Activation of the IκB Kinase Complex and Nuclear Factor-κB Contributes to Mutant Huntingtin Neurotoxicity (original) (raw)
Abstract
Transcriptional dysregulation by mutant huntingtin (Htt) protein has been implicated in the pathogenesis of Huntington's disease (HD). We find that cultured cells expressing mutant Htt and striatal cells from HD transgenic mice have elevated nuclear factor-κB (NF-κB) activity. Furthermore, NF-κB is concentrated in the nucleus of neurons in the brains of HD transgenic mice. In inducible PC12 cells and in HD transgenic mice, mutant Htt activates the IκB kinase complex (IKK), a key regulator of NF-κB. Activation of IKK is likely mediated by direct interaction with mutant Htt, because the expanded polyglutamine stretch and adjacent proline-rich motifs in mutant Htt interact with IKKγ, a regulatory subunit of IKK. Activation of IKK may also influence the toxicity of mutant Htt, because expression of IKKγ promotes aggregation and nuclear localization of mutant Htt exon-1. Moreover, in acute striatal slice cultures, inhibition of IKK activity with an N-terminally truncated form of IKKγ blocks mutant Htt-induced toxicity in medium-sized spiny neurons (MSNs). In addition, blocking degradation of NF-κB inhibitors with a dominant-negative ubiquitin ligase β-transducin repeat-containing protein also reduces the toxicity of mutant Htt in MSNs. Therefore, aberrant NF-κB activation may contribute to the neurodegeneration induced by mutant Htt.
Keywords: Huntington's disease, IKK, polyproline, striatum, polyQ, intrabody
Introduction
Huntington's disease (HD) is a neurodegenerative disorder caused by expansion of polyglutamine (polyQ) in exon-1 (HDx1) of huntingtin (Htt) (Huntington's Disease Collaborative Research Group, 1993). Neuronal death in HD has been variously attributed to polyQ toxicity, activation of caspases, interference with transcription, and sequestration of cellular proteins (Li, 1999; Tobin and Signer, 2000; Wanker, 2000; Zoghbi and Orr, 2000). Several proteins that interact with HDx1 have been identified (Cattaneo et al., 2001), and although the function of some of these proteins in the etiology of HD is unclear, the transcriptional coactivator cAMP response element (CRE)-binding protein (CBP), as well as proteins containing WW domains, have been implicated in HD pathology (Passani et al., 2000; Steffan et al., 2000; Nucifora et al., 2001). Htt binding blocks CBP-mediated gene expression, and histone deacetylase inhibitors reduce polyglutamine-induced cell death in Drosophila (Steffan et al., 2001). Conversely, CRE-mediated gene expression, which requires CBP, is enhanced in HD transgenic mice (Obrietan and Hoyt, 2004). CBP participates in many signaling pathways, including NF-κB (Chen et al., 2002; Zhong et al., 2002). Consequently, mutant Htt dysregulation of CBP can influence nuclear factor-κB (NF-κB)-mediated gene expression.
NF-κB is sequestered in the cytoplasm by a family of inhibitory proteins (IκBs) (Ghosh et al., 1998). IκBs are phosphorylated by a signal-activated kinase complex known as I-κB kinase (IKK) (Ghosh and Karin, 2002). This complex contains two catalytic subunits, IKKα and IKKβ, and a regulatory module, IKKγ (Karin and Lin, 2002). Phosphorylated IκBs are ubiquitinated by an F-box E3-ligase, β-transducin repeat-containing protein (β-TrCP) (Spencer et al., 1999) and subsequently degraded by proteosomes. Liberated NF-κB can bind DNA and promote gene expression (Pahl, 1999).
In the nervous system, cytokines, neurotrophic factors, neurotransmitters, injury, seizure, and proteins implicated in neurodegenerative disorders can all activate the NF-κB pathway (Mattson and Camandola, 2001). Injection of 3-nitropropionic acid (3NP) and kainate into the striatum, which mimics pathology caused by mutant Htt, results in activation of NF-κB and expression of the proapoptotic gene products, myc and p53 (Qin et al., 1999). On the other hand, injection of 3NP into the striatum of mice lacking the p50 subunit of NF-κB results in diminished NF-κB activity and increased cell death (Mattson and Camandola, 2001).
Akt/protein kinase B, an activator of IKK, has been shown to phosphorylate mutant Htt and appears to have a neuroprotective effect (Datta et al., 1999; Humbert et al., 2002). Furthermore, striatal cells from HD knock-in (KI) mice display elevated Akt activity; however, expression of mutant Htt in these cells is not toxic (Gines et al., 2003). Akt also phosphorylates ataxin-1, which binds to 14-3-3 protein and enhances its stability, aggregation, and neurotoxicity (Chen et al., 2003; Emamian et al., 2003). Thus, as a major integrator of cell signaling, NF-κB may regulate the pathogenesis of polyglutamine-induced neurodegeneration. Here we show that mutant Htt binds IKKγ, activates the IKK complex, and elevates NF-κB-dependent gene expression. Furthermore, HDx1-mediated toxicity is significantly reduced by genetic inhibition of IKK and NF-κB activity.
Materials and Methods
Cells and transgenic animals. PC12 cells, which express wild-type (WT) HDx1-enhanced green fluorescent protein (EGFP) with 25 glutamine repeats (25Qs) or mutant HDx1-EGFP with 103Qs in response to ecdysone, were kindly provided by Dr. E. Schweitzer (University of California Los Angeles). These cells were cultured on a collagen I substrate (Fisher Scientific, Tustin, CA) in DMEM with 5% horse serum, 5% fetal bovine serum, 2 mm glutamine, and standard penicillin-streptomycin antibiotics. Immortalized striatal cells from WT and HD KI mice, provided by Dr. M. E. MacDonald (Massachusetts General Hospital, Boston, MA), were cultured as described (Gines et al., 2003). Human embryonic kidney (HEK)-293 cells were cultured as described previously (Khoshnan et al., 2002). The HD transgenic mouse line R6/2 and a knock-in line, in which a 155 polyglutamine stretch was inserted in HDx1 of mouse Htt, have been described (Davies et al., 1997; Lin et al., 2001). Colonies were established and maintained in our animal facility.
Gene reporter assays and transfections. The pNF-κB-luciferase with five enhancer elements and the control plasmid without NF-κB binding sites, pCIS-CK-luciferase, were used for gene reporter assays (Stratagene, La Jolla, CA). To normalize for transfection efficiency between samples, a β-galactosidase (β-gal) construct expressed from the elongation factor (EF)-1α promoter (Invitrogen, Carlsbad, CA) was included in all gene reporter assays. PC12 cells were transfected with control or NF-κB-luciferase plasmids using lipofectamine-plus. On the following day, cells were left untreated or stimulated for 8 hr with 1 μg/ml ecdysone to induce expression of HDx1. Mock treatment or NGF (Sigma, St. Louis, MO) was added (50 ng/ml) and incubated for an additional 6 hr. Cells were suspended in lysis buffer on ice for 10 min and cleared by centrifugation. Luciferase activity was measured by addition of the substrate (Promega, Madison, WI) to equal amounts of protein from each sample. Striatal cells from WT and HD KI mice were transfected with an NF-κB reporter plus β-gal and anti-polyQ (MW2) or anti-polyproline [antipolyP (MW7)] recombinant intrabodies (Khoshnan et al., 2002). On the following day, cells were starved for 2 hr and treated with 5 ng/ml recombinant interleukin (IL)-1β (R & D Systems, Minneapolis, MN) and incubated for an additional 6 hr. Cells were harvested and cleared lysate was used to measure luciferase as described above. All gene reporter assays were corrected for transfection efficiency by normalizing the units of β-gal in the extracts, using the β-gal assay (Invitrogen). β-gal values from the samples were divided by the β-gal value from control, 25polyQ, noninduced PC12 cells (no expression of HDx1) and multiplied by each corresponding luciferase reading. For striatal cells, β-gal values were divided by the value obtained for WT cells without any treatment. Results are shown as relative luciferase units and are representative of at least three independent experiments. Data points are the average of triplicate measurements.
To examine the effects of deleted F-box (ΔF)-βTrCP [obtained from Dr. R. Deshaies (California Institute of Technology) and originally provided by Spencer et al. (1999)] and deleted N terminus (DN)-IKKγ on mutant HDx1-induced NF-κB, PC12 cells were transfected with ΔF-βTrCP or DN-IKKγ plus β-gal and NF-κB reporter in six-well plates. Induction of HDx1 with ecdysone and NGF treatment was as described above. Equal amounts of each sample were used to measure luciferase activity. Binding of NF-κB to its consensus oligonucleotide site was assayed in striatal neurons with the BD TransFactor Kit specific for the p65 subunit following the manufacturer's instructions (BD Biosciences, Mountain View, CA). Briefly, striatal cell lines from WT and HD KI mice were starved for 2 hr and treated with 5 ng/ml IL-1β for 15 or 30 min. Nuclear extracts were obtained using commercial reagents (BD Biosciences). Protein concentration was determined by the BCA method (Pierce, Rockford, IL). Equal amounts of each nuclear fraction were added to wells coated with an NF-κB consensus or mutated control oligonucleotide and incubated at room temperature for 1 hr. Wells were washed extensively and incubated with primary rabbit antibody specific for the p65 subunit of NF-κB. Bound p65 was detected with a goat anti-rabbit antibody conjugated to horseradish peroxidase. After addition of the substrate, 3,3-5,5-tetramethylybenzidine, color development was measured at 655 nm using an ELISA plate reader. Samples were done in triplicate, and the results shown are representative of three independent experiments.
Transfection of HEK-293 cells with mutant HDx1-EGFP alone (Kazantsev et al., 1999) or with one of the following HA-tagged constructs, full-length (F), DN, and deleted C terminus (DC) of IKKγ (provided by Dr. E. Zandi, University of Southern California) (Rothwarf et al., 1998) or ΔF-βTrCP, was done using lipofectamine-plus reagents (Invitrogen). For histochemistry, cultured cells were fixed in 4% paraformaldehyde for 30 min, permeabilized with 70% methanol for 1 hr, and washed with PBS. Cells were stained with anti-HA tag for IKKγ or anti-myc for ΔF-βTrCP (Cell Signaling, Beverly, MA) followed by a goat anti-mouse antibody conjugated to Alexa 594 (Molecular Probes, Eugene, OR). Toto-3, a dimeric cyanine, was used to stain nuclei (Molecular Probes). Cells were washed in PBS, mounted on microscope slides, and examined with a confocal microscope. Mutant HDx1 toxicity was assessed by counting the number of condensed GFP-positive bodies, which are remnants of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)-positive (TUNEL+) cells (Khoshnan et al., 2002), using a florescence microscope. For TUNEL staining, transfected cells grown on coverslips were air dried and fixed 16 hr after transfection as above and permeabilized in ethanol/acetic acid (2:1). Apoptotic cells were labeled with terminal deoxynucleotidyl transferase using digoxigenin-labeled nucleotides and detected by anti-digoxigenin antibody and rhodamine-conjugated secondary according to the instructions provided by the manufacturer (Chemicon, Temecula, CA).
Immunohistochemistry of tissue sections. Brains were taken from paraformaldehyde-fixed WT and R6/2 HD transgenic mice, and cryosections were permeabilized in 70% methanol for 1 hr at -20. After blocking in 3% BSA and 10% normal goat serum, slides were incubated with anti-p65 (Santa Cruz Biotechnology, Santa Cruz, CA) and the neuronal marker anti-neuronal-specific nuclear protein (NeuN) antibodies (Chemicon). p65-positive cells were detected with a goat anti-rabbit secondary antibody conjugated to FITC and goat anti-mouse Alexa Fluor 594 (Molecular Probes). Toto-3 was used to stain nuclei. Sections were examined with a confocal microscope. Total neurons from 16 coronal sections containing cortical and striatal areas of four animals each of WT and HD mice were quantified, and the average percentage of cells with nuclear p65 is presented.
Protein-protein interaction. In vitro binding of IKKγ to mutant HDx1 was performed using glutathione _S_-transferase (GST)-pull down assays. GST-HDx1 with 20 or 51 polyQs, with and without polyproline domains, was obtained from Dr. E. Wanker (Max Plank Institute, Berlin, Germany). These constructs were expressed in Escherichia coli and purified with glutathione beads (Amersham Biosciences, Piscataway, NJ) according to manufacturer's instructions. IKKγ constructs were transcribed and translated in rabbit reticulocytes in the presence of [35S]methionine (Promega). Briefly, 5 μg of GST-HDx1 bound to beads was incubated with 10 μl of each labeled _in vitro_-translated IKKγ product in 500 μl of Tris-based buffer containing 10% glycerol and 5 mm DTT and rocked at room temperature for 2 hr. Beads were washed five times in the same buffer. Bound IKKγ was examined by SDS-PAGE, followed by autoradiography. Recombinant anti-HDx1 intrabodies (Khoshnan et al., 2002) were expressed and examined similarly for binding to GST-HDx1. For competition assays, 10-fold excess _in vitro_-translated intrabodies were added to GST-HDx1 plus DC-IKKγ and processed as above.
For coimmunoprecipitation studies, PC12 cells or striatal tissue of WT and HD transgenic mice was lysed by sonication in buffer A (50 mm HEPES, pH 7.6, 250 mm NaCl, 1% Triton X-100, 2 mm MgCl2, 2 mm DTT, 1 mm Na3VO4, and 20 μm β-glycerophosphate) and a mixture of protease inhibitors (Boehringer Mannheim, Mannheim, Germany). Equal amounts of cleared extracts from each sample were incubated with a rabbit anti-IKKγ (Santa Cruz Biotechnology) coupled to protein-A beads and rocked for 3 hr at 4°C. Cells were washed five times with buffer A. Immune complexes were examined by SDS-PAGE followed by Western blotting with monoclonal antibodies targeting Htt (Ko et al., 2001), as described in the figure legends.
Immune complex kinase assays. To measure IKK activity, immunoprecipitated IKK complexes were examined for the ability to phosphorylate GST-IκBα in vitro. The construct for the N-terminal 61 amino acids of GST-IκBα, which contains the IKK phosphorylation sites (provided by Dr. W. Greene, University of California San Francisco), was expressed in E. coli and purified on glutathione beads as described in the manufacturer's instructions (Amersham Biosciences). To obtain IKK complexes, equal amounts of cleared striatal extracts from WT and HD animals, or from PC12 cells induced to express WT or mutant HDx1-EGFP (all in buffer A), were incubated with a mouse anti-IKKα antibody (Santa Cruz Biotechnology) coupled to protein-A beads and rocked for 3 hr at 4°C. Beads were washed five times in buffer B (50 mm HEPES, pH 7.6, 250 mm NaCl, 1 m urea, 0.1% Nonidet P-40, 6 mm EDTA, 6 mMEGTA, 1 mm DTT with 20 μm β-glycerophosphate) and a mixture of protease inhibitors and equilibrated in kinase buffer (20 mm HEPES, pH 7.6, 2 mm MgCl2, 2 mm MnCl2, 10 μm ATP, 10 mm glycerophosphate, 10 mm NaF, 300 μm Na3VO4, 1 mm dithiothreitol DTT). IKK activity was evaluated in vitro with 1 μg of purified GST-IκBα (N-terminal 61 amino acids) in the presence of 10 μCi of [32P]γ-ATP for 30 min at 30°C. Products were examined by SDS-PAGE followed by autoradiography. Duplicate samples were examined by Western blotting using a rabbit anti-IKKγ antibody (Santa Cruz Biotechnology).
Brain slice preparation. All animal experiments were performed in accordance with the Institutional Animal Care and Use Committee and Duke University Medical Center Animal Guidelines. Postnatal day 7 (P7) CD Sprague Dawley rats (Charles River Laboratory, Raleigh, NC) were decapitated, and the brains were surgically removed and placed in ice-cold Neurobasal medium (Invitrogen). The tissue was fixed to a chilled, stainless steel Vibratome stage using cyanoacrylate glue (Krazy-Glue) and covered with Neurobasal medium. Coronal brain slices (250 μm thick) were cut by Vibratome (VT1000S; Leica, Nussloch, Germany) as described previously (Edgerton and Reinhart, 2003). Brain slices were kept at 37°C in 5.0% CO2 for 1 hr.
Biolistic transfection. Gold particles (1.6 μm gold microcarriers; Bio-Rad, Hercules, CA) were used as DNA carriers for transfection as described previously (Lo et al., 1994). Briefly, gold particles were sonicated in 0.05 m spermidine in the presence of plasmid DNA. The gold-DNA mixture was washed three times in 100% ethanol before being loaded into Helios plastic cartridges (Bio-Rad) according to manufacturer's instructions. Slices were transfected using a Bio-Rad Helios gun with a cyan fluorescent protein (CFP)-tagged Htt fragment, together with yellow fluorescent protein (YFP) as a morphometric marker. In a number of experiments, one of the IKKγ constructs was also cotransfected with the Htt-construct and YFP. CFP-tagged Htt fragments were exon-1 N-terminal fragments containing either a short-Q (Q23) or a long-Q (Q148) polyQ domain.
Brain slice neurodegeneration assay. For each condition, transfections were performed on 12 brain slices per experiment. Protein expression and neurodegeneration (loss of processes, shriveling of the soma) were assayed 2-7 d after transfection using a Leica fluorescence microscope with appropriate filters for YFP and CFP. The total area of the striatum was identified, and transfected MSNs were identified by their morphology. CFP fluorescence was used to determine expression of Htt fragments and their aggregation into macro-inclusions. Each experiment involved 3-12 transfected brain slices per condition, and the data are means from six independent experiments for each condition.
Results
Mutant Htt activates the NF-κB pathway
We first used PC12 cells that express WT (25Q) or mutant (103Q) HDx1 in response to ecdysone (Fig. 1_A_). These cells were transfected with a reporter construct containing NF-κB enhancer element fused to luciferase. The inducible nature of HDx1 expression allows the examination of soluble mutant HDx1 function, before macro-aggregates become visible. Induction of mutant HDx1 expression increases transcription from the NF-κB-dependent promoter approximately fivefold over that of noninduced cells, whereas WT HDx1 has minimal effect (Fig. 1_A_). These results are consistent with a recent report demonstrating upregulation of NF-κB expression by mutant HDx1 in inducible PC12 cells (Sugars et al., 2004). To examine whether mutant HDx1 also influences signal-induced NF-κB activation, we used NGF, which is known to activate NF-κB in PC12 cells (Foehr et al., 2000). Mutant (but not WT) HDx1 strongly enhances NGF-induced NF-κB-dependent gene expression (Fig. 1_A_). Because mutant Htt has been shown to variably influence gene expression from different promoters, we performed the same experiments with a control plasmid that lacks an NF-κB enhancer element. Expression of WT or mutant HDx1 does not influence luciferase expression from the control plasmid (Fig. 1_B_), suggesting that the elevated NF-κB values in Figure 1_A_ are mediated by mutant HDx1 expression. Under the conditions tested, most of the HDx1 remains in the detergent-soluble fraction and is detectable by Western blotting (Fig. 1_A_); however, the NF-κB activity diminishes as mutant HDx1 accumulates and aggregates (data not shown). Thus, soluble mutant HDx1 activates endogenous, and augments NGF-induced, NF-κB-dependent gene expression. Moreover, expression of mutant HDx1 promotes degradation of the inhibitory protein IκBα, a hallmark of NF-κB activation. Levels of IκBα drop significantly by 3 hr and return to normal by 7 hr after induction (Fig. 1_C_). These data confirm the gene reporter assay results and suggest that mutant Htt-induced NF-κB is mediated by degradation of IκBα.
Figure 1.

Mutant Htt activates NF-κB-dependent gene expression. A, PC12 cells transfected with an NF-κB reporter plasmid were induced to express a 25 or 103 polyQ-containing HDx1-EGFP. NGF was added 8 hr after Httinduction, and the cells were incubated for an additional 6 hr. Cleared lysates were assayed for luciferase activity to assess NF-κB-dependent gene expression. The luciferase values were normalized for transfection efficiency using the β-gal reporter gene under transcriptional control of the EF-1α promoter. β-gal values from samples were divided by the β-gal values from control, 25polyQ, noninduced HDx1 (no expression of HDx1) and multiplied by the corresponding luciferase readings. Results are shown as relative luciferase units and are representative of at least three independent experiments, and data points are the average of triplicate measurements. The Western blot at the bottom of A shows the level of soluble HDx1 detected using an anti-Htt antibody. The same blots were probed with anti-tubulin to show equivalent loading in each lane. B, Expression of WT or mutant HDx1 does not influence luciferase expression from the control plasmid, which lacks an NF-κB enhancer element. The experiment was done as in A, using a plasmid reporter without the NF-κ Benhancers. Luciferase units were ∼100-fold less than the activity observed with the NF-κB reporter. C, NF-κB activation by mutant Htt includes degradation of IκBα. PC12 cells were treated with ecdysone to induce HDx1 for the indicated time and examined by Western blotting with anti-IκBα antibody. The same membrane was stained with anti-tubulin antibody for loading accuracy. D, Full-length mutant Htt enhances IL-1β-mediated NF-κB activation. Striatal cells from WT and HD KI mice were transfected with NF-κB or the control reporter and the indicated plasmids. Transfected cells were starved on the following day for 2 hr and treated with 5 ng/ml IL-1β for an additional 6 hr. Luciferase activity was measured in equal amounts of cleared lysate from each sample and normalized to β-gal units from WT striatal cells without IL-1β.
To examine whether full-length mutant Htt also influences NF-κB activity, we used immortalized striatal cell lines obtained from WT and HD KI mice (Gines et al., 2003). Using the reporter with NF-κB enhancer, no significant difference in basal NF-κB activity is observed between striatal cells from WT and HD KI mice (Fig. 1_D_) (compare C lanes in WT and mutant). These data are consistent with a recent report showing that inducible expression of full-length WT or mutant Htt in PC12 cells has no effect on basal NF-κB activation (Sugars et al., 2004). We therefore asked whether WT and mutant cells respond differently to NF-κB-inducing agents. KI striatal cells display a stronger response than WT cells to the NF-κB-inducing cytokine IL-1β (Fig. 1_D_). To verify that mutant Htt mediates the enhanced response to IL-1β, we used recombinant single chain intrabodies, which are known to interfere with the function of mutant HDx1 (Khoshnan et al., 2002) (see below). Two intrabodies, MW2 targeting the expanded polyQ and MW7 recognizing the polyP motifs of HDx1, reduce the IL-1β-mediated NF-κB activation in striatal cells from KI mice (Fig. 1_D_). MW7 is somewhat more potent than MW2 at inhibiting the effects of full-length mutant Htt on NF-κB.
Activated NF-κB functions primarily in the nucleus, where it regulates gene expression. To confirm the authenticity of the gene reporter assays, we measured nuclear p65 binding to a consensus NF-κB oligonucleotide. Extracts from mutant Htt KI striatal cells show elevated nuclear p65 binding in response to IL-1β treatment when compared with the equivalent samples from WT striatal cells (Fig. 2_A_). Specificity of binding was confirmed using a mutated consensus NF-κB oligonucleotide, which failed to show p65 binding in this assay (data not shown). Thus, cells expressing full-length mutant Htt respond more vigorously than cells with WT Htt to IL-1β-induced NF-κB nuclear localization. We also examined whether there is elevated nuclear NF-κB in HD mouse brains. Brain sections from 8-week-old R6/2 HD mice and age-matched controls were stained with an antibody that recognizes the p65 subunit. p65 is concentrated in the nucleus of a majority of the NeuN-positive neurons in the cortex and striatum of HD mice, whereas most of the neurons of the age-matched WT mice contain cytoplasmic p65 (Fig. 2_B,C_). Thus, mutant Htt promotes nuclear localization of NF-κB in vivo as well as in cell culture.
Figure 2.
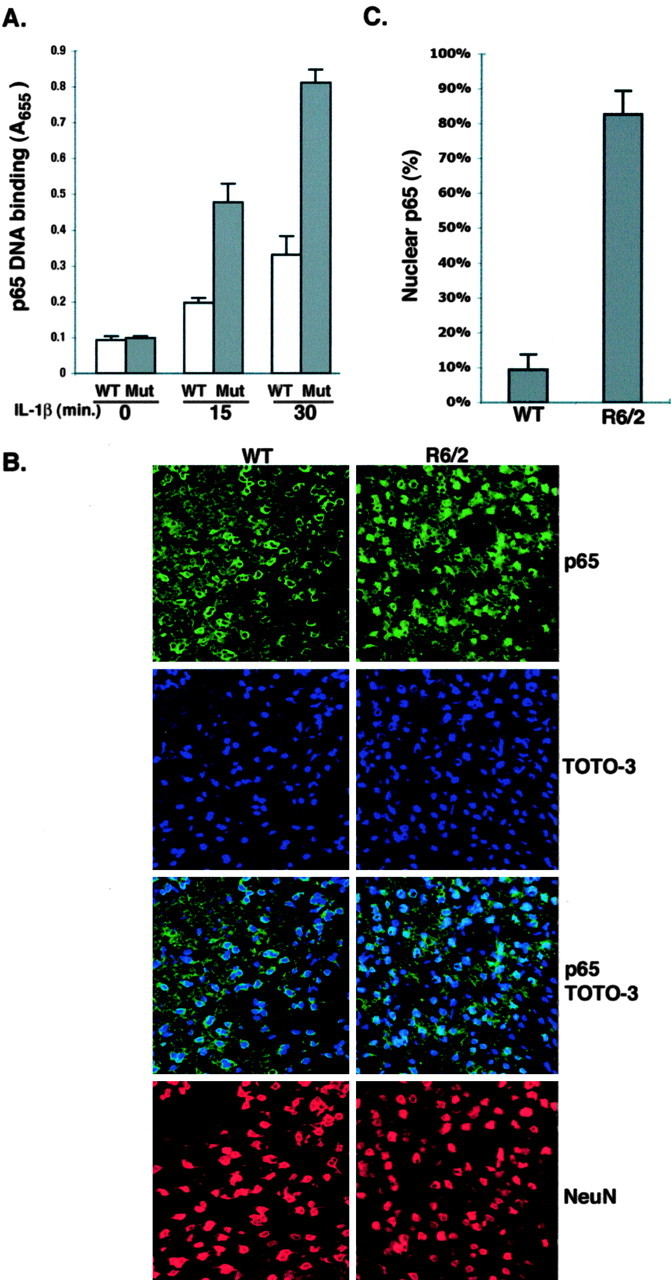
Mutant Htt promotes activation and nuclear localization of NF-κB. A, Nuclear proteins from control and IL-1β-treated striatal cells from mutant Htt KI mice were tested for binding to the oligonucleotide recognized by NF-κB. Similar results showing that nuclear extracts from KI striatal cells have elevated p65 were found in three experiments. B, The nuclear localization of the NF-κB p65 subunit is elevated in HD transgenic brain. WT and R6/2 HD transgenic mouse brains were stained with a rabbit anti-p65 antibody, and an anti-NeuN antibody was included to stain neurons. Toto-3 was used to stain nuclei. Representative confocal micrographs of cortical regions of WT and R6/2 HD mice are presented. C, Quantification of nuclear p65 staining shows the average percentage of positive neurons per microscopic field from 16 brain sections of four animals each for WT and HD mice.
Mutant Htt activates the IKK complex
Signal-induced phosphorylation of serine residues 32 and 36 of IκBα, which is mediated by the IKK complex, is essential for the ubiquitination and proteosome-mediated degradation of IκBα (Ghosh and Karin, 2002). Because IκBα degradation is promoted with mutant HDx1 induction, we examined whether the IKK complex is activated by mutant HDx1 expression. Immunoprecipitated endogenous IKK complexes from extracts of control or PC12 cells induced to express mutant HDx1 were analyzed for their kinase activity by measuring phosphorylation of the recombinant substrate, GST-IκBα. The kinase activity of IKK from cells expressing mutant HDx1 is significantly higher than that from non-HDx1-induced cells (Fig. 3_A_). As expected, NGF treatment also activates the IKK complex, and consistent with our gene reporter assay results, kinase activity appears to be elevated somewhat further when both NGF and HDx1 are present (4.1-fold compared with 2.4- and 3.2-fold in the ecdysone and NGF-treated cells, respectively). Expression of WT HDx1 has no effect on IKK activity (data not shown).
Figure 3.

Mutant HDx1 activates the IKK complex. A, IKK complexes isolated from PC12 cells expressing mutant HDx1 have elevated kinase activity. IKK complexes from control or cells expressing HDx1 were immunoprecipitated with anti-IKKα antibody coupled to protein-A beads. Kinase activity of the precipitated complexes was determined by incubation with GST-IκBα (the N-terminal 62 amino acids) and [32P]γATP. Phosphorylated GST-IκBα was detected by SDS-PAGE and autoradiography (top panels). KA, Kinase activity. The bottom panels show Western blots of duplicate samples for IKKα. WB, Western blot. Similar results were obtained when IKK complexes were isolated from striatal or cortical extracts of HD transgenic R6/2 mice (B) or striatal HD knock-in mice (C). Bottom panels in B and C show Western blotting of precipitated IKK complexes with an anti-IKKα antibody. D, Mutant HDx1 coprecipitates with endogenous IKK complex. IKK complexes were immunoprecipitated with an anti-IKKγ antibody from control or PC12 cells induced to express HDx1 with 25 or 103 polyQ. IKKα was detected by Western blotting (bottom blot). Similar complexes were examined for the presence of HDx1 (arrow) using an anti-Htt antibody (top blot). E, Mutant Htt coimmunoprecipitates with IKK complexes isolated from R6/2 striatal extracts. IKK complexes were obtained with anti-IKKα and analyzed for mutant HDx1 as in A.
To investigate whether IKK activation by mutant HDx1 also occurs in vivo, we used two lines of HD transgenic mice, R6/2 (Davies et al., 1997) and a mutant Htt KI (Lin et al., 2001). IKK complexes immunoprecipitated from striatal or cortical extracts of WT and HD mice were assayed in vitro for IKK activity. Consistent with the PC12 cell results, IKK activity is higher in complexes isolated from striatal (9-fold) as well as cortical (3.8-fold) extracts of 2-month-old R6/2 mice compared with age-matched WT controls (Fig. 3_B_). With KI HD mice, a significant increase in IKK activity is detected in the striatum at 1 year of age (4.8-fold higher than WT) (Fig. 3_C_).
Because IKK activity is elevated in cell culture and in animal models expressing mutant Htt, we tested whether HDx1 associates with IKK. Complexes isolated with an anti-IKK antibody from PC12 cells were examined for the presence of HDx1 by Western blotting. Soluble mutant HDx1 coprecipitates with IKK, whereas minimal WT HDx1 is observed (Fig. 3_D_). Thus, IKK physically forms a complex with mutant HDx1. This interaction also occurs in the brain of HD mice. IKK complexes isolated from brain extracts of R6/2 mice contain soluble HDx1, which is recognized by anti-Htt antibody (Fig. 3_E_). Therefore, activation of IKK by mutant HDx1 may be, in fact, the result of Htt binding to the complex.
Mutant Htt interacts with IKKγ
The activity of the IKK complex is regulated by IKKγ, a glutamine-rich nonkinase subunit of the complex (Rothwarf et al., 1998; Ghosh and Karin, 2002). A homolog of IKKγ that participates in tumor necrosis factor-α receptor signaling interacts with Htt in a yeast two-hybrid assay (Hattula and Paranen, 2000). Therefore, we asked whether the binding of mutant HDx1 to the IKK complex is mediated by IKKγ. A GST pull-down assay using HDx1 fused to GST and [35S]methionine-labeled IKKγ was used. The stringency of the assay was such that there was no binding to GST alone. In vitro translated-IKKγ preferentially binds to GST-HDx1 containing expanded polyQ (Fig. 4_A_). Although deletion of the C-terminal 119 amino acids of IKKγ (DC-IKKγ) has no apparent effect on this binding, removal of the first 134 amino acids (DN-IKKγ) diminishes binding to HDx1 (Fig. 4_A_). Interestingly, mutant HDx1 without its polyP domains does not bind efficiently to IKKγ (Fig. 4_B_). Although we cannot rule out that the absence of proline may alter the solubility of mutant HDx1 and thus artifactually reduce binding to IKKγ, the importance of the polyP domains is further supported by antibody experiments. We performed competition assays using _in vitro_-translated, anti-Htt recombinant intrabodies (Khoshnan et al., 2002). Coincubation of DC-IKKγ plus mutant HDx1 with 10-fold excess of intrabodies specific for polyQ (MW2) or polyP (MW7) competes with binding of IKKγ to HDx1 (Fig. 4_B_). Moreover, these intrabodies inhibit the HDx1-induced NF-κB-dependent gene expression in PC12 cells (Fig. 4_C_). Although anti-polyQ intrabody has a modest inhibitory effect, anti-polyP (MW7) significantly minimizes mutant HDx1-induced NF-κB activation. Thus, IKKγ binding to mutant HDx1 requires the polyQ expansion and is influenced by the polyP domains. Moreover, this interaction likely plays a role in mutant HDx-1-induced IKK and NF-κB activation.
Figure 4.
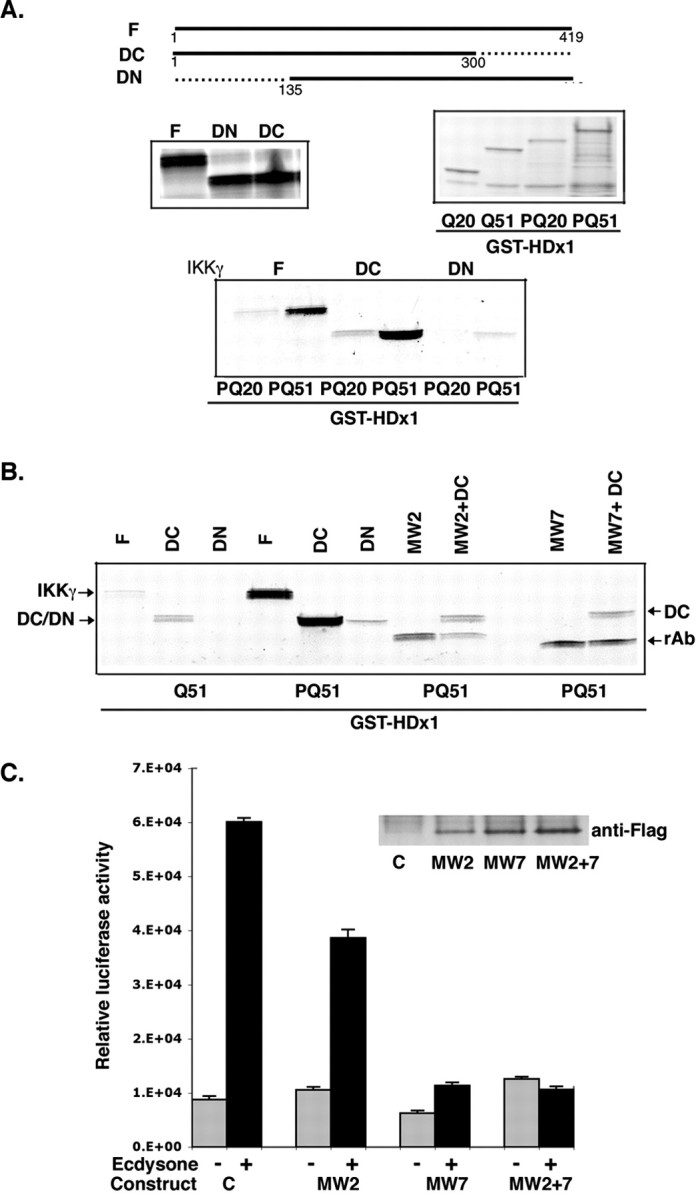
Mutant Htt directly interacts with IKKγ. A, Full-length (F) and C-terminal-truncated IKKγ (DC) bind to mutant HDx1 (51polyQ). IKKγ constructs were expressed by in vitro translation in the presence of [35S]methionine (middle left panel). HDx1 with 20 (PQ20) or 51 (PQ51) polyQ fused to GST was expressed in E. coli and captured on GST beads (middle right panel). Each _in vitro_-translated IKKγ product was incubated with each GST-HDx1. The beads were washed, and bound IKKγ was detected by SDS-PAGE followed by autoradiography (bottom panel). B, Binding of IKKγ to mutant HDx1 requires the Htt polyQ and polyP domains. Recombinant anti-Htt antibodies MW2 and MW7 were translated in vitro and tested for binding to GST-HDx1 (lanes MW2 and MW7). rAb, Recombinant antibody band. Addition of 10-fold excess of MW2 or MW7 to the reactions competes for binding of GST-HDx1 to IKKγ (compare intensity of lanes MW2+DC and MW7+DC with lane DC). C, Expression of anti-HDx1 recombinant antibodies reduces NF-κB-dependent gene expression. PC12 cells expressing mutant HDx1 were transfected using lipofectamine with NF-κB reporter and the indicated Flag-tagged, recombinant, anti-Htt intrabody and β-gal plasmids. Twenty-four hours after transfection, cells were treated with ecdysone. Samples were processed as in Figure 1 A. Transfections were normalized to β-gal units from control, noninduced HDx1. Results are shown as relative luciferase units and are representative of at least three independent experiments. Data points are the average of triplicate measurements. Expression of recombinant anti-Htt intrabody was confirmed by Western blotting (inset).
Inhibition of the NF-κB pathway reduces HDx1-induced toxicity in cell culture and brain slices
Because binding of mutant HDx1 to IKKγ influences IKK activity and NF-κB-mediated transcription, it was important to determine whether IKKγ can modify the toxicity of mutant HDx1. Confocal microscope examination of HEK-293 cells expressing mutant HDx1 and IKKγ at an early time point before widespread toxicity shows that full-length IKKγ promotes aggregation of mutant HDx1 (Fig. 5_A_). Intracellular, microscopically visible aggregates are apparent, and importantly, some of these aggregates localize in the nucleus of transfected cells. C-terminally truncated IKKγ, which also binds to mutant HDx1 (Fig. 4 A), has a similar effect (data not shown). On the other hand, DN-IKKγ, which does not bind to mutant HDx1, has no apparent effect on HDx1 aggregation (Fig. 5_A_). Thus, binding to IKKγ appears to promote aggregation and nuclear localization of mutant HDx1. Overexpression of full-length IKKγ appears to have little effect on toxicity at 48 hr after transfection, however, because the overall number of cells killed by mutant HDx1 does not appear to change (Fig. 5_A_). Toxicity was assessed by counting the number of condensed GFP+ bodies at 48 hr after transfection, which are remnants of dead cells (Khoshnan et al., 2002) (see Fig. 7_B_). Importantly, DN-IKKγ strongly reduces the toxicity of mutant HDx1 (Fig. 5_A_, graph). The effect of DN-IKKγ on mutant HDx1 toxicity was confirmed by TUNEL assay. Compared with cultures with or without full IKKγ, the number of TUNEL+ cells is significantly reduced in cells cotransfected with DN-IKKγ and mutant HDx1 (Fig. 5_B_). DN-IKKγ lacks the binding domain essential for interaction with IKKα and -β and interferes with IKK activity (May et al., 2000; Poyet et al., 2000). DN-IKKγ also blocks basal and NGF-induced NF-κB-dependent gene expression by mutant HDx1 in inducible PC12 cells (Fig. 5_C_). Therefore, inhibition of mutant HDx1 toxicity by DN-IKKγ may be mediated through blockage of the NF-κB pathway.
Figure 5.
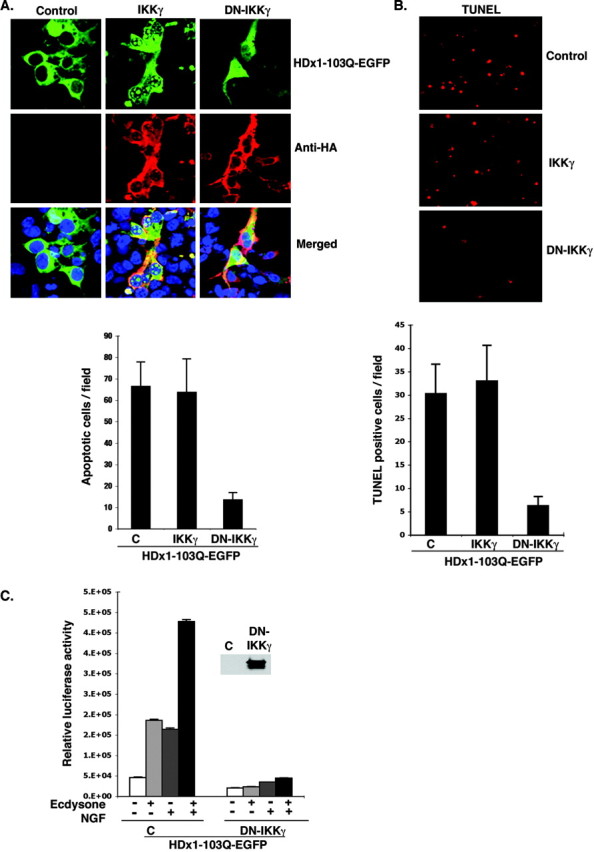
DN-IKKγ reduces the toxicity of mutant HDx1. A, IKKγ promotes aggregation and nuclear localization of Htt. HEK-293 cells were transfected with mutant HDx1-EGFP plus the indicated IKK constructs using lipofectamine and incubated for 16 hr. Cells were fixed and stained for IKKγ with anti-HA antibody (red), and cell nuclei were stained with Toto-3. Slides were examined with a confocal microscope. Conditions were such that mutant HDx1 did not cause aggregation on its own. The graph shows the average number of apoptotic bodies per microscopic field for each sample evaluated 2 d after transfection. Data are average counts of apoptotic cells from three experiments and six random microscope fields for each sample. B, DN-IKKγ expression reduces the toxicity of mutant HDx1. HEK-293 cells were transfected as in A for 16 hr. Apoptotic cells were identified by TUNEL assay as described in Materials and Methods. The top panel shows representative microscopic fields with TUNEL+ cells in each sample. The graph shows the average number of TUNEL+ cells in 16 microscopic fields from three experiments. C, DN-IKKγ blocks HDx1-induced NF-κB activation. Mutant HDx1 cells were cotransfected with NF-κB reporter and HA-tagged DN-IKKγ and β-gal plasmids and processed as described in Figure 1 A. Transfections were normalized to β-gal units from control, noninduced HDx1. Results are shown as relative luciferase units and are representative of at least three independent experiments. Data points are the average of triplicate measurements. Expression of DN-IKKγ was confirmed by Western blotting using an anti-HA antibody (inset).
Figure 7.
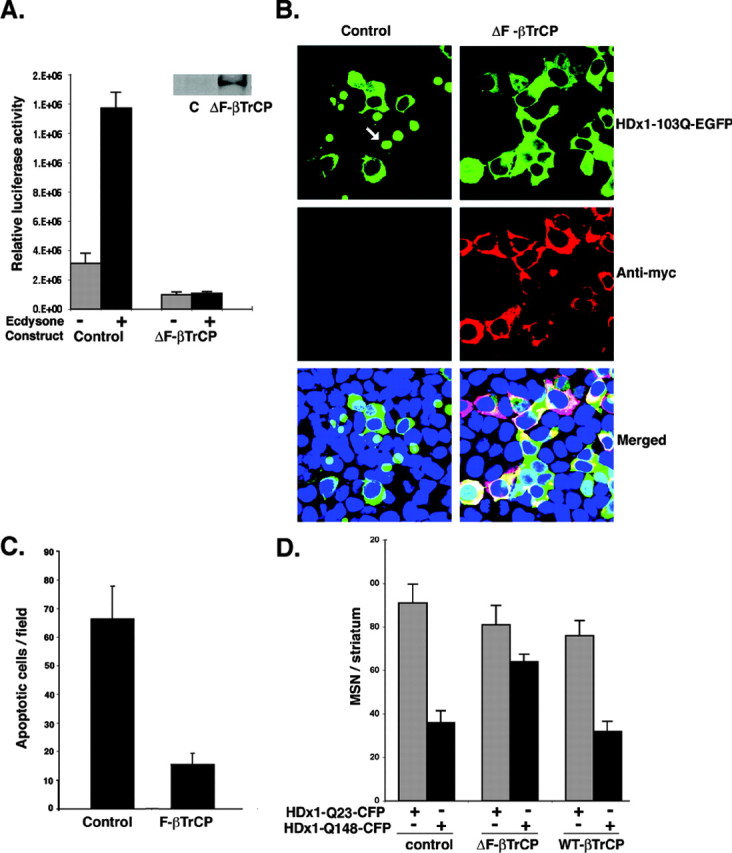
ΔF-βTrCP reduces the toxicity of mutant Htt. A, NF-κB activation by mutant HDx1 is blocked by ΔF-βTrCP. Mutant HDx1-expressing PC12 cells were cotransfected with NF-κB reporter and myc-tagged ΔF-βTrCP and processed for luciferase activity as described in Figure 1 A. Transfections were normalized to β-gal units from control, noninduced HDx1. Results are shown as relative luciferase units and are representative of at least three independent experiments. Data points are the average of triplicate measurements. Expression of ΔF-βTrCP was confirmed by Western blotting using anti-myc antibody (inset). B, Mutant HDx1 and ΔF-βTrCP are colocalized in living HEK-293 cells. Cells were transfected with mutant HDx1 plus control or ΔF-βTrCP constructs as described for Figure 5. Anti-myc antibody was used to detect ΔF-βTrCP. The different stains were merged with a DNA stain to visualize nuclei. C, Apoptotic bodies in transfected HEK-293 cells are reduced by ΔF-βTrCP. Sample processing and quantification were similar to that described for Figure 5_A_. D, Expression of ΔF-βTrCP reduces the toxicity of mutant HDx1 in MSNs. Brain slices were transfected and the data evaluated as described for Figure 6 B.
To extend these results to a more _in vivo_-like model, we used a novel, acute, P7 rat brain slice assay. A biolistic method is used to cotransfect neurons with three constructs: HDx1-CFP to visualize HDx1 expression and associated neurodegeneration, YFP to monitor the full morphology of transfected cells, and IKKγ or DN-IKKγ. Individual, transfected MSNs are identified by location and morphology and monitored daily for 7 d after transfection. Neuronal viability was quantified by counting the number of CFP+ neurons in each slice at 7 d. Expression of mutant HDx1 leads to the formation of Htt aggregates (shown in cyan) 1-2 d after transfection and to cell death within 4-7 d. YFP apoptotic bodies are seen initially, followed by complete loss of YFP fluorescence. In contrast, neurons transfected with WT HDx1 do not contain Htt inclusions and retain their normal dendritic structure (Fig. 6_A_). Coexpression of full-length IKKγ or DN-IKKγ has no obvious effect on MSNs expressing WT HDx1 (Fig. 6_A,B_). Furthermore, expression of IKKγ has no significant effect on neurodegeneration induced by expression of mutant HDx1 (Fig. 6_A,B_); however, the coexpression of DN-IKKγ with mutant HDx1 is significantly neuroprotective (Fig. 6_A,B_). The neuroprotective function of DN-IKKγ is essentially independent of aggregate formation by mutant Htt, because rescued neurons still contain Htt macro-aggregates (Fig. 6_A_). These brain slice results indicate that DN-IKKγ also reduces the neurotoxicity of mutant HDx1 in MSNs residing in a more intact, three-dimensional setting.
Figure 6.
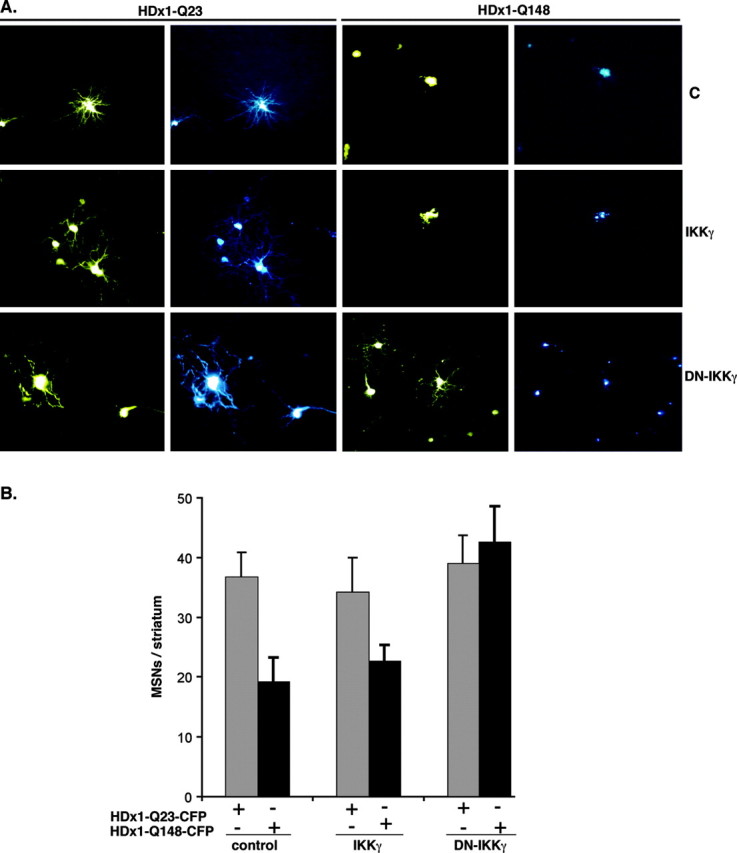
DN-IKKγ protects striatal MSNs against mutant HDx1. A, Living MSNs can be monitored for morphology and Htt expression. Coronal brain slices were cotransfected by biolistic procedures with HDx1-23Q or HDx1-148 and IKKγ or DN-IKKγ. The morphology of transfected cells was monitored by YFP fluorescence (yellow), and Htt expression was monitored by CFP fluorescence (cyan). B, Expression of DN-IKKγ is neuroprotective for MSNs in brain slices. MSN survival was scored 3 d after transfection. Each experiment involved 3-12 transfected brain slices per condition, and the data are means from six independent experiments for each condition.
Because ubiquitination of IκB is a crucial step in liberating NF-κB, we predicted that blocking this step should have an effect similar to that observed with DN-IKKγ. The E3-ubiquitin ligase βTrCP specifically promotes degradation of IκBα (Spencer et al., 1999). A dominant-negative form of βTrCP (ΔF-βTrCP), which has been shown to block degradation of phosphorylated IκBs, was cotransfected with the NF-κB reporter in PC12 cells. Expression of ΔF-βTrCP abolishes basal and mutant HDx1-induced NF-κB activity (Fig. 7_A_). As predicted, ΔF-βTrCP expression also blocks the toxicity of mutant HDx1 in cultured cells (Fig. 7_B,C_). Cells expressing ΔF-βTrCP and mutant HDx1 remain intact, whereas cells transfected with mutant HDx1 and control vector become condensed and eventually detach from the culture dish (Fig. 7_B_, arrow). Moreover, expression of ΔF-βTrCP in the brain slice assays significantly reduces the toxicity of mutant HDx1 in MSNs (Fig. 7_D_). WT-βTrCP does not influence the toxicity of mutant HDx1 in MSNs (Fig. 7_D_). Collectively, these data show that blocking IKK activity or degradation of IκB, both of which result in inhibition of NF-κB, can reduce the toxicity of mutant HDx1.
Discussion
Transcriptional dysregulation has been implicated in HD pathology, and mutant Htt can sequester transcription factors and reduce gene expression (Cattaneo et al., 2001; Ross, 2002; Yu et al., 2002) or increase the transcriptional activity of specific signaling pathways (Yohrling et al., 2003; Obrietan et al., 2004). We find that soluble mutant Htt interacts with the IKK complex, promotes phosphorylation and degradation of IκBα, and subsequently enhances NF-κB activity. Results from the HD cell culture model are supported by findings that IKK complexes from striatal and cortical extracts of HD transgenic mice also have elevated kinase activity, and activated NF-κB is concentrated in the nuclei of cortical and striatal neurons of HD transgenic mice. Most importantly, interference with the NF-κB pathway by expression of DN-IKKγ or ΔF-βTrCP reduces the toxicity of mutant Htt in cell culture and protects striatal MSNs in a brain slice model of HD. These findings implicate the NF-κB pathway in the neurotoxicity of mutant Htt.
Current paradigms of HD pathology include increased sensitivity to NMDA-mediated excitotoxicity and activation of caspases (Cepeda et al., 2001; Zeron et al., 2002). Although the signaling cascades that contribute to enhanced sensitivity of striatal neurons in HD are poorly understood, derangement of Ca2+ efflux from the endoplasmic reticulum by binding of mutant Htt to the inositol (1,4,5)-triphosphate receptor may be a contributing factor (Tang et al., 2003). In neurons and other cell types, elevated Ca2+ influx is a major activator of the IKK complex and subsequently NF-κB (Lilienbaum and Israel, 2003). We find that striatal cells from HD KI mice expressing full-length mutant Htt respond more vigorously to IL-1β-induced NF-κB activation (Fig. 1_C,D_). IL-1β can increase NMDA-mediated neurotoxicity by activation of tyrosine kinases and subsequent phosphorylation of NMDA receptor (Viviani et al., 2003). It is relevant that brain lysates of R6/2 HD mice have significantly higher levels of IL-1β than WT controls (Ona et al., 1999). In addition, inhibitors of IL-1β converting enzymes are effective against the toxicity of mutant Htt in tissue culture and animal models (Chen et al., 2000). Mutant HDx1 also enhances NGF-induced IKK and NF-κB activation (Figs. 1_A_, 3_A_). Therefore, a potential acquired function of mutant Htt is to intensify the effects of extracellular signals that function thorough the IKK complex.
A novel aspect of the present data is the mechanism by which mutant Htt stimulates NF-κB. Mutant Htt activates the IKK complex both in cell culture and in mouse models of HD (Fig. 3). Our evidence indicates that activation of the IKK complex is likely caused by binding of mutant HDx1 to IKKγ. Optimal HDx1 binding requires interaction with the first N-terminal 134 amino acids of IKKγ (Fig. 4), a domain that is essential for interaction with IKKβ and IKKα, two catalytic units of the IKK complex (May et al., 2000). It is plausible that mutant Htt binding to this region of IKKγ triggers oligomerization, a step that is essential for signal-induced IKK activation (Poyet et al., 2000; Tegethoff et al., 2003). It cannot be ruled out, however, that Htt-IKKγ binding mediates IKK interaction with other regulators responsible for phosphorylation and activation of the complex (Ghosh and Karin, 2002). Interaction of Htt with IKKγ requires polyQ expansion as well as the proline-rich motifs of HDx1. A similar scenario has been reported for the interaction of CBP and several WW-rich motif proteins implicated in HD pathology (Faber et al., 1998; Steffan et al., 2001). We find that recombinant intrabodies targeted to the polyP or polyQ motifs can inhibit Htt-IKKγ binding and NF-κB-mediated gene expression. The anti-polyP intrabody, which is more potent, also reduces mutant HDx1 aggregation and protects cells from the toxicity of mutant Htt (Khoshnan et al., 2002). Therefore, the toxicity of mutant HDx1 may be reduced by interference with its binding to the IKK complex.
Expression of IKKγ also promotes aggregation and nuclear localization of mutant HDx1 before the widespread onset of toxicity in cell culture. Considering the propensity of both mutant HDx1 and IKKγ to oligomerize, their interaction may lead to enhanced aggregation of both proteins. Although the role of macro-inclusions in HD pathology is controversial, nuclear localization is thought to be a determinant of pathology (Saudou et al., 1998). IKKγ, which enters the nucleus (Anest et al., 2003; Yamamoto et al., 2003), is associated with nuclear HDx1 aggregates (Fig. 5_A_). Thus, IKKγ-induced nuclear localization of mutant HDx1 may influence Htt function. Conversely, binding of mutant Htt to the IKK complex may regulate nuclear IKKγ function.
The inhibition of mutant HDx1 toxicity by DN-IKKγ in cell culture and in MSNs is likely attributable to deregulation of IKK and subsequent inhibition of NF-κB activity. We cannot rule out the possibility, however, that DN-IKKγ inhibits mutant HDx1 toxicity by an as yet unknown mechanism. Because both IKKγ and mutant Htt interact with CBP and regulate CBP-mediated gene expression (Nucifora et al., 2001; Verma et al., 2004), binding of IKKγ to mutant Htt could influence CBP sequestration and Htt toxicity; however, the similar effects of DN-IKKγ and ΔF-βTrCP in blocking cell death in cell culture and in MSNs support a role for NF-κB in mutant Htt toxicity. Because IKK activity and βTrCP are also essential for processing of the NF-κB precursors p100 and p105 to the p52 and p50 subunits, respectively, DN-IKKγ and ΔF-βTrCP may also interfere with the generation of active NF-κB complexes and reduce the available NF-κB pool (Xiao et al., 2001; Fong and Sun, 2002).
Mutant Htt-induced activation of IKK can influence other signaling pathways that intersect with the NF-κB pathway. For instance, Akt, a modulator of IKK activity and a substrate for NF-κB, regulates the toxicity of mutant Htt and ataxin-1 (Humbert et al., 2002; Meng et al., 2002; Chen et al., 2003; Emamian et al., 2003). In addition, the wingless and NF-κB pathways share the E3-βTrCP ubiquitin ligase, which is essential for degradation of both IκB and β-catenin (Winston et al., 1999). Thus, the inhibition of mutant HDx1-induced toxicity by ΔF-βTrCP may also be mediated by a reduction in β-catenin turnover. Mutant Htt can either stabilize or promote degradation of β-catenin (Carmichael et al., 2002; Gines et al., 2003). Mutant Htt also induces expression of cyclin D1, which is downstream of β-catenin (Gines et al., 2003), whereas signal-induced IKK phosphorylation of β-catenin promotes cyclin D1 expression (Albanese et al., 2003). Thus, the interaction between mutant Htt and the IKK complex can influence a number of signaling pathways and represents an interesting target for intervention in polyglutamine-induced toxicity.
Footnotes
Support for this project was provided by the Hereditary Disease Foundation and by a grant (NS045165-01A1) from the National Institute of Neurological Disorders and Stroke awarded to P.H.P and A. K. We thank E. Schweitzer for providing ecdysone inducible PC12 cells, M. McDonald and S. Gines for mouse striatal cells, W. Greene for the GST-IκBα construct, E. Zandi for IKKγ plasmids, R. Deshaies for ΔF-βTrCP, E. Wanker for GST-HDx1, D. Housman and A. Kazantsev for HDx1-103Q-EGFP, B. Kerr for dissection of brain tissue from HD mice, M. Henson, D. Dunn, and D. Lo for helpful discussions and work with the brain slice experiments, and D. McDowell for administrative assistance.
Correspondence should be addressed to Ali Khoshnan, Biology Division 216-76, California Institute of Technology, Pasadena, CA 91125. E-mail: khoshnan@caltech.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247999-10$15.00/0
References
- Albanese C, Wu K, D'Amico M, Jarrett C, Joyce D, Hughes J, Hulit J, Sakamaki T, Fu M, Ben-Ze'ev A, Bromberg JF, Lamberti C, Verma U, Gaynor RB, Byers SW, Pestell RG (2003) IKKalpha regulates mitogenic signaling through transcriptional induction of cyclin D1 via Tcf. Mol Biol Cell 14: 585-589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS (2003) A nucleosomal function for IκB kinase-α in NF-κB-dependent gene expression. Nature 423: 659-663. [DOI] [PubMed] [Google Scholar]
- Carmichael J, Sugars KL, Bao YP, Rubinsztein DC (2002) Glycogen synthase kinase-3β inhibitors prevent cellular polyglutamine toxicity caused by the Huntington's disease mutation. J Biol Chem 277: 33791-33798. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S (2001) Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci 24: 182-188. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Itri JN, Flores-Hernandez J, Hurst RS, Calvert CR, Levine MS (2001) Differential sensitivity of medium- and large-sized striatal neurons to NMDA but not kainate receptor activation in the rat. Eur J Neurosci 14: 1577-1589. [DOI] [PubMed] [Google Scholar]
- Chen HK, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, Aitken A, Skoulakis EM, Orr HT, Botas J, Zoghbi HY (2003) Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell 113: 457-468. [DOI] [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC (2002) Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J 21: 6539-6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM (2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6: 797-801. [DOI] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13: 2905-2927. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90: 537-548. [DOI] [PubMed] [Google Scholar]
- Edgerton JR, Reinhart PH (2003) Distinct contributions of small and large conductance Ca2+-activated K+ channels to rat Purkinje neuron function. J Physiol (Lond) 548: 53-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT (2003) Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron 38: 375-387. [DOI] [PubMed] [Google Scholar]
- Faber PW, Barnes GT, Srinidhi J, Chen J, Gusell JF, MacDonald ME (1998) Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet 7: 1463-1474. [DOI] [PubMed] [Google Scholar]
- Foehr ED, Lin X, O'Mahony A, Geleziunas R, Bradshaw RA, Greene WC (2000) NF-κB signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J Neurosci 20: 7556-7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong A, Sun SC (2002) Genetic evidence for the essential role of beta-transducin repeat containing protein in the inducible processing of NF-κB2/p100. J Biol Chem 21: 22111-22114. [DOI] [PubMed] [Google Scholar]
- Gines S, Ivanova E, Seong IS, Saura CA, MacDonald ME (2003) Enhanced Akt signaling is an early pro-survival response that reflects _N_-methyl-d-aspartate receptor activation in Huntington's disease knock-in striatal cells. J Biol Chem 278: 50514-50522. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M (2002) Missing pieces in the NF-κB puzzle. Cell [Suppl] 109: 81-96. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB (1998) NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 16: 225-260. [DOI] [PubMed] [Google Scholar]
- Hattula K, Peranen J (2000) FIP-2, a coiled-coil protein, links Huntingtin to Rab8 and modulates cellular morphogenesis. Curr Biol 10: 1603-1606. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F (2002) The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell 2: 831-837. [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971-983. [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A (2002) NF-κB at the crossroads of life and death. Nat Immunol 3: 221-227. [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D (1999) Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci USA 96: 11404-11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnan A, Ko J, Patterson PH (2002) Effects of intracellular expression of anti-huntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc Natl Acad USA 99: 1002-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Ou S, Patterson PH (2001) New anti-huntingtin monoclonal antibodies: implications for huntingtin conformation and its binding proteins. Brain Res Bull 56: 319-329. [DOI] [PubMed] [Google Scholar]
- Li XJ (1999) The early cellular pathology of Huntington's disease. Mol Neurobiol 20: 111-124. [DOI] [PubMed] [Google Scholar]
- Lilienbaum A, Israel A (2003) From calcium to NF-κB signaling pathways in neurons. Mol Cell Biol 23: 2680-2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ (2001) Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet 10: 137-144. [DOI] [PubMed] [Google Scholar]
- Lo DC, McAllister AK, Katz LC (1994) Neuronal transfection in brain slices using particle-mediated gene transfer. Neuron 13: 1263-1268. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Camandola S (2001) NF-κB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 107: 247-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May MJ, D'Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S (2000) Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the I-κB kinase complex. Science 289: 1550-1554. [DOI] [PubMed] [Google Scholar]
- Meng F, Liu L, Chin PC, D'Mello SR (2002) Akt is a downstream target of NF-κB. J Biol Chem 277: 29674-29680. [DOI] [PubMed] [Google Scholar]
- Nucifora FCJ, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA (2001) Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291: 2423-2428. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Hoyt KR (2004) CRE-mediated transcription is increased in Huntington's disease transgenic mice. J Neurosci 24: 791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Fried-lander RM (1999) Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature 399: 263-267. [DOI] [PubMed] [Google Scholar]
- Pahl HL (1999) Activators and target genes of Rel/NF-κB transcription factors. Oncogene 18: 6853-6866. [DOI] [PubMed] [Google Scholar]
- Passani LA, Bedford MT, Faber PW, McGinnis KM, Sharp AH, Gusella JF, Vonsattel JP, MacDonald ME (2000) Huntingtin's WW domain partners in Huntington's disease post-mortem brain fulfill genetic criteria for direct involvement in Huntington's disease pathogenesis. Hum Mol Genet 9: 2175-2182. [DOI] [PubMed] [Google Scholar]
- Poyet JL, Srinivasula SM, Lin JH, Fernandes-Alnemri T, Yamaoka S, Tsichlis PN, Alnemri ES (2000) Full text activation of the IκB kinases by RIP via IKKγ/NEMO-mediated oligomerization. J Biol Chem 275: 37966-37977. [DOI] [PubMed] [Google Scholar]
- Qin ZH, Chen RW, Wang Y, Nakai M, Chuang DM, Chase TN (1999) Nuclear factor κB nuclear translocation upregulates c-Myc and p53 expression during NMDA receptor-mediated apoptosis in rat striatum. J Neurosci 19: 4023-4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA (2002) Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron 35: 819-822. [DOI] [PubMed] [Google Scholar]
- Rothwarf DM, Zandi E, Natoli G, Karin M (1998) IKK-gamma is an essential regulatory subunit of the I-κB kinase complex. Nature 395: 297-300. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95: 55-66. [DOI] [PubMed] [Google Scholar]
- Spencer E, Jiang J, Chen ZJ (1999) Signal-induced ubiquitination of IkappaBalpha by the F-box protein Slimb/beta-TrCP. Genes Dev 13: 284-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM (2000) The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 97: 6763-6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in _Drosophila_Nature 413: 739-743. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Brown R, Cook LJ, Swartz J, Rubinsztein DC (2004) Decreased cAMP response element-mediated transcription: an early event in exon 1 and full-length cell models of Huntington's disease that contributes to polyglutamine pathogenesis. J Biol Chem 279: 4988-4999. [DOI] [PubMed] [Google Scholar]
- Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I (2003) Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 39: 227-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegethoff S, Behlke J, Scheidereit C (2003) Tetrameric oligomerization of I-κB kinase gamma (IKKγ) is obligatory for IKK complex activity and NF-κB activation. Mol Cell Biol 23: 2029-2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin AJ, Signer ER (2000) Huntington's disease: the challenge for cell biologists. Trends Cell Biol 10: 531-536. [DOI] [PubMed] [Google Scholar]
- Verma UN, Yamamoto Y, Prajapati S, Gaynor RB (2004) Nuclear role of Ikappa B kinase-gamma/NF-kappa B essential modulator (IKK gamma/NEMO) in NF-kappa B-dependent gene expression. J Biol Chem 279: 3509-3515. [DOI] [PubMed] [Google Scholar]
- Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M (2003) Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 23: 8692-8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanker EE (2000) Protein aggregation and pathogenesis of Huntington's disease mechanisms and correlations. Biol Chem 81: 937-942. [DOI] [PubMed] [Google Scholar]
- Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW (1999) The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev 13: 270-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G, Harhaj EW, Sun SC (2001) NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell 2: 401-409. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB (2003) Histone H3 phosphorylation by IKKα is critical for cytokine-induced gene expression. Nature 423: 655-659. [DOI] [PubMed] [Google Scholar]
- Yohrling GJ, Farrell LA, Hollenberg AN, Cha JH (2003) Mutant huntingtin increases nuclear corepressor function and enhances ligand-dependent nuclear hormone receptor activation. Mol Cell Neurosci 23: 28-38. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Nguyen HP, Li XJ (2002) Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum Mol Genet 11: 905-914. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA (2002) Increased sensitivity to _N_-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron 14: 849-860. [DOI] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S (2002) The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol Cell 3: 625-636. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT (2000) Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23: 217-247. [DOI] [PubMed] [Google Scholar]