The Basolateral Amygdala Interacts with the Medial Prefrontal Cortex in Regulating Glucocorticoid Effects on Working Memory Impairment (original) (raw)
Abstract
Previous findings indicate that the basolateral complex of the amygdala (BLA) interacts with other brain regions in regulating stress hormone effects on memory functions. Lesions of the BLA or infusions of a β-adrenoceptor antagonist into the BLA block glucocorticoid effects on both memory consolidation and retrieval when administered either systemically or directly into the hippocampus. The present experiments examined BLA and β-adrenoceptor involvement in regulating glucocorticoid effects on spatial working memory, a task that depends on the medial prefrontal cortex (mPFC). Male Sprague Dawley rats with bilateral sham- or NMDA-induced lesions of the BLA received either corticosterone (1.0 or 3.0 mg/kg, i.p.) systemically or the specific glucocorticoid receptor agonist 11β,17β-dihydroxy-6,21-dimethyl-17α-pregna-4,6-trien-20yn-3-one (RU 28362; 3.0 or 10.0 ng in 0.5 μl) into the mPFC shortly before testing on a delayed alternation task in a T-maze. Both glucocorticoid treatments induced comparable impairments in working memory performance in sham-lesioned controls. Although lesions of the BLA alone did not affect working memory, BLA lesions blocked the impairment induced by either corticosterone or RU 28362. Likewise, systemic injections of the centrally acting β-adrenoceptor antagonist propranolol (2.0 mg/kg, i.p.) given before testing prevented corticosterone-induced working memory impairment. These findings indicate that BLA activity is essential for enabling glucocorticoid effects in the mPFC on working memory and suggest that stress hormone-induced modulation of working memory involves noradrenergic activation.
Keywords: β-adrenoceptor, corticosterone, delayed alternation task, glucocorticoid receptor, norepinephrine, propranolol, RU 28362, stress hormone
Introduction
It is well established that glucocorticoid hormones (i.e., corticosterone and cortisol) released from the adrenal cortex during stressful or emotionally arousing experiences influence many aspects of cognitive performance (Lupien and McEwen, 1997; de Kloet et al., 1999; Roozendaal, 2002). Acutely administered glucocorticoids dose-dependently enhance long-term memory consolidation (Roozendaal, 2000; Buchanan and Lovallo, 2001) but impair retrieval of previously learned information (de Quervain et al., 1998, 2000). Complex actions of glucocorticoids on multiple, and often interacting, brain systems underlie such stress hormone effects on memory (Lupien and Lepage, 2001; Roozendaal, 2002). Extensive evidence from our laboratory indicates that the basolateral complex of the amygdala (BLA) (consisting of the basal, lateral, and accessory basal nuclei) is a critical component of the neural circuitry mediating emotional arousal and stress hormone effects on cognitive functions (McGaugh and Roozendaal, 2002; Roozendaal, 2002). Evidence that lesions or pharmacological inactivation of the BLA block the modulatory effects of systemic or intrahippocampal glucocorticoid administration on both memory consolidation and retrieval (Roozendaal and McGaugh, 1996, 1997; Quirarte et al., 1997; Roozendaal et al., 1999a, 2003; Kim et al., 2001) suggests that BLA activity is essential for enabling glucocorticoid effects in other brain regions to modulate memory processes (Roozendaal, 2002).
Stress exposure or glucocorticoid administration also impairs working memory, a dynamic process whereby information is updated continuously (Arnsten and Goldman-Rakic, 1998; Diamond et al., 1999; Wolf et al., 2001). Evidence from lesion, pharmacological, and clinical studies indicates that working memory relies on the integrity of the prefrontal cortex (Brito et al., 1982; Fuster, 1991; Taylor et al., 1999; Levy and Farrow, 2001). Importantly, the BLA projects extensively to several regions of the prefrontal cortex (McDonald, 1991). Because it has been reported that the BLA and the medial prefrontal cortex (mPFC) interact in influencing performance on affectively motivated tasks (Timms, 1977; Perez-Jaranay and Vives 1991; Garcia et al., 1999; Quirk and Gehlert, 2003), the first experiment sought to determine whether BLA functioning is also required to enable glucocorticoid effects on working memory performance. To evaluate this possibility, rats with sham or neurotoxic BLA lesions received either systemic injections of corticosterone or intra-mPFC infusions of the glucocorticoid receptor (GR) agonist 11β,17β-dihydroxy-6,21-dimethyl-17α-pregna-4,6-trien-20yn-3-one (RU 28362) shortly before testing on a delayed alternation task in a T-maze, a task commonly used to investigate spatial working memory functions in rodents (Arnsten, 2000).
Other evidence indicates that glucocorticoids increase norepinephrine turnover in the brain (McEwen, 1987) and that noradrenergic activity of the BLA is critically involved in mediating glucocorticoid effects on memory consolidation and retrieval (Quirarte et al., 1997; Roozendaal et al., 1999a, 2002; B. Roozendaal, E. L. Hahn, S. V. Nathan, D. J.-F. de Quervain and J. L. McGaugh, unpublished observation). Like neurotoxic lesions of the BLA, β-adrenoceptor antagonists administered either systemically or infused into the BLA block glucocorticoid effects on memory consolidation and retrieval. It has further been reported that high levels of corticosterone increase norepinephrine levels in the mPFC (Thomas et al., 1994). Because optimal levels of norepinephrine are necessary for normal mPFC function and working memory performance (Arnsten, 2000; Birnbaum et al., 2000), glucocorticoid effects on working memory impairment may involve an activation of noradrenergic neurotransmission. Therefore, a second experiment examined the effects of systemic administration of the centrally acting β-adrenoceptor antagonist propranolol on glucocorticoid-induced working memory impairment.
Materials and Methods
Subjects. Male adult Sprague Dawley rats (270-320 gm at time of surgery) from Charles River Breeding Laboratories (Wilmington, MA) were housed individually in a temperature-controlled (22°C) vivarium room and maintained on a standard 12 hr light/dark cycle (7 A.M.-7 P.M. lights on). The animals were fed a diet of rat chow (15-17 gm per rat per day) immediately after behavioral testing. Food rewards during cognitive testing were highly palatable miniature chocolate chips, thus minimizing the need for dietary regulation. Water was available ad libitum. Training and testing were performed during the light phase of the cycle between 10 A.M. and 3 P.M., at the rat nadir of the circadian cycle for corticosterone. All experimental procedures were in compliance with the National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the University of California, Irvine.
Surgery. Animals were adapted to the vivarium for at least 1 week before surgery. They were anesthetized with sodium pentobarbital (50 mg/kg of body weight, i.p.) and given atropine sulfate (0.4 mg/kg, i.p.) to maintain respiration. They were subsequently injected with 3.0 ml of saline to facilitate clearance of these drugs and prevent dehydration. The skull was positioned in a stereotaxic frame (Kopf Instruments, Tujunga, CA), and bilateral neurotoxic lesions of the BLA were made with NMDA (Sigma, St. Louis, MO; 1.25 mg/100 μl of phosphate buffer, pH 7.4). The NMDA solution was backfilled into a 30 gauge needle that was attached by a polyethylene tube to a 10 μl Hamilton microsyringe driven by a mini-pump (Sage Instruments, Boston, MA). The needle was placed into the BLA [coordinates: anteroposterior (AP), -2.8 mm from bregma; mediolateral (ML), ±5.0 mm from midline; dorsoventral (DV), -8.6 mm from skull surface (Paxinos and Watson, 1997)], and NMDA (2.5 μg/0.2 μl of phosphate buffer) was infused over a 25 sec period. The injection needle was retained in place for an additional 3 min to optimize diffusion. Sham operations used the same procedure except that the needle was lowered only to the level of the caudate-putamen (coordinates: AP, -2.8 mm; ML ± 5.0 mm; DV, -6.5 mm) and removed after 3 min without infusion. After lesioning of the BLA, some rats received bilateral cannula placement in the prelimbic region of the mPFC for local drug infusions. These rats were placed in a different stereotaxic apparatus, and two stainless steel guide cannulas (11 mm; 23 gauge) were implanted with the cannula tips 1.5 mm above the mPFC (coordinates: AP, +3.7 mm; ML, ±0.7 mm; DV, -2.4 mm). The cannulas were fixed to the skull with two anchoring screws and dental cement. Stylets (11 mm long, 00-insect dissection pins) inserted into each cannula to maintain patency were removed only for drug infusions. After surgery the rats were retained in an incubator until they recovered from anesthesia and then returned to their home cages. Rats were allowed to recover from surgery 7 d before initiation of training.
Working memory testing. On each day of training, the rats were transported from the vivarium to the laboratory, and training began 60 min later. Rats were initially habituated to a T-maze (dimensions, 100 × 90 cm) for 5 d until they were readily eating chocolate chips placed in food wells at the end of each arm. After habituation, the rats were trained on a delayed alternation task that has been described previously (Murphy et al., 1996b; Birnbaum et al., 1999; Arnsten, 2000). Rats were placed in the start box of the maze, the gate was opened, and the animal was allowed to run to a choice point in the maze. On the first trial, food was placed in both arms and the rat was rewarded for entering either arm. Then the rat was placed back in the start box for the intertrial delay. On the subsequent trials, the rat was rewarded only for entering the arm not chosen on the previous trial. If the correct choice was made, the rat was rewarded and then placed back in the start box for the intertrial delay. If the incorrect choice was made, the rat was placed back in the start box without the food reward. Between each trial, the choice point was wiped clean with 10% alcohol to remove any olfactory cues. Each day consisted of 11 trials, although the first trial was not included in the analyses. Accuracy of response and response time were scored, with response time being the time from when the start gate was opened to when the rat made its choice and reached one food well. Rats were tested once daily, five times per week.
The intertrial delay was initially set at “0” sec (∼2-3 sec). Delays were increased in 5 sec increments as needed to stabilize performance at ∼80% accuracy. For the first experiment with systemic corticosterone injections, the average intertrial delay was 13.7 ± 1.6 sec for the first drug treatment and was increased gradually over the study (∼10 weeks) to an average of 30.8 ± 2.1 sec. Sham- and BLA-lesioned rats did not differ in their delays at either the start (p = 0.90) or the end (p = 0.86) of the experiment. Sham- and BLA-lesioned rats receiving intra-mPFC infusions of RU 28362 also did not differ in their intertrial delays at either the start (p = 0.20) or the end (p = 0.71) of the experiment. Training took place until a rat scored between 70 and 90% correct for 3 consecutive days. Once a stable performance was achieved, drug treatment was administered either 30 min (for systemic corticosterone) or 60 min (for intra-mPFC RU 28362) before testing and randomized across rats. There was a minimum of 1 week between drug treatments.
Drug treatment. For systemic glucocorticoid administration, the adrenocortical hormone corticosterone (Sigma; 1.0 or 3.0 mg/kg, i.p.) or vehicle was administered in a volume of 2.0 ml/kg 30 min before behavioral testing in the T-maze. Corticosterone was first dissolved in 100% ethanol and then diluted in 0.9% saline to reach its appropriate concentration. The final concentration of ethanol was 5%. The vehicle solution contained 5% ethanol in saline only. The doses of corticosterone were selected on the basis of previous experiments indicating that these doses induce plasma corticosterone levels resembling moderate to severe stress (de Quervain et al., 1998). The specific GR agonist RU 28362 (Roussel UCLAF, Romainville, France; 3.0 or 10.0 ng in 0.5 μl) was used for glucocorticoid infusions into the mPFC. Receptor binding studies have shown that this compound has selective and high affinity for GRs (Teutsch et al., 1981). RU 28362 was first dissolved in 100% ethanol and subsequently diluted in 0.9% saline to reach a final ethanol concentration of 1%. Bilateral infusions of RU 28362 or an equivalent volume or vehicle (1% ethanol in saline) into the mPFC were given 60 min before testing by using a 30 gauge injection needle connected to a 10 μl Hamilton microsyringe with polyethylene (PE-20) tubing. The injection needle protruded 1.5 mm beyond the tip of the cannula, and a 0.5 μl injection volume was infused over a period of 35 sec by an automated syringe pump (Sage Instruments). The injection needles were retained within the cannulas for an additional 20 sec after drug infusion to maximize diffusion and prevent backflow of drug into the cannulas.
To examine the influence of a β-adrenoceptor antagonist on corticosterone-induced working memory impairment, the data obtained with vehicle treatment and the most effective dose of corticosterone (1.0 mg/kg) of the sham-lesioned rats of the first experiment were used in a subsequent experiment. For this experiment, the centrally acting β-adrenoceptor antagonist dl-propranolol hydrochloride (Sigma; 2.0 mg/kg, i.p.) was infused alone or together with corticosterone (1.0 mg/kg) 30 min before behavioral testing. All drug solutions were freshly prepared before each experiment.
Histology. The rats were anesthetized with an overdose of sodium pentobarbital and perfused intracardially with 0.9% saline followed by 4% formaldehyde solution (w/v). After decapitation, the brains were removed and immersed in fresh 4% formaldehyde. At least 24 hr before sectioning, the brains were transferred to a 20% sucrose (w/v) solution for cryoprotection. Coronal slices of 40 μm were cut on a freezing microtome, mounted on gelatin-coated slides, and stained with cresyl violet. The sections were examined under a light microscope, and the size and location of the BLA lesions and injection needle tips in the mPFC were determined according to the standardized atlas plates of Paxinos and Watson (1997) by an observer blind to drug treatment condition. Rats with improper BLA lesions or injection needle placements were excluded from analysis.
Statistics. Working memory data were analyzed with separate two-way ANOVAs for accuracy and response time with either systemic or intra-mPFC glucocorticoid treatment (three levels) as repeated measure and BLA lesion (two levels) as between-subject variable. Accuracy and response time of the last experiment were analyzed with one-way repeated measures ANOVAs for groups (four levels). Paired and unpaired t tests determined the source of the detected significance, with statistical significance set at p < 0.05.
Results
BLA lesions block working memory impairment induced by systemic corticosterone
Accuracy
To examine the role of the BLA in mediating glucocorticoid effects on working memory, systemic injections of corticosterone (1.0 or 3.0 mg/kg) or vehicle were given in a randomized manner to rats with either sham or BLA lesions. A two-way ANOVA for accuracy showed no significant drug effect (F(2,48) = 1.91; p = 0.15) but did show a significant lesion (F(1,48) = 7.32; p = 0.01) and interaction effect between both factors (F(2,48) = 4.57; p = 0.02). As shown in Figure 1_A_, the average accuracy of sham-lesioned rats given vehicle was 82.3%, and both doses of corticosterone significantly impaired performance in sham-lesioned rats (1.0 mg/kg, p < 0.01; 3.0 mg/kg, _p_ < 0.05, as compared with vehicle). The average accuracy of BLA-lesioned rats given vehicle was 80.0% and did not differ from that of sham-lesioned rats given vehicle (_p_ = 0.48). Importantly, corticosterone administration did not impair performance in BLA-lesioned rats, and accuracy of BLA-lesioned rats injected with the lower dose of corticosterone (1.0 mg/kg) was significantly better than that of sham-lesioned rats given a corresponding dose of corticosterone (_p_ < 0.01). Furthermore, accuracy of BLA-lesioned rats given corticosterone did not differ from that of BLA-lesioned rats given vehicle (_p_ > 0.49).
Figure 1.

Bilateral lesions of the BLA blocked the impairing effects induced by systemic injections of corticosterone (1.0 or 3.0 mg/kg, i.p.) on delayed alternation performance in rats. A, Results represent mean + SEM percent correct choices. B, Results represent mean + SEM response time in seconds. *p < 0.05; **p < 0.01 compared with the corresponding vehicle group; ♦ ♦p < 0.01 compared with the corresponding sham lesion-corticosterone group (n = 13 per group).
Response time
Two-way ANOVA for response time showed no significant drug (F(2,48) = 1.38; p = 0.26), lesion (F(1,48) = 0.15; p = 0.70), or interaction effect (F(2,48) = 0.05; p = 0.95) (Fig. 1_B_).
Histology
Figure 2_A_ shows a representative BLA lesion, and Figure 2_B_ shows a sham lesion control. Histological examination of the lesioned BLA indicated that the lesioned area was characterized by pyknosis and loss of neurons, accompanied by extensive gliosis. In most animals, the BLA lesion was not complete, leaving most of the anterior part intact, but typically ≥50% of the nucleus was damaged bilaterally. The data of three animals were excluded because of only unilateral damage to the BLA. In several animals whose data were included in the analyses, minor damage was seen in the cortex adjacent to the BLA, but the central nucleus of the amygdala was intact in all animals. Figure 2_C_ shows the extent of minimum and maximum BLA lesions of rats included in the experiment.
Figure 2.
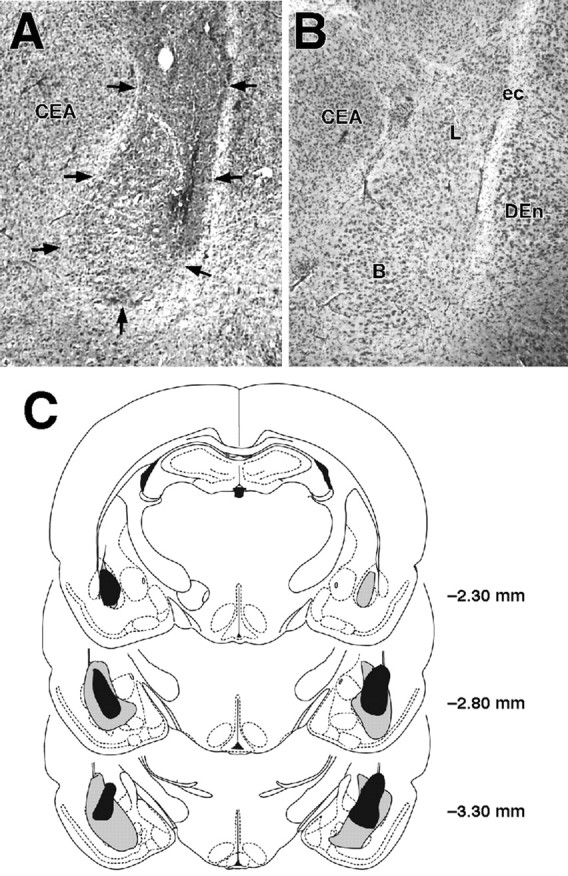
Lesions of the BLA. A, Representative NMDA-induced lesion of the BLA. Arrows denote lesion borders. B, Sham BLA lesion. C, Smallest (black area) and largest (gray area) lesions from rats used in the experiment. Adapted from Paxinos and Watson (1997). B, Basal nuclei; CEA, central amygdala; DEn, dorsal endopiriform nucleus; ec, external capsula; L, lateral nuclei.
BLA lesions block working memory impairment induced by RU 28362 administration into the medial prefrontal cortex
To examine whether the BLA interacts with the mPFC in regulating glucocorticoid effects on working memory, local infusions of the GR agonist RU 28362 (3.0 or 10.0 ng) or vehicle were given directly into the mPFC of rats with either sham or BLA lesions. As can be seen in Figure 3, the pattern of effects induced by RU 28362 administration into the mPFC was highly comparable with that found with systemic injections of corticosterone.
Figure 3.

Bilateral lesions of the BLA blocked the impairing effects induced by infusions of the specific GR agonist RU 28362 (3.0 or 10.0 ng in 0.5 μl) into the mPFC on delayed alternation performance in rats. A, Results represent mean + SEM percent correct choices. B, Results represent mean + SEM response time in seconds. **p < 0.01 compared with the corresponding vehicle group; ♦p < 0.05; ♦ ♦p < 0.01 compared with the corresponding sham lesion-RU 28362 group (n = 12 per group).
Accuracy
A two-way ANOVA for accuracy showed no significant drug effect (F(2,44) = 1.50; p = 0.23) but did show a significant lesion (F(1,44) = 14.52; p = 0.001) and interaction effect between both factors (F(2,44) = 4.96; p = 0.01). As shown in Figure 3_A_, the average accuracy of sham-lesioned rats given vehicle into the mPFC was 76.7%, and the lower dose of RU 28362 (3.0 ng) significantly impaired performance in the sham-lesioned rats (p < 0.01, as compared with vehicle). Working memory impairment induced by the higher dose of RU 28362 (10.0 ng) just failed to reach significance (_p_ = 0.06). The average accuracy of BLA-lesioned rats given vehicle was 77.5% and did not differ from that of sham-lesioned rats given vehicle (_p_ = 0.79). RU 28362 infused into the mPFC did not impair performance in BLA-lesioned rats, and accuracy of BLA-lesioned rats treated with RU 28362 was significantly better than that of sham-lesioned rats given a corresponding dose of RU 28362 (3.0 ng, _p_ < 0.01; 10.0 ng, _p_ < 0.05). Furthermore, accuracy of BLA-lesioned rats given RU 28362 did not differ from that of BLA-lesioned rats given vehicle (_p_ > 0.24).
Response time
Two-way ANOVA for response time showed no significant drug (F(2,44) = 0.80; p = 0.46), lesion (F(1,44) = 0.48; p = 0.50), or interaction effect (F(2,44) = 0.21; p = 0.81) (Fig. 3_B_).
Histology
The maximum and minimum extents of the BLA lesions are shown in Figure 4_A_. These lesions were similar to those described in the first experiment. Three rats with improper BLA lesions were excluded from analysis. BLA lesions of two rats were too small, and one additional rat was excluded because of unilateral damage to the BLA. Cannula placement in the mPFC is shown in Figure 4_B_. All injection needle tips of rats included in the analysis were localized in the prelimbic region of the mPFC. One sham-lesioned rat with extensive tissue damage at the injection needle tip site was excluded from further analysis.
Figure 4.

Lesions of the BLA and cannula placement in the mPFC. A, Smallest (black area) and largest (gray area) lesions from rats used in the experiment. B, Injection needle tips in the mPFC of all sham-lesioned (black circles) and BLA-lesioned (black diamonds) rats included in the experiment. Adapted from Paxinos and Watson (1997).
Systemic propranolol injections block corticosterone effects on working memory
Because previous findings indicated that glucocorticoid effects on cognition are intimately linked with noradrenergic activation, this experiment examined whether systemic injections of the centrally acting β-adrenoceptor antagonist propranolol blocked the impairing effects of systemic corticosterone administration on working memory. The data of the sham-lesioned rats from the first experiment given vehicle and the most effective dose of corticosterone (1.0 mg/kg) were used in this experiment, and subsequent treatments included propranolol and propranolol plus corticosterone given in a randomized manner.
Accuracy
A one-way repeated measures ANOVA for accuracy showed a significant group effect (F(3,36) = 4.14; p = 0.01). As shown in Figure 5_A_, propranolol treatment alone did not significantly alter accuracy relative to vehicle (p = 0.32), but propranolol blocked the impairing effect induced by corticosterone. Performance of rats given propranolol together with corticosterone was significantly better than when corticosterone was given alone (p < 0.01).
Figure 5.

The β-adrenoceptor antagonist propranolol (2.0 mg/kg, i.p.) blocked the impairing effects induced by systemic injections of corticosterone (1.0 mg/kg, i.p.) on delayed alternation performance in rats. A, Results represent mean + SEM percent correct choices. B, Results represent mean + SEM response time in seconds. **p < 0.01 compared with the vehicle group; ♦ ♦p < 0.01 compared with the corticosterone group (n = 13).
Response time
A one-way repeated measure ANOVA for response time showed no significant group effect (F(3,36) = 0.31; p = 0.82) (Fig. 5_B_).
Discussion
We demonstrated previously that neurotoxic lesions of the BLA blocked glucocorticoid effects on enhancement of memory consolidation as well as impairment of memory retrieval (Roozendaal and McGaugh, 1996, 1997; Roozendaal et al., 2003). In addition, systemic administration of a β-adrenoceptor antagonist or a selective blockade of noradrenergic neurotransmission in the BLA prevented the modulatory effects of glucocorticoids on these two memory functions (Quirarte et al., 1997; Roozendaal et al., 2002, 2004; Roozendaal, Hahn, Nathan, de Quervain, and McGaugh, unpublished observation). We present here the novel finding that a comparable mechanism appears to underlie glucocorticoid effects on working memory: lesions of the BLA or systemic administration of a β-adrenoceptor antagonist blocked glucocorticoid-evoked impairment of working memory in rats. Furthermore, because BLA lesions prevented working memory impairment induced by GR agonist infusions into the mPFC, the findings indicate that the BLA interacts with the mPFC in regulating glucocorticoid influences on working memory.
Glucocorticoid effects on working memory
Both corticosterone administered systemically and the GR agonist RU 28362 infused into the mPFC shortly before testing impaired performance on a delayed alternation task. Glucocorticoid administration selectively affected response accuracy but had no detectable effect on motivational or motor variables that might influence delayed alternation performance, as indicated by unaltered response times. Thus, this pattern of results strongly suggests that elevated glucocorticoid levels, via GR activation, impair working memory. This conclusion is consistent with findings indicating that mild uncontrollable stress impairs performance on a delayed alternation task but does not impair performance on non-mnemonic control tasks that have similar motivational and motor demands (Murphy et al., 1996a). The finding that glucocorticoid administration induced working memory deficits is also consistent with that of studies investigating cortisol effects on working memory tasks in human subjects (Lupien et al., 1999; Young et al., 1999; Wolf et al., 2001). Comparable with the findings of the present study, glucocorticoid impairment of working memory in human subjects was not caused by disruption of other processes influencing behavior, such as attention or logical reasoning.
In accord with the present findings, extensive evidence indicates that the mPFC is involved in working memory (Brito et al., 1982; Arnsten, 2000). Electrophysiological studies have shown increased mPFC neuronal activity in the delay period during which the information needs to be retained (Fuster, 1991). Furthermore, drugs that enhance working memory increase mPFC neuronal activity during this delay period, whereas drugs that impair working memory decrease such neuronal activity (Li et al., 1999). Impaired working memory is also observed in patients with mPFC dysfunction, including attention deficit-hyperactivity disorder and schizophrenia (Benson, 1991; Levy and Farrow, 2001; Barch et al., 2003). It is interesting to note that chronic stress or sustained hypercortisolemia induces dendritic reorganization of mPFC pyramidal neurons (Wellman, 2001) and may contribute to the onset or precipitation of symptoms for these neuropsychiatric conditions (Mazure, 1995). Because the mPFC expresses high mRNA and protein levels for GRs (Meaney and Aitken, 1985; Reul and de Kloet, 1985; Patel et al., 2000; Sanchez et al., 2000), glucocorticoids may impair working memory, at least in part, via a GR-mediated inhibition of mPFC activity during task performance. Consistent with our findings, it was reported that stress-level cortisol treatment, which at high doses preferentially binds to GRs, impairs mPFC-dependent inhibitory control of behaviors in squirrel monkeys (Lyons et al., 2000). Although the mPFC also expresses a low density of mineralocorticoid receptors (MRs), a role of this receptor type in mPFC-dependent working memory has not been investigated; however, evidence indicates that MRs are involved in response selection, integration of sensory inputs, and behavioral exploration in other behavioral tasks (Oitzl and de Kloet, 1992; Oitzl et al., 1994) and therefore may mediate nonspecific influences on working memory performance (Douma et al., 1998). In this regard, it is noteworthy that the hippocampus, which contains high densities of both GRs and MRs, is also involved in some types of spatial working memory (Wan et al., 1994; Seamans et al., 1998; Lee and Kesner, 2003) and that stress exposure or glucocorticoid administration impairs performance on hippocampus-dependent working memory tasks (Diamond et al., 1999).
Role of the BLA in regulating glucocorticoid effects on working memory
A major finding of the present study is that BLA lesions blocked the effects of systemically administered corticosterone or intra-mPFC infusions of RU 28362 on working memory. The finding that BLA lesions alone did not affect working memory is in accord with previous evidence (Aggleton et al., 1989; Wan et al., 1994; Bianchin et al., 1999) and suggests that BLA activity is not implicated in working memory performance per se but rather is involved in modulating stress or emotional arousal effects on working memory in other brain regions [but see Barros et al. (2002)]. The current findings indicate that the BLA regulates stress hormone effects on working memory via interactions with the mPFC. Several studies have reported that the BLA interacts with the mPFC (and other frontal lobe regions) in influencing performance on affectively motivated tasks (Timms, 1977; Schoenbaum et al., 1998; Bechara et al., 1999; Garcia et al., 1999; Quirk and Gehlert, 2003). Electrophysiological studies have shown that there are reciprocal inhibitory influences between both brain regions. Stimulation of the BLA predominantly induces inhibitory responses in the mPFC via an activation of local GABAergic interneurons (Perez-Jaranay and Vives, 1991), whereas, conversely, electrical activation of the mPFC reduces BLA activity (Rosenkranz and Grace, 2002). Furthermore, a recent study reported that acute stress blocks the induction of long-term potentiation in the BLA-mPFC pathway in vivo (Maroun and Richter-Levin, 2003). Hence, by activating BLA neurons (Feldman et al., 1983; Roozendaal, 2000), systemically administered glucocorticoids may potentiate inhibition of mPFC activity. BLA lesions may prevent glucocorticoid-induced working memory impairment by blocking stress hormone effects on the inhibition of mPFC neuronal activity. This suggestion fits well with previous findings indicating that BLA lesions block fear-induced decreases in spontaneous electrical activity in the mPFC (Garcia et al., 1999) and is remarkably similar to findings of in vivo electrophysiological studies indicating that BLA lesions block changes in hippocampal neuroplasticity induced by either high-frequency stimulation of the perforant path or elevated glucocorticoid levels (Ikegaya et al., 1995; Kim et al., 2001). In addition to direct connections between both regions (McDonald, 1991), the inhibitory effects of BLA activation on the mPFC may be mediated via indirect projections involving the nucleus accumbens (Jackson and Moghaddam, 2001; Rosenkranz and Grace, 2001; Stevenson and Gratton, 2003) or the mesocortical dopaminergic system (Davis et al., 1994).
Role of the noradrenergic system in regulating glucocorticoid effects on working memory
Numerous studies have shown that glucocorticoids are intimately linked with central noradrenergic mechanisms and permissively increase noradrenergic neurotransmission during emotionally arousing situations (McEwen, 1987; Stone et al., 1987; Duman et al., 1989; Roozendaal et al., 2002). The current findings indicate that systemic injections of the β-adrenoceptor antagonist propranolol, at a concentration that blocks glucocorticoid effects on memory consolidation and retrieval, also prevented the impairing effect of corticosterone on working memory. Importantly, propranolol administration alone did not alter performance. Thus, the blocking of the corticosterone response was not likely caused by additive effects of the two treatments. Because propranolol was administered systemically, there are many sites where propranolol might have exerted its effect. In line with the observation of the first experiment, one interpretation of our findings is that propranolol may have blocked corticosterone effects on working memory impairment by inhibiting β-adrenoceptors within the BLA. Activation of the BLA depends critically on noradrenergic neurotransmission (Liang et al., 1995; McIntyre et al., 2002). An additional possibility is that propranolol might have acted directly on postsynaptic β-adrenoceptors in the mPFC to protect cognitive performance during high circulating levels of corticosterone. Systemically administered corticosterone rapidly increases norepinephrine levels in the mPFC (Thomas et al., 1994) by stimulating GRs in noradrenergic cell populations in the brainstem (Härfstrand et al., 1987; McEwen, 1987; Roozendaal et al., 1999b) or by locally blocking extraneuronal norepinephrine reuptake mechanisms in the mPFC (Grundemann et al., 1998). In conjunction, excessive levels of norepinephrine induce mPFC dysfunction and working memory impairment, at least in part, via an activation of the intracellular cAMP-protein kinase A signal transduction pathway (Taylor et al., 1999). Several studies indicate that glucocorticoids interact with β- and α1-adrenoceptors in stimulating cAMP accumulation (Stone et al., 1987; Duman et al., 1989); however, because stress and glucocorticoids also increase extracellular levels of several other neurotransmitters in the mPFC, including glutamate and dopamine (Moghaddam, 1993; Thomas et al., 1994; Morrow et al., 2000), glucocorticoid effects on working memory impairment may involve interactions between norepinephrine and other neuromodulatory systems in the mPFC.
Functional implications and conclusions
This series of experiments was designed to extend our understanding of the role of the BLA and the noradrenergic system in regulating glucocorticoid effects on different cognitive functions. Because our findings indicate that BLA influences on other brain regions in regulating stress hormone effects on memory are not restricted to the consolidation and retrieval of long-term memories but extend to working memory, it can be hypothesized that the BLA is part of an integrated network of cortical and subcortical brain regions engaged in regulating glucocorticoid effects on different memory functions (McGaugh, 2002; Roozendaal, 2002). By this hypothesis, glucocorticoid effects on these apparently distinct cognitive processes might be interrelated in terms of neurobiological substrate as well as function (Roozendaal, 2002). Thus, exposure to glucocorticoids or emotionally arousing training experiences, evoking noradrenergic activation, may induce BLA activation, in concert with inhibitory effects on the mPFC and other brain regions, to create a brain state that promotes the long-term storage of these emotionally arousing events and thus preserve significant information. According to this view, the temporary impairment of working memory (and long-term memory retrieval) observed concurrently by this pattern of brain activity should not a priori be considered as a maladaptive response, but if associated with a training experience may serve to enhance memory consolidation (e.g., by blocking retroactive interference). In support of this hypothesis, high doses of norepinephrine infused into the mPFC, known to impair working memory, have been reported to induce enhancement of long-term memory consolidation when administered after a training experience (Liang, 2001). Moreover, acutely administered corticosterone increases the expression of neural cell adhesion molecules in the mPFC, molecules that have been implicated in the stabilization of synaptic contacts and long-term memory consolidation (Sandi and Loscertales, 1999); however, prolonged or uncontrolled activation of this brain state such as that found in patients with hypercortisolemia and several neuropsychiatric disorders may be associated with cognitive impairment (Semple et al., 2000; Elzinga and Bremner, 2002).
Footnotes
This research was supported by United States Public Health Service Grant MH12526 from the National Institute of Mental Health (J.L.M.).
Correspondence should be addressed to Dr. Benno Roozendaal, Center for the Neurobiology of Learning and Memory, University of California, Irvine, CA 92697-3800. E-mail: broozend@uci.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/241385-08$15.00/0
References
- Aggleton JP, Blindt HS, Rawlins JN (1989) Effects of amygdaloid and amygdaloid-hippocampal lesions on object recognition and spatial working memory in rats. Behav Neurosci 103: 962-974. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT (2000) Stress impairs prefrontal cortical function in rats and monkeys: role of dopamine D1 and norepinephrine alpha-1 receptor mechanisms. Prog Brain Res 126: 183-192. [DOI] [PubMed] [Google Scholar]
- Arnsten AFT, Goldman-Rakic PS (1998) Noise stress impairs prefrontal cortical cognitive function in monkeys: evidence for a hyperdopaminergic mechanism. Arch Gen Psychiatry 55: 362-369. [DOI] [PubMed] [Google Scholar]
- Barch DM, Sheline YI, Csernansky JG, Snyder AZ (2003) Working memory and prefrontal cortex dysfunction: specificity to schizophrenia compared with major depression. Biol Psychiatry 53: 376-384. [DOI] [PubMed] [Google Scholar]
- Barros DM, Pereira P, Medina JH, Izquierdo I (2002) Modulation of working memory and of long- but not short-term memory by cholinergic mechanisms in the basolateral amygdala. Behav Pharmacol 13: 163-167. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Damasio AR, Lee GP (1999) Different contribution of the human amygdala and ventromedial prefrontal cortex to decision making. J Neurosci 19: 5473-5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DF (1991) The role of frontal dysfunction in attention deficit/hyperactivity disorder. J Child Neurol 6: S9-S12. [DOI] [PubMed] [Google Scholar]
- Bianchin M, Mello e Souza T, Medina JH, Izquierdo I (1999) The amygdala is involved in the modulation of long-term memory, but not in working or short-term memory. Neurobiol Learn Mem 71: 127-131. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Gobeske KT, Auerbach J, Taylor JR, Arnsten AFT (1999) A role for norepinephrine in stress-induced cognitive deficits: α-1-adrenoceptor mediation in the prefrontal cortex. Biol Psychiatry 46: 1266-1274. [DOI] [PubMed] [Google Scholar]
- Birnbaum SG, Podell DM, Arnsten AFT (2000) Noradrenergic alpha-2 receptor agonists reverse working memory deficits induced by the anxiogenic drug, FG7142, in rats. Pharmacol Biochem Behav 67: 397-403. [DOI] [PubMed] [Google Scholar]
- Brito GNO, Thomas GJ, Davis BJ, Gingold SI (1982) Prelimbic cortex, mediodorsal thalamus, septum and delayed alternation in rats. Exp Brain Res 46: 52-58. [DOI] [PubMed] [Google Scholar]
- Buchanan TW, Lovallo WR (2001) Enhanced memory for emotional material following stress-level cortisol treatment in humans. Psychoneuroendocrinology 26: 307-317. [DOI] [PubMed] [Google Scholar]
- Davis M, Hitchcock JM, Bowers MB, Berridge CW, Melia KR, Roth RH (1994) Stress-induced activation of prefrontal cortex dopamine turnover: blockade by lesions of the amygdala. Brain Res 664: 207-210. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Oitzl MS, Joëls M (1999) Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci 22: 422-426. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ-F, Roozendaal B, McGaugh JL (1998) Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature 394: 787-790. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ-F, Roozendaal B, Nitsch RM, McGaugh JL, Hock C (2000) Acute cortisone administration impairs retrieval of long-term declarative memory in humans. Nat Neurosci 3: 313-314. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Park CR, Heman KL, Rose GM (1999) Exposing rats to a predator impairs spatial working memory in the radial arm water maze. Hippocampus 9: 542-552. [DOI] [PubMed] [Google Scholar]
- Douma BRK, Korte SM, Buwalda B, la Fleur SE, Bohus B, Luiten PGM (1998) Repeated blockade of mineralocorticoid receptors, but not glucocorticoid receptors impairs food rewarded spatial learning. Psychoneuroendocrinology 23: 33-44. [DOI] [PubMed] [Google Scholar]
- Duman RS, Strada SJ, Enna SJ (1989) Glucocorticoid administration increases receptor-mediated and forskolin-stimulated cyclic AMP accumulation in rat brain cerebral cortical slices. Brain Res 477: 166-171. [DOI] [PubMed] [Google Scholar]
- Elzinga BM, Bremner JD (2002) Are the neural substrates of memory the final common pathway in posttraumatic stress disorder (PTSD)? J Affect Disord 70: 1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman S, Papir-Kricheli D, Dafny N (1983) Single-cell and multiunit activity in freely moving rats after corticosterone administration. Exp Neurol 80: 427-438. [DOI] [PubMed] [Google Scholar]
- Fuster JM (1991) The prefrontal cortex and its relation to behavior. Prog Brain Res 87: 201-211. [DOI] [PubMed] [Google Scholar]
- Garcia R, Volumba RM, Baudry M, Thompson RF (1999) The amygdala modulates prefrontal cortex activity relative to conditioned fear. Nature 402: 294-296. [DOI] [PubMed] [Google Scholar]
- Grundemann D, Schechinger B, Rappold GA, Schöming E (1998) Molecular identification of the corticosterone-sensitive extraneuronal catecholamine transporter. Nat Neurosci 1: 349-351. [DOI] [PubMed] [Google Scholar]
- Härfstrand A, Fuxe K, Cintra A, Agnati LF, Wikström AC, Okret S, Yu Z, Goldstein M, Steinbusch H, Verhofstad A, Gustafsson JÅ (1987) Glucocorticoid receptor immunoreactivity in monoaminergic neurons in rat brain. Proc Natl Acad Sci USA 83: 9779-9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegaya Y, Saito H, Abe K (1995) Requirement of basolateral amygdala neuron activity for the induction of long-term potentiation in the dentate gyrus in vivo. Brain Res 671: 351-354. [DOI] [PubMed] [Google Scholar]
- Jackson ME, Moghaddam B (2001) Amygdala regulation of nucleus accumbens dopamine output is governed by the prefrontal cortex. J Neurosci 21: 676-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Lee HJ, Han JS, Packard MG (2001) Amygdala is critical for stress-induced modulation of hippocampal long-term potentiation and learning. J Neurosci 21: 5222-5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Kesner RP (2003) Time-dependent relationship between the dorsal hippocampus and the prefrontal cortex in spatial memory. J Neurosci 23: 1517-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy F, Farrow M (2001) Working memory in ADHD: prefrontal/parietal connections. Curr Drug Targets 2: 347-352. [DOI] [PubMed] [Google Scholar]
- Li B-M, Mao Z-M, Wang M, Mei Z-T (1999) Alpha-2 adrenergic modulation of prefrontal cortical neuronal activity related to spatial working memory in monkeys. Neuropyschopharmacology 21: 601-610. [DOI] [PubMed] [Google Scholar]
- Liang KC (2001) Epinephrine modulation of memory: amygdala activation and regulation of long-term memory storage. In: Memory consolidation: essays in honor of James L. McGaugh (Gold PE, Greenough WT, eds), pp 165-183. Washington, DC: American Psychological Association.
- Liang KC, Chen LL, Huang TE (1995) The role of amygdala norepinephrine in memory formation: involvement of memory enhancing effects of peripheral epinephrine. Chin J Physiol 38: 81-91. [PubMed] [Google Scholar]
- Lupien SJ, Lepage M (2001) Stress, memory, and the hippocampus: can't live with it, can't live without it. Behav Brain Res 127: 137-158. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS (1997) The acute effects of corticosteroids on cognition: integration of animal and human studies. Brain Res Rev 24: 1-27. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, Gillin CJ, Hauger RL (1999) Working memory is more sensitive than declarative memory to the acute effects of corticosteroids: a dose-response study in humans. Behav Neurosci 113: 420-430. [DOI] [PubMed] [Google Scholar]
- Lyons DM, Lopez JM, Yang C, Schatzberg AF (2000) Stress-level cortisol treatment impairs inhibitory control of behavior in monkeys. J Neurosci 20: 7816-7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroun M, Richter-Levin G (2003) Exposure to acute stress blocks the induction of long-term potentiation of the amygdala-prefrontal cortex pathway _in vivo._J Neurosci 23: 4406-4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazure CM (1995) Does stress cause psychiatric illness? In: Progress in psychiatry (Spiegel D, ed), p 270. Washington, DC: American Psychiatric Press.
- McDonald AJ (1991) Organization of amygdaloid projections to the prefrontal cortex and associated striatum in the rat. Neuroscience 44: 1-14. [DOI] [PubMed] [Google Scholar]
- McEwen BS (1987) Glucocorticoid-biogenic amine interaction in relation to mood and behavior. Biochem Pharmacol 36: 1755-1763. [DOI] [PubMed] [Google Scholar]
- McGaugh JL (2002) Memory consolidation and the amygdala: a systems perspective. Trends Neurosci 25: 456-461. [DOI] [PubMed] [Google Scholar]
- McGaugh JL, Roozendaal B (2002) Role of adrenal stress hormones in forming lasting memories in the brain. Curr Opin Neurobiol 12: 205-210. [DOI] [PubMed] [Google Scholar]
- McIntyre CK, Hatfield T, McGaugh JL (2002) Norepinephrine levels in the amygdala following inhibitory avoidance training predict retention score. Eur J Neurosci 16: 1223-1226. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Aitken DH (1985) [3H]dexamethasone binding in rat frontal cortex. Brain Res 328: 176-180. [DOI] [PubMed] [Google Scholar]
- Moghaddam B (1993) Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem 60: 1650-1657. [DOI] [PubMed] [Google Scholar]
- Morrow BA, Roth RH, Elsworth JD (2000) TMT, a predator odor, elevates mesoprefrontal dopamine metabolic activity and disrupts short-term working memory in the rat. Brain Res Bull 52: 519-523. [DOI] [PubMed] [Google Scholar]
- Murphy BL, Arnsten AFT, Goldman-Rakic PS, Roth RH (1996a) Increased dopamine turnover in the prefrontal cortex impairs spatial working memory performance in rats and monkeys. Proc Natl Acad Sci USA 93: 1325-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy BL, Arnsten AFT, Jentsch JD, Roth RH (1996b) Dopamine and spatial working memory in rats and monkeys: pharmacological reversal of stress-induced impairment. J Neurosci 16: 7768-7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oitzl MS, de Kloet ER (1992) Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav Neurosci 106: 62-71. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, Fluttert M, de Kloet ER (1994) The effect of corticosterone on reactivity to spatial novelty is mediated by central mineralocorticoid receptors. Eur J Neurosci 6: 1072-1079. [DOI] [PubMed] [Google Scholar]
- Patel PD, Lopez JF, Lyons DM, Burke S, Wallace M, Schatzberg AF (2000) Glucocorticoid and mineralocorticoid receptor mRNA expression in squirrel monkey brain. J Psych Res 34: 383-392. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1997) The rat brain in stereotaxic coordinates, Ed 3. San Diego: Academic. [DOI] [PubMed]
- Perez-Jaranay JM, Vives F (1991) Electrophysiological study of the response of medial prefrontal cortex neurons to stimulation of the basolateral nucleus of the amygdala in the rat. Brain Res 564: 97-101. [DOI] [PubMed] [Google Scholar]
- Quirarte GL, Roozendaal B, McGaugh JL (1997) Glucocorticoid enhancement of memory storage involves noradrenergic activation in the basolateral amygdala. Proc Natl Acad Sci USA 94: 14048-14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Gehlert DR (2003) Inhibition of the amygdala: key to pathological states? Ann NY Acad Sci 985: 263-272. [DOI] [PubMed] [Google Scholar]
- Reul JMHM, de Kloet ER (1985) Two receptor systems for corticosterone in the rat brain: microdistribution and differential occupation. Endocrinology 117: 2505-2512. [DOI] [PubMed] [Google Scholar]
- Roozendaal B (2000) Glucocorticoids and the regulation of memory consolidation. Psychoneuroendocrinology 25: 213-238. [DOI] [PubMed] [Google Scholar]
- Roozendaal B (2002) Stress and memory: opposing effects of glucocorticoids on memory consolidation and memory retrieval. Neurobiol Learn Mem 78: 578-595. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McGaugh JL (1996) Amygdaloid nuclei lesions differentially affect glucocorticoid-induced memory enhancement in an inhibitory avoidance task. Neurobiol Learn Mem 65: 1-8. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, McGaugh JL (1997) Basolateral amygdala lesions block the memory-enhancing effect of glucocorticoid administration in the dorsal hippocampus of rats. Eur J Neurosci 9: 76-83. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Nguyen BT, Power AE, McGaugh JL (1999a) Basolateral amygdala noradrenergic influence enables enhancement of memory consolidation induced by hippocampal glucocorticoid receptor activation. Proc Natl Acad Sci USA 96: 11642-11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, Williams CL, McGaugh JL (1999b) Glucocorticoid receptor activation in the rat nucleus of the solitary tract facilitates memory consolidation: involvement of the basolateral amygdala. Eur J Neurosci 11: 1317-1323. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Quirarte GL, McGaugh JL (2002) Glucocorticoids interact with the basolateral amygdala β-adrenoceptor-cAMP/PKA system in influencing memory consolidation. Eur J Neurosci 15: 553-560. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Griffith QK, Buranday J, de Quervain DJ-F, McGaugh JL (2003) The hippocampus mediates glucocorticoid-induced impairment of spatial memory retrieval: dependence on the basolateral amygdala. Proc Natl Acad Sci USA 100: 1328-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roozendaal B, de Quervain DJ-F, Schelling G, McGaugh JL (2004) A systemically administered β-adrenoceptor antagonist blocks corticosterone-induced impairment of contextual memory retrieval in rats. Neurobiol Learn Mem, in press. [DOI] [PubMed]
- Rosenkranz JA, Grace AA (2001) Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J Neurosci 9: 637-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA (2002) Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons _in vivo_J Neurosci 22: 324-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez MM, Young LJ, Plotsky PM, Insel TR (2000) Distribution of corticosteroid receptors in the Rhesus brain: relative absence of glucocorticoid receptors in the hippocampal formation. J Neurosci 20: 4657-4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandi C, Loscertales M (1999) Opposite effects on NCAM expression in the rat frontal cortex induced by acute vs. chronic corticosterone treatments. Brain Res 828: 127-134. [DOI] [PubMed] [Google Scholar]
- Schoenbaum G, Chiba AA, Gallagher M (1998) Orbitofrontal cortex and basolateral amygdala encode expected outcomes during learning. Nat Neurosci 1: 155-159. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Floresco SB, Phillips AG (1998) D1 receptor modulation of hippocampal-prefrontal cortical circuits integrating spatial memory with executive functions in the rat. J Neurosci 18: 1613-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple WE, Goyer PF, McCormick R, Donovan B, Muzic Jr RF, Rugle L, McCutcheon K, Lewis C, Liebling D, Kowaliw S, Vapenik K, Semple MA, Flener CR, Schulz SC (2000) Higher brain blood flow at amygdala and lower frontal cortex blood flow in PTSD patients with comorbid cocaine and alcohol abuse compared with normals. Psychiatry 63: 65-74. [DOI] [PubMed] [Google Scholar]
- Stevenson CW, Gratton A (2003) Basolateral amygdala modulation of the nucleus accumbens dopaminergic response to stress: role of the medial prefrontal cortex. Eur J Neurosci 17: 1287-1295. [DOI] [PubMed] [Google Scholar]
- Stone EA, McEwen BS, Herrera AS, Carr KD (1987) Regulation of α and β components of noradrenergic cAMP response in cortical slices. Eur J Pharmacol 141: 347-356. [DOI] [PubMed] [Google Scholar]
- Taylor JR, Birnbaum S, Ubriani R, Arnsten AFT (1999) Activation of cAMP-dependent protein kinase A in prefrontal cortex impairs working memory performance. J Neurosci 19: RC23(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teutsch G, Costerousse G, Deraedt R, Benzoni J, Fortin M, Philibert D (1981) 17alpha-alkynyl-11beta, 17-dihydroxyandrostane derivates: a new class of potent glucocorticoids. Steroids 38: 651-665. [DOI] [PubMed] [Google Scholar]
- Thomas DN, Post RM, Pert A (1994) Central and systemic corticosterone differentially affect dopamine and norepinephrine in the frontal cortex of the awake and freely moving rat. Ann NY Acad Sci 764: 467-469. [DOI] [PubMed] [Google Scholar]
- Timms RJ (1977) Cortical inhibition and facilitation of the defense reaction. J Physiol (Lond) 266: 98P-99P. [PubMed] [Google Scholar]
- Wan R-Q, Pang K, Olton DS (1994) Hippocampal and amygdaloid involvement in nonspatial and spatial working memory in rats: effects of delay and interference. Behav Neurosci 108: 866-882. [DOI] [PubMed] [Google Scholar]
- Wellman CL (2001) Dendritic reorganization in pyramidal neurons in medial prefrontal cortex after chronic corticosterone administration. J Neurobiol 49: 245-253. [DOI] [PubMed] [Google Scholar]
- Wolf OT, Convit A, McHugh PF, Kandil E, Thorn EL, De Santi S, McEwen BS, de Leon MJ (2001) Cortisol differentially affects memory in young and elderly men. Behav Neurosci 115: 1002-1011. [DOI] [PubMed] [Google Scholar]
- Young AH, Sahakian BJ, Robbins TW, Cowen PJ (1999) The effects of chronic administration of cortisone on cognitive function in normal male volunteers. Psychopharmacology 145: 260-266. [DOI] [PubMed] [Google Scholar]