Stimulation of Medial Prefrontal Cortex Decreases the Responsiveness of Central Amygdala Output Neurons (original) (raw)
Abstract
In extinction of auditory fear conditioning, rats learn that a tone no longer predicts the occurrence of a footshock. Recent lesion and unit recording studies suggest that the medial prefrontal cortex (mPFC) plays an essential role in the inhibition of conditioned fear following extinction. mPFC has robust projections to the amygdala, a structure that is known to mediate the acquisition and expression of conditioned fear. Fear conditioning potentiates the tone responses of neurons in the basolateral amygdala (BLA), which excite neurons in the central nucleus (Ce) of the amygdala. In turn, the Ce projects to the brainstem and hypothalamic areas that mediate fear responses. The present study was undertaken to test the hypothesis that the mPFC inhibits conditioned fear via feedforward inhibition of Ce output neurons. Recording extracellularly from physiologically identified brainstem-projecting Ce neurons, we tested the effect of mPFC prestimulation on Ce responsiveness to synaptic input. In support of our hypothesis, mPFC prestimulation dramatically reduced the responsiveness of Ce output neurons to inputs from the insular cortex and BLA. Thus, our findings support the idea that mPFC gates impulse transmission from the BLA to Ce, perhaps through GABAergic intercalated cells, thereby gating the expression of conditioned fear.
Keywords: amygdaloid complex, extinction, fear conditioning, intrinsic connections, brainstem, network, emotion
Introduction
Much evidence suggests that the amygdala is necessary for the acquisition and expression of learned fear associations (LeDoux, 2000; Davis and Whalen, 2001) (but see Cahill et al., 1999). This has been demonstrated most clearly for auditory fear conditioning (Quirk et al., 1995; Paré and Collins, 2000; Blair et al., 2001; Maren, 2001; Walker and Davis, 2002), in which rats learn to freeze to a tone [conditioned stimulus (CS)] that predicts a footshock [unconditioned stimulus (US)]. Cortical and thalamic afferents communicate tone and shock information to the basolateral complex of the amygdala (BLA), in which tone-shock associations are thought to be formed (Quirk et al., 1995; Repa et al., 2001; Lamprecht et al., 2002). In turn, the BLA sends glutamatergic projections to the central nucleus (Ce) (Smith and Paré, 1994; Paré et al., 1995; Pitkänen et al., 1997), which is the main source of amygdala outputs to the brainstem and hypothalamic sites (Holstege et al., 1996) that mediate the behavioral and autonomic correlates of fear (LeDoux et al., 1988; Bellgowan and Helmstetter, 1996; De Oca et al., 1998; Davis, 2000).
Once acquired, conditioned fear associations are not always expressed. For instance, repeated presentation of the tone CS in the absence of the US causes conditioned fear responses to rapidly diminish, a phenomenon termed extinction. Behavioral studies have demonstrated that extinction does not erase the CS-US association, but it forms a new memory that inhibits conditioned responses (Pavlov, 1927; Rescorla and Heth, 1975; Bouton and King, 1983; Quirk, 2002).
The following three lines of evidence suggest that long-term memory for extinction depends on the medial prefrontal cortex (mPFC), especially the infralimbic (IL) subregion of mPFC: (1) lesions of IL disrupt recall of extinction (Morgan et al., 1993; Quirk et al., 2000); (2) tone responses of IL neurons are correlated with recall of extinction (Milad and Quirk, 2002); and (3) electrical stimulation of IL reduces freezing to conditioned tones (Milad and Quirk, 2002). Thus, mPFC appears to be essential for inhibition of conditioned fear responses after extinction (but see Gewirtz et al., 1997).
How might mPFC inhibit conditioned fear responses? mPFC sends robust projections to the amygdala (McDonald et al., 1996) and could exert feedforward inhibition of Ce output neurons. However, mPFC also projects directly to the hypothalamic and brainstem targets of the Ce (Hurley et al., 1991; Fisk and Wyss, 2000; Floyd et al., 2000, 2001) and thus could inhibit fear responses independently of the amygdala. Because of its divergent projections, inhibition of Ce would be an efficient way of dampening multiple fear responses after extinction (Royer and Paré, 2002). It is not known, however, whether mPFC neurons can influence Ce excitability. To address this, we recorded the responses of Ce output neurons to cortical and basolateral nucleus stimulation in anesthetized rats and cats. Consistent with an amygdala route of fear inhibition, we show that mPFC stimulation decreases the responsiveness of Ce output neurons to afferent inputs.
Materials and Methods
Experiments were conducted in two different species for scientific and technical reasons. Rats were used because most behavioral and unit data on the involvement of mPFC in extinction were obtained in this species. However, because of the small size of the rat amygdala, some of the experimental manipulations detailed below could only be performed in the larger cat amygdala. Nevertheless, it should be noted that the rat and cat amygdala are identical in terms of cell types, transmitters, physiological properties, and connectivity (Paré and Smith, 1998; Paré et al., 2003). Moreover, the mPFC has similar projections in rats and cats (Musil and Olson, 1988; Sesack et al., 1989).
Rat experiments. All procedures were approved by the Institutional Animal Care and Use Committee of the Ponce School of Medicine, in compliance with the Guide for the Care and Use of Laboratory Animals (Department of Health and Human Services publication 86-23). Experiments were performed in 10 male Sprague Dawley rats (∼300 gm) anesthetized with a mixture of ketamine and xylazine (74 and 11 mg/kg, i.p.). The level of anesthesia was assessed by continuously monitoring the electroencephalogram (EEG) and electrocardiogram (ECG), and supplemental doses were administered to maintain a synchronized EEG and stable heart rate. The body temperature was maintained at 37-38°C with a heating pad.
The bone overlying the amygdala, insula, mesencephalon, and mPFC was removed bilaterally, and the dura mater was opened. To physiologically identify brainstem-projecting Ce neurons, two concentric stimulating electrodes were inserted stereotaxically, just dorsal to the substantia nigra, where Ce axons form a compact bundle en route to more caudal sites (Hopkins and Holstege, 1978). Concentric stimulating electrodes were inserted in the insula to synaptically activate Ce neurons (McDonald, 1998). Stimulating electrodes were also inserted in the mPFC, at the border between the IL and prelimbic subdivisions.
An array of four tungsten microelectrodes (0.4 mm spacing) was aimed stereotaxically at the Ce nucleus. During the experiments, the microelectrode arrays were lowered in steps of 4 μm by a piezoelectric micromanipulator. While moving the microelectrodes, electrical stimuli (0.3 msec, 1 mA, 1 Hz) were delivered to the insula or brainstem to increase the likelihood of encountering responsive cells. At regular intervals, stimuli were also delivered to the mPFC. When responsive neurons with a high signal-to-noise ratio (≥3) were encountered, the stimulus intensity and duration was decreased just above threshold (typically 0.05-0.2 msec pulses of 0.1-0.5 mA), and the type of response (orthodromic vs antidromic) was determined (for the criteria used to distinguish antidromic and orthodromic responses, see Results). The evoked activity of responsive neurons was observed on a digital oscilloscope, digitized, and stored on disk for offline analysis.
To study the influence of mPFC inputs on the responsiveness of Ce neurons, one to three conditioning stimuli (at 300 Hz, typically 0.05-0.2 msec pulses of 0.1-0.5 mA) were delivered to the mPFC before a single test stimulus to the insula or brainstem. This mPFC stimulation frequency was chosen because it is optimal for producing temporal summation of EPSPs in target cells. The interstimulus interval (ISI) was varied from 2 to 200 msec. At least 20 stimuli were delivered at each ISI.
Cat experiments. All procedures were approved by the Institutional Animal Care and Use Committee of Rutgers State University, in compliance with the Guide for the Care and Use of Laboratory Animals (Department of Health and Human Services publication 86-23). Four adult male cats (2.5-3.5 kg) were preanesthetized with a mixture of ketamine and xylazine (15 and 2 mg/kg, i.m.) and artificially ventilated with a mixture of ambient air, oxygen, and isoflurane. Atropine (0.05 mg/kg, i.m.) was administered to prevent secretions. The end-tidal concentration in CO2 was kept at 3.7 ± 0.2%, and the body temperature was maintained at 37-38°C with a heating pad. The level of anesthesia was assessed by continuously monitoring the EEG and ECG. The bone overlying the amygdala, mPFC, and mesencephalon was removed, and the dura mater was opened.
To synaptically drive Ce neurons, an array of three concentric stimulating electrodes was stereotaxically inserted in the lateral, basolateral, and basomedial nuclei, respectively. In addition, the electrodes were inserted obliquely to avoid damaging the Ce nucleus. In all Ce cells, we used a large range of BLA stimulation intensities (up to 1.5 mA) to test the robustness of mPFC inhibitory effects.
As in rat experiments, brainstem stimulating electrodes were positioned just dorsal to the substantia nigra and the mPFC electrodes at the border between IL and the prelimbic area. The procedures used to activate mPFC inputs to the amygdala were identical to those used in the rat experiments. The procedures used to record and physiologically identify brainstem-projecting Ce neurons and test the effects of mPFC stimuli on their responsiveness were identical to those used in the rat experiments.
Histological identification of recording and stimulating sites. At the end of the experiments, recording sites were marked with electrolytic lesions (0.5 mA for 5 sec). After this, the animals were given an overdose of sodium pentobarbital (rat, 100 mg/kg, i.p.; cat, 50 mg/kg, i.v.) and perfused with a cold saline solution (0.9%), followed by a solution containing 2% paraformaldehyde and 1% glutaraldehyde in 0.1 m PBS, pH 7.4. The brains were later sectioned on a vibrating microtome (at 100 μm) and stained with thionin or neutral red to verify the position of the recording electrodes.
The microelectrode tracks were reconstructed by combining micrometer readings with the histology. In doing so, shrinkage resulting from fixation was taken into account. The amount of shrinkage was estimated by measuring the postfixation separation of tracts left by tungsten rods inserted 10 mm apart before fixation, as described previously (Paré et al., 1997).
Analysis. Analyses were performed offline with commercial software (IGOR; WaveMetrics, Lake Oswego, OR) and home-made software running on Macintosh computers (Apple Computers, Cupertino, CA). Spikes were detected using a window discriminator after filtering (0.3-10 kHz) of the raw waves. Peristimulus histograms of unit discharges were computed. All values are expressed as means ± SE.
Results
Rat experiments
Histological and physiological identification of recorded cells
Because brainstem-projecting neurons are concentrated in the medial sector of Ce (CeM) (Hopkins and Holstege, 1978; Veening et al., 1984), microelectrodes were aimed at this subnucleus. Histological reconstructions of electrode tracks confirmed that most recorded amygdala neurons (n = 137) were obtained in the CeM (n = 64) (Fig. 1_A1_) and the ventrally adjacent basolateral (BL) nucleus (n = 59) (Fig. 1_A2_). To activate CeM neurons orthodromically, stimulating electrodes were placed in the insular cortex (Fig. 1_B1_), which projects to the CeM directly and indirectly via the BLA (McDonald, 1998). In addition, because we aimed to study the effects of mPFC inputs (Fig. 1_B2_) on CeM projection neurons, we attempted to identify recorded neurons by antidromic invasion. To this end, stimulating electrodes were inserted just dorsal to the substantia nigra (Fig. 1_B3_), in which CeM axons form a compact bundle en route to more caudal brainstem sites (Hopkins and Holstege, 1978). Figure 1 shows photomicrographs of recording and stimulating sites.
Figure 1.
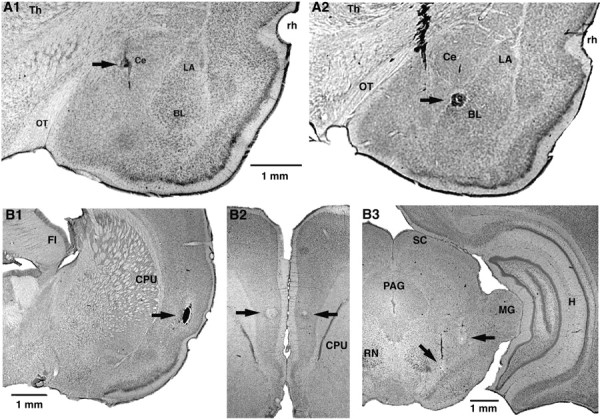
Histological identification of recording (A) and stimulating (B) sites. A, Electrolytic lesions (arrows) made along two different electrode tracks during which we recorded several CeM neurons antidromically responsive to brainstem stimuli. In A1, the lesion marks the site where the first neuron backfired from the brainstem was encountered. In A2, the lesion marks the site where the first neuron antidromically responsive to mPFC stimuli was recorded. B, Tip of stimulating electrodes in the insula (B1), mPFC (B2), and brainstem (B3). The scale bar in A1 is also valid for A2. The scale bar in B1 is also valid for B2. CPU, Striatum; FI, fimbria; H, hippocampus; LA, lateral nucleus of the amygdala; MG, medial geniculate nucleus; OT, optic tract; PAG, periaqueductal gray; RN, red nucleus; rh, rhinal sulcus; SC, superior colliculus; Th, thalamus.
Criteria for identifying antidromic spikes were fixed response latency, high-frequency (300 Hz) following, and (when possible) collision with spontaneously occurring or orthodromically evoked spikes. Using these criteria, we determined that 59 of 64 CeM neurons (92%) could be antidromically activated from the brainstem, indicating that they were CeM output neurons. In BL, 22 of 59 neurons (37%) could be antidromically activated from the ipsilateral mPFC, consistent with the prominent projection of BL to mPFC (McDonald, 1991).
Figure 2 shows fixed latency and collision tests demonstrating antidromic activation of CeM (Fig. 2_A_) and BL (Fig. 2_B_) neurons from the brainstem and mPFC, respectively. The average antidromic response latency of CeM neurons to brainstem stimuli was 23.6 ± 7.3 msec. Latencies were bimodally distributed with peaks at 15 and 30 msec, in agreement with a previous observation in the rabbit (Pascoe and Kapp, 1985). A similar antidromic response latency of 20.9 ± 1.5 msec was observed for BL neurons after stimulation of mPFC.
Figure 2.

Examples of antidromic activation of CeM and BL neurons in the rat. A1, Consistent with antidromic activation, brainstem stimulation fired this CeM neuron at a fixed latency (filled circle, 10 overlapped sweeps). A2, A3, Collision test. Collision was observed when spontaneous action potentials occurred within twice the antidromic response latency (A2, 3 overlapped sweeps) but not with longer intervals (A3, 3 overlapped sweeps). B1, Antidromic activation (filled circle) of a BL neuron in response to stimulation of the ipsilateral (ipsi) mPFC. B2, Collision with spikes evoked by suprathreshold stimulation of the contralateral (contra) mPFC. C, Histogram shows the number of neurons (_x_-axis) in either CeM or BL that were antidromically activated by brainstem (right, gray bars) or mPFC (left, white bars) stimuli, respectively, as a function of depth (_y_-axis). Note abrupt transition from antidromic activation of neurons in response to brainstem or mPFC stimuli at the CeM→BL border (arrows on the right), as determined in histological verification of two electrode tracks.
As the electrode descended through CeM and then BL, there was an abrupt transition in the ability to antidromically activate cells, first from the brainstem and then from mPFC (Fig. 2_C_). Histological analysis of electrode tracks showed this transition to be at the border between CeM dorsally and BL ventrally, consistent with the known projection sites of these nuclei.
Effect of mPFC stimuli on the synaptic and antidromic responsiveness of CeM cells
Although the CeM receives strong excitatory inputs from the insula (McDonald, 1998), it proved surprisingly difficult to synaptically activate CeM neurons from this site. Of the 59 physiologically identified projection cells recorded in CeM, only four were also synaptically activated from the insular cortex. In all four cases, however, mPFC prestimulation produced a robust inhibition of CeM responses to insula stimuli. An example of mPFC-induced inhibition of insula-evoked activity is shown in Figure 3_A_. mPFC prestimulation reduced the probability of synaptic activation of this CeM neuron to zero. The inhibitory effect of mPFC stimulation peaked at interstimulus intervals of 10-20 msec, gradually decreased with longer ISIs and vanished at ISIs ranging between 40 and 100 msec.
Figure 3.

Inhibition of a CeM output neuron by prestimulation of mPFC in the rat. A, CeM output neuron was synaptically driven by stimulation of the insular cortex (INS). Prestimulation of mPFC reduced the responsiveness of this CeM neuron to insular cortex stimulation (each trace shows 10 overlapping sweeps). Interval between mPFC and insula stimuli is indicated on the left. Inhibition lasted for up to 40 msec in this cell (inset). In the inset, responsiveness was calculated from 20 trials at each ISI. B, Inhibition of antidromic responses of a CeM output neuron. Each trace is the response to a single stimulus. The left column shows responses to brainstem (BS) stimuli applied in isolation. Prestimulation of mPFC (right column) reduced the probability of antidromic invasion of this CeM neuron from 80 to 20% (calculated from 20 trials). The ISI was 20 msec in this case.
In addition to studying the effects of mPFC prestimulation on synaptically evoked spikes, we tested whether mPFC stimuli could block brainstem-evoked antidromic responses of CeM neurons. In one of the tested cells (n = 59), mPFC prestimulation reduced the probability of antidromic spiking (from 80 to 20%) (Fig. 3_B_). The contrasting effectiveness of mPFC stimuli in inhibiting synaptic versus antidromic discharges suggests that the inhibitory inputs activated by mPFC end at a location that is electrotonically remote from the soma (see Discussion). However, it is important to stress that the effect of mPFC stimulation on CeM neurons was always inhibitory; mPFC stimulation never activated CeM cells nor facilitated their orthodromic or antidromic responsiveness.
Cat experiments
In the experiments described above, mPFC could have inhibited the synaptic responsiveness of CeM neurons in several ways. One possibility is feedforward inhibition of CeM cells via the recruitment of GABAergic intercalated (ITC) cells (Nitecka and Frotscher, 1989; McDonald and Augustine, 1993; Paré and Smith, 1993a) that project to the Ce (Paré and Smith, 1993b; Royer et al., 1999). Indeed, there is a prominent projection from mPFC to ITC cells (Sesack et al., 1989; McDonald et al., 1996; Freedman et al., 2000; Pinto and Sesack, 2002). Another possibility is inhibition of BLA responses to cortical inputs via the activation of local inhibitory interneurons within the BLA (Rosenkranz and Grace, 2001), leading to a disfacilitation of CeM neurons. We reasoned that the latter possibility would seem unlikely if mPFC stimulation could also inhibit the orthodromic activation of CeM neurons to direct electrical stimulation of the BLA.
However, one potential difficulty with conducting such experiments in rats is the possibility of current spread from the BLA to the adjacent Ce. Thus, to minimize this possibility, we opted to perform these experiments in the cat amygdala (Fig. 4_A,B_), which is larger but has identical internuclear connections to rats (Paré and Smith, 1998).
Figure 4.
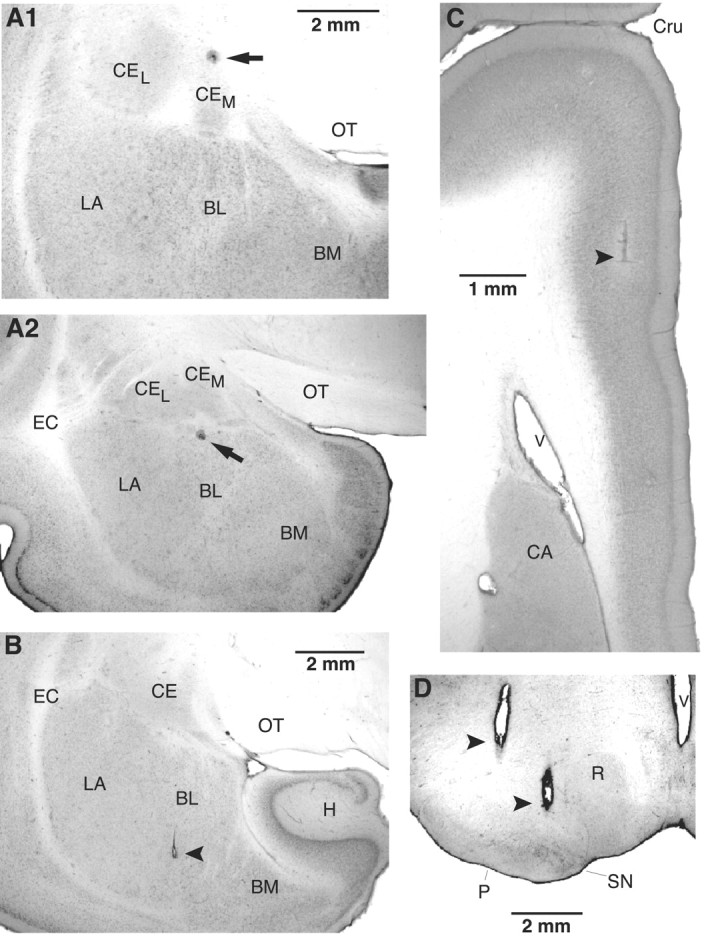
Histological identification of recording (A) and stimulating (B-D) sites. A, Electrolytic lesions made along two different electrode tracks during which we recorded several CeM neurons antidromically responsive to brainstem stimuli. In the top (A1), the lesion marks the site where the first neuron backfired from the brainstem was encountered. In A2, the lesion marks the site where the first neuron antidromically responsive to mPFC stimuli was encountered. B-D, Tip of stimulating electrodes in the BL (B), mPFC (C), and brainstem (D). The scale bar in A1 is also valid for A2. BM, Basomedial nucleus; CA, caudate nucleus; CEL, central lateral nucleus; CEM, central medial nucleus; Cru, cruciate sulcus; EC, external capsule; H, hippocampus; LA, lateral nucleus of the amygdala; OT, optic tract; P, cerebral peduncle; R, red nucleus; SN, substantia nigra; V, ventricle.
Histological and physiological identification of recorded cells A total of 192 neurons were recorded from the cat amygdala. Histological verification of microelectrode tracks (Fig. 4_A_) showed that 144 of these cells were recorded in the CeM, 21 were recorded in the CeL or amygdalostriatal transition area, and 27 were recorded in the BLA. As in rats, we observed an abrupt transition in the ability of brainstem and mPFC stimuli to evoke antidromic discharges as the electrodes advanced from the CeM to the BL nucleus (Fig. 4_A_).
Consistent with the presence of glutamatergic projections from BLA to Ce (Smith and Paré, 1994; Paré et al., 1995), most CeM neurons could be synaptically activated by electrical stimuli delivered in one or more nuclei of the BLA (Fig. 4_B_). In particular, 58 CeM neurons were synaptically activated by stimulation of the lateral nucleus (latency, 13.01 ± 0.58 msec), 92 from the basolateral nucleus (latency, 8.34 ± 0.30 msec), and 82 from the basomedial nucleus (latency, 8.81 ± 0.38 msec). In fact, most CeM neurons showed no spontaneous spiking and could only be identified via responses to stimulation.
In addition, 69 of these orthodromically responsive CeM neurons responded antidromically to stimulation of the brainstem (latency, 14.25 ± 0.99 msec), confirming that they were CeM output neurons. As was observed in rats, antidromic response latencies were bimodally distributed (modes of 8 and 17 msec).
Effect of mPFC stimuli on the synaptic and antidromic responsiveness of CeM cells
mPFC prestimulation (Fig. 4_C_) inhibited BLA-induced synaptic responses in 51 of 69 CeM output cells (74%). In only one CeM cell did mPFC stimulation evoke spiking, albeit at a long and variable latency (∼70-90 msec), suggestive of a polysynaptic response.
An example of mPFC-induced inhibition in the cat is shown in Figure 5. As in the rat, mPFC prestimulation produced a dramatic reduction in BLA-evoked spiking. In the example of Figure 5, the BLA stimulation intensity was adjusted just above threshold so that orthodromic spikes could be evoked in essentially all control trials (Fig. 5_A_, top trace, B, 0 msec ISI). However, in all tested cells (n = 51), mPFC prestimuli also produced a robust inhibition of Ce orthodromic spiking when we used the highest intensity BLA stimuli that our equipment could deliver (1.5 mA). As was observed in rats, the mPFC-evoked inhibition gradually decreased with increasing mPFC-BLA ISIs, disappearing at ISIs ranging from 60 to 120 msec.
Figure 5.
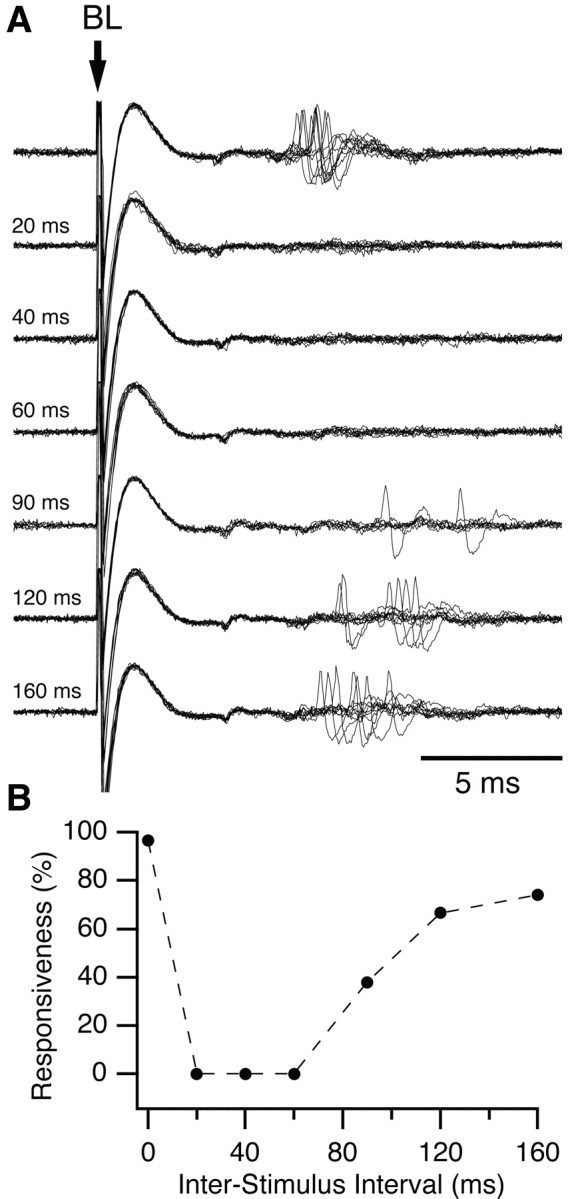
Inhibition of a CeM output neuron by prestimulation of mPFC in the cat. A, BL stimulation activated this CeM output neuron at a latency of ∼7 msec. Prestimulation of ipsilateral mPFC eliminated synaptic responses to BL stimulation. Interval between mPFC and BL stimuli is indicated on the left. B, Graph plotting responsiveness to BL stimuli (_y_-axis) as a function of mPFC-BL ISIs (_x_-axis). Responsiveness was calculated from 20 trials at each ISI. The inhibitory effect of mPFC stimulation lasted ∼120 msec in this neuron.
A similar time course of inhibition was observed for BLA-evoked field potentials in the CeM (Fig. 6). BLA stimulation evoked a prominent negative potential that peaked at 10 msec latency. This field response was significantly reduced by mPFC prestimulation (peak reduction of 61%; p < 0.001; t test). This experiment was repeated in three different animals, and similar results were obtained (maximal reductions of 50% or higher).
Figure 6.

Inhibition of BL-evoked field responses in CeM by mPFC prestimulation. Inset shows CeM field response to BL stimulation, both without and with mPFC prestimulation (60 msec ISI). Graph plots BL-evoked field potential amplitude (negative component, _y_-axis) as a function of mPFC-BL ISI (_x_-axis). Note the reduction in negative component amplitude with mPFC prestimulation. mPFC stimuli applied in isolation evoked a positive field potential in CeM (data not shown).
As was observed in rats, mPFC stimulation was more effective in inhibiting orthodromic than antidromic Ce discharges. Nevertheless, mPFC stimulation inhibited brainstem-evoked antidromic spikes in 7 of 69 cells (10%).
Exactly 81% of central lateral nucleus (CeL) neurons (17 of 21 cells) could also be orthodromically activated from one or more BLA nuclei. In all cases, mPFC prestimulation produced a robust inhibition of BLA-evoked discharges in CeL cells, as was observed in the CeM. These results suggest that GABAergic CeL neurons projecting to CeM (Paré et al., 1993b) are unlikely to mediate the inhibitory effects of mPFC on CeM neurons.
Discussion
Descending projections of the central amygdala are thought to mediate behavioral components of conditioned fear reactions, such as freezing and startle potentiation (LeDoux et al., 1988; Helmstetter, 1992; Davis, 2000). Although it has been proposed previously that mPFC reduces conditioned fear responses via its projections to the amygdala (al Maskati and Zbrozyna, 1989; Morgan et al., 1993; Milad and Quirk, 2002; Rosenkranz and Grace, 2002; Royer and Paré, 2002), this has never been directly tested.
In the present study, we observed that stimulation of the mPFC dramatically reduced the responsiveness of CeM neurons to insula and BLA inputs. The inhibitory effect of mPFC stimulation occurred in CeM neurons that were antidromically activated from the brainstem. Thus, our findings provide direct physiological support for the hypothesis that the mPFC reduces fear responses by reducing amygdala output (Garcia et al., 1999; LeDoux, 2000; Milad and Quirk, 2002).
In the following account, we consider the circuits that might mediate mPFC inhibition of CeM neurons and discuss the relevance of our findings for the extinction of conditioned fear responses.
Possible circuits mediating mPFC inhibition of the amygdala
Given that mPFC projections to the amygdala are excitatory (Smith et al., 2000), mPFC inhibition of CeM must involve inhibitory interneurons within the amygdala. A previous study showed that mPFC stimulation inhibited the response of BLA projection neurons to inputs from cortical area Te3 (Rosenkranz and Grace, 2001). It was suggested that this inhibition resulted from a preferential projection of mPFC to BLA inhibitory interneurons as opposed to projection cells. Although mPFC activation of local interneurons in BLA is possible, we believe that this explanation is unlikely for the following reasons: (1) prefrontal inputs to the BLA terminate almost entirely on dendritic spines (Brinley-Reed et al., 1995; Smith et al., 2000), suggesting that the main targets of mPFC axons are projection neurons rather than interneurons; and (2) mPFC input to parvalbumin-expressing cells, a common type of inhibitory interneuron in BLA, are extremely sparse (Smith et al., 2000).
An alternative explanation for the findings of Rosenkranz and Grace (2001), however, is that antidromic activation of BLA neurons by mPFC stimulation fired local recurrent collaterals ending on inhibitory interneurons in BLA. This possibility is supported by the fact that as many as 37% of BL neurons could be backfired from mPFC in the present study. Accordingly, stimulation of any site that receives a substantial projection from BLA might be expected to inhibit subsequent synaptic activation of BLA because of local recurrent inhibition.
Moreover, even if mPFC axons activated inhibitory local-circuit cells of the BLA, this effect could not account for the inhibition of CeM responses to direct electrical stimulation of the BLA. Indeed, BLA interneurons do not project to the Ce nucleus. This contention is supported by electron microscopic studies in which it was found that BLA axons only form asymmetric synapses in the Ce (Smith and Paré, 1994; Paré et al., 1995).
Also remote is the possibility that, by exciting BLA inhibitory interneurons, mPFC stimuli reduced the number of BLA projection cells activated by direct electrical stimuli. Indeed, if this was the case, one would predict that applying high-intensity electrical stimuli in the BLA would overwhelm the more subtle intra-BLA synaptic inhibition and abolish mPFC effects in the Ce. At odds with this idea, however, we observed that mPFC could inhibit Ce orthodromic spiking evoked by the highest intensity BLA stimuli that our equipment could deliver.
An alternative possibility is that the inhibitory effect of mPFC stimulation is mediated by ITC cells, which send GABAergic projections to the CeM (Fig. 7) (Paré and Smith, 1993b). ITC cells are a likely candidate because they receive a robust projection from mPFC (Sesack et al., 1989; McDonald et al., 1996; Freedman et al., 2000) and inhibit CeM output neurons (Royer et al., 1999). Moreover, activation of mPFC by local picrotoxin injections produced a threefold increase in the number of c-Fos immunoreactive ITC neurons with little change in the BLA (Berretta et al., 2003). Also consistent with the idea that ITC cells mediated the inhibitory effect of mPFC, physiological data suggest that ITC axons form synapses with the dendrites but not the soma of Ce neurons (Delaney and Sah, 2001). This could explain why it was easier to inhibit synaptic as opposed to antidromic discharges of CeM cells in the present study. Finally, at least some cortical inputs can fire ITC cells, as was shown with electrical stimulation of the perirhinal area (Collins and Paré, 1999). Interestingly, ITC cells exhibit NMDA-dependent long-term potentiation and depression (Royer and Paré, 2002), raising the possibility that they might store a more persistent memory of extinction learning.
Figure 7.

Scheme depicting the IL-amygdala interactions hypothesized to underlie the phenomenon described in this study. IL inhibits CeM projection neurons via GABAergic ITC cells, thereby reducing CeM responses to inputs from BL or cortex. For clarity, several intramygdala projections have been omitted. BS, Brainstem; IC, internal capsule; OT, optic tract; Pu, putamen; rh, rhinal fissure.
Relevance to extinction of conditioned fear
Consistent with our present findings, stimulation of mPFC prevents blood pressure increases induced by exposure to conditioned tones (Zbrozyna and Westwood, 1991) or electrical stimulation of the amygdala (al Maskati and Zbrozyna, 1989). Is there any evidence, however, that mPFC neurons naturally increase their activity at times when conditioned fear responses are reduced? Recording from behaving rats during auditory fear conditioning, it was reported recently that neurons in the IL region of the mPFC increased their responses to tones after extinction, when rats were recalling extinction training (Milad and Quirk, 2002). Rats showing the largest IL tone responses exhibited the least freezing to the tone. A similar finding has been reported for field potentials in mPFC (Herry and Garcia, 2002). IL appears to be essential for recall of extinction memory, because rats with lesions of IL act as if they never received extinction training (Quirk et al., 2000).
IL activity is not only necessary for recall of extinction, it appears to be sufficient. Pairing fear-conditioned tones with brief (300 msec) electrical stimulation of IL at tone onset (designed to mimic extinction-induced tone responses) reduced freezing to the tone (Milad and Quirk, 2002). In contrast, stimulation of nearby prelimbic cortex or IL shortly before or after tone onset had no effect (Vidal-Gonzalez et al., 2002), suggesting that IL stimulation acts by gating the response of downstream structures to conditioned stimuli. Our present findings suggest that the Ce nucleus of the amygdala is a critical downstream structure (Fig. 7). Given its divergent projections to multiple fear-expression centers, inhibition of CeM projection neurons by mPFC would be an efficient way of dampening multiple fear responses after extinction (Royer and Paré, 2002). Inhibition of fear expression via ITC cells could explain how fear behavior might be extinguished, despite the persistence of conditioned tone responses in lateral amygdala neurons throughout extinction (Repa et al., 2001). However, the possibility that mPFC also regulates fear expression via its direct projections to hypothalamus and brainstem (Floyd et al., 2000, 2001) cannot be ruled out.
In conclusion, we showed that stimulation of mPFC reduces the responsiveness of CeM output neurons to synaptic activation from BLA and insular cortex. Activation of CeM output neurons by BLA is a critical link in the expression of conditioned fear responses. Our findings suggest that mPFC gates the transmission from BLA to CeM output neurons and thereby gates the expression of conditioned fear responses. These findings support a growing number of lesion and recording studies implicating mPFC in the storage and expression of extinction memory.
Footnotes
This work was supported by National Science Foundation Grant IBN-0208712 to D.P. and National Institutes of Health Grants R01-MH58883 and S06-GM08239 to G.J.Q.
Correspondence should be addressed to Denis Paré, Center for Molecular and Behavioral Neuroscience, Aidekman Research Center, Rutgers, The State University of New Jersey, 197 University Avenue, Newark, NJ 07102. E-mail: pare@axon.rutgers.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238800-08$15.00/0
References
- al Maskati HA, Zbrozyna AW ( 1989) Stimulation in prefrontal cortex area inhibits cardiovascular and motor components of the defence reaction in rats. J Auton Nerv Syst 28: 117-125. [DOI] [PubMed] [Google Scholar]
- Bellgowan PS, Helmstetter FJ ( 1996) Neural systems for the expression of hypoalgesia during nonassociative fear. Behav Neurosci 110: 727-736. [DOI] [PubMed] [Google Scholar]
- Berretta S, Pantazopoulos P, Pantazopoulos H, Paré D ( 2003) Medial prefrontal (mPFC) stimulation increases c-Fos expression in intercalated neurons of the amygdala. Soc Neurosci Abstr, in press. [DOI] [PMC free article] [PubMed]
- Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE ( 2001) Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem 8: 229-242. [DOI] [PubMed] [Google Scholar]
- Bouton ME, King DA ( 1983) Contextual control of the extinction of conditioned fear: tests for the associative value of the context. J Exp Psychol Anim Behav Process 9: 248-265. [PubMed] [Google Scholar]
- Brinley-Reed M, Mascagni F, McDonald AJ ( 1995) Synaptology of prefrontal cortical projections to the basolateral amygdala: an electron microscopic study in the rat. Neurosci Lett 202: 45-48. [DOI] [PubMed] [Google Scholar]
- Cahill L, Weinberger NM, Roozendaal B, McGaugh JL ( 1999) Is the amygdala a locus of “conditioned fear?” Some questions and caveats. Neuron 23: 227-228. [DOI] [PubMed] [Google Scholar]
- Collins DR, Paré D ( 1999) Spontaneous and evoked activity of intercalated amygdala neurons. Eur J Neurosci 11: 3441-3448. [DOI] [PubMed] [Google Scholar]
- Davis M ( 2000) The role of the amygdala in conditioned and unconditioned fear and anxiety. In: The amygdala (Aggleton JP, ed), pp 213-288. New York: Oxford UP.
- Davis M, Whalen PJ ( 2001) The amygdala: vigilance and emotion. Mol Psychiatry 6: 13-34. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Sah P ( 2001) Pathway-specific targeting of GABA(A) receptor subtypes to somatic and dendritic synapses in the central amygdala. J Neurophysiol 86: 717-723. [DOI] [PubMed] [Google Scholar]
- De Oca BM, DeCola JP, Maren S, Fanselow MS ( 1998) Distinct regions of the periaqueductal gray are involved in the acquisition and expression of defensive responses. J Neurosci 18: 3426-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisk GD, Wyss JM ( 2000) Descending projections of infralimbic cortex that mediate stimulation-evoked changes in arterial pressure. Brain Res 859: 83-95. [DOI] [PubMed] [Google Scholar]
- Floyd NS, Price JL, Ferry AT, Keay KA, Bandler R ( 2000) Orbitomedial prefrontal cortical projections to distinct longitudinal columns of the periaqueductal gray in the rat. J Comp Neurol 422: 556-578. [DOI] [PubMed] [Google Scholar]
- Floyd NS, Price JL, Ferry AT, Keay KA, Bandler R ( 2001) Orbitomedial prefrontal cortical projections to hypothalamus in the rat. J Comp Neurol 432: 307-328. [DOI] [PubMed] [Google Scholar]
- Freedman LJ, Insel TR, Smith Y ( 2000) Subcortical projections of area 25 (subgenual cortex) of the macaque monkey. J Comp Neurol 2000 421: 172-188. [PubMed] [Google Scholar]
- Garcia R, Vouimba RM, Baudry M, Thompson RF ( 1999) The amygdala modulates prefrontal cortex activity relative to conditioned fear. Nature 402: 294-296. [DOI] [PubMed] [Google Scholar]
- Gewirtz JC, Falls WA, Davis M ( 1997) Normal conditioned inhibition and extinction of freezing and fear-potentiated startle following electrolytic lesions of medial prefrontal cortex in rats. Behav Neurosci 111: 712-726. [DOI] [PubMed] [Google Scholar]
- Helmstetter FJ ( 1992) The amygdala is essential for the expression of conditional hypoalgesia. Behav Neurosci 106: 518-528. [DOI] [PubMed] [Google Scholar]
- Herry C, Garcia R ( 2002) Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci 22: 577-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege G, Bandler R, Saper CB ( 1996) The emotional motor system. Prog Brain Res 107: 3-6. [DOI] [PubMed] [Google Scholar]
- Hopkins DA, Holstege G ( 1978) Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the cat. Exp Brain Res 32: 529-547. [DOI] [PubMed] [Google Scholar]
- Hurley KM, Herbert H, Moga MM, Saper CB ( 1991) Efferent projections of the infralimbic cortex of the rat. J Comp Neurol 308: 249-276. [DOI] [PubMed] [Google Scholar]
- Lamprecht R, Farb CR, LeDoux JE ( 2002) Fear memory formation involves p190 RhoGAP and ROCK proteins through a GRB2-mediated complex. Neuron 36: 727-738. [DOI] [PubMed] [Google Scholar]
- LeDoux JE ( 2000) Emotion circuits in the brain. Annu Rev Neurosci 23: 155-184. [DOI] [PubMed] [Google Scholar]
- LeDoux JE, Iwata J, Cicchetti P, Reis DJ ( 1988) Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci 8: 2517-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S ( 2001) Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 24: 897-931. [DOI] [PubMed] [Google Scholar]
- McDonald AJ ( 1991) Organization of amygdaloid projections to the prefrontal cortex and associated striatum in the rat. Neuroscience 44: 1-14. [DOI] [PubMed] [Google Scholar]
- McDonald AJ ( 1998) Cortical pathways to the mammalian amygdala. Prog Neurobiol 55: 257-332. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Augustine JR ( 1993) Localization of GABA-like immunore-activity in the monkey amygdala. Neuroscience 52: 281-294. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F, Guo L ( 1996) Projections of the medial and lateral prefrontal cortices to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience 71: 55-75. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ ( 2002) Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70-74. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Romanski LM, LeDoux JE ( 1993) Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett 163: 109-113. [DOI] [PubMed] [Google Scholar]
- Musil SY, Olson CR ( 1988) Organization of cortical and subcortical projections to medial prefrontal cortex in the cat. J Comp Neurol 272: 219-241. [DOI] [PubMed] [Google Scholar]
- Nitecka L, Frotscher M ( 1989) Organization and synaptic interconnections of GABAergic and cholinergic elements in the rat amygdaloid nuclei: single- and double-immunolabeling studies. J Comp Neurol 279: 470-488. [DOI] [PubMed] [Google Scholar]
- Paré D, Collins DR ( 2000) Neuronal correlates of fear in the lateral amygdala: multiple extracellular recordings in conscious cats. J Neurosci 20: 2701-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paré D, Smith Y ( 1993a) Distribution of GABA immunoreactivity in the amygdaloid complex of the cat. Neuroscience 57: 1061-1076. [DOI] [PubMed] [Google Scholar]
- Paré D, Smith Y ( 1993b) The intercalated cell masses project to the central and medial nuclei of the amygdala in cats. Neuroscience 57: 1077-1090. [DOI] [PubMed] [Google Scholar]
- Paré D, Smith Y ( 1998) Intrinsic circuitry of the amygdaloid complex: common principles of organization in rats and cats. Trends Neurosci 21: 240-241. [DOI] [PubMed] [Google Scholar]
- Paré D, Smith Y, Paré JF ( 1995) Intra-amygdaloid projections of the basolateral and basomedial nuclei in the cat: _Phaseolus vulgaris_-leucoagglutinin anterograde tracing at the light and electron microscopic level. Neuroscience 69: 567-583. [DOI] [PubMed] [Google Scholar]
- Paré D, Lebel E, Lang EJ ( 1997) Differential impact of miniature synaptic potentials on the soma and dendrites of pyramidal neurons _in vivo_J Neurophysiol 78: 1735-1739. [DOI] [PubMed] [Google Scholar]
- Paré D, Royer S, Smith Y, Lang EJ ( 2003) Contextual inhibitory gating of impulse traffic in the intra-amygdaloid network. Ann NY Acad Sci 985: 78-91. [DOI] [PubMed] [Google Scholar]
- Pascoe JP, Kapp BS ( 1985) Electrophysiological characteristics of amygdaloid central nucleus neurons during Pavlovian fear conditioning in the rabbit. Behav Brain Res 16: 117-133. [DOI] [PubMed] [Google Scholar]
- Pavlov I ( 1927) Conditioned reflexes. London: Oxford UP.
- Pinto A, Sesack SR ( 2002) Prefrontal cortex projection to the rat amygdala: ultrastructural relationship to dopamine D1 and D2 receptors. Soc Neurosci Abstr 28: 587.6. [Google Scholar]
- Pitkänen A, Savander V, LeDoux JE ( 1997) Organization of intraamygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci 20: 517-523. [DOI] [PubMed] [Google Scholar]
- Quirk GJ ( 2002) Memory for extinction of conditioned fear is long-lasting and persists following spontaneous recovery. Learn Mem 9: 402-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Repa C, LeDoux JE ( 1995) Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: parallel recordings in the freely behaving rat. Neuron 15: 1029-1039. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K ( 2000) The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 20: 6225-6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE ( 2001) Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat Neurosci 4: 724-731. [DOI] [PubMed] [Google Scholar]
- Rescorla RA, Heth CD ( 1975) Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process 1: 88-96. [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA ( 2001) Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J Neurosci 21: 4090-4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA ( 2002) Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons _in vivo_J Neurosci 22: 324-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Paré D ( 2002) Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience 115: 455-462. [DOI] [PubMed] [Google Scholar]
- Royer S, Martina M, Paré D ( 1999) An inhibitory interface gates impulse traffic between the input and output stations of the amygdala. J Neurosci 19: 10575-10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Deutch AY, Roth RH, Bunney BS ( 1989) Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol 290: 213-242. [DOI] [PubMed] [Google Scholar]
- Smith Y, Paré D ( 1994) Intra-amygdaloid projections of the lateral nucleus in the cat: PHA-L anterograde labeling combined with postembedding GABA and glutamate immunocytochemistry. J Comp Neurol 342: 232-248. [DOI] [PubMed] [Google Scholar]
- Smith Y, Paré JF, Paré D ( 2000) Differential innervation of parvalbumin-immunoreactive interneurons of the basolateral amygdaloid complex by cortical and intrinsic inputs. J Comp Neurol 416: 496-508. [PubMed] [Google Scholar]
- Veening JG, Swanson LW, Sawchenko PE ( 1984) The organization of projections from the central nucleus of the amygdala to brainstem sites involved in central autonomic regulation: a combined retrograde transport-immunohistochemical study. Brain Res 303: 337-357. [DOI] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Milad MR, Quirk GJ ( 2002) Anatomical and temporal specificity in the ability of infralimbic cortex stimulation to simulate extinction memory. Soc Neurosci Abstr 28: 83.13. [Google Scholar]
- Walker DL, Davis M ( 2002) The role of amygdala glutamate receptors in fear learning, fear-potentiated startle, and extinction. Pharmacol Biochem Behav 71: 379-392. [DOI] [PubMed] [Google Scholar]
- Zbrozyna AW, Westwood DM ( 1991) Stimulation in prefrontal cortex inhibits conditioned increase in blood pressure and avoidance bar pressing in rats. Physiol Behav 49: 705-708. [DOI] [PubMed] [Google Scholar]