bock E (2000) Neural cell adhesion molecule-stimulated neurite outgrowth depends on activation of protein kinase C and the Ras-mitogen-activated protein kinase pathway. J Neurosci 20: 2238–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins AR, Dias MM, Vasconcelos TM, Caldo H, Costa MC, Chimelli L, Larson RE (1999) Microwave-stimulated recovery of myosin-V immunoreactivity from formalin-fixed, paraffin-embedded human CNS. J Neurosci Methods 92: 25–29. [DOI] [PubMed] [Google Scholar]
- Martins VR, Graner E, Garcia-Abreu J, de Souza SJ, Mercadante AF, Veiga SS, Zanata SM, Neto VM, Brentani RR (1997) Complementary hydropathy identifies a cellular prion protein receptor. Nat Med 3: 1376–1382. [DOI] [PubMed] [Google Scholar]
- Martins VR, Linden R, Prado MA, Walz R, Sakamoto AC, Izquierdo I, Brentani RR (2002) Cellular prion protein: on the road for functions. FEBS Lett 512: 25–28. [DOI] [PubMed] [Google Scholar]
- Monnet C, Gavard J, Mege RM, Sobel A (2004) Clustering of cellular prion protein induces ERK1/2 and stathmin phosphorylation in GT1–7 neuronal cells. FEBS Lett 576: 114–118. [DOI] [PubMed] [Google Scholar]
- Morse WR, Whitesides JG, III, LaMantia AS, Maness PF (1998) p59fyn and pp60c-src modulate axonal guidance in the developing mouse olfactory pathway. J Neurobiol 36: 53–63. [DOI] [PubMed] [Google Scholar]
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O (2000) Signal transduction through prion protein. Science 289: 1925–1928. [DOI] [PubMed] [Google Scholar]
- Nicolet CM, Craig EA (1989) Isolation and characterization of STI1, a stress-inducible gene from _Saccharomyces cerevisiae_Mol Cell Biol 9: 3638–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicotera P, Orrenius S (1998) The role of calcium in apoptosis. Cell Calcium 23: 173–180. [DOI] [PubMed] [Google Scholar]
- Pan T, Wong BS, Liu T, Li R, Petersen RB, Sy MS (2002) Cell-surface prion protein interacts with glycosaminoglycans. Biochem J 368: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt LF, Tomaselli KJ (1991) Extracellular matrix molecules and their receptors: functions in neural development. Annu Rev Neurosci 14: 531–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger R, Edenhofer F, Lasmezas CI, Weiss S (1997) The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat Med 3: 1383–1388. [DOI] [PubMed] [Google Scholar]
- Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC (2003) Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem 278: 40877–40881. [DOI] [PubMed] [Google Scholar]
- Ruegg UT, Burgess GM (1989) Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol Sci 10: 218–220. [DOI] [PubMed] [Google Scholar]
- Sakudo A, Lee DC, Li S, Nakamura T, Matsumoto Y, Saeki K, Itohara S, Ikuta K, Onodera T (2005) PrP cooperates with STI1 to regulate SOD activity in PrP-deficient neuronal cell line. Biochem Biophys Res Commun 328: 14–19. [DOI] [PubMed] [Google Scholar]
- Sales N, Rodolfo K, Hassig R, Faucheux B, Di Giamberardino L, Moya KL (1998) Cellular prion protein localization in rodent and primate brain. Eur J Neurosci 10: 2464–2471. [DOI] [PubMed] [Google Scholar]
- Sales N, Hassig R, Rodolfo K, Di Giamberardino L, Traiffort E, Ruat M, Fretier P, Moya KL (2002) Developmental expression of the cellular prion protein in elongating axons. Eur J Neurosci 15: 1163–1177. [DOI] [PubMed] [Google Scholar]
- Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M (2005) Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol 169: 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, Prusiner SB (2001) Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol 314: 1209–1225. [DOI] [PubMed] [Google Scholar]
- Schmitt-Ulms G, Hansen K, Liu J, Cowdrey C, Yang J, DeArmond SJ, Cohen FE, Prusiner SB, Baldwin MA (2004) Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat Biotechnol 22: 724–731. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Lehmann S, Moulder KL, Harris DA (1995) Sulfated glycans stimulate endocytosis of the cellular isoform of the prion protein, PrPC, in cultured cells. J Biol Chem 270: 30221–30229. [DOI] [PubMed] [Google Scholar]
- van der Spuy J, Kana BD, Dirr HW, Blatch GL (2000) Heat shock cognate protein 70 chaperone-binding site in the co-chaperone murine stress-inducible protein 1 maps to within three consecutive tetratricopeptide repeat motifs. Biochem J 345: 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanata SM, Lopes MH, Mercadante AF, Hajj GN, Chiarini LB, Nomizo R, Freitas AR, Cabral AL, Lee KS, Juliano MA, de Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR (2002) Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J 21: 3307–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]