Postsynaptic Density 95 controls AMPA Receptor Incorporation during Long-Term Potentiation and Experience-Driven Synaptic Plasticity (original) (raw)
Abstract
The regulated delivery of AMPA-type glutamate receptors (AMPARs) to synapses is an important mechanism underlying synaptic plasticity. Here, we ask whether the synaptic scaffolding protein PSD-95 (postsynaptic density 95) participates in AMPAR incorporation during two forms of synaptic plasticity. In hippocampal slice cultures, the expression of PSD-95–green fluorescent protein (PSD-95–GFP) increases AMPAR currents by selectively delivering glutamate receptor 1 (GluR1)-containing receptors to synapses, thus mimicking long-term potentiation (LTP). Mutational analysis shows that the N terminal of PSD-95 including the first two PDZ [PSD-95/Discs large (Dlg)/zona occludens-1 (ZO-1)] domains is necessary and sufficient to mediate this effect. Further supporting a role in synaptic plasticity, wild-type PSD-95 occludes LTP and dominant negative forms block LTP. Moreover, we demonstrate that PSD-95 also participates in AMPAR delivery during experience-driven plasticity in vivo. In the barrel cortex from experience-deprived animals, the expression of PSD-95–GFP selectively increases AMPAR currents, mimicking experience-driven plasticity. In nondeprived animals, PSD-95–GFP produces no additional potentiation, indicating common mechanisms between PSD-95-mediated potentiation and experience-driven synaptic strengthening. A dominant negative form of PSD-95 blocks experience-driven potentiation of synapses. Pharmacological analysis in slice cultures reveals that PSD-95 acts downstream of other signaling pathways involved in LTP. We conclude that PSD-95 controls activity-dependent AMPAR incorporation at synapses via PDZ interactions not only during LTP in vitro but also during experience-driven synaptic strengthening by natural stimuli in vivo.
Keywords: hippocampus, synaptic plasticity, AMPA receptors, receptor trafficking, PSD-95, synaptic scaffolding proteins, barrel cortex, sensory experience
Introduction
Unraveling the molecular mechanisms of synaptic plasticity will likely provide insight into functional circuit formation and behavioral plasticity (Scannevin and Huganir, 2000; Sheng and Lee, 2001; Malinow and Malenka, 2002; Bredt and Nicoll, 2003). One mechanism proposed to occur during long-term potentiation (LTP) in the hippocampus (Shi et al., 1999, 2001; Hayashi et al., 2000) and experience-driven plasticity in barrel cortex (Takahashi et al., 2003) is the delivery of AMPA-type glutamate receptors (AMPARs) to synapses. Although there is keen interest in the molecular events leading to the delivery and stabilization of these receptors during synaptic plasticity, they are still poorly understood.
Recent findings suggest that the postsynaptic density protein PSD-95, a member of the membrane associated guanylate kinases (MAGUKs) family, may have a role in synaptic maturation and function. MAGUKs share a common domain organization, consisting of three N-terminal PSD-95/Discs large (Dlg)/zona occludens-1(ZO-1) (PDZ) domains, an Src homology (SH3) domain, and an enzymatically inactive guanylate kinase (GK) domain. They can interact with ion channels, membrane receptors, cytoskeletal components, and intracellular signaling molecules and may organize signaling complexes (Garner et al., 2000; Sheng and Sala, 2001). Prolonged expression of PSD-95–green fluorescent protein (PSD-95–GFP) promotes the maturation of presynaptic and postsynaptic components at excitatory synapses, increases the number of spines, and enhances glutamatergic transmission in dissociated neurons (El-Husseini et al., 2000). Results from transient expression in slice cultures suggest that PSD-95 can control AMPAR content at synapses (Schnell et al., 2002; Beique, 2003). In addition, the developmental appearance of PSD-95 at hippocampal and cortical synapses parallels closely the synaptic appearance of glutamate receptor 1 (GluR1) (Martin et al., 1998; Petralia et al., 1999; Sans et al., 2000; Aoki et al., 2001). However, these studies have not established whether PSD-95 plays a role in activity-dependent AMPAR delivery during synaptic plasticity.
AMPARs are heteromultimers composed of subunits GluR1–GluR4 (Hollmann and Heinemann, 1994; Dingledine et al., 1999) and subunit-specific rules govern their incorporation to synapses in hippocampus and cortex. AMPARs containing subunits with long cytoplasmic tails (e.g., GluR1) require plasticity-inducing activity (Shi et al., 1999; Hayashi et al., 2000; Takahashi et al., 2003); their delivery enhances synaptic transmission and is blocked by the expression of the cytoplasmic tail (C-tail) of GluR1 (Shi et al., 2001; Takahashi et al., 2003). In contrast, AMPARs composed of only subunits with short C-tails (i.e., GluR2 and GluR3) are continuously delivered and replace existing AMPARs at synapses; thus maintaining transmission (Malinow et al., 2000; Shi et al., 2001).
Here, we test whether PSD-95 is necessary and sufficient to drive the incorporation of AMPARs to synapses during two different forms of synaptic plasticity. We study the effect of PSD-95 on the subunit-specific delivery of AMPARs at basal conditions and during LTP in slice cultures. To test the role of PSD-95 on synaptic strengthening driven by natural stimuli in the intact brain, we chose the developing barrel cortex as a model system, in which principal whiskers map topographically onto contralateral primary sensory cortex (Woolsey and Van der Loos, 1970; Welker, 1971, 1976; Welker and Woolsey, 1974). Sensory experience can easily be modified by trimming whiskers. The period between postnatal day 10 (P10) and P15 is marked for experience-dependent sensory map plasticity and spine motility in neurons of cortical layer II/III (Lendvai et al., 2000; Stern et al., 2001). In addition, the same rules governing AMPAR trafficking in vitro apply to connections between layer IV and layer II/III neurons. In particular, experience through the whiskers strengthens AMPAR-mediated transmission by driving GluR1-containing receptors into synapses (Takahashi et al., 2003). Sensory deprivation, although not reducing the number of spines and excitatory synapses (Micheva and Beaulieu, 1996; Vees et al., 1998), blocks GluR1-dependent AMPAR delivery (Takahashi et al., 2003).
Our results indicate that increased levels of PSD-95 mimic and occlude LTP and experience-driven plasticity in vivo. In addition, dominant negative forms of PSD-95 block AMPAR delivery during these forms of plasticity. We conclude that PSD-95 is a critical factor driving AMPAR incorporation during LTP and experience-driven synaptic strengthening.
Materials and Methods
GFP-tagged constructs and expression strategies. Rat PSD-95 fused to enhanced GFP (EGFP) was obtained from Dr. S. Okabe (Tokyo Medical and Dental University, Tokyo, Japan) (Okabe et al., 1999). Point mutations in PSD-95 were introduced by PCR, or using the Quick Change mutagenesis kit (Stratagene, Cedar Creek, TX). Truncated PSD-95 was generated by deleting the C-terminal part of PSD-95, and inserting EGFP in frame at the _Xma_I site between PDZ2 and PDZ3. All PSD-95 constructs were subcloned into pSinRep5 and expressed using Sindbis virus (Invitrogen, Carlsbad, CA) for ∼16–36 hr. For coexpression, we used particle-mediated biolistic gene transfer. GFP-tagged AMPAR subunits and the GluR1 C-tail-GFP have been described previously (Shi et al., 1999, 2001); red fluorescent protein (DsR-T1) was obtained from Dr. B. Glick (University of Chicago, Chicago, IL). All constructs were subcloned into cytomegalovirus-promoter driven expression vectors. Plasmid DNAs were coated onto 1.6 μm gold microcarriers at a 1:1 copy ratio as described previously (McAllister, 2000). Biolistic gene transfer was performed using a Helios gene gun (Bio-Rad, Hercules, CA) with helium pressure set to 180–200 psi; expression was allowed for ∼36 hr. We assume cotransfection for two reasons: First, the vast majority of neurons cotransfected with DsR-T1 and PSD-95–GFP exhibit red and green fluorescence; second, the electrophysiological effects could not be explained by the expression of either one of the constructs alone.
Slice cultures and pharmacological treatments. Hippocampal slices were prepared from P6 or P7 rat pups as described previously (Shi et al., 1999, 2001; Hayashi et al., 2000) and maintained in culture for 6–10 d. For pharmacological experiments, drugs were added to the culture medium just before infection with Sindbis and were maintained during the expression time (every 12 hr), but omitted during recordings. Different treatments were: Addition of 10 mm MgCl2 (Sigma, St. Louis, MO), 20 μm KN-93 (Sigma), 20 μm PD98059 (Calbiochem, San Diego, CA) or 20 μm SB203580 (Calbiochem).
Two-photon laser-scanning microscopy and image analysis. Images were collected using a custom-built two-photon microscope based on an Olympus Fluoview laser-scanning microscope (Olympus America, Melville, NY). The light source was a mode-locked Ti:sapphire laser (Mira 900F, Coherent, Santa Clara, CA) tuned to a wavelength of 910 nm. The microscope was equipped with a 40×, 0.75 numerical aperture water immersion lens. The subcellular localization of GFP-tagged constructs was determined using methods similar to those established previously (Barria and Malinow, 2002; Piccini and Malinow, 2002). Briefly, dual-wavelength images were acquired in Z-steps of 0.5 μm. Images were analyzed offline using custom-written software in MatLab (MathWorks, Natick, MA). First, spines and adjacent dendritic regions were identified using the red channel (DsR-T1). Subsequently, background-subtracted and leak-corrected (red to green, ∼10%; green to red, ∼5%) integrated red and green fluorescence was calculated for these structures. The ratio of green to red signal was obtained for each spine and adjacent dendrite (volume-corrected green signal). This measure was used to obtain spine/dendrite ratios subsequently and to determine whether constructs were accumulated in spines. A ratio of 1 indicates passive distribution and a ratio >1 indicates accumulation of the GFP-tagged construct in spines.
In vivo infection of cortical neurons and sensory deprivation. Surgery was performed in accordance with the animal care and use guidelines of the Cold Spring Harbor Laboratory. Neonatal rats (P11–P12) were anesthetized using a ketamine/xylazine mixture (ketamine, 0.56 mg/g; xylazine, 0.03 mg/gm body weight). The skin overlying the skull was cut and pushed to the side. A small window was opened in the skull 2 mm posterior and 4.5 mm lateral to the anterior fontanel. These coordinates reliably target the barrel cortex in neonatal rats (Chen et al., 2000a; Lendvai et al., 2000; Takahashi et al., 2003). Glass pipettes (tip diameter, ∼10–12 μm) were used to penetrate the dura and to pressure-inject (∼15 psi) recombinant Sindbis virus into the barrel cortex while avoiding damage to surface blood vessels. The skull was closed, and the skin was repositioned and maintained with cyanoacrylate glue. Sensory deprivation was initiated by trimming (to <1 mm) all large whiskers (columns 1–4, α–δ) contralateral to the injection site immediately after surgery; this was repeated every 12 hr. Animals were allowed to recover and subsequently returned to their mother and littermates.
Slice preparation. Two days after virus injection, rats were killed by decapitation and their brains were removed quickly and transferred to ice-cold dissection buffer containing the following (in mm): 110 cholinechloride, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 25 NaHCO3, 1.25 NaH2PO4, 25 glucose, 11.6 ascorbic acid, and 3.1 pyruvic acid bubbled with a mixture of 5% CO2 and 95% O2, pH 7.4. Coronal slices of the barrel cortex (300 μm) were cut using a vibratome (VT1000S, Leica Microsystems, Bannockburn, IL). Slices were transferred to artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 11 glucose, 2.5 CaCl2, and 1.3 MgCl2 bubbled with a mixture of 5% CO2 and 95% O2, pH 7.4, and allowed to recover for 1 hr at 22–25°C before recording. The barrel cortex was identified under low magnification and transillumination, in which large whisker-related barrels are readily seen.
Electrophysiology and data analysis. Simultaneous whole-cell recordings were obtained from pairs of neighboring or nearby (<50 μm apart) control and infected or transfected pyramidal neurons under visual guidance using differential interference contrast and fluorescence microscopy. The recording chamber was perfused with ACSF containing the following (in mm): 119 NaCl, 2.5 KCl, 26 NaHCO3, 1 NaH2PO4, 11 glucose, and 0.1 picrotoxin (Sigma, St. Louis, MO), bubbled with a mixture of 5% CO2 and 95% O2, pH 7.4,. To measure evoked responses for hippocampal slice cultures, 4 mm CaCl2 and 4 mm MgCl2 were added; for cortical slices, 2 mm CaCl2 and 1.3 mm MgCl2 were added; and to prevent bursting caused by recurrent excitation, 1–4 μm 2-chloroadenosine (Sigma) was added to the ACSF. All recordings were performed at 26°C. Patch pipettes (3.5–7 MΩ) were filled with internal solution containing the following (in mm): 115 cesium methanesulfonate, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 Na2ATP, 0.4 Na3GTP, 10 sodium phosphocreatine, and 0.6 EGTA, pH 7.25. For rectification analysis, 0.1 mm spermine (Sigma) was included in the internal solution. Whole-cell recordings were performed using two Axopatch-1D amplifiers (Axon Instruments, Union City, CA), and data were acquired (ITC-18 Computer Interface; Instrutech, Port Washington, NY) and analyzed using custom software written in Igor Pro (WaveMetrics, Lake Oswego, OR). Only neuron pairs with series and input resistances within 25% were included. Synaptic responses were evoked using bipolar electrodes (Frederick Haer, Bowdoinham, ME) giving single voltage pulses (200 μs, 0.5–10 V) at a frequency of 0.2–0.33 Hz. Electrodes were placed over Schaffer collateral fibers ∼200 μm lateral to the recording site in hippocampal cultures, and in cortical layer IV ∼200–300 μm below the recording site in layer II/III in cortical slices. Stimulus intensity was adjusted so responses could typically be evoked in both cells. Average EPSC amplitudes were obtained from 40 to 100 sweeps at each holding potential. The AMPA-mediated EPSC was measured as peak inward current at –60 mV, the NMDA-mediated component was measured as the late component (80–85 msec after stimulus) of the outward current at +40 mV. Rectification was calculated as the ratio of the peak AMPA current in 100 μm d, l-APV (Tocris, Ellisville, MO) at –60 and +40 mV, corrected by the current at 0 mV. Some rectification data, and most LTP data, were obtained by recording from single neurons (rather than paired recordings). LTP was induced by pairing 3 Hz stimulation with depolarization of the postsynaptic neuron to 0 mV for 90 sec; recordings were maintained for at least 35 min after pairing. The EPSC amplitude was normalized to the average amplitude before pairing. The peptide pep2m/G10 (KRMKVAKNAQ, Research Genetics, Huntsville, AL) was dissolved in internal solution (concentration, 2 mm) and included in the patch pipette. AMPAR-mediated miniature currents (minis) were recorded at –60 mV in ACSF containing 4 mm CaCl2, 2 mm MgCl2, and 1 μm TTX (Calbiochem). NMDAR-mediated minis were isolated at –60 mV in ACSF containing 2 mm CaCl2, no Mg2+, 5 μm NBQX (Sigma), and 1 μm TTX. Minis were analyzed using MiniAnalysis software (Synaptosoft, Decatur, GA). All data are reported as means ± SEM. Statistical analysis for paired recordings used the Wilcoxon test. To compare potentiation between experimental groups, we calculated the ratio of infected to uninfected AMPA current (AMPAInf/AMPAUninf) for each pair in each group and compared distributions using a two-sample Kolmogorov–Smirnov test (KS test). The KS test was also used for other large datasets (mini and spine analysis). In cases in which the t test has previously been used (e.g., LTP induction), the paired or unpaired Student's t test (t test) was used (as indicated). Significance was set at p < 0.05.
Results
PSD-95 enhances synaptic transmission by adding AMPARs to synapses
To examine the effects of acutely increased levels of PSD-95, we expressed wild-type (wt) PSD-95–GFP in CA1 pyramidal neurons in hippocampal slices for short periods (16–36 hr). In paired recordings from neighboring infected and uninfected neurons, we find that the AMPA component of synaptic transmission is significantly increased (Fig. 1_A,B_), consistent with previous findings (Schnell et al., 2002; Beique, 2003). We do not detect a difference in the late component of the EPSC at +40 mV mediated by NMDARs (Fig. 1_A,B_). In contrast, the expression of GFP by Sindbis virus had no effect on AMPAR-mediated transmission (36.8 ± 3.0 vs 33.2 ± 2.8 pA, control vs infected; n = 47; p = 0.20) (Fig. 2_F_). To elucidate the synaptic basis for this change, we recorded AMPA-mediated miniature EPSCs. We detected an increase in both amplitude and frequency (shown as interevent interval) of AMPA minis (Fig. 1_C–E_). We wanted to rule out the possibility that our method of assessing NMDA currents over-looked amplitude or kinetic differences attributable to a subunit switch in NMDARs (cf. Losi et al., 2003). Thus, we recorded evoked NMDA currents in the presence of NBQX and found that rise and decay times were not altered by the expression of PSD-95–GFP (supplemental Fig. 1_A,B_). We also found no significant changes in average mini amplitude or frequency (shown as interevent-interval) and the kinetics of NMDA minis (Fig. 1_F_). To determine whether structural modifications are concomitant with changes in AMPAR-mediated transmission we used two-photon imaging. Unlike the long-term expression of PSD-95–GFP in dissociated neurons (El-Husseini et al., 2000), but consistent with results from short-term expression (Marrs et al., 2001), we did not observe a change in spine density (supplemental Fig. 1_C_). However, we detected a small but significant increase in spine size (supplemental Fig. 1_D_). Spines may get bigger when AMPARs are added to synapses by PSD-95, because spine size has been correlated with AMPAR content (Nusser et al., 1998; Matsuzaki et al., 2001). Together, our results indicate that the acute expression of PSD-95–GFP does not result in increased synapse number but augments the number of AMPAR at synapses that already contain and those that did not contain AMPARs.
Figure 1.

Expression of PSD-95–GFP specifically potentiates AMPA-mediated EPSCs. A, Synaptic currents recorded simultaneously from nearby pyramidal neurons held at –60 and +40 mV, one uninfected and one expressing PSD-95–GFP. B, The AMPA component of EPSCs was significantly increased from 28.5 ± 2.5 to 64.8 ± 4.9 pA (control and infected neurons, respectively; n = 51), where as the late NMDA component was not altered (13.1±1.3 and 13.5±1.8 pA, control and infected neurons, respectively; n = 33). Calibration: 20 pA, 50 msec. C, Miniature EPSCs recorded at –60 mV from a pair of CA1 neurons. D, E, Cumulative histograms of amplitude and interevent interval for AMPA minis (n = 6 cells/group). The mini amplitude increases, whereas the interevent-interval decreases significantly (t test for each binned data point). F, Overlaid average miniature NMDA currents recorded from a pair of CA1 neurons recorded at –60 mV in 0 mm Mg 2+ and 5 μm CNQX. G, Plots of amplitude (23.3 ± 1.1 and 23.4 ± 1.0 pA, control and infected, respectively) and interevent interval (10.4 ± 1.2 and 10.1 ± 1.2 sec, control and infected, respectively) for average NMDA minis indicate no significant change (n = 10 cells/group; t test). H, Time to half decay of average NMDA minis (32.3 ± 2.9 and 34.3 ± 3.6 msec, control and PSD-95–GFP-expressing neurons, respectively) also shows no difference (n = 10 cells/group; t test).
Figure 2.
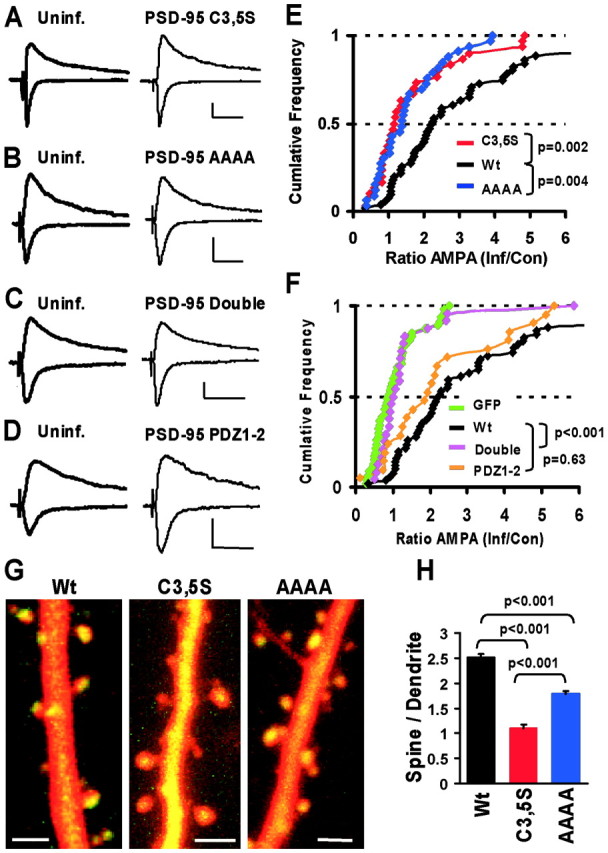
Membrane localization and PDZ interactions are necessary and sufficient for potentiation of AMPA currents. A–D, Synaptic currents measured in paired recordings from nearby control and infected neurons expressing mutant forms of PSD-95–GFP at –60 and +40 mV. Calibration: 20 pA, 50 msec. E, F, Cumulative distributions of AMPA-EPSC ratios (AMPAInf/AMPAUninf) in paired recordings are used to compare AMPA current potentiation by different mutants (p values from KS tests). E, The small potentiating effect of PSD-95-C3, 5S (n = 30) and PSD-95-AAAA (n = 33) was highly significantly different from wt PSD-95 (p = 0.002 and p = 0.004, respectively). F, Like GFP (n = 47), PSD-95-Double (n = 24) did not potentiate AMPA currents and was significantly different from wt PSD-95 (p < 0.001), whereas PSD-95–PDZ1–2 (n = 21) potentiated AMPA–EPSCs similar to wt-PSD-95 (p = 0.63). G, Subcellular distribution of mutant forms of PSD-95 compared with wt PSD-95–GFP. Dual-wavelength two-photon images of CA1 pyramidal neurons in slice cultures (9 d in vitro) coexpressing DsR-T1 and GFP-tagged PSD-95 constructs for ∼40–48 hr. Scale bars, 2μm. H, Spine/dendrite ratio shows that wt-PSD-95 is strongly accumulated in spines (ratio, 2.51 ± 0.07; n = 312 spines), C3, 5S is passively distributed (ratio, 1.11 ± 0.06; n = 285 spines), and the AAAA mutant is accumulated in spines (ratio, 1.78 ± 0.06; n = 323 spines), but less than wt (from three neurons each; KS tests).
Membrane localization and PDZ interactions are necessary and sufficient for synaptic potentiation by PSD-95
To determine which properties of PSD-95 are important for AMPAR delivery to synapses we generated mutant and truncated forms. PSD-95 undergoes palmitoylation at two N-terminal cysteines, enabling its targeting to the cell membrane (Topinka and Bredt, 1998). These cysteines have also been implicated in multimerization and ternary complex formation (Hsueh and Sheng, 1999). To prevent association with the cell membrane and to compromise multimerization and synaptic clustering (Arnold and Clapham, 1999; Craven et al., 1999) we replaced cysteines 3 and 5 with serines (PSD-95C3, 5S). We also generated a construct carrying point mutations in the first two PDZ domains (R70 to A, G71 to A, K165 to A, and K168 to A; PSD-95AAAA), which should impair binding to proteins with PDZ ligands (e.g., channels and membrane proteins as well as intracellular signaling molecules) (Kornau et al., 1995; Niethammer et al., 1996; Sheng and Sala, 2001) (supplemental Fig. 2_A_). A third construct carried a combination of all these mutations (Double) and is impaired in both synaptic localization and binding to PDZ partners.
Mutations at either the palmitoylation or PDZ-interaction sites significantly reduced the potentiating effects of PSD-95 (Fig. 2_A,B,E_). Together, the two mutations completely removed the potentiating effects (Fig. 2_C,F_). To determine whether mutated regions were sufficient to mediate potentiation, we generated a truncated form of PSD-95, containing the N terminal and the first two PDZ domains fused to GFP (PDZ1–2). Expression of this construct potentiated AMPA-EPSCs similarly to wt PSD-95 (Fig. 2_D,F_), indicating that the truncated construct is sufficient to mediate this effect. NMDAR-mediated transmission was not altered by the expression of mutant or truncated forms of PSD-95 (Fig. 2_A–D_, supplemental Fig. 2_B_). Moreover, the differences in AMPA-current potentiation were not caused by differences in expression levels between wt and mutant forms of PSD-95–GFP (see supplemental information).
On the basis of these findings, we reasoned that mutants of PSD-95 that carry either a mutation in the N terminal (C3,5S) or in the first two PDZ domains (AAAA) would be good candidates for dominant negative constructs. To gain more information about these mutant forms of PSD-95, we determined their subcellular distribution compared with wt PSD-95–GFP. We performed dual-wavelength two-photon imaging of neurons coexpressing a cytoplasmic marker (DsR-T1) and GFP-tagged forms of PSD-95 and computed a volume-corrected spine/dendrite ratio for the GFP signal (see Materials and Methods) as a measure of spine accumulation. As expected, wt PSD-95–GFP was strongly targeted to spines and often accumulated in one region of the spine, presumably the PSD (Fig. 2_G_). In contrast, PSD-95C3,5S–GFP did not accumulate in spines, but was passively distributed (Fig. 2_G,H_; spine/dendrite ratio, ∼1). PSD-95AAAA–GFP still accumulated in spines, although less than wt PSD-95–GFP (Fig. 2_G,H_). Our results indicate that these mutant forms of PSD-95 display aberrant localization. Together, our results indicate that spine localization and PDZ interactions are necessary properties of PSD-95 to mediate AMPA-current potentiation.
PSD-95 mimics LTP by delivering GluR1-containing receptors to synapses
AMPARs are delivered to synapses by two distinct routes: regulated or constitutive pathways. To test whether PSD-95 participates in the activity-dependent pathway, we coexpressed PSD-95–GFP with recombinant GluR1–GFP. Recombinant GluR1 forms homomeric receptors that show complete inward rectification and require LTP or increased calcium calmodulin-dependent-protein kinase II (CaMKII) activity to drive them into synapses (Shi et al., 1999; Hayashi et al., 2000). Compared with nontransfected neurons, AMPA currents in neurons coexpressing GluR1–GFP and PSD-95–GFP were significantly increased in amplitude and more rectified (Fig. 3_A,B_). This property is the electrophysiological signature indicating that recombinant receptors were incorporated into synapses. As an independent test we coexpressed PSD-95–GFP and a mutant form of GluR1 [GluR1(T887A)], which carries a mutation in the C-tail preventing PDZ-ligand interactions. This mutant receptor cannot be delivered to synapses by activity (e.g., increased CaMKII-activity or LTP) and it blocks LTP (Hayashi et al., 2000). Coexpression of GluR1(T887A)–GFP and PSD-95–GFP resulted in a block of AMPA-current potentiation and no significant change in rectification (Fig. 3_C,D_). To test whether PSD-95 drives endogenous GluR1-containing receptors to synapses, we coexpressed PSD-95–GFP with the C-tail of GluR1 (amino acids 809–889). This fragment of GluR1 prevents LTP (Shi et al., 2001) and also prevented AMPA-current potentiation by PSD-95 (Fig. 3_E,F_). As a control, we coexpressed PSD-95–GFP with DsRed, which resulted in AMPA-current potentiation, but no change in rectification (supplemental Fig. 3_A,B_). In summary, our results indicate that the expression of PSD-95 can drive recombinant and endogenous GluR1-containing receptors to synapses, thus mimicking LTP.
Figure 3.

Expression of PSD-95 mimics LTP by driving GluR1 to synapses. A, C, E, AMPA-EPSCs recorded simultaneously from nearby CA1 neurons at –60 and +40 mV in 100μm APV. Calibration: 10 pA, 40 msec. A, Coexpression of PSD-95–GFP and GluR1–GFP resulted in potentiated and more rectified AMPA current. B, The relative AMPA-EPSC amplitude (100 ± 9.0 vs 159 ± 13.5%; n = 35) and rectification (2.3 ± 0.2; n = 43 vs 3.9 ± 0.3; n = 40; t test) were significantly increased in control versus transfected neurons. C, Coexpression of PSD-95–GFP and GluR1(T887A)–GFP resulted in no change in AMPA-current amplitude or rectification. D, Relative AMPA-EPSCs (100±10.1 and 95.6±8.8%; n = 29) and rectification (2.0±0.1, n = 29 vs 2.3 ± 0.2, n = 27; t test) were not significantly different in control versus transfected neurons. E, Co-expression of PSD-95–GFP and GluR1–C-tail–GFP did not change AMPA-EPSC amplitude. F, Relative AMPA-EPSCs were not significantly different in control versus transfected neurons (100 ± 13.4 vs 108.3 ± 11.7%; n = 31).
PSD-95 does not affect cycling of GluR2-containing receptors in the constitutive pathway
We also tested whether PSD-95 influenced AMPAR trafficking through the constitutive pathway. Interfering with cycling of GluR2/3-containing receptors could lead to their accumulation at synapses and hence, increased AMPA transmission. We addressed this in two ways: first by testing whether PSD-95 can deliver recombinant GluR2 [GluR2(R607Q), which forms homomeric, rectifying receptors (Shi et al., 2001)] to synapses. Second, we assayed whether PSD-95 increased the pool of cycling, endogenous GluR2/3-containing receptors by using a peptide that interferes with cycling by preventing interaction of GluR2 with _N_-ethylmaleimide-sensitive factor (NSF) (Nishimune et al., 1998; Osten et al., 1998; Zhu et al., 2000).
To measure how much recombinant GluR2 is constitutively inserted at synapses, we expressed GluR2(R607Q)–GFP together with a control vector. Consistent with previous results (Shi et al., 2001), AMPA-mediated transmission was not potentiated, but rectification was increased (Fig. 4_A,C,D_). We then coexpressed PSD-95–GFP with GluR2(RQ)–GFP and observed potentiation and a small change in rectification (Fig. 4_B–D_), which is expected whether PSD-95 drives receptors into synapses that do not readily participate in the cycling pathway. The selective delivery of GluR1/2 should produce a rectification smaller than GluR2(RQ) alone (≪3.7); equal delivery of GluR1/2 and GluR2(RQ) should produce rectification similar to that without PSD-95 expression (∼3.7), whereas selective delivery of GluR2(RQ) should produce a larger rectification (≫3.7). Thus, finding that neurons coexpressing GluR2(RQ) and PSD-95 show a rectification of ∼2.7 supports the delivery of endogenous GluR1/2 by PSD-95.
Figure 4.

PSD-95 does not affect the constitutive pathway of AMPAR delivery. A, B, AMPA-EPSCs recorded simultaneously from nearby CA1 neurons at –60 and +40 mV in 100μm APV. Calibration: 30 pA, 40 msec. A, Expression of GluR2(R607Q)–GFP did not alter AMPA-EPSC amplitude, but rectification was increased. B, PSD-95–GFP was coexpressed with GluR2(RQ)–GFP. AMPA-EPSC amplitude was strongly increased, but rectification was only slightly increased. C, Relative AMPA-EPSCs were not significantly different in control versus GluR2(RQ)-transfected neurons (100 ± 17.2 and 84.9 ± 7.6%; n = 15), but significantly increased in control versus neurons coexpressing PSD-95–GFP and GluR2(RQ)–GFP (100±11.2 vs 164.8± 22.3%; n = 17). D, Rectification was strongly increased in control versus GluR2(RQ)–GFP-transfected neurons (2.2 ± 0.2, n = 16 vs 3.7 ± 0.4, n = 13; t test) and slightly increased in control versus PSD-95–GFP and GluR2(RQ)–GFP-expressing neurons (2.0 ± 0.2, n = 16 vs 2.7 ± 0.2, n = 16; t test). Importantly, rectification in neurons coexpressing PSD-95 and GluR2(RQ) was significantly smaller than in neurons expressing GluR2(RQ) alone (t test). E, Changes in synaptic AMPA currents recorded during infusion of peptide pep2m (2 mm) into two nearby neurons. EPSC amplitudes decreased in the uninfected neuron, but there was little change in a neuron expressing PSD-95–GFP. Calibration: 40 pA, 20 msec. F, The residual AMPA-EPSC 25 min after infusion of pep2m is significantly smaller in control than infected neurons (55.8 ± 10.7 vs 84 ± 9.5% of initial value; n = 11; t test).
As an independent test, we infused the peptide pep2m/G10 into neurons, which reduces the number of synaptic AMPARs by blocking GluR2–NSF interaction and thereby GluR2/3 cycling. We observed that the amplitude of AMPA-mediated EPSCs decreased more strongly in control neurons than in PSD-95–GFP-expressing neurons (Fig. 4_E_). On average, the decrease of AMPA currents after 25 min of peptide infusion was significantly larger in control than infected neurons (Fig. 4_F_). This result is consistent with a larger fraction cycling GluR2/3 receptors at synapses in control than in PSD-95-expressing neurons. Moreover, it suggests that there are more noncycling GluR1/2 receptors at synapses in PSD-95 expressing neurons. Together, our findings indicate that PSD-95 does not deliver recombinant, homomeric GluR2(RQ), or endogenous GluR2/3 receptors to synapses but acts on GluR1/2 receptors.
PSD-95-induces synaptic potentiation that occludes LTP
Our findings indicate that synaptic potentiation by PSD-95 mimics LTP by delivering GluR1-containing receptors to synapses. Because the generation of LTP is a saturable process (Bliss and Lomo, 1973), it follows that whether PSD-95-induced potentiation precisely mimics LTP, it should occlude LTP. To test this, we examined LTP in neurons that had been expressing PSD-95–GFP for ∼16–24 hr. Indeed, after a pairing protocol, transmission mostly returned to baseline levels after 30–35 min in these PSD-95-expressing neurons (Fig. 5_A,B_). The initial, transient increase in transmission is likely not mediated by GluR1 delivery, because a similar transient potentiation is seen in neurons expressing the GluR1 C-tail (Shi et al., 2001) but could be caused by changes in the phosphorylation state of AMPARs already at the synapse (Barria et al., 1997; Lee et al., 2000). As a control, we recorded from noninfected neurons in the same group of slice cultures and confirmed that these cells showed robust LTP (Fig. 5_A,B_). To rule out the possibility that the induction or expression of LTP was compromised by infection with Sindbis virus, we also recorded from neurons that expressed GFP. These neurons showed stable LTP 30–35 min after induction (1.96 ± 0.29-fold of baseline; n = 8), which was similar to that seen in uninfected neurons (1.88 ± 0.22-fold of baseline; n = 13; p = 0.72; t test). The amount of LTP in GFP-expressing neurons was significantly different from that observed in PSD-95–GFP-expressing neurons (1.96 ± 0.29-fold, n = 8 vs 1.17 ± 0.20-fold, n = 12; GFP and PSD-95–GFP, respectively; p = 0.026; t test). Because potentiation caused by the expression of PSD-95 resembles LTP, the absence of LTP (and thus the lack of additional AMPAR delivery during LTP) is most easily explained by, and consistent with, occlusion.
Figure 5.

Expression of wt PSD-95 occludes and expression of putative dominant negative forms of PSD-95 blocks LTP. A, C, E, Time courses of relative changes in AMPA-mediated EPSCs by pairing-induced LTP for control and infected neurons. Induced (paired) pathways are shown in large and control (unpaired) pathways in small symbols for infected and control neurons. The time of delivery of the pairing protocol is indicated by the bar. A, Expression of PSD-95–GFP-occluded LTP. Transmission onto infected neurons returned almost to baseline 30–35 min after pairing (1.17 ± 0.20; n = 12) and was significantly different from the potentiation observed in control neurons (1.88 ± 0.22; n = 13; p = 0.028; t test). _C_i Expression of PSD-95C3,5S–GFP blocked LTP. In PSD-95C3,5S–GFP-expressing neurons, transmission returned almost to baseline levels 30–35 min after pairing (1.17 ± 0.16; n = 10). This was significantly different from potentiation in the control neurons (1.89 ± 0.23; n = 7; p = 0.02; t test). E, Expression of PSD-95AAAA–GFP blocked LTP. In infected neurons, transmission returned close to baseline levels 30–35 min after pairing (1.21 ± 0.15; n = 11), which was significantly different from control neurons (2.10 ± 0.29; n = 11; p = 0.014; t test). B, D, F, Example traces of EPSCs in uninfected and infected neurons before and ∼32–35 min after pairing. Traces are the averages of 30 sweeps. Calibration: 40 pA, 20 msec.
Dominant negative forms of PSD-95 block LTP
To address whether endogenous PSD-95 plays a role in LTP, we tested the effects of putative dominant negative mutants on LTP. Because PSD-95C3, 5S and PSD-95AAAA retain only one of the two main properties necessary for AMPA-current potentiation, they would be expected to act as dominant negative constructs. In neurons expressing these mutants under basal conditions, AMPAR-mediated transmission was not depressed, which is consistent with GluR1 delivery not normally occurring in our slice culture conditions (Shi et al., 1999). However, when LTP was induced, neurons expressing either PSD-95C3, 5S or PSD-95AAAA showed only an initial, transient increase in synaptic transmission, which returned to baseline levels 30–35 min after induction (Fig. 5_C–F_). We again tested noninfected neurons in the same group of slices, and as expected, control neurons exhibited robust LTP (Fig. 5_C–F_). For both mutant forms, the residual potentiation observed 30–35 min after induction was significantly different in infected and uninfected neurons. In summary, this lack of LTP in neurons that express dominant negative mutants of PSD-95 is most easily explained by a block of LTP.
Probing the role of PSD-95 in an in vivo model for synaptic plasticity
To substantiate our findings in slice cultures, we wanted to determine whether PSD-95 also participates in synaptic plasticity driven by natural stimuli in vivo. We chose to study experience-driven AMPAR delivery to synapses between layer IV and layer II/III pyramidal neurons in the barrel cortex of young rats, because this form of plasticity shares molecular mechanisms with LTP: Activity driven by experience through the whiskers strengthens AMPAR-mediated transmission by delivering GluR1-containing receptors to synapses (Takahashi et al., 2003). We injected Sindbis virus encoding wt or mutant PSD-95–GFP into rat barrel cortex and allowed expression in vivo for 2 d while animals were either allowed normal experience or were deprived of sensory input (trimming all principal whiskers contralateral to the injected hemisphere) (Fig. 6_A_). After this, we prepared slices of barrel cortex and obtained paired recordings from infected and control neighboring layer II/III pyramidal neurons to compare EPSCs evoked by stimulation in layer IV (Fig. 6_A_). As in hippocampal slices, the expression of GFP using Sindbis virus did not affect synaptic responses or plasticity in vivo (Takahashi et al., 2003), so observed effects are not caused by the viral infection of neurons.
Figure 6.

Experimental approach, and effects of sensory deprivation in barrel cortex. A, Sketch of the experimental protocol. Sindbis virus was injected at P11 or P12 and rats were allowed or deprived of sensory experience by trimming all contralateral whiskers for 2 d. Cortical slices were obtained and the barrel cortex was identified by trans-illumination. Paired recordings from neighboring layer II/III pyramidal neurons, infected and control, were obtained and EPSCs elicited by stimulation of layer IV. B, C, Sensory deprivation decreased the AMPA/NMDA ratio at synapses. B, EPSCs recorded in uninfected layer II/III pyramidal neurons at holding potentials of –60 and +40 mV from animals with intact or deprived whiskers. Calibration: 10 pA, 40 msec. C, The AMPA/NMDA ratio was significantly lower in deprived (1.5 ± 0.2; n = 16) than in nondeprived animals (2.6 ± 0.3; n = 20; t test).
To confirm that experience indeed drives the delivery of AMPARs, we measured the ratio of AMPAR- to NMDAR-mediated transmission in uninfected neurons from animals with intact or deprived whiskers. The AMPA/NMDA ratio of EPSCs in layer II/III pyramidal neurons from animals deprived for 2 d was significantly reduced when compared with that from animals with intact whiskers (Fig. 6_B,C_). In contrast, ipsilateral deprivation does not affect AMPAR trafficking (Takahashi et al., 2003).
Expression of PSD-95 in barrel cortex occludes experience-driven AMPAR delivery
We predict that increasing levels of PSD-95 should increase AMPA-mediated transmission and prevent further delivery of AMPARs by sensory experience (occlusion). To test this hypothesis, we expressed wt PSD-95GFP in cortical neurons for 2 d while animals had normal whisker-driven experience. In subsequent paired recordings from infected and control layer II/III pyramidal neurons, AMPA currents were slightly, but not significantly, increased in infected versus control neurons, whereas NMDA currents were not changed (Fig. 7_A,C_). This result would be expected whether the expression of PSD-95–GFP either produced no effect on synapses or whether experience-driven AMPAR delivery occluded potentiation by PSD-95.
Figure 7.

Expression of wt PSD-95 mimics and occludes experience-driven AMPAR delivery in barrel cortex. A, B, Paired recordings of EPSCs from layer II/III pyramidal neurons held at –60 and +40 mV, one neuron uninfected and one expressing wt PSD-95–GFP. Calibration: 20 pA, 40 msec. A, In intact whisker animals, there was no difference in the AMPAR- or NMDAR-mediated EPSC in a control and a neuron expressing PSD-95–GFP. B, In deprived animals, the AMPA-EPSC was potentiated by the expression of PSD-95–GFP, whereas the NMDA current was unaffected. C, Summary of relative changes in AMPAR- and NMDAR-mediated EPSCs. The AMPA component was not significantly different between control and infected neurons in intact animals (100 ± 17.7 and 112.7 ± 12.8%; n = 19) but potentiated during contralateral deprivation (100 ± 18.9 and 168.5 ± 25.4%, control and infected, respectively; n = 20). In all cases, NMDA currents were not significantly changed (Intact, 100 ± 21.0 and 83.1 ± 13.0%, control and infected, n = 17; Deprived, 100 ± 19.7 and 93.8 ± 13.4%, control and infected, n = 15).
Expression of PSD-95 mimics experience-dependent delivery of AMPARs
A second prediction from our in vitro work is that increasing levels of synaptic PSD-95 should mimic the experience-driven delivery of AMPAR, because it is sufficient to deliver GluR1-containing receptors to synapses in slice cultures under conditions when GluR1 delivery does not normally occur (Hayashi et al., 2000). We expressed PSD-95–GFP in barrel cortex for 2 d while trimming whiskers contralateral to the injected cortical hemisphere. In contrast to slices prepared from nondeprived animals, AMPAR-mediated transmission was significantly potentiated in infected compared with control neurons (Fig. 7_B,C_). This effect was specific, because NMDA components of EPSCs were not significantly altered (Fig. 7_B,C_). Thus, the expression of PSD-95–GFP affects AMPA currents in barrel cortex, and this conforms with observations in slice cultures in which PSD-95 drives AMPARs to synapses. In summary, our data from intact and deprived animals suggest that the expression of PSD-95–GFP mimics and occludes experience-driven synaptic delivery of AMPAR in barrel cortex in vivo.
Expression of dominant negative PSD-95 blocks experience-dependent delivery of AMPARs
To address whether endogenous PSD-95 mediates the observed effects, we tested whether a dominant negative construct of PSD-95 could prevent experience-driven AMPAR delivery in the barrel cortex. Again, animals with intact whiskers were infected with a virus expressing PSD-95AAAA–GFP for 2 d, and paired recordings were obtained from layer II/III pyramidal neurons. If this construct had dominant negative function in vivo, it should block experience-driven synaptic strengthening, and transmission onto infected neurons should be less than onto control neurons (Takahashi et al., 2003). As predicted, we found that AMPAR-mediated EPSCs were significantly depressed, but NMDAR-mediated transmission was not altered (Fig. 8_A,C_).
Figure 8.

Expression of dominant negative PSD-95 blocks experience-driven AMPAR delivery to synapses in barrel cortex. A, B, EPSCs evoked in layer IV and recorded simultaneously from nearby layer II/III pyramidal neurons held at –60 and +40 mV. Calibration: 10 pA, 40 msec. A, In intact whisker animals, the AMPAR- but not the NMDAR-mediated EPSC was depressed in a neuron expressing PDS-95AAAA–GFP. B, In deprived animals, there was no change in AMPA or NMDA currents in the control versus the infected neuron. C, Summary of relative changes in AMPAR- and NMDAR-mediated EPSCs. The AMPA component was significantly depressed by PSD-95AAAA when whiskers were intact (100 ± 11.1 and 54.5 ± 7.0%, control and infected; n = 18), but no change was seen during contralateral deprivation (100 ± 14.3 and 100.3 ± 15.2%, control and infected; n = 20). In all cases, NMDA currents were not significantly changed (Intact, 100 ± 16.7 and 93.5 ± 15.1%, control and infected, n = 14; Deprived, 100 ± 16.4 and 90.9 ± 11.4%, control and infected, n = 16).
To demonstrate that PSD-95AAAA-GFP selectively prevents plasticity driven by experience through whiskers and did not reduce AMPA currents by another mechanism, we tested its effect in deprived whisker animals. Because contralateral deprivation prevents experience-driven AMPAR delivery, a dominant negative construct is expected to have no effect. Indeed, in slices from deprived animals, AMPAR- as well as NMDAR-mediated transmission onto infected neurons was not altered (Fig. 8_B,C_). Together, our findings indicate that the expression of PSD-95AAAA–GFP acts as dominant negative construct and interferes with endogenous PSD-95 function to block experience-driven AMPAR delivery to cortical synapses in vivo.
PSD-95 acts downstream of signaling cascades involved in LTP
To address the mechanism by which PSD-95 drives the synaptic incorporation of AMPARs, we performed a series of pharmacological experiments in hippocampal slice cultures. We first examined whether spontaneous synaptic activity is required for PSD-95-mediated potentiation. Incubating slices in high Mg2+, which reduces spontaneous activity (Zhu et al., 2000), while expressing PSD-95–GFP did not prevent the potentiation of AMPA currents (Fig. 9_A,E_). Another possibility is that PSD-95 affects other signaling cascades implicated in LTP (e.g., the activation of CaMKII, which is required for the induction of LTP) (Malinow et al., 1989). Active CaMKII can mimic LTP and drives GluR1-containing receptors to synapses (Pettit et al., 1994; Hayashi et al., 2000). In addition, PSD-95 is a substrate for CaMKII-phosphorylation in the postsynaptic density (Yoshimura et al., 2000). We incubated slice cultures in the cell-permeable CaMKII antagonist KN-93 (Sumi et al., 1991) at a concentration that blocks LTP in hippocampal slices from young rats (Zhu et al., 2000) and found that this treatment did not prevent AMPA-current potentiation (Fig. 9_B,E_).
Figure 9.

PSD-95 acts downstream of synaptic activity and other signaling pathways involved in LTP. A–D, AMPA-EPSCs measured in paired recordings from CA1 neurons held at –60 mV to compare amplitudes in uninfected and PSD-95–GFP-expressing neurons. Slices had been subjected to different pharmacological treatments (10 mm Mg 2+, 20 μm KN-93, 20 μm PD98059, 20 μm SB203580). Calibration: 30 pA, 40 msec. E, Summary and comparison of changes in AMPA-EPSCs. *Significant potentiation within one experimental group. In high Mg 2+, PSD-95–GFP increased AMPA currents from 100 ± 17.2 to 204.7 ± 28.3% (n = 22; p = 0.003), in KN-93 from 100 ± 12.0 to 184.2 ± 15.1% (n = 27; p = 0.001), in PD98059 from 100 ± 10.1 to 227.7 ± 16.1% (n = 39; p < 0.0005), and in SB203580 from 100 ± 15.5 to 196.1 ± 11.6% (n = 40; p < 0.0005). Comparison between different treatments was done for AMPAInf/AMPAUninf ratios from each group using KS tests (see Materials and Methods; p values above bars); no significant differences were detected. F, Model for the role of PSD-95 in activity-dependent AMPAR delivery to synapses. Strong synaptic activity leads to signaling events that trigger changes in several proteins, including PSD-95. Synaptic accumulation, multimerization and formation of PDZ interactions by PSD-95 result in the recruitment of extrasynaptic GluR1-containing receptors to synapses, possibly in a complex with PSD-95 and other proteins. Interestingly, an increased level of PSD-95 can drive this process, whereas an increased level of GluR1 does not.
PSD-95 has been shown to interact with regulators of small GTPases of the ras family [i.e., synGAP, a ras inactivator (Chen et al., 1998; Kim et al., 1998) and SPAR, a rapGAP (Pak et al., 2001)]. Ras and rap downstream effectors are mitogen-activated protein kinases (MAPKs), which have been implicated in synaptic plasticity (Sweatt, 2001) and AMPAR trafficking (Zhu et al., 2002). We therefore tested whether PSD-95 affected signaling through these pathways. We used a cell-permeable blocker of MEK (MAP kinase kinase), the enzyme downstream of p42/44-MAPK activation (PD98059) (Dudley et al., 1995) and a blocker of p38-MAPK (SB203580; Lee et al., 1999) at concentrations that block ras and rap downstream signaling in slice cultures (Zhu et al., 2002). Incubation of slice cultures in PD98059 or in SB203580 did not alter the potentiation of AMPA currents by PSD-95 (Fig. 9_C–E_). Our data indicate that PSD-95 acts independently or downstream of the tested signaling pathways implicated in the generation of LTP. Together with our findings that the expression of PSD-95 precisely mimics and dominant negative forms block LTP, we favor a model in which PSD-95 acts downstream of known signaling mediating LTP (Fig. 9_F_). It appears that the increased availability of PSD-95 at the synapse is sufficient to recruit GluR1-containing AMPARs.
Discussion
The molecular mechanisms controlling the delivery and subsequent stabilization of AMPARs during synaptic plasticity are still poorly understood. Recent findings have suggested PSD-95 as candidate molecule in these processes (El-Husseini et al., 2000; Schnell et al., 2002; Beique, 2003). However, particularly in the light of findings from PSD-95 mutant mice that exhibit enhanced LTP (Migaud et al., 1998), it remained controversial whether and how PSD-95 participates in synaptic plasticity. Here, we show that PSD-95 controls AMPAR delivery during synaptic strengthening by LTP in vitro and during experience-driven synaptic plasticity in vivo. We use three criteria to establish this: First, expression of wt PSD-95 mimics, and second wt PSD-95 occludes AMPAR delivery during synaptic strengthening; third, dominant negative forms of PSD-95 block the incorporation of AMPAR during plasticity.
Several findings indicate that expression of PSD-95 mimics key aspects of LTP and experience-driven synaptic potentiation: (1) AMPAR-mediated transmission is selectively enhanced. Interestingly, AMPARs appear to be incorporated at silent and non-silent synapses. (2) PSD-95 drives recombinant GluR1-containing receptors into synapses. (3) PSD-95-mediated potentiation is blocked by recombinant mutant GluR1 [GluR1(T887A)] that also blocks LTP. (4) The GluR1 C-tail prevents potentiation by PSD-95. (5) PSD-95 does not deliver recombinant GluR2(RQ) or endogenous GluR2/3 receptors to synapses. (6) PSD-95 potentiates AMPAR-mediated transmission in animals deprived of whiskers.
In addition, we demonstrate that the expression of PSD-95 occludes synaptic potentiation. Occlusion occurs when a manipulation fully mimics a process and thereby prevents more of the same process. This criterion is satisfied because the expression of PSD-95 (1) mimics AMPAR delivery during LTP and experience-driven plasticity and (2) prevents further LTP or synaptic strengthening in animals with intact whiskers and sensory experience. To address whether PSD-95 normally participates in the delivery of AMPAR during synaptic plasticity, we used dominant negative mutants to block the function of endogenous PSD-95. We reasoned that constructs lacking either membrane targeting and synaptic clustering or PDZ interactions could act as dominant negatives because they retain only some of the properties of PSD-95 necessary to drive potentiation (this study and Schnell et al., 2002). Both dominant negative constructs of PSD-95 blocked LTP in slice cultures. PSD-95AAAA also blocked experience-driven plasticity, because it specifically depressed AMPA- but not NMDA-mediated currents in animals with intact, but not with deprived whiskers. When LTP is occluded or blocked in slice cultures, the initial increase in transmission (within <15 min of induction) appears not or less affected than the later phase (at ∼30 min). This is expected, because experiments that interfere with GluR1 delivery during LTP also selectively affect this late phase (Hayashi et al., 2000; Shi et al., 2001). The initial, transient increase in transmission is likely caused by other events: e.g., changes in the phosphorylation state of AMPARs already at the synapse (Barria et al., 1997; Lee et al., 2000), or movement of AMPARs into synapses that is independent of GluR1-mediated interactions.
Results from slice cultures are fully compatible with those in the barrel cortex in vivo. Similar to the barrel cortex from deprived animals, hippocampal slice cultures are essentially deprived of their natural input. In slice cultures, LTP is necessary and sufficient for the synaptic delivery of GluR1-containing receptors (Hayashi et al., 2000; Shi et al., 2001); in the barrel cortex, whisker-driven activity is necessary and sufficient to strengthen layer IV to II/III transmission and deliver GluR1 (Takahashi et al., 2003). Therefore, dominant negative forms of PSD-95 are not expected to depress basal AMPA transmission in slice cultures (as they do not) but should block LTP (as they do), whereas these constructs should depress AMPA transmission in the barrel cortex of intact whisker animals (as they do). Dominant negative constructs induced a small potentiation (<30%) in slice cultures, which is not enough to occlude LTP (typically, ∼100% increase), but is consistent with a block of LTP. Furthermore, PSD-95AAAA specifically blocked experience-driven AMPAR delivery but had no effect on basal AMPA transmission in the barrel cortex of deprived animals. Accordingly, this explains why PSD-95 strongly potentiates AMPA currents in slice cultures, whereas it only potentiates AMPA currents in whisker-deprived animals.
What are the implications for AMPAR trafficking in vivo? Our study supports conserved mechanisms mediating GluR1 delivery during LTP and by natural stimuli in vivo. Interfering with GluR1 delivery by expressing the GluR1 C-tail or interfering with endogenous PSD-95 by expressing PSD-95AAAA has a very similar effect (∼60 and ∼50% AMPA-depression, respectively) in animals with intact whiskers. This, as well as immunohistochemical detection of these proteins in the developing cortex (Martin et al., 1998; Munoz et al., 1999; Aoki et al., 2001) suggests that both GluR1 and PSD-95 are expressed sufficiently in layer II/III pyramidal neurons at this point. Interestingly, increasing PSD-95 does not induce significant potentiation in intact-whisker animals (only ∼13%). This suggests that between P12 and P14, the delivery of GluR1-containing AMPAR is close to maximally driven by whisker experience. However, this does not exclude the possibility that other mechanisms of synaptic maturation and potentiation operate during this developmental period (e.g., presynaptic or other postsynaptic mechanisms). These mechanisms likely contribute in infected and uninfected neurons and may not be affected by the manipulation of PSD-95.
Early studies had proposed that PSD-95 is important for the localization and clustering of NMDARs at synapses (Kim et al., 1996; Kornau et al., 1997). Indeed, in immature dissociated cerebellar granule cells, the expression of PSD-95 drives a subunit switch in NMDARs from NR2B- to NR2A-containing receptors at synapses (Losi et al., 2003). In contrast, in hippocampus we find no evidence for such a switch. This result could be explained by differences in age and preparation, because there may already be more NR2A subunits at synapses in our hippocampal cultures before we express PSD-95 (compare decay times in Losi et al., 2003, and Barria and Malinow, 2002). Our findings support the idea that PSD-95 assembles scaffolding complexes at synapses that are ultimately required to drive the delivery of AMPARs.
How are our results reconcilable with previous studies on PSD-95? Mice with a disruption of the PSD-95 gene showed a shift of NMDAR-dependent synaptic plasticity toward enhanced LTP (Migaud et al., 1998). However, these animals had altered PSD-95 expression throughout development and expressed low levels of a truncated form of PSD-95. When this truncated form of PSD-95 is expressed in slice cultures, it retains the synaptic potentiating effects of full-length PSD-95 (this study and Schnell et al., 2002). It is possible that chronic, aberrant expression of this fragment together with a reduction in full-length PSD-95 contributes to the phenotype in mutant mice. Because the truncated PSD-95 is unlikely to maintain all functions of the full-length protein, there is a potential for compensatory mechanisms (e.g., from other MAGUKs). This makes it difficult to conclude from studies in the mutant mouse whether and how PSD-95 contributes to synaptic plasticity. Our findings are in line with and expand on previous reports that proposed a role for PSD-95 in recruitment of AMPAR to synapses (Schnell et al., 2002; Beique, 2003).
Could our results be attributable to effects normally mediated by other MAGUKs? We cannot completely rule out this possibility. However, several observations argue against this view. First, expression of other MAGUKs produced no or smaller potentiation [e.g., synapse-associated protein (SAP)-97 and SAP-102 (I. Ehrlich, Y. Hayashi, and R. Malinow, unpublished results; Schnell et al., 2002). Second, the developmental appearance of GluR1 at synapses is mirrored by PSD-95, whereas it runs counter to the developmental decrease of SAP-102 (Petralia et al., 1999; Sans et al., 2000). Third, the onset of sensory-driven activity correlates strongly with the synaptic accumulation of PSD-95 but not SAP-102 in the visual system (Yoshii et al., 2003). Fourth, increased levels of PSD-95 in barrel cortex have been correlated with whisker-mediated learning (Skibinska et al., 2001). We thus favor the view that our results are attributable to effects normally mediated by PSD-95. Nevertheless, in other forms of plasticity, (e.g., TTX-induced plasticity in dissociated cultured neurons) other MAGUKs or other mechanisms may contribute to synaptic potentiation (Ehlers, 2003).
How does PSD-95 enhance transmission? Our mutational analysis suggests that synaptic targeting and perhaps multimerization of PSD-95 as well as its ability to interact with PDZ ligands play a major role. AMPA-EPSCs are potentiated because PSD-95 recruits nonsynaptic GluR1-containing receptors to synaptic sites without affecting the cycling pool of AMPARs. Because PSD-95 is not known to bind directly to GluR1, there must be intervening proteins. One possibility is an interaction of GluR1 via protein 4.1N and cytoskeletal elements with the PSD-95 signaling complex (Shen et al., 2000; Lisman and Zhabotinsky, 2001). Other compelling candidates are members of the transmembrane AMPAR regulatory protein (TARP) family, one of which is stargazin, which can bind AMPARs and PSD-95 (Chen et al., 2000b; Tomita et al., 2003). TARPs may regulate AMPAR trafficking to the cell membrane and then, through interaction with PSD-95, recruit AMPAR to synaptic sites (Chen et al., 2000b; Schnell et al., 2002; Tomita et al., 2003). However, it is not yet clear whether AMPAR–TARP–PSD-95 association is driven by stimuli that induce synaptic plasticity. Several of our findings suggest that other proteins or mechanisms may be necessary to drive GluR1 into synapses. First, the GluR1 C-tail and GluR1(T887A) prevent potentiation by PSD-95 (and during LTP), indicating the requirement of one or several other proteins in the delivery pathway, because TARPs are not thought to interact with the GluR1 C-tail directly (Chen et al., 2000b; Tomita et al., 2003). Second, PSD-95 drives GluR1-containing but not GluR2/3-containing receptors into synapses, but to date little specificity has been shown in TARPs binding to GluRs and MAGUKs (Chen et al., 2000b; Tomita et al., 2003). Thus, it is possible that other proteins or mechanisms mediate subunit specificity.
Our pharmacological results suggest that PSD-95 does not act by enhancing known signaling pathways implicated in synaptic plasticity, but functions downstream of them. Interestingly, an increase in the availability of PSD-95 at the synapse results in potentiation, whereas an increase in the level of GluR1 does not (Shi et al., 1999; Hayashi et al., 2000). From these findings, we propose a model (Fig. 9_E_) in which plasticity-inducing activity (LTP or experience-driven activity) triggers signaling events leading to changes in effector proteins, including PSD-95. Synaptic accumulation, multimerization and PDZ interactions by PSD-95 result in the recruitment of GluR1-containing receptors to synapses, possibly in a complex with PSD-95 and other proteins. Consistent with our findings, PSD-95 levels increase at synapses during developmental AMPAfication (Sans et al., 2000) or during sensory- or learning-induced plasticity (Skibinska et al., 2001; Yoshii et al., 2003). It will be interesting to determine whether and how PSD-95 can be locally elevated at synaptic sites under physiological conditions to induce synaptic plasticity.
Note. A study published during the review of this manuscript (Stein et al., 2003) shows results that are similar to some presented here.
Footnotes
This work was supported by the German Academic Exchange Service (I.E.), the Deutsche Forschungsgemeinschaft (I.E.), the National Institutes of Health (R.M.), and the Alle Davis and Maxine Harrison Endowment (R.M.). We thank Dr. S. Okabe (Tokyo Medical and Dental University, Tokyo, Japan) and Dr. B. Glick (University of Chicago, Chicago, IL) for providing cDNA, Dr. Y. Hayashi (Massachusetts Institute of Technology, Cambridge, MA) for the initial batch of PSD-95–GFP virus, N. Dawkins-Pisani for technical assistance, B. Burbach for help with two-photon imaging, M. Reigl and C. Kopec for programming, S. Rumpel and T. Takahashi for help with in vivo injections, C. Aizenman, A. Holtmaat, and K. Zito for comments on a previous version of this manuscript, and members of the Malinow laboratory for helpful discussion.
Correspondence should be addressed to Dr. Roberto Malinow, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724. E-mail: Malinow@cshl.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240916-12$15.00/0
References
- Aoki C, Miko I, Oviedo H, Mikeladze-Dvali T, Alexandre L, Sweeney N, Bredt DS (2001) Electron microscopic immunocytochemical detection of PSD-95, PSD-93, SAP-102, and SAP-97 at postsynaptic, presynaptic, and nonsynaptic sites of adult and neonatal rat visual cortex. Synapse 40: 239–257. [DOI] [PubMed] [Google Scholar]
- Arnold DB, Clapham DE (1999) Molecular determinants for subcellular localization of PSD-95 with an interacting K+ channel. Neuron 23: 149–157. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R (2002) Subunit-specific NMDA receptor trafficking to synapses. Neuron 35: 345–353. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR (1997) Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276: 2042–2045. [DOI] [PubMed] [Google Scholar]
- Beique J-CA, R (2003) PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol (Lond) 546: 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Lomo T (1973) Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 232: 331–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA (2003) AMPA receptor trafficking at excitatory synapses. Neuron 40: 361–379. [DOI] [PubMed] [Google Scholar]
- Chen BE, Lendvai B, Nimchinsky EA, Burbach B, Fox K, Svoboda K (2000a) Imaging high-resolution structure of GFP-expressing neurons in neocortex in vivo. Learn Mem 7: 433–441. [DOI] [PubMed] [Google Scholar]
- Chen HJ, Rojas-Soto M, Oguni A, Kennedy MB (1998) A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 20: 895–904. [DOI] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA (2000b) Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 408: 936–943. [DOI] [PubMed] [Google Scholar]
- Craven SE, El-Husseini AE, Bredt DS (1999) Synaptic targeting of the postsynaptic density protein PSD-95 mediated by lipid and protein motifs. Neuron 22: 497–509. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7–61. [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR (1995) A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci USA 92: 7686–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6: 231–242. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290: 1364–1368. [PubMed] [Google Scholar]
- Garner CC, Nash J, Huganir RL (2000) PDZ domains in synapse assembly and signalling. Trends Cell Biol 10: 274–280. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287: 2262–2267. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S (1994) Cloned glutamate receptors. Annu Rev Neurosci 17: 31–108. [DOI] [PubMed] [Google Scholar]
- Hsueh YP, Sheng M (1999) Requirement of N-terminal cysteines of PSD-95 for PSD-95 multimerization and ternary complex formation, but not for binding to potassium channel Kv1.4. J Biol Chem 274: 532–536. [DOI] [PubMed] [Google Scholar]
- Kim E, Cho KO, Rothschild A, Sheng M (1996) Heteromultimerization and NMDA receptor-clustering activity of Chapsyn-110, a member of the PSD-95 family of proteins. Neuron 17: 103–113. [DOI] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau LF, Huganir RL (1998) SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 20: 683–691. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269: 1737–1740. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Seeburg PH, Kennedy MB (1997) Interaction of ion channels and receptors with PDZ domain proteins. Curr Opin Neurobiol 7: 368–373. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL (2000) Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405: 955–959. [DOI] [PubMed] [Google Scholar]
- Lee JC, Kassis S, Kumar S, Badger A, Adams JL (1999) p38 mitogen-activated protein kinase inhibitors–mechanisms and therapeutic potentials. Pharmacol Ther 82: 389–397. [DOI] [PubMed] [Google Scholar]
- Lendvai B, Stern EA, Chen B, Svoboda K (2000) Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature 404: 876–881. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Zhabotinsky AM (2001) A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 31: 191–201. [DOI] [PubMed] [Google Scholar]
- Losi G, Prybylowski K, Fu Z, Luo J, Wenthold RJ, Vicini S (2003) PSD-95 regulates NMDA receptors in developing cerebellar granule neurons of the rat. J Physiol (Lond) 548: 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126. [DOI] [PubMed] [Google Scholar]
- Malinow R, Schulman H, Tsien RW (1989) Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245: 862–866. [DOI] [PubMed] [Google Scholar]
- Malinow R, Mainen ZF, Hayashi Y (2000) LTP mechanisms: from silence to four-lane traffic. Curr Opin Neurobiol 10: 352–357. [DOI] [PubMed] [Google Scholar]
- Marrs GS, Green SH, Dailey ME (2001) Rapid formation and remodeling of postsynaptic densities in developing dendrites. Nat Neurosci 4: 1006–1013. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Furuta A, Blackstone CD (1998) AMPA receptor protein in developing rat brain: glutamate receptor-1 expression and localization change at regional, cellular, and subcellular levels with maturation. Neuroscience 83: 917–928. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H (2001) Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4: 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK (2000) Biolistic transfection of neurons. Sci STKE 2000: PL1. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Beaulieu C (1996) Quantitative aspects of synaptogenesis in the rat barrel field cortex with special reference to GABA circuitry. J Comp Neurol 373: 340–354. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG (1998) Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396: 433–439. [DOI] [PubMed] [Google Scholar]
- Munoz A, Woods TM, Jones EG (1999) Laminar and cellular distribution of AMPA, kainate, and NMDA receptor subunits in monkey sensory-motor cortex. J Comp Neurol 407: 472–490. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Kim E, Sheng M (1996) Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci 16: 2157–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimune A, Isaac JT, Molnar E, Noel J, Nash SR, Tagaya M, Collingridge GL, Nakanishi S, Henley JM (1998) NSF binding to GluR2 regulates synaptic transmission. Neuron 21: 87–97. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P (1998) Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron 21: 545–559. [DOI] [PubMed] [Google Scholar]
- Okabe S, Kim HD, Miwa A, Kuriu T, Okado H (1999) Continual remodeling of postsynaptic density and its regulation by synaptic activity. Nat Neurosci 2: 804–811. [DOI] [PubMed] [Google Scholar]
- Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB (1998) The AMPA receptor GluR2 C terminus can mediate a reversible, ATP-dependent interaction with NSF and alpha- and beta-SNAPs. Neuron 21: 99–110. [DOI] [PubMed] [Google Scholar]
- Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M (2001) Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron 31: 289–303. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Esteban JA, Wang YX, Partridge JG, Zhao HM, Wenthold RJ, Malinow R (1999) Selective acquisition of AMPA receptors over postnatal development suggests a molecular basis for silent synapses. Nat Neurosci 2: 31–36. [DOI] [PubMed] [Google Scholar]
- Pettit DL, Perlman S, Malinow R (1994) Potentiated transmission and prevention of further LTP by increased CaMKII activity in postsynaptic hippocampal slice neurons. Science 266: 1881–1885. [DOI] [PubMed] [Google Scholar]
- Piccini A, Malinow R (2002) Critical postsynaptic density 95/disc large/zonula occludens-1 interactions by glutamate receptor 1 (GluR1) and GluR2 required at different subcellular sites. J Neurosci 22: 5387–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos II J, Hell JW, Wenthold RJ (2000) A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci 20: 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL (2000) Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci 1: 133–141. [DOI] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA (2002) Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA 99: 13902–13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Liang F, Walensky LD, Huganir RL (2000) Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1N-linked actin cytoskeletal association. J Neurosci 20: 7932–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Lee SH (2001) AMPA receptor trafficking and the control of synaptic transmission. Cell 105: 825–828. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sala C (2001) PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci 24: 1–29. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105: 331–343. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R (1999) Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 284: 1811–1816. [DOI] [PubMed] [Google Scholar]
- Skibinska A, Lech M, Kossut M (2001) PSD95 protein level rises in murine somatosensory cortex after sensory training. NeuroReport 12: 2907–2910. [DOI] [PubMed] [Google Scholar]
- Stein V, House DR, Bredt DS, Nicoll RA (2003) Postsynaptic density-95 mimics and occludes hippocampal long-term potentiation and enhances long-term depression. J Neurosci 23: 5503–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Maravall M, Svoboda K (2001) Rapid development and plasticity of layer 2/3 maps in rat barrel cortex in vivo. Neuron 31: 305–315. [DOI] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H (1991) The newly synthesized selective Ca 2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun 181: 968–975. [DOI] [PubMed] [Google Scholar]
- Sweatt JD (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76: 1–10. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Svoboda K, Malinow R (2003) Experience strengthens transmission by driving AMPA receptors into synapses. Science 299: 1585–1588. [DOI] [PubMed] [Google Scholar]
- Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA, Bredt DS (2003) Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol 161: 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topinka JR, Bredt DS (1998) N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron 20: 125–134. [DOI] [PubMed] [Google Scholar]
- Vees AM, Micheva KD, Beaulieu C, Descarries L (1998) Increased number and size of dendritic spines in ipsilateral barrel field cortex following unilateral whisker trimming in postnatal rat. J Comp Neurol 400: 110–124. [PubMed] [Google Scholar]
- Welker C (1971) Microelectrode delineation of fine grain somatotopic organization of (SmI) cerebral neocortex in albino rat. Brain Res 26: 259–275. [PubMed] [Google Scholar]
- Welker C (1976) Receptive fields of barrels in the somatosensory neocortex of the rat. J Comp Neurol 166: 173–189. [DOI] [PubMed] [Google Scholar]
- Welker C, Woolsey TA (1974) Structure of layer IV in the somatosensory neocortex of the rat: description and comparison with the mouse. J Comp Neurol 158: 437–453. [DOI] [PubMed] [Google Scholar]
- Woolsey TA, Van der Loos H (1970) The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex: the description of a cortical field composed of discrete cytoarchitectonic units. Brain Res 17: 205–242. [DOI] [PubMed] [Google Scholar]
- Yoshii A, Sheng MH, Constantine-Paton M (2003) Eye opening induces a rapid dendritic localization of PSD-95 in central visual neurons. Proc Natl Acad Sci USA 100: 1334–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura Y, Aoi C, Yamauchi T (2000) Investigation of protein substrates of Ca(2+)/calmodulin-dependent protein kinase II translocated to the postsynaptic density. Brain Res Mol Brain Res 81: 118–128. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R (2000) Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci 3: 1098–1106. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R (2002) Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110: 443–455. [DOI] [PubMed] [Google Scholar]