Dopamine Neurons Mediate a Fast Excitatory Signal via Their Glutamatergic Synapses (original) (raw)
Abstract
Dopamine neurons are thought to convey a fast, incentive salience signal, faster than can be mediated by dopamine. A resolution of this paradox may be that midbrain dopamine neurons exert fast excitatory actions. Using transgenic mice with fluorescent dopamine neurons, in which the axonal projections of the neurons are visible, we made horizontal brain slices encompassing the mesoaccumbens dopamine projection. Focal extracellular stimulation of dopamine neurons in the ventral tegmental area evoked dopamine release and early monosynaptic and late polysynaptic excitatory responses in postsynaptic nucleus accumbens neurons. Local superfusion of the ventral tegmental area with glutamate, which should activate dopamine neurons selectively, produced an increase in excitatory synaptic events. Local superfusion of the ventral tegmental area with the D2 agonist quinpirole, which should increase the threshold for dopamine neuron activation, inhibited the early response. So dopamine neurons make glutamatergic synaptic connections to accumbens neurons. We propose that dopamine neuron glutamatergic transmission may be the initial component of the incentive salience signal.
Keywords: accumbens, addiction, dopamine, schizophrenia, slice, glutamate, ventral tegmental area, yellow fluorescent protein, incentive salience
Introduction
Mesoaccumbens dopamine (DA) neurons in the ventral tegmental area (VTA) project to medium-spiny neurons in the nucleus accumbens (nAcc), where they are thought to convey information associated with reward. Errors in the prediction of reward are encoded by a transient increase in DA neuron activity after unpredicted reward or by a decrease after failure of a predicted reward (Schultz, 1998), whereas reward uncertainty appears to be encoded by a crescendo of DA neuron activity between the conditioned stimulus and reward (Fiorillo et al., 2003). DA responses, which are typically slow (Benoit-Marand et al., 2001) and modulatory, do not mediate direct excitatory or inhibitory actions (Mogenson et al., 1988). More precise timing requires a fast ion-channel-mediated signal. One possibility is that DA neurons have glutamatergic actions. Studies in intact animals over many years have shown that stimulation of DA neurons in the substantia nigra (SN) excites striatal neurons. Although these responses have been attributed to the activation of passing fibers (Wilson, 1998), Wilson et al. (1982) reported that SN stimulation reliably elicits a delayed, apparently monosynaptic, excitatory response, which they argued originated from DA neurons; however, this could not be tested directly in the intact brain.
Several other lines of evidence suggest that DA neurons may be glutamatergic. Kaneko et al. (1990) showed by immunostaining that phosphate-activated glutaminase (PAG) is present in most DA neurons; PAG is the enzyme principally responsible for the production of neurotransmitter glutamate (Kaneko, 2000). Plaitakis and Shashidharan (2000) showed that DA neurons express the glutamate transporter excitatory amino acid transporter 3 (EAAT3). Most recently, the third vesicular glutamate transporter isoform (vGLUT3) was found in DA neurons, as well as other monoaminergic neurons (Fremeau et al., 2002; Gras et al., 2002). Therefore, DA neurons have many of the hallmarks of glutamatergic neurons. Arguably, the strongest evidence that DA neurons are glutamatergic is that when they are grown in culture with their expected target neurons, they made fast, excitatory synaptic connections, both in micro-coculture (Joyce and Rayport, 2000) and in triple-explant slice culture (Plenz and Kitai, 1996). Here, we have used transgenic mice with fluorescent DA neurons (Zhuang et al., 2004) to make a brain slice preparation encompassing the mesoaccumbens DA projection and asked whether DA neurons make glutamatergic connections in the intact circuitry of the brain.
Materials and Methods
DA transporter positive neuron-specific enhanced–yellow fluorescent protein reporter mice. Animal procedures followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were performed under protocols approved by the Institutional Animal Care and Use Committees of Columbia University and New York State Psychiatric Institute. DA transporter positive neuron-specific enhanced–yellow fluorescent protein expression (DAT-YFP) mice were obtained by breeding DAT-Cre recombinase +/– mice with ROSA26-YFP +/– mice (Zhuang et al., 2004). Pups were genotyped by PCR at approximately postnatal day 7 (P7), and double heterozygote (DAT-YFP) mice were used for experiments. The primer sequences for DAT-Cre genotyping were5′GTTGATGAGGGTGGAGTTGGTC,5′GCCGCATAACCAGTGAAACAGC, and 5′TCCATAGCCAATCTCTCCAGTC; for ROSA26-YFP genotyping they were 5′AAAGTCGCTCTGAGTTGTTAT, 5′GCGAAGAGTTTGTCCTCAACC, and 5′GGAGCGGGAGAAATGGATATG. Some DAT-YFP mice were obtained by breeding DAT-Cre homozygous males with ROSA26-YFP homozygous females. Because there was no appreciable difference between the results obtained from the mice from these two breeding strategies, the results were combined.
Slice preparation. P10–P20 DAT-YFP mice were anesthetized with ketamine, then decapitated. The brain was removed into cold, high-glucose artificial CSF (high-glucose ACSF) containing the following (in mm): 75 NaCl, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 0.7 CaCl2, 2 MgCl2, and 100 glucose, pH 7.4, and saturated with 95% O2 and 5% CO2. We made 500 μm horizontal slices using an Advanced Vibroslice (model NVSLM1; World Precision Instruments, Sarasota, FL). The horizontal slice encompassing the mesoaccumbens projection was separated at the midline to obtain two usable hemislices. Optimal slices included mainly the shell region of the nAcc. After 1 hr of preincubation at room temperature, slices were placed in the recording chamber (∼1 ml capacity) on the stage of an upright fluorescence microscope (model BX-61WI; Olympus, Tokyo, Japan) and held down with nylon mesh. The recording chamber was continuously perfused with ACSF containing the following (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 25 glucose, pH 7.4, saturated with 95% O2 and 5% CO2 at ∼1 ml/min. YFP was visualized with a 100 W mercury lamp with a bandpass filters designed for visualizing YFP (filter set XF104-2; Omega Optical, Brattleboro, VT). Images were captured with a Photometrics Sensys camera (Roper Scientific, Tucson, AZ) and IP-Lab software (Scanalytics, Fairfax, VA). Figures were assembled in Adobe Photoshop 7 and Illustrator 10 (Adobe Systems, San Jose, CA). All experiments were performed at room temperature (20–22°C).
Electrochemical recordings. Disk carbon fiber electrodes of 5 μm in diameter with a freshly cut surface were placed in the nAcc slice to a depth of ∼100 μm. For cyclic voltammetry (CV), a triangular voltage wave (–400 to +1000 mV at 300 V/sec vs Ag/AgCl) was applied to the electrode every 100 msec using a waveform generator (Model 39; Wavetek, Norwich, Norfolk, UK). Current was recorded with an Axopatch 200B amplifier (Axon Instruments, Union City, CA), with a low-pass Bessel filter set at 10 kHz, digitized at 25 kHz (Instrunet Board; GW Instruments, Somerville, MA), and acquired with the Superscope II program (GW Instruments). The background-subtracted CV served to calibrate the electrodes and to identify the released substance. For amperometry, a constant voltage of +400 mV was applied via the Axopatch 200B. Amperometric traces were filtered with a digital Hamming filter (125 Hz cutoff frequency). Slices were stimulated with a bipolar electrode with 300 μm tip separation that could be moved from the nAcc, along the medial forebrain bundle (MFB), to the VTA. Two trains (three pulses, 100 Hz) of pulses (400 μA, 1 msec) were delivered every 5 sec with an Iso-Flex stimulus isolator triggered by a Master-8 pulse generator (A.M.P.I., Jerusalem, Israel). Experiments were performed in ACSF that included 10 μm gabazine (SR-95531; Sigma-RBI, St. Louis, MO). In some experiments, 10 μm nomifensine (Sigma-RBI) was included in bath solution to enhance DA release. Stimulus artifacts were reduced by subtraction of traces clamped at 0 mV. The DA clearance rate was calculated using the Michaelis–Menten equation. The clearance rate was evaluated as the first-order rate constant k (_V_max/_K_m) (Budygin et al., 2002).
Whole-cell patch-clamp recording. Medium-spiny neurons in nAcc were identified using visible light differential interference contrast image enhanced with a CCD camera (model OLY-150; Olympus). GABAA responses were blocked with 10 μm gabazine in the external solution. Recording pipettes were fabricated from standard wall borosilicate glass capillary with filament (Warner Instruments, Hamden, CT). Pipette resistances were 5–9 MΩ. The internal solution contained the following (in mm): 140 K-gluconate, 2 MgCl2, 0.1 CaCl2, 10 HEPES, 1 EGTA, 2 ATP-Na2, and 0.1 GTP-Na2, pH 7.3. To block unclamped Na+ currents, 5 mm lidocaine _N_-ethyl bromide (QX-314; Sigma-RBI) was added to the internal solution. The liquid junction potential was ∼15 mV and was corrected offline. Cells were voltage-clamped at –85 mV, unless otherwise noted, using an Axopatch 200 amplifier (Axon Instruments). Series resistances were 20–45 MΩ and not compensated online to avoid adding excessive baseline noise. However, because recorded responses were small and not very fast, both online and offline compensation (performed in Axograph 4.6, Axon Instruments) gave identical results. Synaptic responses were evoked with bipolar tungsten electrodes placed over the VTA, made by putting together two tungsten single electrodes (0.5 MΩ, World Precision Instruments) with a tip separation of 500 μm. A train of three pulses of 500 μsec duration at 1 kHz (quasi-single pulse, 1–2.5 mA) or a single 1.2 msec (6–7 mA) pulse was applied at 0.2 Hz, unless otherwise noted. Occasionally, this stimulation generated antidromic action currents, so QX-314 was added to the internal solution of the recording pipette. Responses were filtered at 5 kHz by a four-pole Bessel filter. Cells with <10 pA synaptic response (mean amplitude) or with high failure rates were discarded. To confirm that the responses were glutamatergic, 40 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; Tocris Cookson, Ballwin, MO) or 10 μm _R_(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH-23390; Sigma-RBI) were added to the bath solution. We evaluated the effects of drugs after >10 min of application. Typically, CNQX required 2–3 min to the onset of blockade.
Local drug application. A 10 mm stock solution of (–)-quinpirole (Sigma-RBI) was made in water and a 5 mm stock solution of (–)-sulpiride (Sigma-RBI) was made in ACSF with 10 mm Advasep7 (CyDex, Overland Park, KS). These stock solutions were diluted in ACSF containing 10 μm gabazine to make a 10 or 50 μm quinpirole or a 50 μm sulpiride working solution, respectively. Glutamate (1–5 mm) was dissolved in ACSF containing 10 μm gabazine. Fast Green (Sigma) was used to verify localized application of drug. The same patch pipettes used for recording were used for local drug application, but filled with 0.22 μm-filtered working solution. Drugs were applied by continuous positive pressure of ∼0.4–0.8 kg/cm2 generated by a pneumatic pressure system (Miniframe PPS-2; Medical Systems, Greenvale, NY). Applied pressure was adjusted for each pipette before use to ensure a constant application of drug. Total applied drug solution was 20–100 μl (quinpirole, sulpiride) or 0.6–1.5 μl (glutamate), and perfusion with normal ACSF (1 ml/min) was maintained throughout experiments to wash away drug as it diffused from the site of application.
Data analysis. All data were acquired using Pulse Control 4.7 (Richard J. Bookman, University of Miami, Miami, FL, http://chroma.med.miami.edu/cap/; InstruTech, Port Washington, NY) running under Igor Pro 4.04 (WaveMetrics, Lake Oswego, OR) on a PowerMac G3 (Apple Computers, Cupertino, CA). The sampling interval was 100 μsec. Recorded data were analyzed offline in Igor Pro and Axograph 4.6. Response delay was defined as the time from the beginning of stimulus artifact to the onset of the response (5% of the peak amplitude); when responses had more than one peak, the onset of the first peak was regarded as the onset of response. The rise time of the synaptic response was defined as the time from 20 to 80% of peak amplitude. To evaluate the effects of local glutamate application, amplitude histograms of synaptic events were made under control conditions and during drug application using a bin size of 2 pA. Subtraction of the control from the drug application histogram gave a histogram of putative evoked events. The size of individual synaptic events was determined by integration from 10 msec before the onset of the events to 50 msec after the onset. The significance of differences was determined by t test, unless otherwise noted, with p < 0.05 regarded as statistically significant. Comparison of the effects of stimulus frequency and glutamate application were done by one-way repeated-measures ANOVA. Data are reported as means ± SEM (unless otherwise noted).
Results
Visualization of the mesoaccumbens projection in the slice
We used transgenic DAT-YFP mice (Fig. 1_A_) (Zhuang et al., 2004). These mice were obtained from crosses of DAT-Cre mice, in which the gene for the Cre recombinase was knocked in to the DAT locus, with ROSA26-YFP reporter mice (Srinivas et al., 2001). We made thick horizontal slices (500 μm) from young mice with the hope of encompassing the relatively coplanar mesoaccumbens projection in a single slice. In such slices made from DAT-YFP mice, YFP fluorescence was identified in the DA neuron cell groups and their projections (Fig. 1_B,C_). DA neuron axons could be seen exiting the VTA, traveling in a rostrolateral direction in the MFB, and then diverging as they reached the nAcc; other axons projected to cortex (Fig. 1_B_). In the nAcc and cortex, the axons gave rise to a cloud of fluorescent terminals.
Figure 1.
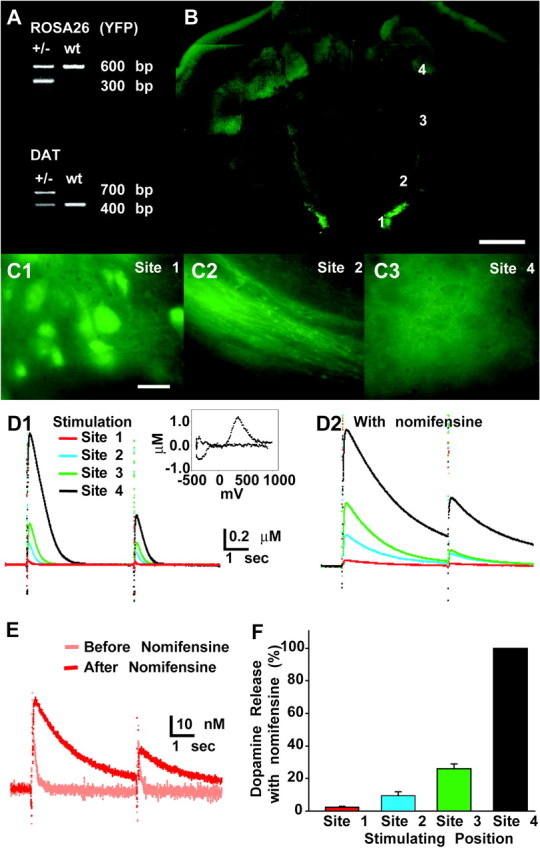
DA release in brain slices from DAT-YFP mice. A, PCR genotyping of DAT-YFP mice. The presence of ROSA26-YFP cassette gave a 300 bp band, whereas the wild-type (wt) band was 600 bp; the presence of a DAT-Cre cassette gave a 700 bp band, whereas the wt band was 400 bp. B, Fluorescence image of a complete 500 μm horizontal slice encompassing the mesoaccumbens projection at P20 (all experiments were done on hemislices, split at the midline). Scale bar, 1 mm. C, Expanded image from another slice (P19) of DA neuron cell bodies in the VTA (C1), DA axons in MFB, (C2) and DA neuron terminal distribution in the nAcc (C3). The approximate location of each expanded image in the slice is indicated by the numbers in B. Scale bar, 20μm. D, E, Amperometric recordings of evoked DA release in nAcc (all recordings made at site 4 in B). Stimulation was applied at the four sites, extending from the VTA to the nAcc, as indicated in B. All traces are from the same slice. D1, DA release without nomifensine. A cyclic voltammogram recorded at site 4 is shown in the inset. D2, DA release in the presence of nomifensine (10μm; 10 min). Calibration same as in D1. E, Expanded amperometric traces of VTA-evoked DA release before and after nomifensine. F, Summary of normalized evoked DA release in the nAcc versus the site of stimulation with nomifensine (5 hemislices from 3 mice).
Electrochemical measures of dopamine release in the mesoaccumbens projection
DAT-YFP mice are heterozygous for Cre in the DAT locus and so have reduced DAT function. At best, the mutant allele produces ∼20% wild-type DAT expression, so DAT-YFP mice should have ∼60% wild-type DAT levels (Zhuang et al., 2004). Although DAT knock-out mice show profound alterations in their DA system and behavior, alterations are much subtler in full heterozygotes (Giros et al., 1996). To see whether the DA release was affected by the reduced DAT expression in DAT-YFP mice, we recorded DA release in the nAcc using electrochemical techniques. We started with local stimulation in the nAcc (three pulses at 100 Hz) and recorded DA release by CV; the background-subtracted voltammogram showed that the released substance was mainly DA (Fig. 1_D_, inset). We then switched to amperometry to increase temporal resolution and detection sensitivity. Local stimulation of the nAcc (site 4, see below) generated 1.2 ± 0.2 μm DA release (n = 5 slices), with a calculated DA clearance rate of 5.1 ± 0.2 sec–1 (n = 5 slices). These values were close to the release and clearance rate reported in the rat nAcc of 1.37 μm and 10.5 per second (Jones et al., 1995; Phillips et al., 2002), as well as in the wild-type mouse nAcc shell of 0.5 μm and 4.0 sec–1, respectively (Budygin et al., 2002), showing that DA dynamics were nearly normal in DAT-YFP mice.
To assess the integrity of the mesoaccumbens projection in the slice, we stimulated VTA DA neuron axons at four sites, with site 1 the VTA and site 4 the nAcc. As we moved the stimulating electrode caudally from the nAcc along the fluorescent DA neuron axons in the MFB to the VTA (stimulation sites are numbered in Fig. 1_B_), DA release decreased from ∼1 μm atsite4to30nm at site 1, which was 2.5 ± 0.3% of the release in the nAcc (at site 4); sites 3 and 2 were intermediate at 26 ± 3 and 9.5 ± 2.5%, respectively (Fig. 1_F_). Bath application of the DAT inhibitor nomifensine (10 μm, 10 min) dramatically increased the signal (Fig. 1_D,E_), showing further that the recorded signal was mainly DA, and that substantial DAT function remained. These observations indicate that only part of the mesoaccumbens projection was intact; so our additional results will likely provide only a minimum estimate of the magnitude of the DA neuron signal in the intact animal.
Synaptic responses in nAcc medium-spiny neurons
Medium-spiny neurons were easily distinguished from neighboring cholinergic interneurons by size (Kawaguchi, 1993), and their identity was confirmed at the initiation of whole-cell recording by their characteristic very negative resting potential (–84.3 ± 2.2 mV; n = 28 cells). In total, we recorded from 228 slices and recorded VTA-evoked EPSCs in 76 (33%) of the slices. In slices with intact connections, EPSCs were observed in ∼12% of recorded cells. We used the GABAA antagonist gabazine (10 μm) in the bath solution and the open Na+-channel blocker lidocaine _N_-ethyl bromide (QX-314) in the intracellular solution, to block GABAergic inputs and antidromic activation of medium-spiny neurons, respectively. VTA stimulation generated an early and a late response (Fig. 2_A_). The early response had a constant latency of 19.9 ± 0.8 msec (n = 24 cells) (Fig. 2_A_, left histogram) and amplitude 25.1 ± 1.9 pA (n = 24 cells).
Figure 2.
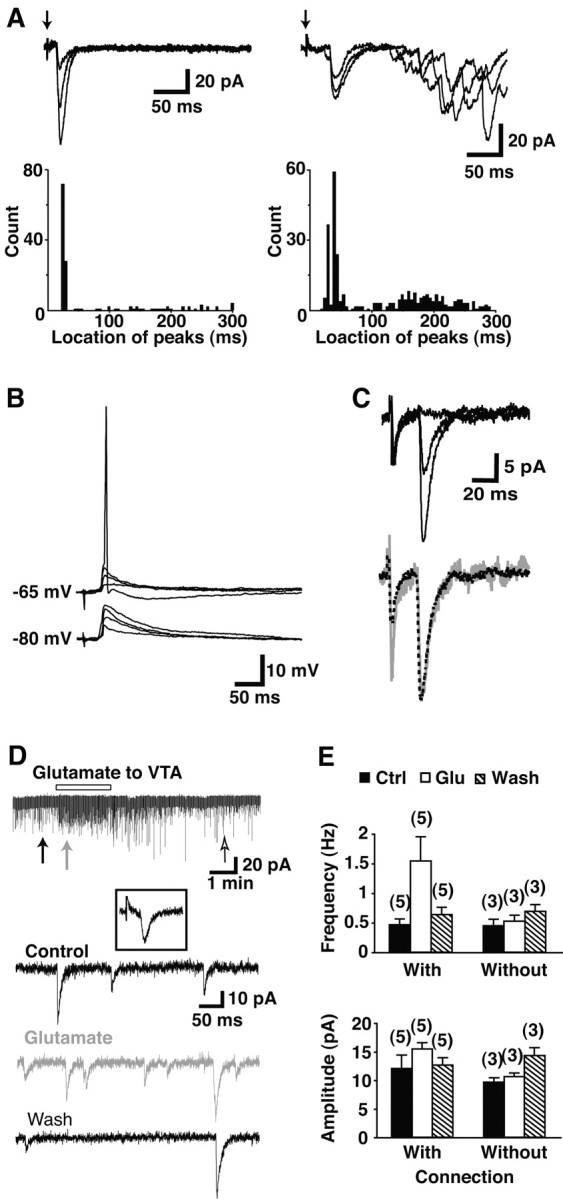
Synaptic responses evoked in nAcc medium-spiny neurons by VTA stimulation. A–C, Electrical stimulation of the VTA. A, In each of two hemislices, local field stimulation was delivered to the VTA with a bipolar electrode (arrow indicates stimulus artifact), whereas recording from an nAcc neuron clamped to –85 mV; three superimposed traces are shown. In one hemislice, just an early response was seen (left), whereas in another both an early and a late response were seen (right). A peak location histogram for each cell is shown below the traces (n = 100 traces, left; n = 120 traces, right). B, Under current clamp, VTA stimulation evoked EPSPs capable of firing the nAcc neuron when it was in the up state, here mimicked by a 20 mV depolarization (4 traces are superimposed). C, The early EPSC did not change shape with increasing stimulus intensity. nAcc responses were evoked by 0.7, 0.9, and 1 mA stimulation (top traces, each the average of 10 traces). When the 0.9 mA-induced response (gray line) was scaled to the same peak amplitude as the 1 mA-induced trace (black dotted line), the traces exactly superimposed. D, E, Chemical stimulation of the VTA. VTA neurons were activated by the local application of glutamate (1 mm), pressure-ejected from a patch pipette. D, Time course and representative traces before (control), during local application of glutamate, and after (wash). Downward spikes are synaptic events. Arrows indicate sampling points for the representative traces. The inset shows the early response in the same cell induced by the electrical stimulation of the VTA, confirming that there was an intact VTA–nAcc synaptic connection. The calibration is the same as for the representative traces. E, Comparison of the effects of the local application of glutamate on the frequency (top) and average amplitude of synaptic events (bottom) between cells with intact VTA-nAcc synaptic connections (left) and those without (right). The numbers of the recorded cells are indicated in parentheses.
In vivo, nAcc medium-spiny neurons alternate between a down state of approximately –80 mV and an up state of approximately –60 mV (Wilson and Groves, 1981; Wilson and Kawaguchi, 1996). Under current clamp and without QX-314, when we depolarized cells to mimic the up state, the early response was often strong enough to drive cells to fire (Fig. 2_B_). The early response showed a progressive increase with increasing stimulation intensity, and in some cases, it had two peaks (Fig. 2_A_, right); both peaks had almost the same activation threshold (see below). When the stimulus intensity was increased further, the early response plateaued at a stimulus intensity of 2.4 ± 0.4 mA (n = 5 cells) (Fig. 2_C_). Increasing stimulus intensity from approximately half maximum (47.6 ± 5.4%; 1.3 ± 0.2 mA) to maximum had no effect on the location of the peak of the early response (28.4 ± 0.8 msec for half maximum vs 27.8 ± 0.6 msec for maximum; p = 0.24), or in peak onset (21.1 ± 2.2 msec for half maximum vs 20.9 ± 2.1 msec for maximum; p = 0.15), rise time (1.9 ± 0.3 msec for half maximum vs 2.0 ± 0.4 msec for maximum; p = 0.65), or decay time constant (12.1 ± 1.4 msec for half maximum vs 11.7 ± 0.6 msec for maximum; p = 0.78) (Fig. 2_C_, bottom). This argues that the early response arises from a relatively homogeneous population of cells.
The early response had a reversal potential of 2.1 ± 2.5 mV (n = 4 cells) (Fig. 3_A,B_), and was completely blocked by 40 μm CNQX (n = 5 cells) (Fig. 3_C_). There have been reports of excitatory effects of D1 receptor activation (Lavin and Grace, 2001) and of D1-mediated excitatory responses elicited by MFB stimulation (Gonon, 1997). However, the early response was not affected by application of the D1 antagonist SCH-23390 (10 μm); the peak amplitude of the early response in the presence of SCH-23390 was 101.0 ± 9.1% of control (n = 4 cells). These results argue strongly that the early response is mediated by glutamatergic synaptic transmission.
Figure 3.
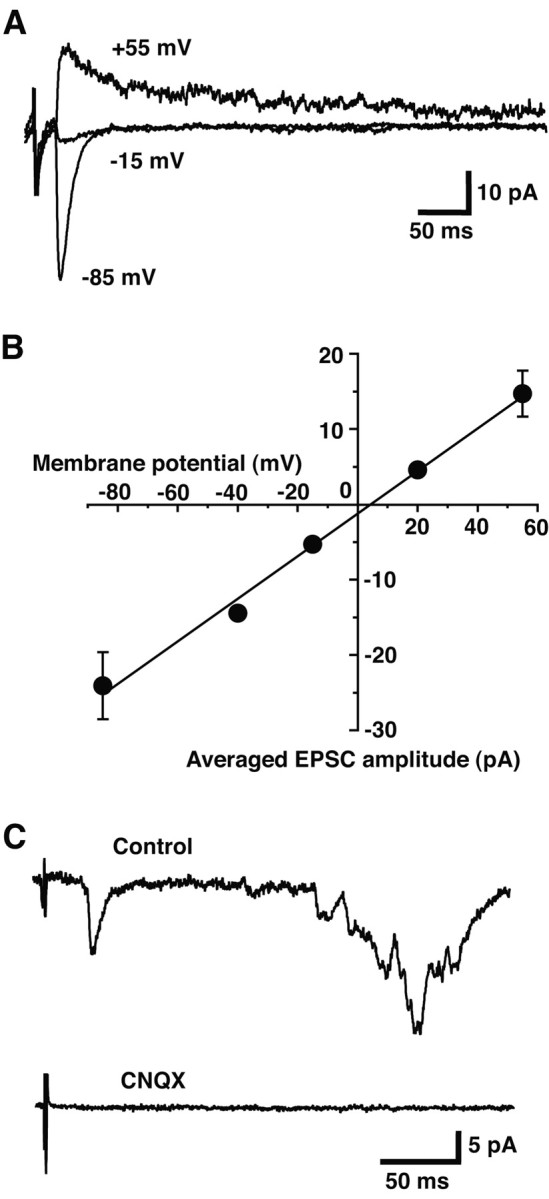
Recorded synaptic responses are glutamatergic. A, EPSCs were evoked by VTA stimulation while holding the postsynaptic nAcc neuron at –85, –15, and +55 mV (average of 10 traces is shown). B, Current–voltage plot shows a reversal potential of 2.1 mV, characteristic of a glutamatergic response (line is a least-squares linear regression fit; r = 0.99). Points at –85, –15, and +55 mV were from four cells; additional points at –40 and +20 mV were added from one cell. C, Both early and late responses were completely blocked by CNQX (40μm; 5 min; average of 10 traces).
To examine unitary synaptic events, we activated DA neurons by the local application of glutamate (1–5 mm) to the VTA. Glutamate should act via AMPA as well as NMDA receptors (once cells are depolarized), because both receptors mediate synaptic inputs onto DA neurons (Mereu et al., 1991). We used electrical stimulation to assess whether there was an intact mesoaccumbens connection (compare with Fig. 2_D_). Glutamate infusion increased the frequency of fast synaptic events (Fig. 2_D_) only in nAcc neurons with an early response (inset). On average, glutamate infusion increased the frequency of synaptic events from control, 0.47 ± 0.16 to 1.54 ± 0.41 Hz; after recovery, the frequency dropped to 0.63 ± 0.12 Hz (n = 5 cells; p < 0.05; ANOVA) (Fig. 2_E_, top). There was no significant change in the average amplitude of events (n = 5 cells; p = 0.49; ANOVA) (Fig. 2_E_, bottom). These unitary synaptic evens likely arise from asynchronous firing of DA neurons activated by the local glutamate application. Because slices lacking responses to electrical stimulation never showed response to chemical stimulation, our results were not attributable to the action of glutamate that diffused away from the site of application in the VTA (Fig. 4_E_) (p = 0.30 for frequency; p = 0.15 for amplitude; ANOVA). It is possible that DA neurons excite cholinergic neurons; however, cholinergic neurons are unlikely to mediate fast excitatory events because medium-spiny neurons do not express nicotinic acetylcholine receptors (Zhou et al., 2002).
Figure 4.
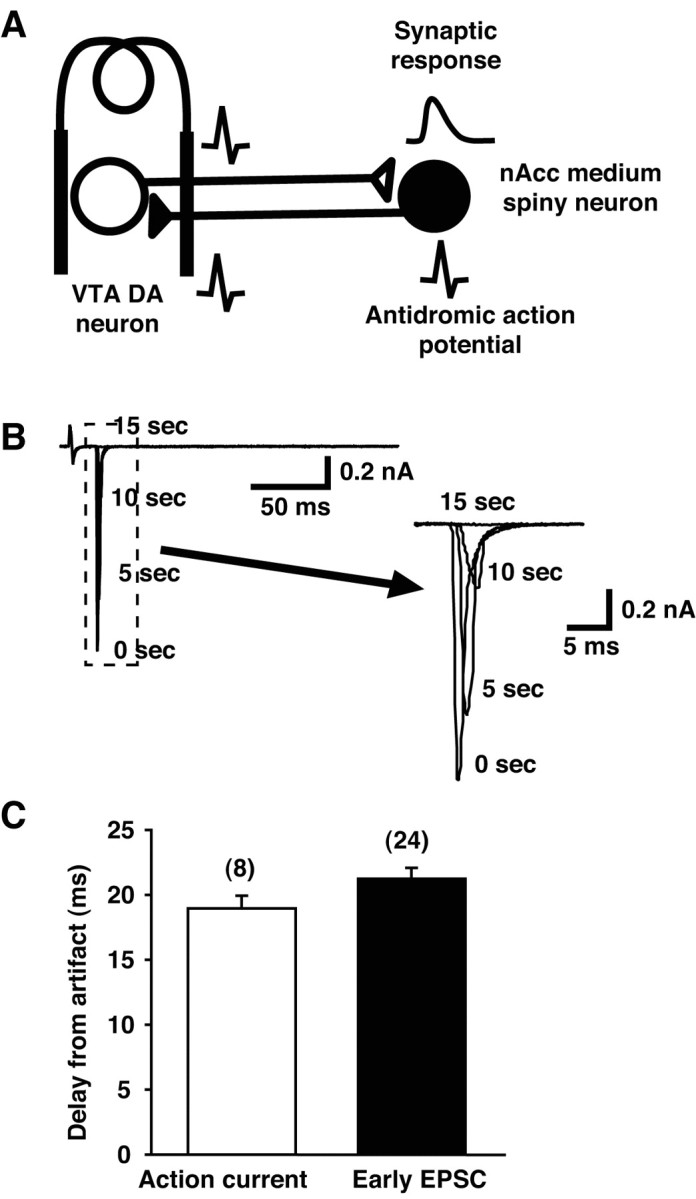
Latency of early response is short. A, DA neurons in the VTA and nAcc medium-spiny neurons make reciprocal synaptic connections, so stimulation of the VTA generates orthodromic action potentials in DA neuron axons and antidromic action potentials in nAcc medium-spiny neuron axons. B, A sample of antidromic action current recorded in nAcc medium-spiny neuron. The action current was rapidly and progressively blocked by the QX-314 in the recording pipette. Times after starting stimulation (0, 5, 10, and 15 sec) are indicated; an expanded view is shown to the right. C, The delay to the antidromic action currents and the early EPSC were nearly identical (numbers of recorded cells are in parentheses), indicating that the early response is monosynaptic.
These glutamate-evoked events were in the range of 8–12 pA and had an average amplitude of 10.3 ± 0.1 pA. Integrating the events gave a charge carried per event of 86.7 ± 5.8 fC (n = 5 cells). In contrast the charge associated with the electrically evoked early response was 331.2 ± 37.8 fC (n = 24 cells). This suggests that ∼3.8 synaptic connections contributed to the early response.
The late response was also completely blocked by CNQX (Fig. 3_C_). However, in contrast with the early response, the late response had multiple peaks and fluctuated considerably in latency (Fig. 2_A_, right). The mean latency was 169.8 ± 19.5 msec (n = 10 cells) and amplitude 35.8 ± 2.8 pA (n = 10 cells) (Fig. 2_B_, right). Typically, the late response disappeared after 6–7 min of stimulation. The long latency, variability, and fragility of the late response indicate that it is mediated polysynaptically, presumably disappearing because the intermediate neurons mediating the polysynaptic connection failed to fire.
Dopamine neurons do not elicit a distinct postsynaptic signal
The CNQX blockade of the excitatory responses apparently eliminated the entire DA neuron signal. Because DA effects are principally thought to be modulatory, a direct postsynaptic action of DA would be unlikely. However, a subtle postsynaptic action might have been missed. Therefore, we followed the baseline current of the postsynaptic cells after the application of D1 or D2 antagonists. Neither the D1 antagonist SCH-23390 (10 μm bath application) nor the D2 antagonist sulpiride (50 μm local application in the nAcc) caused any significant change in the baseline current (for SCH-23390 application, 34 ± 12 pA; control, 21 ± 6 pA; n = 4 cells; p = 0.21; for sulpiride application, 52 ± 39 pA; control, 44 ± 30 pA; n = 5 cells; p = 0.42). Local application of sulpiride did not have any effect on the early response (29.9 ± 7.8 pA for control; 27.5 ± 6.5 pA for sulpiride; n = 5 cells; p = 0.31), suggesting that there was little or no ongoing activation of presynaptic D2 receptors (unlike the situation in culture) (Sulzer et al., 1998). However, we cannot completely exclude the possibility that there might still be a slow DA effect that went undetected because so many DA neuron axons were transected during slice preparation, the postsynaptic nAcc cells were subject to washout, and we focused on single-spike firing (and not bursting).
Early excitatory response is monosynaptic
To determine whether the early EPSC was monosynaptic, we first compared the latency of the early EPSC to that of antidromic action currents evoked in nAcc neurons projecting to the VTA (Fig. 4_A_). Because the axons of both VTA DA neurons and nAcc medium-spiny neurons are thin and mostly unmyelinated (Siggins, 1978; Chang et al., 1981) and take almost the same course (Usuda et al., 1998), the conduction times and velocities should be comparable (Ryan et al., 1986; Rodriguez and Gonzalez-Hernandez, 1999). Indeed in the rat in vivo, the conduction velocity of DA neuron axons is 0.52 m/sec, giving a latency range of 7–20 msec (Rodriguez and Gonzalez-Hernandez, 1999), whereas antidromically activated action potentials traveling in medium-spiny neuron axons have a conduction velocity of 0.65 m/sec, giving a latency range of 4–20 msec (Ryan et al., 1986). Therefore, if the early EPSC was generated monosynaptically, the antidromic action currents and the early response should have nearly identical latencies. Antidromic action currents were distinguished by their faster time course and larger amplitude (Fig. 4_B_), and as would be expected showed a rapid block with inclusion of QX-314 in the patch pipette. After four stimuli, and ∼15 sec from the onset of stimulation (Fig. 4_B_), action currents were completely blocked. The delay of the first-generated antidromic action current (we measured the first, because the delay increased with progressive Na+ channels blockade) (Fig. 4_B_, expanded traces) was 18.9 ± 1.0 msec (n = 8 cells). This was not significantly different from the delay of the early EPSC, which was 19.9 ± 0.8 msec (p = 0.12) (Fig. 4_C_). In P21 mice, the distance from the VTA to the nAcc is ∼4 mm, and compensating for the fact that our experiments were done at room temperature using a Q10 of 1.6 (Berg-Johnsen and Langmoen, 1992), the conduction velocity we recorded (∼0.2 m/sec) is consistent with previous measurements. Therefore, the latency of the early EPSC is attributable exclusively to axonal conduction time, leaving no time for an interposed polysynaptic connection.
Second, we asked whether the early response would show the characteristic reliability of monosynaptic connections when evoked at high frequency. If the response was generated polysynaptically, peak location and amplitude should fluctuate with increasing stimulus frequency because of failures of synaptic transmission at intermediate connections and failures in activating intervening cells (Kandel et al., 1967; Berry and Pentreath, 1976). We found that when we increased stimulus frequency from 0.1 to 10 Hz (sample records under 0.2 and 10 Hz stimulation are shown in Fig. 5_A_) there was no significant change (Fig. 5_B_) in latency (19.6 ± 1.2 msec at 0.2 Hz, n = 7 cells vs 19.4 ± 1.3 msec at 10 Hz, n = 5 cells; p = 0.39; ANOVA) or in peak amplitude (21.0 ± 3.7 pA at 0.2 Hz vs 19.8 ± 3.6 pA at 10 Hz; p = 0.79; ANOVA). Moreover, there was no increase in the variability of the latency measured as the SD of peak location (3.0 ± 0.3 msec at 0.2 Hz vs 3.0 ± 0.3 msec at 10 Hz; p = 0.71; ANOVA), nor was there any increase in the failure rate between 0.2 Hz (10.5 ± 2.8%) and 10 Hz (8.6 ± 3.8%) stimulation (p = 0.72). Together, these results show that the early EPSC is monosynaptic.
Figure 5.
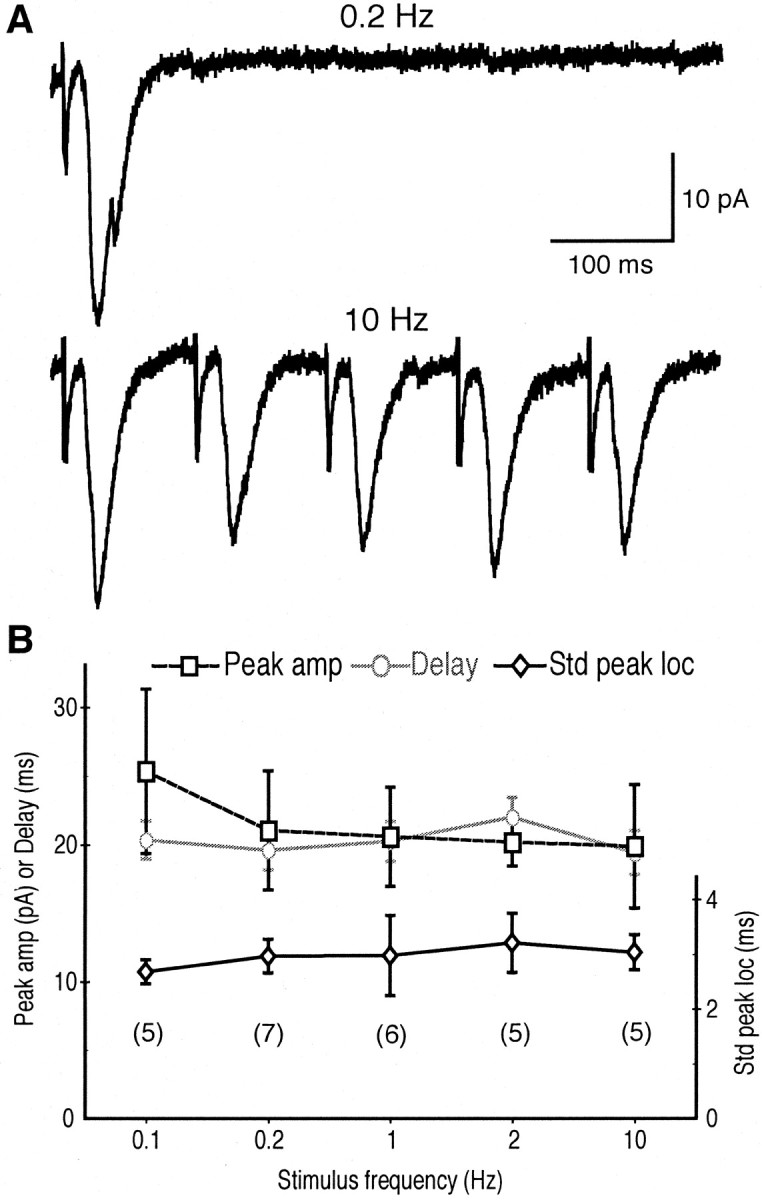
Reliability of early response is consistent with monosynapticity. A, Representative traces of the early response evoked at 0.2 Hz (top) and 10 Hz (bottom) from the same nAcc cell (average of 10 traces). B, Effects of increasing stimulus frequency on the average peak amplitude of the early EPSC (squares; left axis), the latency (circles; left axis), and SD of peak location (diamonds; right axis), showing no frequency dependence, as expected for a monosynaptic response. The numbers of cells recorded are indicated under the data points.
Origin of the late response
In contrast, the late response was clearly polysynaptic, because it had a much longer latency (>100 msec), fluctuated extensively, and failed with repeated stimulation. The late response could result from the antidromic activation of cortical neurons, which might in turn drive other cortical excitatory neurons projecting to the striatum (Wilson, 1998), and by analogy to the nAcc. Alternatively, DA neurons could excite cortical neurons projecting to the striatum/nAcc directly (Seamans and Yang, 2004). To test for cortical mediation of the late response, we split the mesoaccumbens slice at the midline and removed the cortex and hippocampus from one side, whereas the other side was left intact as a control. In two animals, data were successfully obtained from both hemislices (Fig. 6_A_); removal of the cortex and hippocampus eliminated the late response. Results obtained in unpaired hemislices were confirmatory, so in the summary graph, data from unpaired slices were included (Fig. 6_B_). The late response was never seen after the removal of the cortex, compared with 42% of the time in intact hemislices. Therefore, we conclude that the late response is polysynaptically generated via the cortex.
Figure 6.
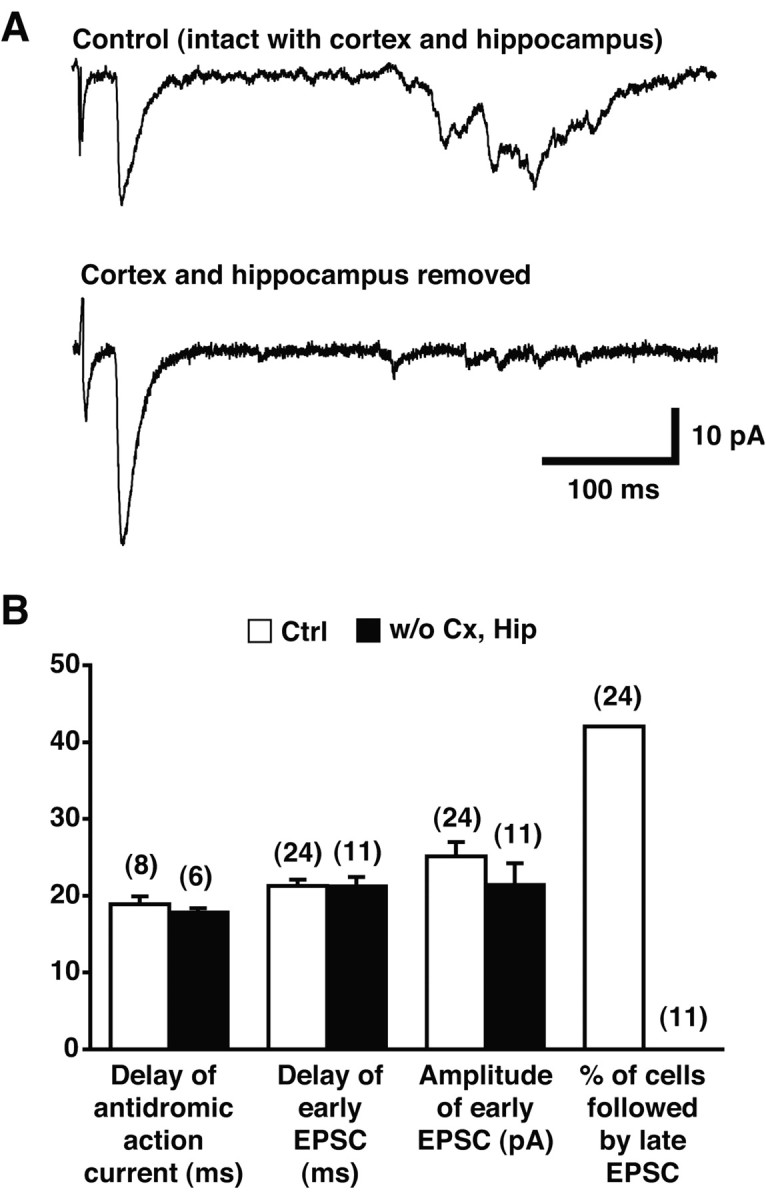
Late response requires intact cortical circuits. A, Representative traces in control hemislice (top) and the paired hemislice in which the cortex and hippocampus were removed (bottom; average of 10 traces). B, The delay of the antidromic action current, the delay of the early response, and the peak amplitude of the early response were not different between control (white columns) and after removal (black columns), whereas the late response was never seen in slices after removal. The numbers of recorded cells are indicated in parentheses. The graph includes results from paired and nonpaired hemislices.
As expected, the early response was not affected by the removal of the cortex and hippocampus. There were no significant changes in latency (21.3 ± 0.8 msec in control, n = 24 cells vs 21.2 ± 1.2 msec after removal, n = 11 cells; p = 0.99), in peak amplitude (25.1 ± 1.9 pA in control, n = 24 cells vs 21.4 ± 2.8 pA after removal, n = 11 cells; p = 0.30), or in the delay of antidromic action currents (18.9 ± 1.0 msec in control, n = 8 cells vs 17.8 ± 0.6 msec after removal, n = 6 cells; p = 0.36). This provided additional confirmation that the early response is monosynaptic.
The monosynaptic response originates from DA neurons
The VTA is made up of DA and GABA neurons, both of which project to the nAcc (Steffensen et al., 1998). In addition, nearby passing fibers of the internal capsule or basal cerebral peduncle may synapse in the nAcc (Wilson et al., 1982, 1998). Arguments can be made that all passing fiber projections are excitatory; however, they would not be activated by the local application of glutamate. Because the VTA is made up of DAergic and GABAergic neurons, and GABA transmission was blocked (by gabazine), local glutamate application in the VTA (Fig. 2_D,E_) should have only activated DA neuron connections. To test further that DA neurons give rise to the excitatory responses in the nAcc, we took advantage of the fact that in the VTA only DA neurons bear D2 receptors (Meador-Woodruff et al., 1989; Sesack et al., 1994), which are inhibitory (Lacey et al., 1987; Missale et al., 1998). Moreover, immunocytochemical studies have confirmed that myelinated passing fibers near the VTA do not bear D2 receptors (Sesack et al., 1994). So we would predict that if and only if the early response originates from DA neurons would local application of the D2 agonist quinpirole inhibit the early response.
We applied quinpirole just to the VTA using a patch pipette (Fig. 7_A_), while rapidly perfusing the slice (1 ml/min) to remove the quinpirole as it diffused away. Because the application pipette was located just above the slice and the drug would be both diluted and partially washed away before reaching the cells, the experiment required higher concentrations of quinpirole than for bath applications. Local superfusion of the VTA with 10 μm quinpirole reduced the amplitude of the early response to 84.0 ± 0.9% of control (n = 3 cells; p < 0.05) (Fig. 7_C_), whereas 50 μm quinpirole reduced the amplitude to 43.6 ± 6.0% (n = 6 cells; p < 0.01) (Fig. 7_B,C_). Recovery was complete for 10 μm (104.4 ± 5.7%; n = 3 cells) and nearly complete for 50 μm (94.2 ± 11.1%; n = 6 cells) (Fig. 7_B_). Together with the demonstration of monosynapticity, these results indicate that the early excitatory response is glutamatergic and originates from VTA DA neurons.
Figure 7.
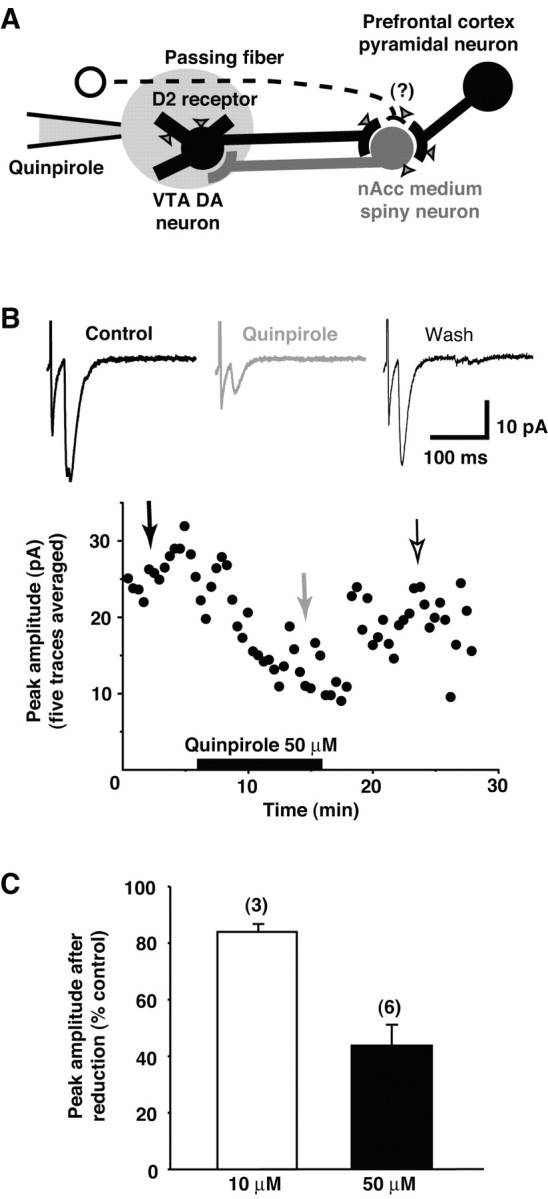
Local application of quinpirole in VTA inhibits early response. A, Local application of quinpirole to the VTA should reduce the early response if it arises from DA neurons, because D2 receptors (indicated by triangles) are present solely on DA neurons in the VTA. B, Local application of quinpirole selectively reduced the amplitude of the early response. The time course and representative traces (at time points indicated by arrows) during local application of quinpirole (50 μm) are shown for a representative experiment. Each point in the graph is the average of five traces; electrophysiological records are the average of 10 traces. C, Increasing the quinpirole concentration increased the inhibitory effect, further confirming that the early response arises from DA neurons. The abscissa indicates the quinpirole concentration. The numbers of recorded cells are shown in parentheses.
Discussion
We used transgenic mice with fluorescent DA neurons to guide the preparation of horizontal slices encompassing the mesoaccumbens projection. Even with an optimized slice preparation, evoked DA release showed that only a minority of the projection remained intact. Nonetheless, stimulation of the DA neuron cell bodies evoked a fast, excitatory response that we argue is mediated by monosynaptic glutamatergic connections made by DA neurons projecting to the nAcc. The key observations were that the fast, excitatory response met all criteria for a monosynapticity, and that it was elicited by focal glutamate application and inhibited by the focal application of quinpirole (a D2 agonist). We also observed a slower, more variable polysynaptic, excitatory, late response mediated by cortical neurons.
Slice encompassing the mesoaccumbens projection
Encompassing the VTA–nAcc pathway in a single slice proved challenging. Despite using thick (500 μm) slices from young mice, neither DA release nor excitatory responses were seen if the angle was off, even slightly. Amperometry showed that DA release diminished profoundly as the stimulating electrode was moved caudally along the MFB, to ∼3% with VTA stimulation, suggesting that the majority of DA neuron axons were cut while sectioning. Therefore, the excitatory actions we recorded likely underestimate the strength of the glutamatergic connection in intact animals, just as they underestimate VTA-evoked DA release.
We did not study the nigrostriatal projection, because it spreads too widely to be encompassed in the plane of a slice. However, our results could be extended to SN-DA neurons, which exhibit a similar response to behavioral stimulation (Schultz, 1998) and also express PAG (Kaneko et al., 1990), EAAT3 (Plaitakis and Shashidharan, 2000), and vGLUT3 (Fremeau et al., 2002). Indeed, 6-hydroxy-DA lesioning of the MFB results in a loss of 17% of asymmetric synapses in the striatum (Ingham et al., 1998), arguing that the nigrostriatal projection also has excitatory actions. Moreover, this morphological data suggests that a significant number of excitatory terminals in the striatum (and presumably also the nAcc) arise from DA neurons.
Another potential concern was that we used P10–P20 mice (most P13–P20), in which the DA system was still immature. However, the presynaptic machinery required for DA release already exists at P1 in the rat (Leroux-Nicollet et al., 1990). DA release itself is detectable at P5–P6 and reaches mature levels at approximately P20 (Restani et al., 1990). In the nAcc, D1 and D2 receptors are present at birth, then increase rapidly after P7 to reach adult levels by P21 (Tarazi and Baldessarini, 2000). The last to appear are D2 autoreceptors on DA neuron terminals, which become functional at P5 (Andersen and Gazzara, 1994). In the rat, DAT density in the VTA and SN is near adult levels at P5; however, in target areas, DAT density is only approximately half of adult levels, but then steadily increases to reach adult levels by ∼4 weeks of age (Coulter et al., 1996). Therefore, although the DA system was immature when we recorded, all the elements were in place, and most were nearly mature.
Origin of the early EPSC
Given that the early excitatory response evoked by VTA stimulation was monosynaptic and glutamatergic, the crucial issue was whether it arose from the DA neurons. Under GABA blockade, chemical stimulation of the VTA should activate only DA neuron connections, and we saw an increase in medium-sized excitatory events after local glutamate application in the VTA. These events could be summed to produce a response superimposable on the electrically evoked early response, arguing that both excitatory responses arose from the same set of connections. To pursue this specificity issue further we took advantage of the fact that the only elements bearing D2 receptors in the VTA are DA neurons, so local application of quinpirole should act exclusively on DA neurons. We saw the predicted result, namely that selective inhibition of DA neurons reduced the early response.
It is also possible that part of the early EPSC results from the activation of passing fibers. This seems unlikely because increasing stimulus currents did not change the shape of the early response, as would be expected if a second population of synapses was activated with higher-intensity stimulation. If passing fibers were intact and activated, the most likely pathway would be cortical efferent fibers in the cerebral peduncle and internal capsule that send axon branches to the nAcc (Siggins, 1978; Wilson, 1998). However, these axons are myelinated and thicker than DA axons, and so should show dramatically faster conduction times. Antidromic activation of these fibers should generate a monosynaptic response in medium-spiny neurons with a much shorter latency (1.5–4 msec) (Wilson et al., 1982, 1998). We observed no such short-latency responses. The other set of passing fibers near the VTA are afferent thalamic projections in the medial lemniscus. However, the thalamus is dorsal to the level of the mesoaccumbens slice, and the fibers run perpendicular to the plane of section (Paxinos and Franklin, 2001), and so would be cut by the sectioning. Therefore, it seems unlikely that the residual response after quinpirole is caused by a passing fiber component. More likely, it was caused by a partial effect of quinpirole, because the local superfusion may not have reached DA neurons deep in the slice, some DA neurons may express fewer D2 receptors (Cragg and Greenfield, 1997), or the overall magnitude of the quinpirole-mediated inhibition may be insufficient to block the activation of the neurons completely, because D2 activation of K+ channels in DA neurons is not robustly inhibitory (Lacey et al., 1987). Based on these arguments, we conclude that the early response originates from VTA DA neurons.
Origin of the late EPSC
We found that the late response required intact cortical connections. There are two ways VTA stimulation could activate cortical neurons. One is antidromic activation of cortical efferent fibers that synapse on other cortical neurons and in turn synapse on nAcc neurons. Consistent with this, wider field stimulation of the entire VTA and SN (by moving the electrode laterally to stimulate the dense cortical efferent fibers more effectively) increased the chance of observing the late EPSC, and it became more reliable (Chuhma et al., 2003). The more interesting possibility is that VTA DA neurons directly excite cortical neurons, possibly via glutamatergic transmission (Seamans and Yang, 2004) or direct excitatory effects of DA (Lavin and Grace, 2001) on cortical neurons, which in turn excite nAcc neurons. Because different populations of DA neurons project to the nAcc and cortex, the DA neuron glutamatergic signal in cortex may have a distinctly different role. Seamans and Yang (2004) have suggested that the glutamatergic transmission from VTA DA neurons may be the salience signal in the prefrontal cortex.
Possible roles of glutamate transmission by DA neurons
DA receptors are metabotropic and so exert principally modulatory actions. For timing-precise signaling, DA neurons apparently require a faster mode of transmission, one that we have now shown may be mediated by glutamate corelease. If so, how do the DA and glutamate signals interact? Goto and O'Donnell (2001) found that stimulation of the VTA evokes a fast-onset, prolonged depolarization of nAcc medium-spiny neurons in vivo, in effect putting the postsynaptic neurons into the up state. Separate application of D1 or D2 antagonist had no impact on the response, whereas their combined application reduced the duration of the up state, but not the amplitude. Together with our results, this suggests a scenario in which glutamate initiates the transition to the up state, the duration of which is controlled by DA. With single-spike firing (3–4 Hz), which we have focused on here, DA neuron activity serves mainly to provide a basal tone, because there is no build-up of released DA (Benoit-Marand et al., 2000), as DA release is limited by rapid DAT-mediated reuptake. Moreover, a majority of DA receptors are located extrasynaptically (Yung et al., 1995) and DA overflow would then be required (Gonon, 1997). In contrast, DA levels would increase significantly with burst firing (∼15 Hz), in response to salience stimuli, and overflow to enhance glutamate action. So we would propose that the salience signal is in fact initiated by DA neuron glutamatergic transmission. This would drive postsynaptic neurons into their up state, the duration of which would be modulated by DA, in effect increasing the signal-to-noise ratio or the duration of gating of cortical signals (O'Donnell, 2003).
Given the many roles of DA neurons in neuropsychiatric disorders, much will need to be reconsidered if a major component of the DA neurons signal is in fact glutamatergic. For instance, both abused (psychostimulants) and therapeutic (antipsychotics) drugs are likely to exert differential control of the DA and glutamate signals, so subtleties in their action may have to do with their disrupting the normal relationship between the DA and glutamatergic signals of DA neurons.
Footnotes
This work was supported by the Japan Society for the Promotion of Science (N.C.), the Parkinson's Disease Foundation (X.Z.), the National Alliance for Research on Schizophrenia and Depression (N.C., X.Z., J.M., R.H.), and the National Institute on Drug Addiction (D.S., S.R.). We thank Frank Costantini for the generous gift of the ROSA26-YFP reporter mice.
Correspondence should be addressed to Dr. Stephen Rayport, Psychiatry–Neuroscience, Columbia University, New York, NY 10032. E-mail: sgr1@columbia.edu.
J. Masson's present address: Faculté de Médecine, Pitié-Salpetrière, 91 boulevard de l'Hôpital, 75634 Paris cedex 13, France.
DOI:10.1523/JNEUROSCI.4317-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/240972-10$15.00/0
References
- Andersen SL, Gazzara RA (1994) The development of D2 autoreceptor-mediated modulation of K+-evoked dopamine release in the neostriatum. Brain Res Dev Brain Res 78: 123–130. [DOI] [PubMed] [Google Scholar]
- Benoit-Marand M, Jaber M, Gonon F (2000) Release and elimination of dopamine in vivo in mice lacking the dopamine transporter: functional consequences. Eur J Neurosci 12: 2985–2992. [DOI] [PubMed] [Google Scholar]
- Benoit-Marand M, Borrelli E, Gonon F (2001) Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics _in vivo_J Neurosci 21: 9134–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg-Johnsen J, Langmoen IA (1992) Temperature sensitivity of thin unmyelinated fibers in rat hippocampal cortex. Brain Res 576: 319–321. [DOI] [PubMed] [Google Scholar]
- Berry MS, Pentreath VW (1976) Criteria for distinguishing between monosynaptic and polysynaptic transmission. Brain Res 105: 1–20. [DOI] [PubMed] [Google Scholar]
- Budygin EA, John CE, Mateo Y, Jones SR (2002) Lack of cocaine effect on dopamine clearance in the core and shell of the nucleus accumbens of dopamine transporter knock-out mice. J Neurosci 22: RC222(1–4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HT, Wilson CJ, Kitai ST (1981) Single neostriatal efferent axons in the globus pallidus: a light and electron microscopic study. Science 213: 915–918. [DOI] [PubMed] [Google Scholar]
- Chuhma N, Zhang H, Sulzer D, Rayport S (2003) The intact mesoaccumbens dopamine projection in the brain slice: dopamine release and origin of the excitatory polysynaptic components. Soc Neurosci Abstr 29: 461.20. [Google Scholar]
- Coulter CL, Happe HK, Murrin LC (1996) Postnatal development of the dopamine transporter: a quantitative autoradiographic study. Brain Res Dev Brain Res 92: 172–181. [DOI] [PubMed] [Google Scholar]
- Cragg SJ, Greenfield SA (1997) Differential autoreceptor control of somatodendritic and axon terminal dopamine release in substantia nigra, ventral tegmental area, and striatum. J Neurosci 17: 5738–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorillo CD, Tobler PN, Schultz W (2003) Discrete coding of reward probability and uncertainty by dopamine neurons. Science 299: 1898–1902. [DOI] [PubMed] [Google Scholar]
- Fremeau Jr RT, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH (2002) The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci USA 99: 14488–14493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG (1996) Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379: 606–612. [DOI] [PubMed] [Google Scholar]
- Gonon F (1997) Prolonged and extrasynaptic excitatory action of dopamine mediated by D1 receptors in the rat striatum _in vivo_J Neurosci 17: 5972–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, O'Donnell P (2001) Network synchrony in the nucleus accumbens _in vivo_J Neurosci 21: 4498–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B, El Mestikawy S (2002) A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci 22: 5442–5451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham CA, Hood SH, Taggart P, Arbuthnott GW (1998) Plasticity of synapses in the rat neostriatum after unilateral lesion of the nigrostriatal dopaminergic pathway. J Neurosci 18: 4732–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Garris PA, Kilts CD, Wightman RM (1995) Comparison of dopamine uptake in the basolateral amygdaloid nucleus, caudate-putamen, and nucleus accumbens of the rat. J Neurochem 64: 2581–2589. [DOI] [PubMed] [Google Scholar]
- Joyce MP, Rayport S (2000) Mesoaccumbens dopamine neuron synapses reconstructed in vitro are glutamatergic. Neuroscience 99: 445–456. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Frazier WT, Waziri R, Coggeshall RE (1967) Direct and common connections among identified neurons in _Aplysia_J Neurophysiol 30: 1352–1376. [DOI] [PubMed] [Google Scholar]
- Kaneko T (2000) Enzymes responsible for glutamate synthesis and degradation. In: Handbook of chemical neuroanatomy, Vol 18: glutamate (Ottersen OP, Storm-Mathisen J, eds), pp 203–230. Amsterdam: Elsevier.
- Kaneko T, Akiyama H, Nagatsu I, Mizuno N (1990) Immunohistochemical demonstration of glutaminase in catecholaminergic and serotoninergic neurons of rat brain. Brain Res 507: 151–154. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y (1993) Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci 13: 4908–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA (1987) Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol (Lond) 392: 397–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin A, Grace AA (2001) Stimulation of D1-type dopamine receptors enhances excitability in prefrontal cortical pyramidal neurons in a state-dependent manner. Neuroscience 104: 335–346. [DOI] [PubMed] [Google Scholar]
- Leroux-Nicollet I, Darchen F, Scherman D, Costentin J (1990) Postnatal development of the monoamine vesicular transporter in mesencephalic and telencephalic regions of the rat brain: a quantitative autoradiographic study with [3H]dihydrotetrabenazine. Neurosci Lett 117: 1–7. [DOI] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Mansour A, Bunzow JR, Van Tol HH, Watson Jr SJ, Civelli O (1989) Distribution of D2 dopamine receptor mRNA in rat brain. Proc Natl Acad Sci USA 86: 7625–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu G, Costa E, Armstrong DM, Vicini S (1991) Glutamate receptor subtypes mediate excitatory synaptic currents of dopamine neurons in midbrain slices. J Neurosci 11: 1359–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG (1998) Dopamine receptors: from structure to function. Physiol Rev 78: 189–225. [DOI] [PubMed] [Google Scholar]
- Mogenson GJ, Yang CR, Yim CY (1988) Influence of dopamine on limbic inputs to the nucleus accumbens. Ann NY Acad Sci 537: 86–100. [DOI] [PubMed] [Google Scholar]
- O'Donnell P (2003) Dopamine gating of forebrain neural ensembles. Eur J Neurosci 17: 429–435. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates, Ed 2. San Diego: Academic.
- Phillips PE, Hancock PJ, Stamford JA (2002) Time window of autoreceptor-mediated inhibition of limbic and striatal dopamine release. Synapse 44: 15–22. [DOI] [PubMed] [Google Scholar]
- Plaitakis A, Shashidharan P (2000) Glutamate transport and metabolism in dopaminergic neurons of substantia nigra: implications for the pathogenesis of Parkinson's disease. J Neurol 247 [Suppl 2]: II/25–II/35. [DOI] [PubMed] [Google Scholar]
- Plenz D, Kitai ST (1996) Organotypic cortex-striatum-mesencephalon cultures: the nigrostriatal pathway. Neurosci Lett 209: 177–180. [DOI] [PubMed] [Google Scholar]
- Restani P, Corsini E, Galimberti R, Galli CL (1990) Postnatal ontogenesis of dopaminergic and serotoninergic systems in rat caudate nucleus. Pharmacol Res 22: 343–350. [DOI] [PubMed] [Google Scholar]
- Rodriguez M, Gonzalez-Hernandez T (1999) Electrophysiological and morphological evidence for a GABAergic nigrostriatal pathway. J Neurosci 19: 4682–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan LJ, Young SJ, Groves PM (1986) Substantia nigra stimulation evoked antidromic responses in rat neostriatum. Exp Brain Res 63: 449–460. [DOI] [PubMed] [Google Scholar]
- Schultz W (1998) Predictive reward signal of dopamine neurons. J Neurophysiol 80: 1–27. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Yang CR (2004) The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol, in press. [DOI] [PubMed]
- Sesack SR, Aoki C, Pickel VM (1994) Ultrastructural localization of D2 receptor-like immunoreactivity in midbrain dopamine neurons and their striatal targets. J Neurosci 14: 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggins GR (1978) Electrophysiological role of dopamine in the striatum: excitatory or inhibitory? In: Psychopharmacology: a generation of progress (Lipton MA, DiMascio A, Killam KF, eds), pp 143–157. New York: Raven.
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F (2001) Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol 1: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen SC, Svingos AL, Pickel VM, Henriksen SJ (1998) Electrophysiological characterization of GABAergic neurons in the ventral tegmental area. J Neurosci 18: 8003–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Joyce MP, Lin L, Geldwert D, Haber SN, Hattori T, Rayport S (1998) Dopamine neurons make glutamatergic synapses _in vitro_J Neurosci 18: 4588–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarazi FI, Baldessarini RJ (2000) Comparative postnatal development of dopamine D(1), D(2) and D(4) receptors in rat forebrain. Int J Dev Neurosci 18: 29–37. [DOI] [PubMed] [Google Scholar]
- Usuda I, Tanaka K, Chiba T (1998) Efferent projections of the nucleus accumbens in the rat with special reference to subdivision of the nucleus: biotinylated dextran amine study. Brain Res 797: 73–93. [DOI] [PubMed] [Google Scholar]
- Wilson CJ (1998) Basal ganglia. In: The synaptic organization of the brain (Shepherd GM, ed), pp 329–375. New York: Oxford UP.
- Wilson CJ, Groves PM (1981) Spontaneous firing patterns of identified spiny neurons in the rat neostriatum. Brain Res 220: 67–80. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Kawaguchi Y (1996) The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J Neurosci 16: 2397–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ, Chang HT, Kitai ST (1982) Origins of postsynaptic potentials evoked in identified rat neostriatal neurons by stimulation in substantia nigra. Exp Brain Res 45: 157–167. [DOI] [PubMed] [Google Scholar]
- Yung KKL, Bolam JP, Smith AD, Hersch SM, Ciliax BJ, Levey AI (1995) Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience 65: 709–730. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Wilson CJ, Dani JA (2002) Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol 53: 590–605. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Masson J, Gingrich JA, Rayport S, Hen R (2004) Targeted gene expression in dopaminergic and serotonergic neurons of the mouse brain. J Neurosci Methods, in press. [DOI] [PubMed]