Delayed Secondary Phase of Peri-Infarct Depolarizations after Focal Cerebral Ischemia: Relation to Infarct Growth and Neuroprotection (original) (raw)
Abstract
In focal cerebral ischemia, peri-infarct depolarizations (PIDs) cause an expansion of core-infarcted tissue into adjacent penumbral regions of reversible injury and have been shown to occur through 6 hr after injury. However, infarct maturation proceeds through 24 hr. Therefore, we studied PID occurrence through 72 hr after both transient and permanent middle cerebral artery occlusion (MCAo) via continuous DC recordings in nonanesthetized rats. PIDs occurred an average 13 times before reperfusion at 2 hr and then ceased for an average ∼8 hr. After this quiescent period, PID activity re-emerged in a secondary phase, which reached peak incidence at 13 hr and consisted of a mean 52 PIDs over 2-24 hr. This phase corresponded to the period of infarct maturation; rates of infarct growth through 24 hr coincided with changes in PID frequency and peaked at 13 hr. In permanent MCAo, PIDs also occurred in a biphasic pattern with a mean of 78 events over 2-24 hr. Parameters of secondary phase PID incidence correlated with infarct volumes in transient and permanent ischemia models. The role of secondary phase PIDs in infarct development was further investigated in transient MCAo by treating rats with a high-affinity NMDA receptor antagonist at 8 hr after injury, which reduced post-treatment PID incidence by 57% and provided 37% neuroprotection. Topographic mapping with multielectrode recordings revealed multiple sources of PID initiation and patterns of propagation. These results suggest that PIDs contribute to the recruitment of penumbral tissue into the infarct core even after the restoration of blood flow and throughout the period of infarct maturation.
Keywords: spreading depression, peri-infarct depolarization, brain injury, direct current potential, penumbra, middle cerebral artery occlusion, focal cerebral ischemia
Introduction
Spreading depression (SpD) is an electrochemical wave that propagates through neural tissue at 2-5 mm/min, causing prolonged (1-5 min) cellular depolarization, cessation of unit firing, and a hallmark drop in extracellular DC potential of 10-20 mV. SpD was first shown to occur in the context of ischemic injury in 1977 (Branston et al., 1977) and since then has been investigated as a pathogenic mechanism in clinical stroke and traumatic brain injury (Hansen and Lauritzen, 1984; Obrenovitch and Urenjak, 1997; Strong et al., 2002). In experimental focal cerebral ischemia, SpD incidence correlates with infarct size (Dijkhuizen et al., 1999). This relationship may be causal, because eliciting SpD remote from the infarct region [i.e., by potassium application (Busch et al., 1996; Takano et al., 1996) or electrical stimulation (Back et al., 1996)] results in larger infarct volumes. Accordingly, administration of drugs (Gill et al., 1992; Iijima et al., 1992; Mies et al., 1993, 1994; Rawanduzy et al., 1997; Tatlisumak et al., 2000) or physiologic treatments (Chen et al., 1993) that decrease the frequency of SpD associated with ischemic injury is also neuroprotective.
SpD may be triggered by high [K+]o and glutamate in the ischemic core and actively propagates tissue depolarization, ionic imbalances, and glutamate release into adjacent tissue. In the injury penumbra, where blood supply is compromised, SpD waves cause further reduction in tissue PO2 and exacerbate metabolic stress-failure (Back et al., 1994; Somjen, 2001). In this context, SpD waves are referred to as peri-infarct depolarizations (PIDs), reflecting their pathogenic role and similarity to anoxic depolarization. In middle cerebral artery occlusion (MCAo) models, anesthetized rats have exhibited up to eight PIDs over 3 postinjury hours (Gill et al., 1992; Iijima et al., 1992; Mies et al., 1993; Back et al., 1996), and cats and monkeys have displayed up to 20 and 8 PIDs, respectively, through 4 hr after MCA electrocoagulation (Strong et al., 2000).
PIDs in the initial hours after injury have been shown to cause a step-wise increase in the ischemic tissue volume (Busch et al., 1996; Takano et al., 1996). If PID events mediate expansion of the core region of pannecrosis into the ischemic penumbra, they should recur throughout the course of infarct maturation. Because lesion volumes mature through 24 hr in rat MCAo, we established a procedure to perform chronic DC recordings in nonanesthetized freely behaving rats after MCAo injury. In these recordings, negative shifts of the DC potential with characteristics of SpD/PID recurred through ∼40 hr after injury with 10-fold greater incidence than previously reported. Peak rates of infarct growth at 8-18 hr were matched by increased rates of PID occurrence, and inhibition of PIDs over this period with a high-affinity NMDA receptor antagonist also provided neuroprotection. These results demonstrate an association between PIDs and ischemic cell death and suggest a novel target for delayed neuroprotective therapy.
Materials and Methods
Male Sprague Dawley rats (Charles River Laboratories, Raleigh, VA), weighing 275-350 gm, were used in these experiments. For surgical procedures, anesthesia was induced with 5% halothane delivered in oxygen and maintained at 1.5% throughout surgery, and core body temperatures were maintained at 37.0°C by a homeothermic heating blanket. All procedures were approved by the Walter Reed Army Institute of Research Animal Care and Use Committee. All research was conducted in compliance with the Animal Welfare Act, Guide for the Care and Use of Laboratory Animals (National Research Council), and other federal statutes and regulations relating to animals and experiments involving animals. Animals were maintained in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
DC recordings. DC recordings were made with epidural Ag/AgCl electrodes prepared from 0.010 inch diameter Ag wire (Sigmund Cohn, Mt. Vernon, NY). Wire was flamed to produce 0.6-0.8 mm diameter spherical tips and then chloridized. Electrodes were placed in burr holes through the skull made with metal probes, and two screws were placed over the uninjured hemisphere to serve as head-mount anchors. The free ends of the electrode wires were soldered to a multipin connector (March Electronics, West Hempstead, NY), and the assembly was fixed to the skull with cranioplastic cement (Plastics One, Roanoke, VA). Signals were recorded through shielded cables, input to separate channels for DC and AC amplification with a Grass (Grass Instruments, Natick, MA) Model 15 amplifier system (15A12 DC and 15A54 AC amplifiers), digitized at 100 Hz, and collected with EEG recording and analysis software (Polyview or Gamma; Astro-Med, West Warwick, RI). DC recordings were filtered with a 0.1 Hz low-pass cutoff, and AC recordings were bandpass filtered at 0.5-70 Hz.
Recording montages. A two-electrode referential recording montage was used to monitor PID activity. Lead electrodes were placed over frontal and parietal cortices (1.5 mm anterior and 2.5 posterior to bregma, respectively) in the injured hemisphere, medial to the core infarction (1.5 mm lateral to bregma). For topographic mapping experiments, eight electrodes were implanted over the injured hemisphere in a configuration of two columns in the anteroposterior axis located ∼1.5 and 4.5 mm lateral to bregma. Each column consisted of four electrodes located 3.5 mm anterior, 2.0 mm anterior, 2.0 mm posterior, and 5.0 mm posterior to bregma. In both montages, a reference electrode was located posterior to λ.
Middle cerebral artery occlusion. Animals were subjected to MCAo by the intraluminal filament method on the day after electrode implantation (Britton et al., 1997). The right external carotid artery was ligated, cauterized, and cut, and its branches were coagulated. A 35 mm length of 3-0 nylon monofilament (Ethicon, Somerville, NJ) with a rounded tip was then inserted into the internal carotid artery via the proximal end of the external carotid artery stump. The filament was advanced ∼20 mm beyond the carotid artery bifurcation when slight resistance was encountered. Animals subjected to transient MCAo were briefly re-anesthetized at 2 hr and reperfused by retraction of the filament. For sham MCAo surgeries, the same procedures were followed, but the filament was advanced only 10 mm beyond the carotid bifurcation and was left in place until the animal was killed at 24 hr.
Experimental procedures. Animals were provided 20-25 gm of rat feed per day and had water access ad libitum before MCAo; thereafter, food and water were available ad libitum. Animals were individually housed under a 12 hr light/dark cycle. On the day of MCAo surgery, rats were transferred from their cages to custom designed plexiglas recording chambers (Dragonfly, Ridgeley, WV) equipped with multichannel swivel commutators (Plastics One). The multipin connector on the rat skull was connected to the swivel system via a flexible shielded cable, allowing free movement of the animals during recordings, and interfaced with the amplifier system (Tortella et al., 1999).
Before MCAo surgery, rectal temperatures and neurologic function were assessed, and baseline recordings were made continuously for 30 min. Immediately after surgery, animals were returned to recording chambers, and continuous digital records were obtained for the subsequent 24 or 72 hr. Rectal temperatures and neurologic function were assessed at 2 hr after injury and before the animal was killed at the study endpoint. Animals that did not exhibit full neurologic impairment at 2 hr after injury (n = 4), had subarachnoid hemorrhage at the time of killing (n = 2), or died before the study endpoint (n = 5) were excluded from the study. Criteria for suppression of EEG during MCAo, used previously to exclude animals and reduce variability in infarct volumes (Hartings et al., 2003), were not used in this study. Animals were killed at 24 hr after injury with two exceptions. In three permanent MCAo animals, DC recordings were made for 72 hr. Also, infarct data shown in Figure 5 were derived from animals killed at 3, 6, 12, 18, 24, and 72 hr after 2 hr transient MCAo. PIDs were not monitored in these animals.
Figure 5.
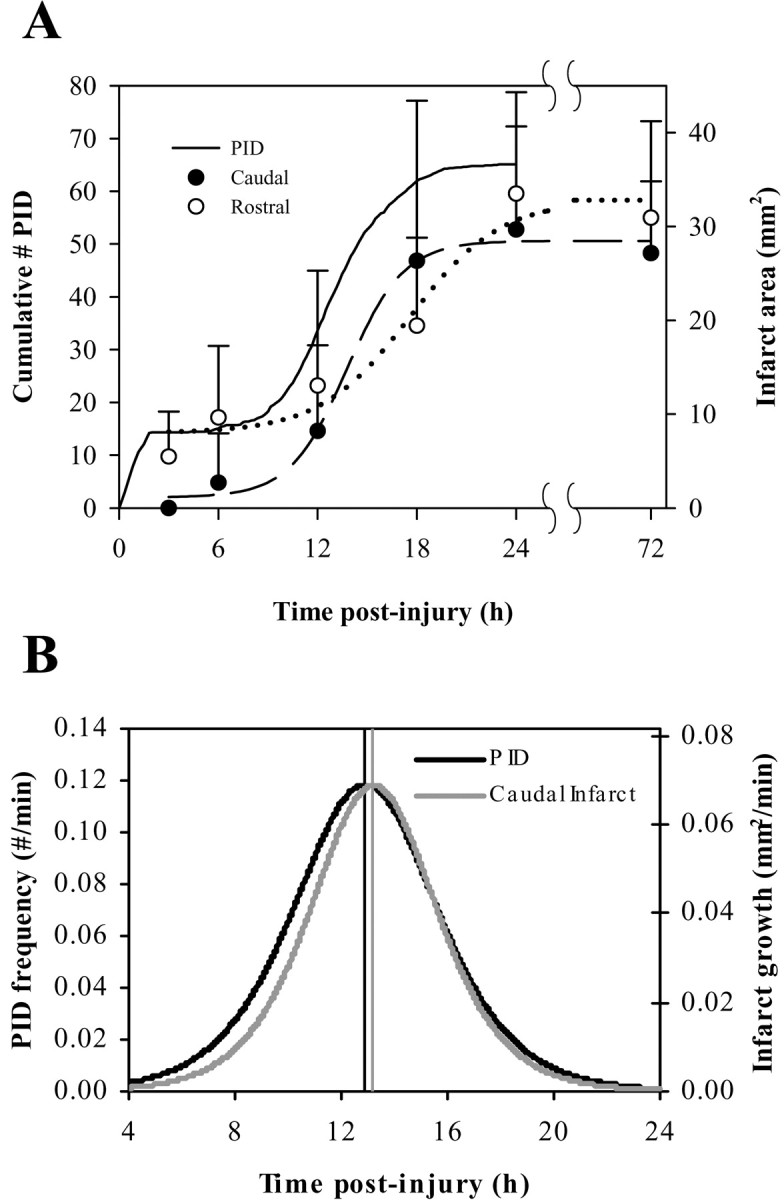
Parallel of infarct growth rates and PID frequency. A, Animals subjected to transient MCAo were killed at 3, 6, 12, 18, 24, and 72 hr after injury (n = 5-8), and infarct areas of coronal sections at 5 mm (○, rostral) and 8 mm (•, caudal) from the rostral pole were quantified. Error bars show SD. The solid line shows the mean cumulative number of PIDs in transient MCAo [i.e., the integral of the histogram function in Fig. 3_A_, or 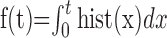 ]. Rostral, caudal, and PID data were fit with the logistic equation f(x) = (a/ 1 0 + (e _b(x_-c))) + d and yielded the following parameters: PID, a = 51.27, b = -0.55, c = 12.90, d = 13.70; rostral, a = 24.80, b = -0.37, c = 17.61, d = 8.00; caudal, a = 26.15, b = -0.63, c = 13.21, d = 1.16. PID data were fit over 3-24 hr (function not shown). B, Derivative functions of the logistic equations fit to PID and caudal infarct data. Vertical lines denote values for parameter c, corresponding to peak rates of PID occurrence and infarct growth.
]. Rostral, caudal, and PID data were fit with the logistic equation f(x) = (a/ 1 0 + (e _b(x_-c))) + d and yielded the following parameters: PID, a = 51.27, b = -0.55, c = 12.90, d = 13.70; rostral, a = 24.80, b = -0.37, c = 17.61, d = 8.00; caudal, a = 26.15, b = -0.63, c = 13.21, d = 1.16. PID data were fit over 3-24 hr (function not shown). B, Derivative functions of the logistic equations fit to PID and caudal infarct data. Vertical lines denote values for parameter c, corresponding to peak rates of PID occurrence and infarct growth.
In two control experiments, to verify identification of SpD/PID events, animals were anesthetized with halothane, placed in a stereotax, and implanted with epidural electrodes over the frontal cortex as described above. A craniotomy was then made over the occipital cortex, and the dura was excised. SpD was provoked by placing a cotton ball soaked in 3 m KCl on the craniotomy, and recordings were made as described above.
Intrathecal conantokin-G treatment. A group of rats subjected to transient MCAo received treatment with intrathecal conantokin-G (Con-G) at 8 hr after injury, and another group serving as controls received saline. Con-G, a 17 amino acid peptide derived from the marine snail Conus geographus, has been shown in voltage-clamp studies of recombinant NMDA receptors to be a selective competitive antagonist of the NR2B subunit (Donevan and McCabe, 2000) and displaces binding of (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK801) in adult rat brain sections (Klein and Castellino, 1999). Treatment was administered via an intrathecal catheter (LoPachin et al., 1981) implanted 2 d before MCAo surgery. Con-G (40 nmol) was given over 1 min by bolus injection in 10 μl of saline, and the same volume of saline alone was administered to control animals. Intrathecal doses of 80 nmol produce Con-G brain levels in the low nanomolar range, as determined by high-performance liquid chromatography (A. J. Williams, personal communication).
Infarct and neurologic assessment. Animals were deeply anesthetized and killed by decapitation at the specified study endpoints. Forebrains were cut in seven 2 mm coronal sections beginning 1 mm from the rostral pole, stained with 2,3,5-triphenyltetrazolium chloride (TTC), and digitally imaged using a computerized analysis system (LOATS, Westminster, MD). Areas of infarction were defined as those completely lacking red TTC stain, and total infarct volumes were computed by integrating infarct areas of sequential brain sections (Williams and Tortella, 2002).
Neurologic function was assessed by testing for four behaviors according to a modification (Tortella et al., 1999) of the procedure described by Bederson et al. (1986): (1) contralateral shoulder adduction (curling) and (2) unequal forelimb flexion when suspended by the tail, (3) open-field circling, and (4) unequal resistance to left versus right lateral displacement. Any animals that did not exhibit all four of these behaviors at 2 hr after injury were excluded from the study.
Data analysis. PIDs were identified as negative shifts in DC potential occurring on both frontal and parietal electrodes (see Results). To generate topographic maps from multielectrode recordings (see Fig. 8), DC offsets were calculated using a moving average and subtracted from the original data. Data were then smoothed using a 3 sec boxcar average. Data points between electrodes were interpolated using a minimum curvature surface algorithm and the output projected on a scaled schematic of the dorsal surface of the right hemisphere. Data are reported as means ± SD unless indicated otherwise.
Figure 8.
Topographic mapping of PID propagation patterns. _A_-C, Representative examples of rostrocaudal (A), caudorostral (B), and lateromedial (C) propagation patterns of individual PID events. _A_-C, Recordings were made from six to eight electrodes located at positions shown on the schematic dorsal-view diagrams of the ischemic (right) hemisphere. Recordings from each electrode location are shown at the left and are color coded to denote the recording site. Time and voltage calibrations are shown for each group of recordings. Topographic maps were constructed for each of the three (A-C) PIDs at three time slices (see Materials and Methods). Vertical white lines in recordings mark the three time slices (i, ii, iii) for each of the corresponding topographic maps. Colors in the maps reflect voltage values according to the color bars shown in each panel. Note that maps were constructed by interpolating voltage values from actual values recorded at electrode locations and do not reflect actual voltage values at sites distant from the electrodes.
Results
Data set and recordings
We report data from 25 animals in which chronic DC recordings were obtained after successful MCAo. Of these, 11 animals were subjected to 2 hr transient MCAo, seven were subjected to permanent MCAo, three received sham MCAo surgery, and four were used to generate topographic DC maps after MCAo. Mean rectal temperatures of injured animals were 37.5 ± 0.6 before MCAo, 38.8 ± 0.5 at 2 hr after injury, and 36.9 ± 0.7 at 24 hr.
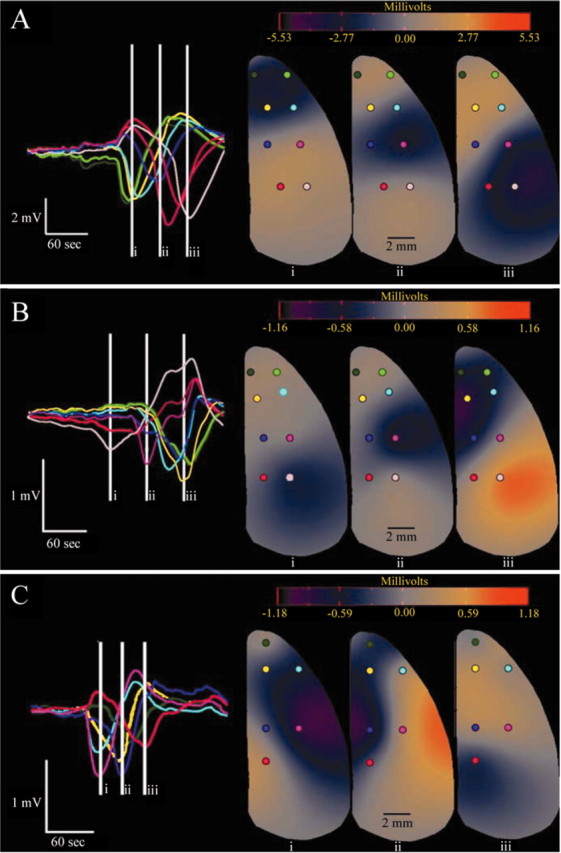
DC recordings were made in freely behaving rats from epidural Ag/AgCl electrodes with the dura mater intact. Figure 1_A_ shows a representative 24 hr example of these recordings. DC potentials showed substantial offset and fluctuations both before and after injury. Most activity was of small amplitude, although some potential shifts were on the order of several millivolts. After MCAo, stereotyped negative deflections of 1.0-2.5 mV amplitude (range, ∼0.5-4.0 mV) and ∼30-60 sec total duration (Fig. 1_A,B_) appeared in both frontal and parietal recording locations, similar to PIDs recorded previously with epidural Ag/AgCl electrodes (Chen et al., 1993). To verify that deflections of this amplitude were SpD/PID events, we used the same epidural recording technique to monitor SpD evoked by cortical application of 3 m KCl in two halothane-anesthetized rats. With this technique, negative DC deflections accompanying depression of the EEG were 3.0-5.5 mV in amplitude (Fig. 2_A_). To further validate our epidural recordings, the dura mater was excised before electrode placement in two additional animals. After MCAo and recovery from anesthesia, larger amplitude 7-10 mV DC shifts were accompanied by EEG depressions characteristic of SpD/PID (Fig. 2_B_). Animals subjected to sham MCAo surgery did not exhibit DC shifts in the 24 hr recovery period. Because minimal damage was induced by electrode placement (Fig. 2_C_), we conclude that the DC shifts illustrated in Figure 1, A and B, reflect PID phenomena induced by MCAo.
Figure 1.

Continuous 24 hr recording of PID activity. A, Twenty-four hour DC recordings from a frontal, medial electrode in transient MCAo. PIDs are evidenced as negative spikes. B, High-resolution example of a PID event. C, Frequency distribution of inter-PID intervals for all transient MCAo animals during the ischemic period (black line) and after reperfusion (second phase; gray line). The ordinal axis represents the proportion of total PID intervals recorded during the respective period.
Figure 2.
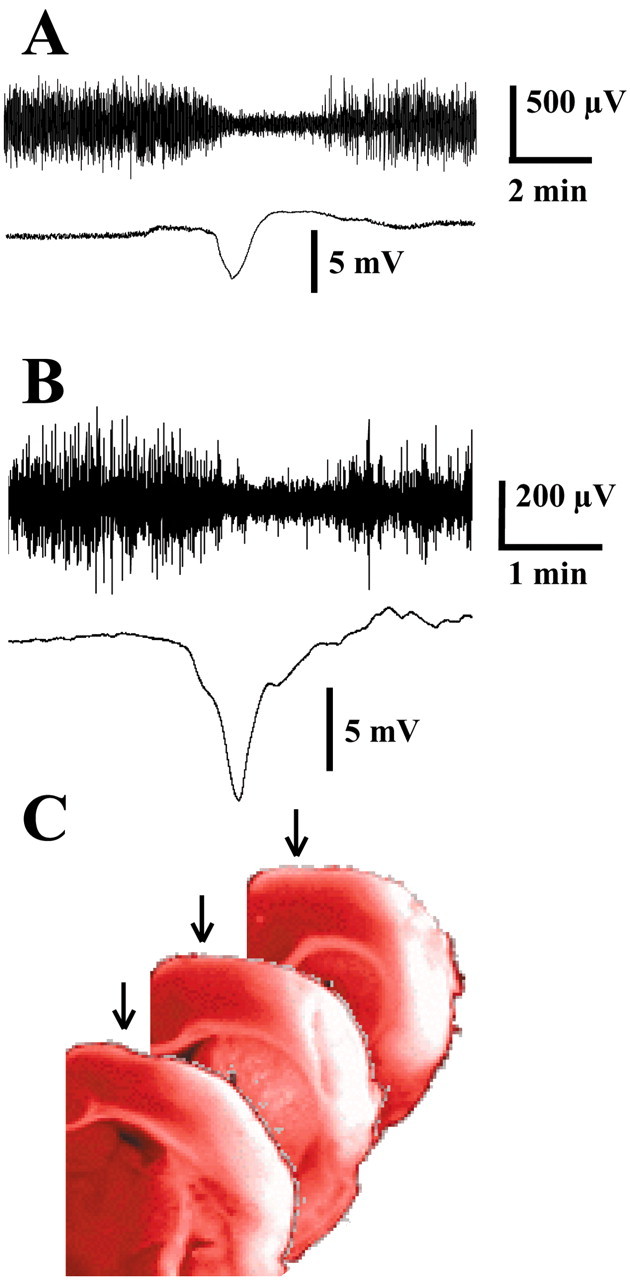
Epidural recordings of DC shifts with Ag/AgCl electrodes. A, AC-coupled EEG and DC potentials associated with SpD induced by application of 3 m KCl to the cortex. B, EEG and DC potentials recorded through epidural electrodes with excised dura mater after MCAo in a freely behaving animal. C, TTC-stained sections of the infarcted right hemisphere of an animal in which 123 PIDs were recorded over 24 hr. Electrodes were located 1.5 mm lateral to midline (arrows) and induced no observable cortical damage.
PID incidence
PID activity in Figure 1_A_ follows a biphasic pattern, which was typical for transient MCAo animals. PIDs began soon after MCAo and recurred periodically over the subsequent 2 hr. After reperfusion, activity ceased and did not recur for the following 6 hr. At ∼8 hr after injury, however, PID activity re-emerged in a delayed secondary phase. During this phase, which lasts 12 hr in Figure 1_A_, PIDs recurred continuously and often in rapid succession. Frequency plots of inter-PID intervals (Fig. 1_C_) show that these events occurred most often with refractory periods of 4-5 min during occlusion, but that shorter 2-3 min intervals (minimum, 1 min 54 sec) were common in the secondary phase.
Figure 3 shows PID times for all animals in raster (dot-gram) and histogram format. Histograms were constructed by accumulating PID events from all animals into 10 min bins. For transient MCAo, the initial PID phase was defined by the 2 hr occlusion period and had a mean of 15.9 ± 4.6 events. After reperfusion, quiescent periods with no PIDs averaged 7 hr 48 min (± 2 hr 43 min). The delayed secondary phase reached a peak population frequency of eight PID per hour at ∼13 hr and consisted of an average 55.2 ± 35.7 events.
Figure 3.

PID incidence over 24 hr after MCAo. A, Raster plots indicate times of PID occurrence relative to induction of transient MCAo for each of 11 animals. PID occurrences were accumulated across all animals into a population histogram with 10 min bins. Total PID counts were divided by the number of animals and multiplied by six to yield measures of mean hourly frequency. B, Same as A, except for permanent MCAo.
Animals subjected to permanent MCAo also showed a secondary phase of PID activity enduring through 24 hr (Fig. 3_B_). Peak PID rates were similar for both initial and secondary phases and for permanent and transient MCAo. Animals with permanent MCAo had an average 11.6 ± 4.9 events during the first 2 hr and 75.9 ± 26.2 over 2-24 hr. Seventy-two hour recordings of three permanent MCAo animals established that PIDs were rare after the waning of the secondary phase (data not shown). Two of these animals had one or two events at 37-43 hr, in both cases, close to 24 hr after cessation of the secondary phase.
PID and infarct volume
Rats subjected to transient MCAo had core infarct volumes of 111 mm3 (± 22 SEM) at 24 hr. Infarct volumes for 24 and 72 hr permanent MCAo were 292 mm3 (± 47 SEM) and 288 mm3 (± 41 SEM). For transient MCAo, there were no significant correlations between core infarct volume and the number of PIDs occurring during occlusion, in the secondary phase, or over the entire 24 hr recovery period (Fig. 4_A_). However, earlier times of onset of the secondary phase of PID were predictive of larger infarct volumes (_R_2 = 0.61; p < 0.01) (Fig. 4_A_), as was also the case for greater total durations of the secondary phase (time of last - time of first PID after reperfusion; _R_2 = 0.54; p = 0.01). With permanent MCAo, the numbers of PIDs in the secondary phase (2-24 hr; _R_2 = 0.65) (Fig. 4_B_) and for the full 24 hr (_R_2 = 0.59) were significantly positively correlated with infarct volume (p < 0.05). When permanent and transient MCAo are considered together, the same is true: PIDs in the secondary phase (_R_2 = 0.30) and the full 24 hr (_R_2 = 0.24), but not the first 2 hr (_R_2 = 0.10), correlate with infarct volume (p < 0.05). There were no significant relationships between the number of PIDs during occlusion and any parameters of the secondary phase for either permanent or transient MCAo.
Figure 4.
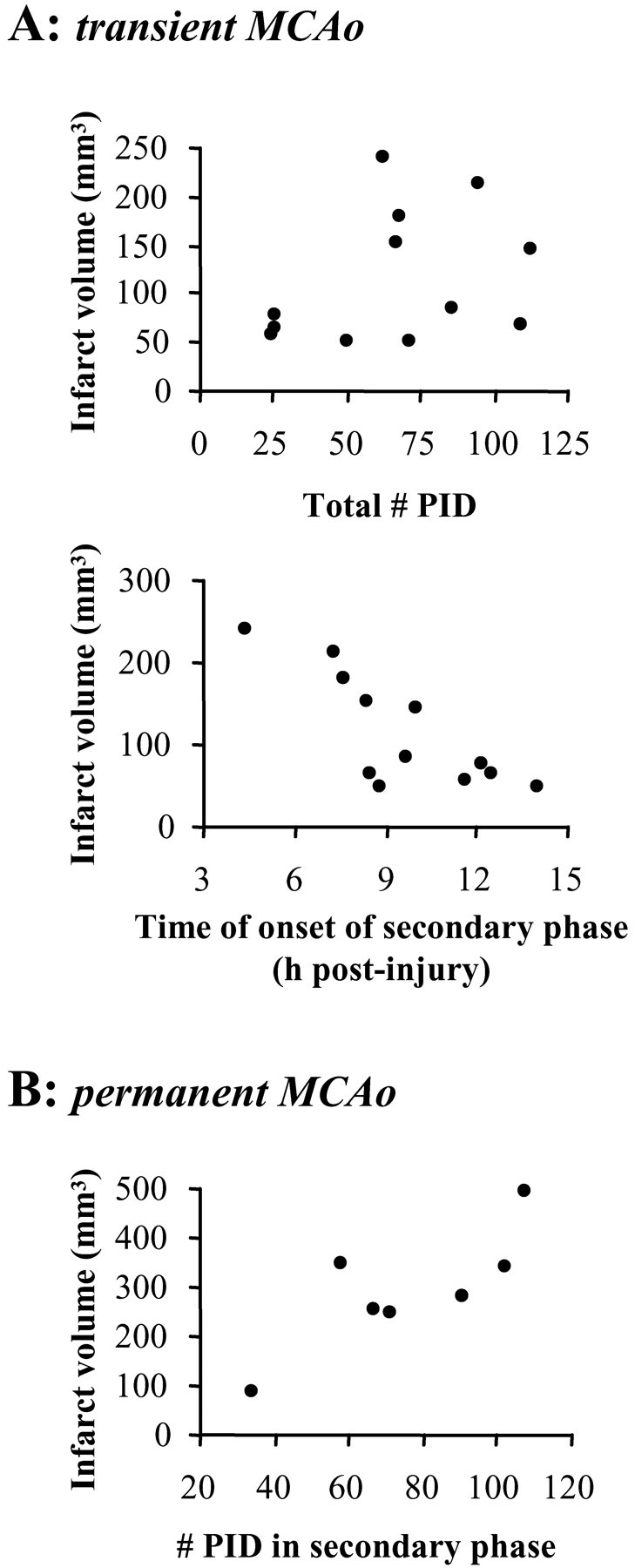
Correlations of PIDs with infarct volume. A, Top, Scatter plot of the total number of PIDs recorded per animal versus corresponding infarct volumes measured at 24 hr after transient MCAo. Bottom, The time of onset of the secondary phase of PIDs correlates with the final infarct volume (see Results). B, In permanent MCAo, the number of PIDs in the secondary phase correlates with the injury volume measured at 24 hr.
PID and infarct development
To explore the timing of infarct growth relative to the secondary phase of PIDs, a separate group of animals was killed at different postinjury times, and infarct areas of coronal sections 5 mm (rostral) and 8 mm (caudal) from the rostral pole were quantified. Figure 5 shows that both infarct growth and the cumulative number of PIDs followed similar nonlinear time courses, with the greatest rates of increase occurring at 10-16 hr. To compare time courses more directly, each data set was modeled by least squares nonlinear regression using the sigmoidal logistic function. The peak rates of change for these model equations, which correspond to times of maximum PID frequency and infarct growth, were 12.90 hr (PID; σ = 0.02), 13.21 hr (caudal; σ = 0.54), and 17.61 hr (rostral; σ = 2.00). Rates of infarct growth at the caudal level changed with a time course nearly identical to that for PID frequency (Fig. 5_B_).
Delayed treatment with NMDA receptor antagonist
The time course of PIDs over 8-18 hr after injury suggests that they contribute to infarct development. To investigate this hypothesis further, we sought to inhibit PIDs over this secondary phase. NMDA receptor-mediated currents are involved in the propagation of SpD/PID, and their antagonism has been shown to reduce PID occurrence in the early phase of rat focal ischemia (Gill et al., 1992; Mies et al., 1993; Dietrich et al., 1994). Therefore, we treated rats subjected to transient MCAo with the selective NR2B NMDA receptor antagonist Con-G (see Materials and Methods) (Donevan and McCabe, 2000), which has a neuroprotection ED50 of 50 nmol in rat MCAo when given 4 hr after injury (Williams et al., 2002). Here, we administered Con-G to rats (n = 11) intrathecally in a 40 nmol bolus dose at 8 hr after injury, and a control group of rats (n = 11) received saline.
Con-G- and saline-treated rats exhibited PIDs with similar frequency during 2 hr ischemia (Con-G, 12.2 ± 5.1; saline,13.0 ± 4.2) and after reperfusion before treatment (Con-G, 3.0 ± 10.4; saline, 5.7 ± 9.6) (Fig. 6_B_). After treatment, however, the Con-G group had a significantly reduced incidence of PIDs (19.1 ± 22.2) compared with controls (44.3 ± 28.5; two-tail Mann-Whitney U test; p < 0.05). Figure 6_D_ shows the mean PID frequency for each group over 24 hr after injury; differences emerge after Con-G administration at ∼8 hr. This reduction of secondary phase PIDs was associated with a 37% decrease in core infarct volume at 24 hr (Fig. 6_C_). The mean infarct volume for Con-G animals was 121.8 (±100.1 mm3), which was significantly less than that for control animals (194.8 ± 72.3 mm3; one-tail Mann-Whitney U test; p < 0.05).
Figure 6.
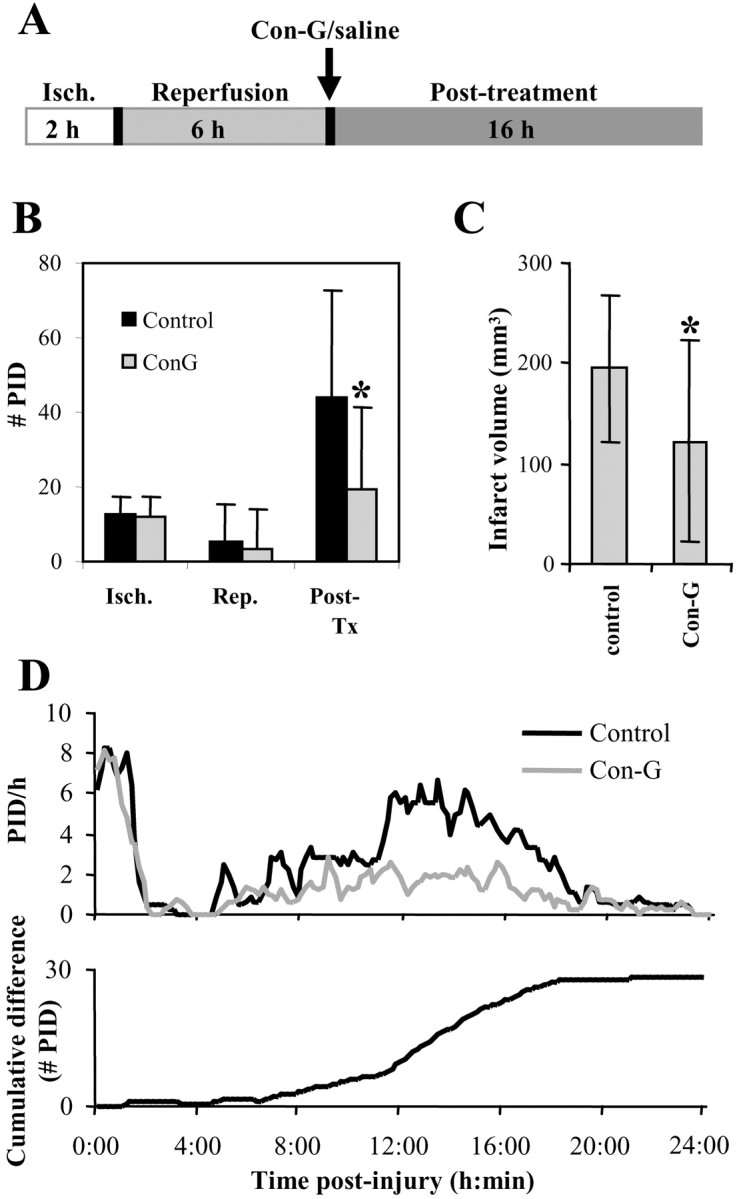
Con-G reduces infarct volumes and PID incidence over 8-24 hr. A, Schematic diagram of Con-G experiments showing the three epochs (ischemia, reperfusion before treatment, and post-treatment) over which PIDs were scored. B, Means and SDs of PID counts for Con-G-treated and saline-treated rats during each time epoch shown in A. C, Means and SDs of core infarct volumes. Asterisks denote significant differences (see Results). D, Top, Mean hourly rates of PID occurrence were computed separately for Con-G-treated and saline-treated rats. The 24 hr postinjury period is shown in 10 min bins, as in Figure 3. Bottom, The difference in mean PID rates for the two groups was computed by subtracting PID frequencies of Con-G rats from those of control rats. This function was then integrated cumulatively to illustrate the time course of the difference in the number of PIDs experienced in control versus Con-G animals. On average, control rats experienced 30 more PIDs than Con-G animals.
Patterns of PID propagation
The relative latency of DC deflections on electrodes over frontal and parietal regions varied for different PID events, implicating more than a single source for their initiation. In some cases, frontal deflection preceded parietal and in others vice versa (Fig. 7_A_). To investigate these patterns, time differences were measured between peak negativities for each wave, with positive latencies indicating earlier appearance on the frontal electrode. Figure 7 shows latency values for consecutive PIDs in four animals and illustrates several trends that emerged. First, latencies in the secondary phase were initially quite short, indicating more rapid propagation speed, but grew progressively longer through subsequent events. Second, latencies were not randomly distributed but instead clustered around particular values (frequency distributions) (Fig 7_B,D,E_, insets). Third, the sequential order in which latency values occurred was also nonrandom. Ordered sequences included series of consecutive latencies with similar values or alternations between two values (Fig. 7_C,E_).
Figure 7.

Serial ordering of PID propagation patterns. A, Simultaneous recordings show relative timing of DC negativities at frontal and parietal electrodes. B-E, Frontal-to-parietal latency values for each PID are plotted versus their number in serial order of occurrence for four transient MCAo animals. Positive values indicate previous appearance on frontal electrode. Insets show frequency distributions of latency values for each of the four animals (ordinate, number of events; abscissa, seconds).
To investigate the spatiotemporal patterns of DC potentials underlying these different latencies, topographic maps were constructed from multichannel recordings obtained in four animals. The most common PID pattern observed was a wave of negative DC deflections initiating in frontal regions and propagating caudally (Fig. 8_A_). These rostrocaudal waves corresponded to positive latencies of ∼30-80 sec (Fig. 6). PID waves also initiated in parieto-occipital regions and propagated to frontal areas, yielding negative latencies ranging from 10 to 80 sec (Fig. 8_B_). A third PID pattern consisted of initiation in lateral frontoparietal regions bordering the infarct and propagation to midline (Fig. 8_C_). In this case, the wavefront travels approximately parallel to the medial electrode column, reaching each electrode with relative synchrony. As indicated by the bimodal latency distributions in Figure 7, animals usually exhibited two of these patterns most predominantly.
Discussion
PID frequency
Chronic recordings of DC potential revealed a recurrence of PIDs in rat focal cerebral ischemia through 40 hr after injury. The mean numbers of PID events through 24 hr were 71 and 88 for transient and permanent MCAo, respectively, which represent 9- and 11-fold greater incidence than previously reported in the rat (Gill et al., 1992; Iijima et al., 1992; Mies et al., 1993; Back et al., 1996). This higher incidence is primarily attributable to the discovery of a secondary phase of PID activity during infarct maturation made possible by long-term monitoring in unanaesthetized freely behaving animals. However, even PID incidence over the initial 2 hr occlusion period was higher than previously reported. The lower incidence in previous studies is likely attributable to the continuous use of anesthesia, which is neuroprotective and slows brain metabolism, and particularly halothane, which reduces SpD/PID frequencies (Saito et al., 1997; Kitahara et al., 2001). In addition, maintaining rectal temperatures, which closely reflect brain temperatures (Xue et al., 1992; Zhang et al., 1994), at normothermic (37°C) levels in anesthetized preparations can reduce PID frequency (Chen et al., 1993). In this study, halothane was discontinued immediately after filament insertion, and rectal temperatures increased to an average 38.8°C by 2 hr after occlusion. These physiologic factors may also account for brief intervals between successive PIDs reported here, which were often shorter (<5 min) than refractory periods for mechanically or chemically evoked SpD.
The time course of PID activity showed two distinct phases: an initial phase lasting 2-3 hr after injury and a secondary phase predominating at 8-18 hr. In transient MCAo, PIDs ceased entirely for a variable 2-12 hr period after reperfusion. This cessation, occurring in all animals, may be attributable to, in part, the brief (∼5 min) use of halothane during reperfusion-filament retraction surgery. This is unlikely because animals subjected to permanent occlusion were not re-anesthetized, yet also showed a consistent reduction in PID occurrence beginning ∼3 hr after injury. This preceded a distinct secondary phase, which began at the same time (∼8-10 hr) as in transient MCAo. These data indicate that the period of relative quiescence followed by a secondary phase of PIDs reflects an inherent sequence in the progression of infarct pathology.
PID and infarct development
The number of PIDs in the secondary phase was significantly correlated with core infarct volumes for permanent MCAo and for both permanent and transient MCAo combined. For transient MCAo, the time of onset of the secondary phase was a strong predictor of infarct volume. Infarct growth studies also suggested a contribution of secondary phase PIDs to infarct maturation. Coronal brain sections 5 mm from the rostral pole showed infarcts as early as 1 hr after reperfusion in transient MCAo. However, infarction in more caudal regions began developing after an additional 3 hr delay, and the time course of this development paralleled that of the secondary phase of PIDs over 6-20 hr after injury. These results, together with the predominance of rostrocaudal PID propagation patterns, suggest that PIDs initiated by earlier developing frontotemporal infarction contribute to the caudal expansion of injury.
If PIDs in the secondary phase contribute to infarct expansion, inhibiting them should be associated with reduced injury volume. To test this, rats were administered the high-affinity NMDA receptor antagonist Con-G, which has been shown previously to provide neuroprotection with an 8 hr therapeutic window in rat MCAo (Williams et al., 2002). Compared with control saline-treated rats, those given Con-G at 8 hr after injury had 57% less PIDs over 8-24 hr and also a 37% reduction of infarct volumes. These results are consistent with a pathogenic role for PIDs in the secondary phase. However, as is the case with previous studies on PIDs during the ischemic phase (Iijima et al., 1992; Chen et al., 1993), it is unknown whether reducing PID occurrences provides neuroprotection, or vice versa. Additional discussion of this issue with regard to the use of glutamate receptor antagonists is provided below.
PID propagation patterns
Topographic mapping and measurement of relative latencies of DC negativities revealed several patterns of PID propagation, including caudorostral, lateromedial, and most commonly rostrocaudal. For the latter, negativities appeared first near the frontal pole and propagated caudally to parietal and occipital areas. Latencies between frontal and parietal electrodes separated by 4 mm ranged from ∼30 to 80 sec. These latencies grew progressively longer through the secondary phase, indicating slower propagation that may reflect worsening pathology.
The multiple foci for PID development may reflect different mechanisms of initiation. Nedergaard and Hansen (1993) showed that the initiation of depolarization waves in MCAo can occur with characteristics of either SpD, as provoked by high [K+]o and glutamate, or ischemic depolarization resulting from hypoperfusion in ischemic border zones. These processes may occur in different cortical regions. Additionally, rostrocaudal waves may initiate in the striatum and propagate to the cerebral cortex, rather than initiate within the cortex itself. The rat brain is unique in that the claustrum and nucleus accumbens provide a rostral bridge of nearly continuous gray matter between the striatum and neocortex, and Vinogradova et al. (1991) demonstrated that SpD, occurring in the striatum, can traverse this bridge. Thus, the striatal infarct core might serve as an independent source of PIDs, which propagate to the cortex and contribute to its worsening pathology. This additional source may, in part, account for the high incidence of PIDs in MCAo of the rat compared with other species as well as provide another “apparent” site of cortical PID initiation.
PID: the principal mechanism of penumbral cell death?
Previous studies have established that during SpD, input resistances of participating neurons are reduced 50-90%, [Ca2+]o falls to <0.1 mm, and [K+]o increases to >50 mm. In normally perfused tissue, these changes associated with SpD are not likely to provoke cellular damage but rather may provide neuroprotective conditioning against subsequent ischemic injury (Matsushima et al., 1996; Yanamoto et al., 1998). In regions of the ischemic penumbra, however, the metabolic challenges of SpD-like depolarizations, properly termed PIDs, deplete ATP (Busch et al., 1996), decrease PO2 (Back et al., 1994), induce permanent cell swelling (Kempski et al., 2000), and are associated with a transient or permanent reduction of local blood flow (Strong et al., 2003). In vitro, it has been shown that such SpD-like depolarizations cause neuronal damage when coupled with oxygen and glucose deprivation (Jarvis et al., 2001). In vivo, PIDs cause a step-wise increase in ischemic lesion volume (Busch et al., 1996; Takano et al., 1996) and eventual permanent depolarization of penumbral areas bordering the core infarction (Higuchi et al., 2002). However, these studies have not targeted time points (>4 hr) over which the majority (80%) of cell death occurs. Thus, the present study is unique in demonstrating that PIDs mediate not only increases in ischemic territory but also persist throughout and predict rates of growth of infarcted territory.
Indeed, whereas the excitotoxic effects of glutamate in vitro are well established, it is possible that cell death in the focal ischemic penumbra occurs primarily as a consequence of the metabolic stress of PIDs, and that glutamate release and excitotoxicity occur only as secondary consequences of these depolarizations (Hossman, 1994; Jarvis et al., 2001). Obrenovitch and Urenjak (1997) have argued, for instance, that the neuroprotective effects of glutamate receptor antagonists in brain injury models do not necessarily imply the occurrence of excitotoxic processes. In reducing PID frequency, administration of glutamate receptor antagonists reduces both infarct volume (Gill et al., 1992; Iijima et al., 1992; Mies et al., 1993) and ATP depletion after rat MCAo (Mies et al., 1994). In these and the present Con-G experiments, neuroprotection may be attributable to blocking the spread of PIDs, which depend in part on glutamate receptors, and not because of direct effects on glutamate-induced currents per se. An implication of this perspective is that neuroprotective strategies could focus on the blockade of PIDs by targeting propagation mechanisms other than glutamate receptors, thus side-stepping toxic consequences of their antagonism. Given the present results, this hypothesis further suggests that a more delayed therapeutic window might be possible than has been achieved in clinical trials to date.
It has been questioned whether SpD-PID occurs in the human brain. However, SpD-like events were demonstrated electrophysiologically to recur through 3-4 d after injury in trauma patients with intracranial hemorrhage (Strong et al., 2002; but see Mayevsky et al., 1996). Electrophysiological evidence of SpD was also found in response to KCl administration in human striatum and hippocampus in vivo (Sramka et al., 1977), and SpD can be elicited in human brain slices in vitro (Avoli et al., 1991; Gorji et al., 2001). During migraine visual aura, fMRI revealed changes in blood oxygen level-dependent signals with characteristics of SpD (Hadjikhani et al., 2001). Together, these data suggest that SpD-like depolarizations are involved in a number of human diseases including ischemic stroke. Although direct evidence is missing for this condition, the current results present new evidence that inhibition of PIDs might be a necessary component of stroke neuroprotection strategies. Experimentally, PID monitoring throughout the secondary phase represents a novel functional endpoint for neuroprotection investigation and therapeutic assessments.
Footnotes
We thank Dr. Rossana Berti for use of her infarct volume data and Brad Cunningham and Chelyse Stefanik for expert technical support. Our views do not purport or reflect the position of the Department of the Army or the Department of Defense (AR 360-5, paragraph 4-3).
Correspondence should be addressed to Dr. Jed A. Hartings, Division of Neurosciences, Walter Reed Army Institute of Research, 503 Robert Grant Avenue, Silver Spring, MD 20910. E-mail: jed.hartings@amedd.army.mil.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2311602-09$15.00/0
References
- Avoli M, Drapeau C, Louvel J, Pumain R, Olivier A, Villemure JG ( 1991) Epileptiform activity induced by low extracellular magnesium in the human cortex maintained _in vitro_Ann Neurol 30: 589-596. [DOI] [PubMed] [Google Scholar]
- Back T, Kohno K, Hossman K-A ( 1994) Cortical negative DC deflections following middle cerebral artery occlusion and KCl-induced spreading depression: effect on blood flow, tissue oxygenation, and electroencephalogram. J Cereb Blood Flow Metab 14: 12-19. [DOI] [PubMed] [Google Scholar]
- Back T, Ginsberg MD, Dietrich WD, Watson BD ( 1996) Induction of spreading depression in the ischemic hemisphere following experimental middle cerebral artery occlusion: effect on infarct morphology. J Cereb Blood Flow Metab 16: 202-213. [DOI] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuh M, Nishmura MC, Davis RL, Bartkowski H ( 1986) Rat middle cerebral artery occlusion: evaluation of the model and development of a neurological examination. Stroke 17: 472-476. [DOI] [PubMed] [Google Scholar]
- Branston NM, Strong AJ, Symon L ( 1977) Extracellular potassium activity, evoked potential and tissue blood flow: relationships during progressive ischaemia in baboon cerebral cortex. J Neurol Sci 32: 305-321. [DOI] [PubMed] [Google Scholar]
- Britton P, Lu M, Laskosky M, Tortella F ( 1997) Dextromethorphan protects against cerebral injury following transient, but not permanent, focal ischemia in rats. Life Sci 60: 1729-1740. [DOI] [PubMed] [Google Scholar]
- Busch E, Gyngell ML, Mansfred E, Hoehn-Berlage M, Hossman K-A ( 1996) Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J Cereb Blood Flow Metab 16: 1090-1099. [DOI] [PubMed] [Google Scholar]
- Chen Q, Chopp M, Bodzin G, Chen H ( 1993) Temperature modulation of cerebral depolarization during focal cerebral ischemia in rats: correlation with ischemic injury. J Cereb Blood Flow Metab 13: 389-394. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Feng Z-C, Leistra H, Watson BD, Rosenthal M ( 1994) Photothrombotic infarction triggers multiple episodes of cortical spreading depression in distant brain regions. J Cereb Blood Flow Metab 14: 20-28. [DOI] [PubMed] [Google Scholar]
- Dijkhuizen RM, Beekwilder JP, van der Worp HB, van der Sprenkel JWB, Tulleken KAF, Nicolay K ( 1999) Correlation between tissue depolarizations and damage in focal ischemic rat brain. Brain Res 840: 194-205. [DOI] [PubMed] [Google Scholar]
- Donevan SD, McCabe RT ( 2000) Conantokin G is an NR2B-selective competitive antagonist of _N-_methyl-d-aspartate receptors. Mol Pharmacol 58: 614-623. [DOI] [PubMed] [Google Scholar]
- Gill R, Andine P, Hillered L, Persson L, Hagberg H ( 1992) The effect of MK-801 on cortical spreading depression in the penumbral zone following focal ischaemia in the rat. J Cereb Blood Flow Metab 12: 371-379. [DOI] [PubMed] [Google Scholar]
- Gorji A, Scheller D, Straub H, Tegtmeier F, Kohling R, Hohling JM, Tuxhorn I, Ebner A, Wolf P, Werner Panneck H, Oppel F, Speckmann EJ ( 2001) Spreading depression in human neocortical slices. Brain Res 906: 74-83. [DOI] [PubMed] [Google Scholar]
- Hadjikhani N, Sanchez del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RBH, Sorensen AG, Moskowitz MA ( 2001) Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci USA 98: 4687-4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AJ, Lauritzen M ( 1984) The role of spreading depression in acute brain disorders. An Acad Bras Cienc 56: 457-479. [PubMed] [Google Scholar]
- Hartings JA, Williams AJ, Tortella FC ( 2003) Occurrence of nonconvulsive seizures, periodic epileptiform discharges, and intermittent rhythmic delta activity in rat focal ischemia. Exp Neurol 179: 139-149. [DOI] [PubMed] [Google Scholar]
- Higuchi T, Takeda Y, Hashimoto M, Nagano O, Hirakawa M ( 2002) Dynamic changes in cortical NADH fluorescence and direct current potential in rat focal ischemia: relationship between propagation of recurrent depolarization and growth of the ischemic core. J Cereb Blood Flow Metab 22: 71-79. [DOI] [PubMed] [Google Scholar]
- Hossman K-A ( 1994) Glutamate-mediated injury in focal cerebral ischemia: the excitotoxin hypothesis revised. Brain Pathol 4: 23-36. [DOI] [PubMed] [Google Scholar]
- Iijima T, Mies G, Hossmann K-A ( 1992) Repeated negative DC deflections in rat cortex following middle cerebral artery occlusion are abolished by MK-801: effect on volume of ischemic injury. J Cereb Blood Flow Metab 12: 727-733. [DOI] [PubMed] [Google Scholar]
- Jarvis CR, Anderson TR, Andrew RD ( 2001) Anoxic depolarization mediates acute damage independent of glutamate in neocortical brain slices. Cereb Cortex 11: 249-259. [DOI] [PubMed] [Google Scholar]
- Kempski O, Otsuka T, Seiwert T, Heimann A ( 2000) Spreading depression induces permanent cell swelling under penumbra conditions. Acta Neurochir [Suppl] 76: 251-255. [DOI] [PubMed] [Google Scholar]
- Kitahara Y, Taga K, Abe H, Shimoji K ( 2001) The effects of anesthetics on cortical spreading depression elicitation and c-fos expression in rats. J Neurosurg Anesthesiol 13: 26-32. [DOI] [PubMed] [Google Scholar]
- Klein RC, Castellino FJ ( 1999) Inhibition of MK801 binding in adult rat brain sections by conantokin-G and conantokin-T. Neurosci Lett 273: 171-174. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Rudy TA, Yaksh TL ( 1981) An improved method for chronic catheterization of the rat spinal subarachnoid space. Physiol Behav 27: 559-561. [DOI] [PubMed] [Google Scholar]
- Matsushima K, Hogan MJ, Hakim AM ( 1996) Cortical spreading depression protects against subsequent focal cerebral ischemia in rats. J Cereb Blood Flow Metab 16: 221-226. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Doron A, Manor T, Meilin S, Zarchin N, Ouaknine GE ( 1996) Cortical spreading depression recorded from the human brain using a multiparametric monitoring system. Brain Res 740: 268-274. [DOI] [PubMed] [Google Scholar]
- Mies G, Iijima T, Hossmann KA ( 1993) Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat. NeuroReport 4: 709-711. [DOI] [PubMed] [Google Scholar]
- Mies G, Kohno K, Hossmann K-A ( 1994) Prevention of periinfarct direct current shifts with glutamate antagonist NBQX following occlusion of the middle cerebral artery in the rat. J Cereb Blood Flow Metab 14: 802-807. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Hansen AJ ( 1993) Characterization of cortical depolarizations evoked in focal cerebral ischemia. J Cereb Blood Flow Metab 13: 568-574. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP, Urenjak J ( 1997) Is high extracellular glutamate the key to excitotoxicity in traumatic brain injury? J Neurotrauma 14: 677-698. [DOI] [PubMed] [Google Scholar]
- Rawanduzy A, Hansen A, Hansen TW, Nedergaard M ( 1997) Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. J Neurosurg 87: 916-920. [DOI] [PubMed] [Google Scholar]
- Saito R, Graf R, Hubel K, Fujita T, Rosner G, Heiss W-D ( 1997) Reduction of infarct volume by halothane: effect on cerebral blood flow or perifocal spreading depression-like depolarizations. J Cereb Blood Flow Metab 17: 857-864. [DOI] [PubMed] [Google Scholar]
- Somjen GG ( 2001) Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 81: 1065-1096. [DOI] [PubMed] [Google Scholar]
- Sramka M, Brozek G, Bures J, Nadvornik P ( 1977) Functional ablation by spreading depression: possible use in human stereotactic neurosurgery. Appl Neurophysiol 40: 48-61. [PubMed] [Google Scholar]
- Strong AJ, Smith SE, Whittington DJ, Meldrum BS, Parsons AA, Krupinski J, Hunter AJ, Patel S ( 2000) Factors influencing the frequency of fluorescence transients as markers of peri-infarct depolarizations in focal cerebral ischemia. Stroke 31: 214-222. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Fabricius M, Boutelle MG, Hibbins SJ, Hopwood SE, Jones R, Parkin MC, Lauritzen M ( 2002) Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke 33: 2738-2743. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Hopwood SE, Boutelle MG, Parkin MC, Bezzina L, Yih-Huei S, Dunn AK ( 2003) Measuring dynamic changes in perfusion in the penumbra with high spatial and temporal resolution using laser speckle imaging: comparison with indicator clearance. J Cereb Blood Flow Metab 23[Suppl] 1: 300. [Google Scholar]
- Takano K, Latour LL, Formato JE, Carano RAD, Helmer KG, Hasegawa Y, Sotak CH, Fisher M ( 1996) The role of spreading depression in focal ischemia evaluated by diffusion mapping. Ann Neurol 39: 308-313. [DOI] [PubMed] [Google Scholar]
- Tatlisumak T, Takano K, Meiler MR, Fisher M ( 2000) A glycine site antagonist ZD9379 reduces number of spreading depressions and infarct size in rats with permanent middle cerebral artery occlusion. Acta Neurochir 76: 331-333. [DOI] [PubMed] [Google Scholar]
- Tortella FC, Britton P, Williams A, Lu XCM, Newman AH ( 1999) Neuroprotection (focal ischemia) and neurotoxicity (electroencephalographic) studies in rats with AHN649, a 3-amino analog of dextromethorphan and low-affinity _N-_methyl-d-aspartate antagonist. J Pharmacol Exp Ther 291: 399-408. [PubMed] [Google Scholar]
- Vinogradova LV, Koroleva VI, Bures J ( 1991) Re-entry waves of Leao's spreading depression between neocortex and caudate nucleus. Brain Res 538: 161-164. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Tortella FC ( 2002) Neuroprotective effects of the sodium channel blocker RS100642 and attenuation of ischemia-induced brain seizures in the rat. Brain Res 932: 45-55. [DOI] [PubMed] [Google Scholar]
- Williams AJ, Ling G, McCabe RT, Tortella FC ( 2002) Intrathecal conantokin-g is neuroprotective in a rat model of focal cerebral ischemia. NeuroReport 13: 1-4. [DOI] [PubMed] [Google Scholar]
- Xue D, Huang ZG, Smith KE, Buchan AM ( 1992) Immediate or delayed mild hypothermia prevents focal cerebral infarction. Brain Res 587: 66-72. [DOI] [PubMed] [Google Scholar]
- Yanamoto H, Hashimoto N, Nagata I, Kikuchi H ( 1998) Infarct tolerance against temporary focal ischemia following spreading depression in rat brain. Brain Res 784: 239-249. [DOI] [PubMed] [Google Scholar]
- Zhang RL, Chopp M, Chen H, Garcia JH, Zhang ZG ( 1994) Postischemic (1 hour) hypothermia significantly reduces ischemic cell damage in rats subjected to 2 hours of middle cerebral artery occlusion. Stroke 24: 1235-1240. [DOI] [PubMed] [Google Scholar]