Chronic Exposure to Rotenone Models Sporadic Parkinson's Disease in Drosophila melanogaster (original) (raw)
Abstract
Parkinson's disease (PD) is a movement disorder characterized by the selective degeneration of nigrostriatal dopaminergic neurons. Both familial and sporadic cases present tremor, rigidity, slowness of movement, and postural instability. Although major insights into the genes responsible for some rare hereditary cases have arisen, the etiology of sporadic cases remains unknown. Epidemiological studies have suggested an association with environmental toxins, mainly mitochondrial complex I inhibitors such as the widely used pesticide rotenone. In recent years, Drosophila melanogaster has been used as a model of several neurodegenerative diseases, including a genetic model of PD. Here, we studied the neurodegenerative and behavioral effects of a sublethal chronic exposure to rotenone in Drosophila. After several days, the treated flies presented characteristic locomotor impairments that increased with the dose of rotenone. Immunocytochemistry analysis demonstrated a dramatic and selective loss of dopaminergic neurons in all of the brain clusters. The addition of l-dopa (3,4-dihydroxy-l-phenylalanine) into the feeding medium rescued the behavioral deficits but not neuronal death, as is the case in human PD patients. In contrast, the antioxidant melatonin (_N_-acetyl-5-methoxytryptamine) alleviated both symptomatic impairment and neuronal loss, supporting the idea that this agent may be beneficial in the treatment of PD. Therefore, chronic exposure to pesticides recapitulates key aspects of PD in Drosophila and provides a new in vivo model for studying the mechanisms of dopaminergic neurodegeneration.
Keywords: dopamine, rotenone, melatonin, neurodegeneration, Drosophila melanogaster, Parkinson's disease
Introduction
Parkinson's disease (PD) is the most frequent neurodegenerative movement disorder (Olanow and Tatton, 1999). Its pathological hallmarks are a progressive degeneration of nigrostriatal dopaminergic neurons and the presence of neuronal cytoplasmic inclusions containing ubiquitinated proteins, the Lewy bodies. Both familial and sporadic cases of PD are characterized by tremor, rigidity, slowness of movement, and postural instability. However, these symptoms patently appear at a late stage of the disease when >70% of the nigrostriatal dopaminergic neurons are already dead (Lees, 1992). No treatment is currently available that could delay or stop this dopamine neuron degeneration.
The specific molecular mechanisms leading to neuronal death in PD are still not understood (Dauer and Przedborski, 2003; Giasson and Lee, 2003). Recent insights have led to the discovery of genes responsible for hereditary cases of PD or related disorders, in particular α_-synuclein_ and parkin. However, these familial forms of the disease are rare. More than 95% of late-onset idiopathic PD cases are sporadic, and their etiology remains unknown. Postmortem studies implicated mitochondrial impairments (Schapira et al., 1990), and epidemiological studies (Di Monte, 2003) suggested an association with environmental toxins, in particular mitochondrial complex I inhibitors such as rotenone. Rotenone is a commonly used natural pesticide prepared from the roots of certain tropical plants, such as Derris elliptica. This lipophilic compound freely crosses cell membranes and accesses cytoplasm and mitochondria.
In vitro, rotenone has been shown to produce cell apoptosis, accumulation and aggregation of α-synuclein and ubiquitin, oxidative damage, and endoplasmic reticulum stress (Ryu et al., 2002; Sherer et al., 2002). In vivo, chronic systemic exposure to rotenone has been used to model PD in the rat, in which it was shown to induce dopaminergic neurodegeneration, Parkinson-like behavior, and the occurrence of cytoplasmic inclusions similar to the Lewy bodies (Betarbet et al., 2000). However, several observations have questioned the reliability of this model. Significant differences were observed between rats of the same strain, making it difficult to test new pharmacological treatments. It has been reported recently that rotenone leads to nonselective neuronal death in this model (Hoglinger et al., 2003), whereas another study suggested that this may in part depend on the mode of administration of the pesticide (Sherer et al., 2003). Finally, it is unknown whether the movement phenotypes of the rotenone-treated rat result from dopamine deficiency and thus can be used to assess the degree of dopaminergic degeneration.
In recent years, Drosophila melanogaster has been used as a model of several neurodegenerative diseases (Bonini and Fortini, 2003), including a genetic model of PD based on directed expression of human α_-synuclein_ in the Drosophila brain (Feany and Bender, 2000; Auluck et al., 2002). As an advantage, this invertebrate offers a large panel of genetic approaches and allows the rapid screening of potential therapeutic drugs. Here, we studied the neurodegenerative and behavioral effects of a sublethal chronic exposure to rotenone in Drosophila. We found that the treated flies presented a selective loss of dopaminergic neurons in the brain and severe locomotor impairments. Importantly, the addition of 3,4-dihydroxy-l-phenylalanine (l-dopa) to the feeding medium rescued the motor deficits but did not prevent dopaminergic cell death, as in human PD patients. In contrast, we observed that the antioxidant _N_-acetyl-5-methoxytryptamine (melatonin) rescued both rotenone-induced behavioral defects and neuronal loss.
Materials and Methods
Drosophila culture and drug treatment. Flies were maintained at 25°C on a standard cornmeal-agar diet. Because we observed variability in the relative sensitivity of different Drosophila strains to rotenone, all of the experiments presented here were performed with the same y, w strain (The FlyBase Consortium, 2003). For the drug feeding experiments, 20 adults were placed in a vial and incubated at 22°C for 7 d. We used Formula 4-24 instant Drosophila medium (Carolina Biological Supply, Burlington, NC) rehydrated with a solution containing 0-500 μm rotenone. This molecule is rather unstable, so the rotenone powder was kept in the freezer and used within 1 month. We made sure that freshly made solutions were transparent or pale yellow. Dark-colored solutions were discarded. New vials were tested on a small number of flies before sizing up the experiments. When required, l-dopa and melatonin were added in the food at 1 and 5 mm final concentrations, respectively. All chemicals were obtained from Sigma (St. Louis, MO).
Locomotion assay. We determined locomotor ability with a negative geotaxis assay as described previously (Feany and Bender, 2000; Friggi-Grelin et al., 2003; Rival et al., 2004). Twenty females (∼14 d of age) were anesthetized and placed in a vertical plastic column (length, 25 cm; diameter, 1.5 cm). After a 30 min recovery from CO2 exposure, flies were gently tapped to the bottom of the column. After 1 min, flies that reached the top of the column and flies that remained at the bottom were counted separately. Three trials were performed in each experiment at 1 min intervals. The scores are the mean of the numbers of flies at the top (_n_top) and at the bottom (_n_bot), expressed as percentages of the total number of flies (_n_tot). Results are presented as the mean ± SEM of the scores obtained in three independent experiments. For each experiment, a performance index (PI) was calculated, defined as 1/2[(_n_tot + _n_top - _n_bot)/_n_tot]. Statistical analysis was performed on the PIs with the Student's t test.
Immunostaining and cell counting. Fluorescent immunodetection was performed on paraformaldehyde-fixed, whole-mount dissected nervous tissue (Friggi-Grelin et al., 2003). The following antibodies were used: an affinity purified rabbit polyclonal antibody against rat tyrosine hydroxylase (Pel-Freez Biologicals, Rogers, AR) at a 1:25 dilution, rat polyclonal anti-Elav (9F8A9; Developmental Studies Hybridoma Bank, Iowa City, IA) at a 1:5 dilution, and rabbit polyclonal anti-serotonin (Sigma) at a 1:200 dilution. FITC-labeled goat anti-rabbit IgG, tetramethylrhodamine isothiocyanate-labeled goat anti-rabbit IgG, and FITC-labeled goat anti-rat IgG (Jackson ImmunoResearch, West Grove, PA) were used as secondary antibodies at a 1:250 dilution. Tissues were mounted in Vectashield (Vector Laboratories, Burlingame, CA). Confocal microscopy was performed on a Zeiss (Oberkochen, Germany) LSM 510 microscope. The number of dopaminergic and serotoninergic neurons was scored in whole-mount brains by direct observation on a Leica (Nussloch, Germany) DMRB microscope. Control experiments were always performed in parallel with flies incubated in the absence of drug. Statistical analysis was performed with the Student's t test.
Results
Exposure to rotenone causes locomotor deficits
To determine the effects of rotenone in Drosophila, we placed flies in contact with a rotenone-supplemented feeding medium. The pesticide was applied at sublethal doses (i.e., <750 μm). Because this drug is extremely lipophilic, it can cross the gut barrier and possibly the insect external cuticle to get into the brain. Because of these barriers and the powerful excretion system of flies, the final concentration of rotenone in the brain was probably much lower than the initial concentration in the food medium. After 7 d, flies presented major locomotor defects. They had a tendency to stay at the bottom of vials and did not appear to coordinate their legs normally. This phenotype was quantified with a negative geotaxis test (see Materials and Methods). We observed that in the absence of rotenone, >90% of the flies reached the top of the column in 1 min. In contrast, the higher the concentration of rotenone in the medium, the higher the number of flies that were unable to climb (Fig. 1_A_). The half-effect was obtained at ∼25 μm rotenone.
Figure 1.
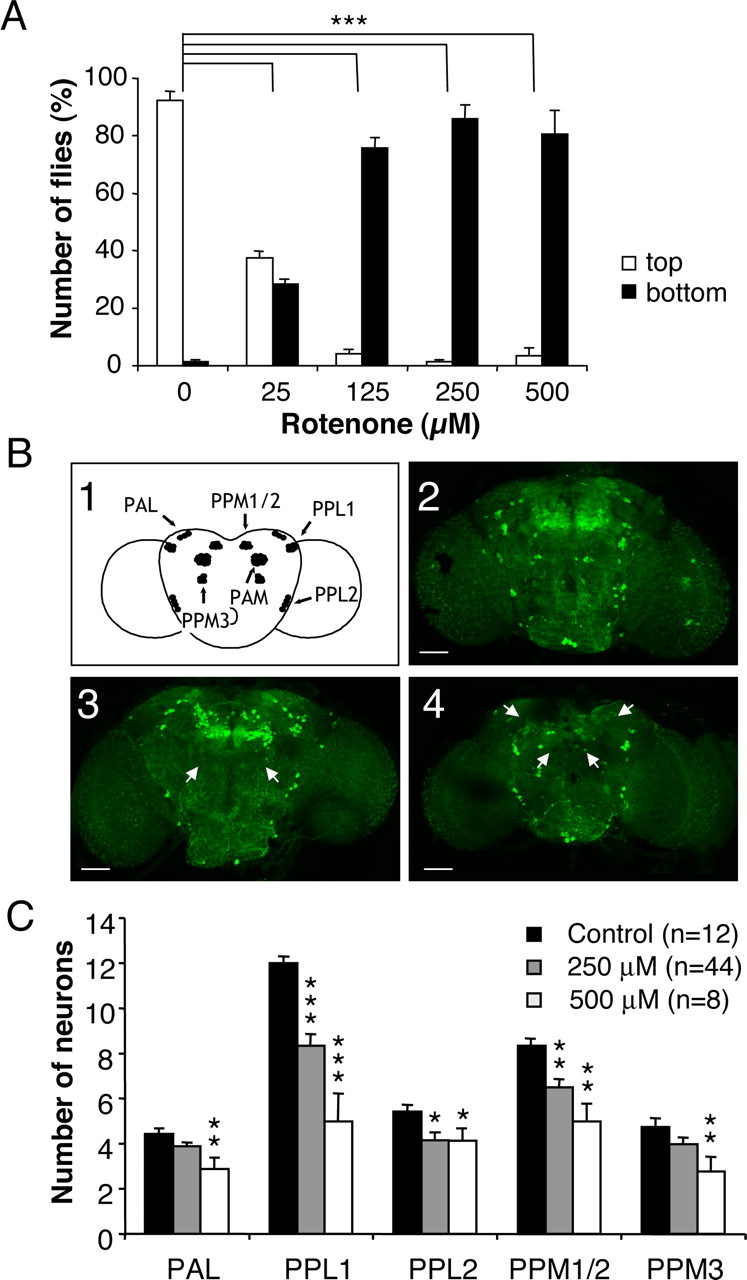
Exposure of Drosophila to rotenone induces severe locomotor deficits and dopaminergic neuron loss. A, Negative geotaxis assay of adult flies exposed previously for 7 d to various amounts of rotenone. White bars indicate the percentage of flies that climbed to the top of the column, and black bars indicate the percentage of flies that remained at the bottom after 1 min. Differences in PI between control and rotenone-treated flies were highly significant (***p < 0.001). B1, Schematic representation of the dopaminergic neuron clusters in Drosophila adult brain in frontal view. _B2_-B4, Tyrosine hydroxylase immunolabeling showing dopaminergic neuron patterns in multifocal confocal views of adult fly brains after 7 d of exposure to 0 (control), 250, and 500 μm rotenone, respectively. White arrows indicate the total absence of certain clusters after rotenone exposure (PPM3 in B3 and PAM and PAL in B4). Scale bars, 50 μm. C, Quantification of the number of neurons in dopaminergic clusters of control brains (black bars) or in brains of flies exposed for 7 d to 250 μm (gray bars) or 500 μm (white bars) rotenone. The density of the neurons in the PAM clusters was too high to allow precise scoring of their number. n indicates the number of brain hemispheres examined in each condition. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with control values.
Rotenone induces dopaminergic neuron degeneration
To investigate whether rotenone administration induced dopaminergic neuron loss, brains of flies exposed to 250 and 500 μm rotenone for 7 d were dissected and immunostained for the dopamine biosynthesis enzyme tyrosine hydroxylase. Six neuronal dopaminergic clusters are normally present in each Drosophila adult brain hemisphere (Budnik and White, 1988; Nässel and Elekes, 1992; Friggi-Grelin et al., 2003); these clusters are schematized in Figure 1_B1_ and can be seen in control brain (Fig. 1_B2_). After exposure to rotenone, confocal projections of whole-mount brains showed abnormalities characterized by the disappearance of part or the totality of dopaminergic cell clusters (Fig. 1_B3,B4_). Although this effect varied in intensity from one fly to another, drastic phenotypes were observed frequently, such as the total disappearance of the PPM3 clusters (Fig. 1_B3_) or the PAL and PAM clusters (Fig. 2_B4_) (see also Fig. 2_A_). We quantified cell loss by individually counting neurons in each cluster (except the PAM cluster, in which the density of neurons is too high to allow precise numbering of the cells). Statistically, neuronal loss affected all of the dopaminergic clusters indiscriminately and appeared generally to increase with the concentration of rotenone (Fig. 1_C_).
Figure 2.
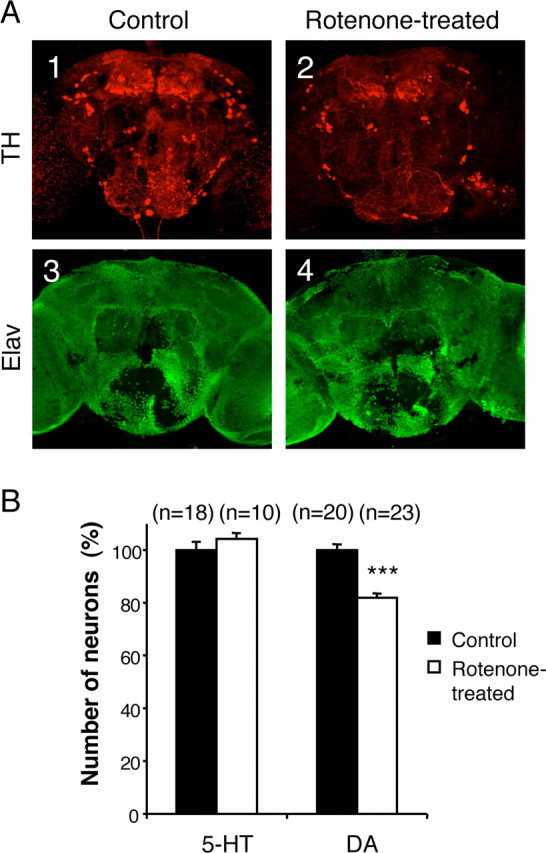
Selectivity of the neuronal degeneration induced by rotenone. A, Coimmunolabeling of adult brains with anti-tyrosine hydroxylase (TH) (A1, A2) and anti-Elav (A3, A4) antibodies after 7 d of exposure to 125 μm rotenone. The pesticide induced obvious dopaminergic neuron loss, whereas the general pattern of the pan-neuronal Elav marker was not modified. B, Total number of serotoninergic (5-HT) and dopaminergic (DA) neurons in fly brains exposed for 7 d to 125 μm rotenone (white bars) compared with control brains (black bars), expressed as a percentage of the mean value of the control. n indicates the number of brain hemispheres examined in each condition. ***p < 0.001.
Neuronal selectivity of the effect of rotenone
Figure 2_A_ shows anti-tyrosine hydroxylase and anti-Elav coimmunostaining of brains from adult flies kept for 7 d in the absence (Fig. 2_A1_-A3, control) or presence (Fig. 2_A2_-A4) of 125 μm rotenone. Exposure to rotenone did not elicit visible changes in the general morphology of the brain or a noticeable decrease in volume. Although the pesticide induced obvious dopaminergic neuron loss, no vacuolization was apparent, and no difference could be observed among the patterns of brain neuron nuclei revealed by anti-Elav immunostaining, indicating that rotenone did not induce widespread neurodegeneration. Moreover, in conditions that induced locomotor deficits and dopamine neuron loss, we observed that the number of serotonin neurons was not altered (Fig. 2 B). Therefore, the degeneration triggered by rotenone appears to affect the dopaminergic neurons selectively.
Correlation between the locomotor phenotype and dopamine deficiency
In humans, the administration of l-dopa, a precursor of dopamine biosynthesis, is the most effective pharmacological symptomatic treatment of PD. We tested this drug on flies by coadministering it together with rotenone in the feeding medium. As shown in Figure 3_A_, 1 mm l-dopa had no effect by itself on fly behavior, but it improved significantly the performances of Drosophila treated with 125 μm rotenone in the negative geotaxis test. These flies were obviously more active than flies treated with rotenone only, and a number of them were able to reach the top of the column in the locomotor test. This result suggests that the locomotor deficits induced by rotenone are caused by dopamine deficiency. Higher concentrations of l-dopa could not be tested, because they had deleterious effects on fly behavior (data not shown). Despite the improvement in locomotor activity, we observed that the loss of dopaminergic neurons was not significantly different in flies fed with rotenone only and in rotenone-fed flies behaviorally rescued with 1 mm l-dopa (Fig. 3_B_).
Figure 3.

l-Dopa improves locomotor phenotype but does not lessen dopaminergic neuron loss. A, Negative geotaxis assay of adult flies exposed previously for 7 d to 125 μm rotenone, 1 mm l-dopa, or both, compared with control flies maintained in the absence of drugs. Representation and statistics are the same as in Figure 1 A. B, Quantification of neurons in control brains and in brains of flies exposed for 7 d to 125 μm rotenone (R) with or without 1 mm l-dopa (LD). The number of dopaminergic neurons and the loss of cells as a result of rotenone exposure were not significantly altered by l-dopa. n indicates the number of brain hemispheres examined in each condition. Statistics are the same as in Figure 1_C_.
Melatonin protects dopaminergic neurons from rotenone
Drosophila were then coexposed to rotenone and melatonin, an antioxidant and free radical scavenger (Coto-Montes and Hardeland, 1999; Reiter et al., 2001). Melatonin clearly improved the movement behavior of rotenone-treated flies, even more evidently than l-dopa. Accordingly, it also improved their score in the negative geotaxis test (Fig. 4_A_). Quantification of the number of dopaminergic cells after 1 week of rotenone feeding revealed that the presence of melatonin significantly rescued the loss of neurons in all of the clusters. After treatment with rotenone plus melatonin, the number of neurons in some clusters was identical to that of the untreated control (Fig. 4_B_).
Figure 4.

Melatonin improves locomotor deficits and protects dopaminergic neurons. A, Negative geotaxis assay of adult flies exposed previously for 7 d to 125 μm rotenone, 5 mm melatonin, or both, compared with control flies maintained in the absence of drugs. Representation is the same as that in Figure 1 A. B, Quantification of dopaminergic neurons in brains of control flies exposed or not exposed for 7 d to 5 mm melatonin (M) and in flies exposed for 7 d to 125 μm rotenone (R) with or without 5 mm melatonin. n indicates the number of brain hemispheres examined in each condition. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with flies exposed to rotenone without melatonin.
Discussion
In this study, we show that chronic exposure of Drosophila to sublethal doses of rotenone, a respiratory chain complex I inhibitor, recapitulates the main symptomatic feature of PD: a selective loss of dopaminergic neurons inducing locomotor deficits. In agreement with previous studies (Betarbet et al., 2000; Di Monte, 2003), these results suggest that rotenone and other complex I inhibitors used as pesticides or herbicides are environmental factors that may contribute to the appearance of sporadic PD in humans.
We noted several differences between this new Drosophila PD model and the model based on the expression of human α_-synuclein_ in the Drosophila brain (Feany and Bender, 2000; Auluck et al., 2002). First, the loss of neurons appears more widespread in the rotenone model. Indeed, α-synuclein was reported to induce 20-50% dopamine cell loss in two brain clusters when rotenone affected all of the dopaminergic clusters, and in some cases, whole clusters were found to disappear. Second, at variance with the α-synuclein model, we could not detect Lewy body-like cytoplasmic inclusions using an anti-ubiquitin antibody (data not shown). However, this was expected, because α-synuclein molecules, which are absent in Drosophila, are the primary constituent of the fibrils forming Lewy bodies (Goedert, 2001). This indicates that the appearance of Lewy bodies is not necessary for dopamine neuron loss. Accordingly, mutations in parkin lead to autosomal recessive juvenile parkinsonism, a human disease symptomatically related to PD that is characterized by the absence of Lewy bodies (Kitada et al., 1998). Third, although expression of α_-synuclein_ can induce progressive retinal degeneration in flies (Feany and Bender, 2000), no such effect could be demonstrated with rotenone. Fourth, a correlation between the locomotor deficits and dopaminergic cell loss was not clearly established in the α-synuclein model (Feany and Bender, 2000; Beal, 2001; Auluck et al., 2002). In the rotenone model, l-dopa significantly rescued the locomotor deficits, arguing for a selective effect of this toxin on dopaminergic neurons.
Little is known about the precise mechanism of rotenone action that can lead to neurodegeneration. Previous studies suggested that pesticides and herbicides could act by aggregating α-synuclein (Uversky et al., 2001; Manning-Bog et al., 2002). Because no homolog of α_-synuclein_ is present in the Drosophila genome, such a mechanism could not be relevant here. The selectivity of rotenone action is likely attributable to a specific sensitivity of Drosophila dopaminergic neurons to reactive oxygen species and oxidative damages. Signs of oxidative damages have been detected frequently in dopaminergic neurons from PD patients, suggesting an implication of oxidative stress in this disease (Beal, 2003; Dauer and Przedborski, 2003; Jenner, 2003). We reported recently a moderate level of dopamine neuron death when glutamate transport is abolished in the Drosophila brain, a condition that increases oxidative stress (Rival et al., 2004). However, in comparison, the neurodegenerative effect of rotenone appears more severe and more selective.
In our model, feeding l-dopa to flies did not reduce cell loss, indicating that this drug acts simply as a dopamine precursor to rescue rotenone-induced endogenous dopamine deficits. Similarly, in humans, l-dopa treatment is not efficient to halt dopaminergic neuron degeneration. In contrast, the prevention of both locomotor deficits and neuron degeneration in Drosophila by the antioxidant melatonin suggests that rotenone-induced cell death results from an oxidative process. However, melatonin could also act by a different mechanism unrelated to its antioxidant properties. Such a protective effect of melatonin has been demonstrated in other experimental models of PD (Dabbeni-Sala et al., 2001; Antolin et al., 2002). This suggests that melatonin could be used to prevent oxidative stress occurring during PD in humans.
In conclusion, chronic exposure of Drosophila to rotenone recapitulates key aspects of sporadic PD and provides a genetically tractable animal model to analyze the molecular mechanisms relating oxidative stress to dopamine neuron death.
Footnotes
This work was supported by a grant from the Fondation de France (S.B.). We thank all members of our group and Alexis Brice for helpful discussions and Olivier Monaco for assistance in digital art preparation.
Correspondence should be addressed to Serge Birman, Centre National de la Recherche Scientifique, Université de la Méditerranée, Developmental Biology Institute of Marseille, Campus de Luminy, Case 907, F-13288 Marseille Cedex 9, France. E-mail: birman@ibdm.univ-mrs.fr.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410993-06$15.00/0
References
- Antolin I, Mayo JC, Sainz RM, del Brio Mde L, Herrera F, Martin V, Rodriguez C (2002) Protective effect of melatonin in a chronic experimental model of Parkinson's disease. Brain Res 943: 163-173. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 295: 865-868. [DOI] [PubMed] [Google Scholar]
- Beal MF (2001) Experimental models of Parkinson's disease. Nat Rev Neurosci 2: 325-334. [DOI] [PubMed] [Google Scholar]
- Beal MF (2003) Mitochondria, oxidative damage, and inflammation in Parkinson's disease. Ann NY Acad Sci 991: 120-131. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3: 1301-1306. [DOI] [PubMed] [Google Scholar]
- Bonini NM, Fortini ME (2003) Human neurodegenerative disease modeling using _Drosophila_Annu Rev Neurosci 26: 627-656. [DOI] [PubMed] [Google Scholar]
- Budnik V, White K (1988) Catecholamine containing neurons in Drosophila melanogaster: distribution and development. J Comp Neurol 268: 400-413. [DOI] [PubMed] [Google Scholar]
- Coto-Montes A, Hardeland R (1999) Antioxidative effects of melatonin in Drosophila melanogaster: antagonization of damage induced by the inhibition of catalase. J Pineal Res 27: 154-158. [DOI] [PubMed] [Google Scholar]
- Dabbeni-Sala F, Di Santo S, Franceschini D, Skaper SD, Giusti P (2001) Melatonin protects against 6-OHDA-induced neurotoxicity in rats: a role for mitochondrial complex I activity. FASEB J 15: 164-170. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39: 889-909. [DOI] [PubMed] [Google Scholar]
- Di Monte DA (2003) The environment and Parkinson's disease: is the nigrostriatal system preferentially targeted by neurotoxins? Lancet Neurol 2: 531-538. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW (2000) A Drosophila model of Parkinson's disease. Nature 404: 394-398. [DOI] [PubMed] [Google Scholar]
- Friggi-Grelin F, Coulom H, Meller M, Gomez D, Hirsh J, Birman S (2003) Targeted gene expression in Drosophila dopaminergic cells using regulatory sequences from tyrosine hydroxylase. J Neurobiol 54: 618-627. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Lee VM (2003) Are ubiquitination pathways central to Parkinson's disease? Cell 114: 1-8. [DOI] [PubMed] [Google Scholar]
- Goedert M (2001) Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2: 492-501. [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Feger J, Prigent A, Michel PP, Parain K, Champy P, Ruberg M, Oertel WH, Hirsch EC (2003) Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J Neurochem 84: 491-502. [DOI] [PubMed] [Google Scholar]
- Jenner P (2003) Oxidative stress in Parkinson's disease. Ann Neurol 53 [Suppl 3]: S26-S38. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605-608. [DOI] [PubMed] [Google Scholar]
- Lees AJ (1992) When did Ray Kennedy's Parkinson's disease begin? Mov Disord 7: 110-116. [DOI] [PubMed] [Google Scholar]
- Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA (2002) The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem 277: 1641-1644. [DOI] [PubMed] [Google Scholar]
- Nässel DR, Elekes K (1992) Aminergic neurons in the brain of blowflies and Drosophila: dopamine- and tyrosine hydroxylase-immunoreactive neurons and their relationship with putative histaminergic neurons. Cell Tissue Res 267: 147-167. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG (1999) Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci 22: 123-144. [DOI] [PubMed] [Google Scholar]
- Reiter RJ, Acuna-Castroviejo D, Tan DX, Burkhardt S (2001) Free radical-mediated molecular damage. Mechanisms for the protective actions of melatonin in the central nervous system. Ann NY Acad Sci 939: 200-215. [PubMed] [Google Scholar]
- Rival T, Soustelle L, Strambi C, Besson MT, Iche M, Birman S (2004) Decreasing glutamate buffering capacity triggers oxidative stress and neuropil degeneration in the Drosophila brain. Curr Biol 14: 599-605. [DOI] [PubMed] [Google Scholar]
- Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA (2002) Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci 22: 10690-10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 54: 823-827. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT (2002) An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered α-synuclein metabolism and oxidative damage. J Neurosci 22: 7006-7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Kim JH, Betarbet R, Greenamyre JT (2003) Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol 179: 9-16. [DOI] [PubMed] [Google Scholar]
- The FlyBase Consortium (2003) The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res 31: 172-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Li J, Fink AL (2001) Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson's disease. FEBS Lett 500: 105-108. [DOI] [PubMed] [Google Scholar]