Glial Reactivity in Ciliary Neurotrophic Factor-Deficient Mice after Optic Nerve Lesion (original) (raw)
Abstract
There is evidence that ciliary neurotrophic factor (CNTF), in addition to its neurotrophic activity, positively regulates astrogliosis after CNS injury. CNTF and its receptor, CNTFRα, are strongly upregulated in activated astrocytes. Application of CNTF upregulates GFAP expression in cultured astrocytes and induces various aspects of gliosis in the intact brain. Here we examined whether inactivation of the CNTF gene results in the expected changes in glial reactivity by analyzing gliosis in the superior colliculus (SC) after optic nerve crush.
Basal expression levels of GFAP and vimentin in unlesioned CNTF-deficient mice were reduced by 66 and 37%, respectively. Absolute numbers of astrocytes were found not to be different. Surprisingly, however, lesion induced robust activation of astrocytes in CNTF-deficient mice; the time course of activation was even accelerated as compared with wild-type animals. At later time points, activation reached the same level. With respect to microglial cells, basal expression of microglial markers was unaltered in CNTF–knock-out animals. Lesion-induced upregulation of Iba-1, ICAM-1, and F4/80 in microglial cells was unaffected in CNTF-deficient animals. Differences were observed with respect to the time course of microglial activation, different markers being affected differentially. We further demonstrate that lesion induces upregulation of CNTF-related cytokines (LIF, NNT-1) and, interestingly, a more pronounced upregulation of cytokine receptor components (LIF receptor β, gp130) and TGFβ in CNTF-deficient animals. Our results thus indicate that CNTF is required for the development and maintenance of the mature astrocyte phenotype and provide evidence that CNTF is part of the complex regulatory network modulating lesional glial reactivity after lesion.
Keywords: gliosis, astrocytes, GFAP, microglia, ciliary neurotrophic factor, leukemia inhibitory factor
Introduction
There is increasing evidence that ciliary neurotrophic factor (CNTF) is of particular importance as a regulator of reactive processes after lesion in the PNS and CNS. CNTF and its specific receptor component, CNTFRα, are dramatically upregulated in reactive astrocytes (Ip et al., 1993; Rudge et al., 1994; Lee et al., 1997; Kirsch et al., 1998). When applied exogenously, CNTF induces astrogliosis in the unlesioned brain (Winter et al., 1995; Levison et al., 1996; Kahn et al., 1997), stimulates nuclear hypertrophy, and increases GFAP content of astrocytes in vitro (Levison et al., 1998). The GFAP promotor contains CNTF-responsive elements (Clatterbuck et al., 1996; Kahn et al., 1997), suggesting a direct influence of CNTF on the regulation of glial reactivity. After ligand binding to CNTFRα, a trimeric receptor is assembled that contains the signaling components leukemia inhibitory factor receptor β (LIFRβ) and glycoprotein 130 (gp130) (Davis and Yancopoulos, 1993; Davis et al., 1993). This activates receptor-associated Janus kinases (JAKs) and tyrosine phosphorylation of transcription factors of the signal transducer and activator of transcription (STAT) family, followed by their dimerization and nuclear translocation (Bonni et al., 1993; Cattaneo et al., 1999; Takeda and Akira, 2000). Activation of the JAK/STAT-pathway in astrocytes and microglial cells after lesions in the nervous system has been shown in several studies (Planas et al., 1996; Acarin et al., 2000; Justicia et al., 2000; Schwaiger et al., 2000). Taken together, these data would suggest that CNTF is involved in regulating glial activation after lesion.
A large number of other cytokines and growth factors have been shown to be involved in mediating astroglial or microglial activation. Interleukin (IL)-1, IL-2, γ-interferon, TGF-β, TNF-α, and others have been found to result in massive astrogliosis when injected directly into the brain or overexpressed in transgenic animals (Giulian et al., 1988; Brosnan et al., 1989; Yong et al., 1991; Chiang et al., 1994; Klein et al., 1997). IL-10, on the other hand, seems to have an inhibitory effect on astrocyte activation (Balasingam and Yong, 1996). Thus an intricate network of stimulatory and inhibitory factors seems to regulate the activation state of astrocytes and microglia in the CNS.
In the experiments reported here, we have examined the role of CNTF in regulating glial reactivity by using CNTF–knock-out animals, for which no lesion-associated phenotype has been described so far. In the optic nerve lesion model, the results of lesioning can be monitored in the target area of retinal ganglion cells, the superior colliculus, which is at some distance from the actual lesion site, thereby minimizing the direct effects of breaching the blood–brain barrier.
We demonstrate that in the absence of CNTF, the glial reaction to lesion in principle is unimpaired. Morphological analyses, however, suggest that at early time points after lesion (3 dpl), astrocyte activation has progressed further in CNTF–knock-out animals. This is accompanied by increased mRNA expression of key components of its receptor system, namely gp130 and LIFRβ. In addition, we find that TGF-β1 is upregulated more strongly in CNTF-deficient mice, suggesting that the balance of signaling pathways, which normally underlie the lesion-induced activation of glial cells, is altered in the absence of CNTF.
| Target gene | Forward primer | Reverse primer |
|---|---|---|
| CNTFRα | ACCCACCACGAAGATCTGTGA | GCAGCCAGGACCAGAGTGAC |
| LIFRβ | GGAAGTCCGTGCTCCATCAA | AAAGGACCACCCTCCTCCATTA |
| gp130 | CACCCAGCCCCTGCTAGACT | TCTCATCCCCACCGTCTACACT |
| GFAP | ATTGCTGGAGGGCGAAGAA | CGGATCTGGAGGTTGGAGAA |
| Vimentin | GGATCAGCTCACCAACGACAA | GTGCTTTCGGCTTCCTCTCTCT |
| Iba-1 | CTTGAAGCGAATGCTGGAGAA | GGAGCCACTGGACACCTCTCT |
| ICAM-1 | GGGAATGTCACCAGGAATGTGT | ATCACGAGGCCCACAATGAC |
| F4/80 | CGCTGCTGGTTGAATACAGAGA | GTCCAGGCAAGGAGGACAGA |
| CD11b | CAGGGACAACCACACCTCTTG | GCAGCGTCATACCAGCACAC |
| CNTF | TCGTTCAGACCTGACTGCTCTT | CAGTGCTTGCCACTGGTACAC |
| LIF | ACTGCCGTGAGCCTCCAG | CTTGTTGCACAGACGGCAAA |
| NNT-1 | TGCGGCTGACCCAGAACTAT | CCAGGCTACGTCGGAGTTCA |
| TGF-β1 | TTTGGAGCCTGGACACACAGTA | GCTTGCGACCCACGTAGTAGA |
| GAPDH | TCCCATTCTTCCACCTTTGATG | GTCCACCACCCTGTTGCTGTA |
Materials and Methods
Animals. Adult male animals were used in all experiments. CNTF–/– breeding animals (Masu et al., 1993) were kindly provided by M. Sendtner (Würzburg, Germany) and were bred on a C57BL/6 background. They were housed on a 12 hr light/dark cycle with ad libitum access to food and water. All experimental procedures were approved by the local animal care committee and were in accordance with the guidelines instituted by German laws for animal experiments. Control mice (C57BL/6) were obtained from Charles River (Wiga, Germany) and were housed under the same conditions for at least 2 weeks before they were used in the experiments.
Optic nerve lesions. Animals were anesthetized with a mixture of 25% Ketavet (Parke-Davis; 100 mg/ml), 6% Rompun (Bayer; 20 mg/ml), and 2.5% Ventranquil (Albrecht; 10 mg/ml) in physiological saline at a dose of 2.5 ml/kg body weight. Access to the orbita was gained by lateral canthotomy. The optic nerve was exposed by cutting the lateral rectus muscle and by blunt dissection of the tissue surrounding the optic nerve. The meninges were left intact. Crush lesioning was performed with forceps of 1 mm tip width for 30 sec. Immediately after the crush, retinal circulation was checked ophthalmoscopically. Only animals that showed unimpaired circulation within 2 min after the nerve was lesioned were included in this study. The eyelid was closed by a suture to prevent drying of the cornea.
Morphological analysis. After various survival times, animals were transcardially perfused first with 0.9% NaCl in 20 mm phosphate buffer (PB), pH 7.2, followed by 4% paraformaldehyde in 0.1 m PB for 10 min. The brains were removed and postfixed in the same fixative for 3 hr at 4°C followed by cryoprotection in 20% sucrose in 0.1 m PB, pH 7.2, at 4°C overnight. Cryostat sections (30 μm, coronal plane) were cut in series through the midbrain at the level of the superior colliculus. Series consisting of five to six sections covering the whole rostrocaudal extent of the superior colliculus were collected into individual wells of a multiwell plate. Sections from wild-type and knock-out animals were marked by needle punches to enable their identification, and one series of sections from a wild-type and a CNTF–knock-out animal was processed free-floating for immunocytochemistry after placing them together into a single well of a 12-well tissue culture plate to ensure equal staining conditions. Immuncytochemical staining was performed with the following antibodies at the dilutions given: anti-S100 (1:10,000; Swant, Bellinzona, Switzerland), anti-GFAP (1:500; Dako, Glostrup, Denmark), anti-F4/80 (1:50), anti-CD11b (1:50; all from Serotec/Biozol, Eching, Germany), anti-Iba-1 (1:2000; a kind gift of Dr. Y. Imai, National Institute of Neuroscience, Tokyo, Japan). All incubations were performed at room temperature (RT) unless noted otherwise. Sections were first treated at RT with methanol containing 0.3% H2O2 to quench endogenous peroxidase activity for 30 min. After one brief washing step in 0.1 m PB, unspecific binding sites were blocked by incubation with 10% normal serum (normal rabbit or normal goat serum, depending on the secondary antibody used; Biotrend, Köln, Germany), 1% Triton X-100 in 0.1 m PB for 1 hr. Incubation with the primary antibodies was performed overnight at 4°C in 0.1 m PB containing 10% blocking solution. After three washes (15 min each) with PB, sections were incubated with biotinylated secondary antibody (Vector Laboratories, Burlingame, CA) diluted 1:250 in PB containing 2% horse serum for 60 min at room temperature. Bound antibodies were detected by the indirect immunoperoxidase method using the ABC-Elite kit (Vector Laboratories) and DAB/H2O2 following the manufacturer's recommendations. Sections were mounted onto glass slides, air dried, and coverslipped in Hyper-Mount (Shandon, Frankfurt, Germany) after dehydration in a graded series of ethanol and toluene.
To quantitate staining intensity, digital images of the stained sections were obtained with an Olympus PD-11 digital camera using standardized illumination conditions. Images obtained in this way were then analyzed with an image analysis program (Scion-Image, Scion Corporation). Mean gray levels and optical density (sum of the gray values in the selection, with background subtracted) on the ipsilateral and contralateral side were measured in 10 circular measuring fields ∼100 μm in diameter placed randomly over the stratum opticum and stratum griseum superficiale of the superior colliculus. Measurements were done in three to six sections of a minimum of three animals of each genotype per time point analyzed. All measurements from one animal were averaged and treated as an independent data point in the subsequent statistical analysis. For evaluating Iba-1 staining, the optical density ratio between the contralateral and ipsilateral side was chosen as a measuring parameter. For evaluation of F4/80 and GFAP staining, a mean gray value (sum of gray values of all pixels in the selection divided by number of pixels) was calculated by subtracting the mean gray value of the ipsilateral from that obtained on the contralateral side. For these markers, calculation of the optical density ratio was not possible because of the absence of stained cells on the ipsilateral side. Statistical analysis was performed by two-tailed t tests or two-way ANOVA using GraphPad Prism (GraphPad Software, San Diego, CA) and the post-test calculators provided at http://www.graphpad.com. Details are given in respective figure legends.
Real-time quantitative RT-PCR. Tissue for PCR analysis of mRNA expression was dissected from brains frozen immediately after dissection. Purification of total RNA was performed by sonication (Branson Sonifier, Danbury, CT) in 1 ml Trizol (Invitrogen, Karlsruhe, Germany) according to the manufacturer's suggestions. After the RNA pellet was dried and redissolved in RNAase-free water, concentration was determined spectrophotometrically. Total RNA (1–2 μg) was then subjected to DNAase I treatment (Invitrogen) and reverse transcribed with oligo-dT as a primer and Moloney murine leukemia virus-reverse transcriptase (Promega, Mannheim, Germany) for 1 hr at 42°C.
Aliquots of cDNA corresponding to ∼6 ng of total RNA were used for PCR analysis with the GeneAmp 5700 and the SybrGreen detection kit (Applied Biosystems, Weiterstadt, Germany). The following primers were selected from published mouse sequences using the Primer3 program available from the Whitehead Institute for Biomedical Research. Primer sequences are given in 5′→3′ direction.
The primers were used at a concentration of 250 nm in the PCR reaction. Cycling conditions were as follows: 10 min at 95°C (to activate the hot-start _Taq_-polymerase supplied with the SybrGreen detection kit), followed by 40 cycles of 1 min at 95°C and 1 min at 60°C (two-step PCR). During amplification, the fluorescence signal, which is proportional to the amount of double-stranded product produced, was monitored. In this way a complete amplification profile for each of the 96 wells of a PCR plate was obtained, which was used for the analysis. At the end of the PCR run, melting curves of the amplified products were obtained, which were used to control specificity of the amplification reaction. Pilot experiments, in which aliquots of the amplified products were separated on agarose gels, were performed to ensure amplification of specific products of the predicted length.
From the amplification curves obtained in this way, a threshold cycle number (Ct) was calculated. Ct corresponds to the cycle number at which the amplification curve reaches a fluorescent signal of a user-defined magnitude. Thresholds were selected so that Ct values lay at the beginning of the linear phase of amplification. Expression levels were normalized to those obtained for the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase. Values from triplicate measurements of each experimental animal were averaged and treated as independent data points in the subsequent statistical analysis by two-tailed, unpaired t tests. Details of relative quantification of gene expression using this method can be found in User Bulletin 2, which can be found by searching for the phrase “relative quantitation of gene expression” at the following site: http://docs.appliedbiosystems.com/search.taf.
Results
Five days after crush lesioning of the optic nerve in mice, astrocytes and microglial cells are activated in the deafferented SC contralateral to the lesion side (Fig. 1_a_–c). Glial activation is restricted to the contralateral side and to the superficial layers of the SC where retinal ganglion cell axons terminate. Activation of astrocytes on the contralateral side could be readily detected by staining with antibodies against GFAP, and no detectable staining was observed ipsilateral to the lesion side and in unlesioned control mice. GFAP-stained cells show the typical morphology of activated astrocytes with numerous thick processes and hypertrophied somata (Fig. 1_a_, inset). Microglial cells can be stained with various markers; some of them, like Iba-1, stain both resting and activated microglial cells (Fig. 1_b_), and others, like F4/80, stain only activated microglial cells (Fig. 1_c_). Staining of midbrain sections with antibodies against Iba-1 shows a regular distribution of ramified microglial cells. After lesion, microglial cells in the contralateral SC are stained more intensely and show the typical activated morphology with decreased ramification and hypertrophy of the soma region (Fig. 1_b_, inset). Activation of astrocytes and microglial cells in the deafferented superior colliculus as evidenced by staining for these markers first becomes detectable 2 d after lesion (data not shown). This is in contrast to the situation in areas directly affected by a lesion in other lesion paradigms such as stab wounds or ischemia, where glial activation becomes apparent within hours after lesion (Raivich et al., 1999).
Figure 1.
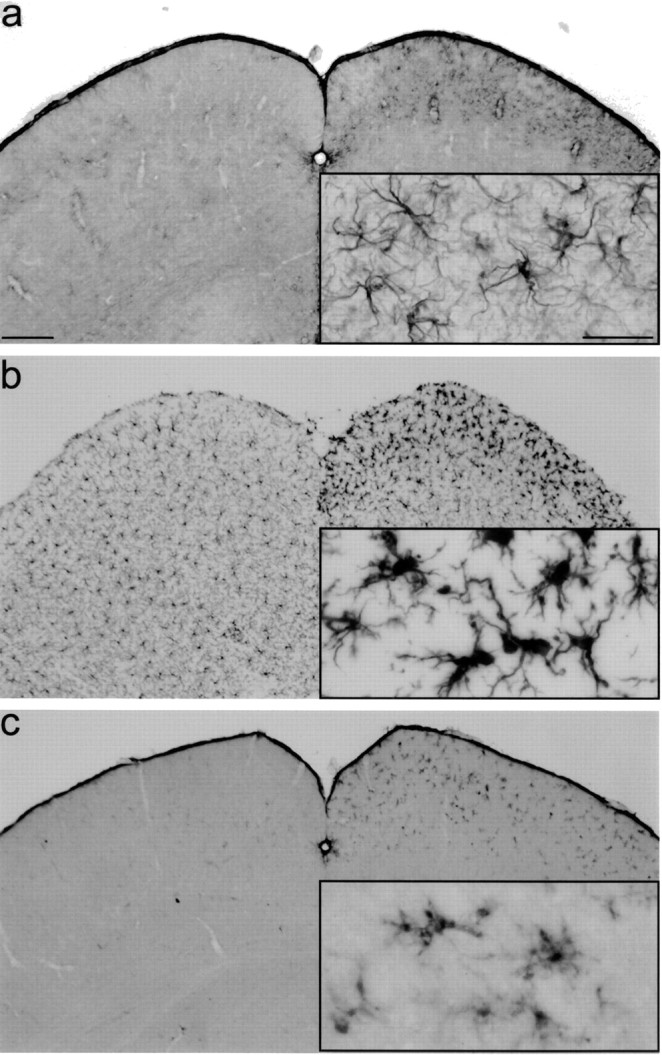
Astroglial and microglial activation after optic nerve lesion in the mouse. Five days after a crush lesion of the left optic nerve, cryosections through the midbrain at the level of the superior colliculus were immunostained with antibodies against GFAP (a) to detect activated astrocytes and Iba-1 (b) or F4/80 (c), both specifically staining microglial cells. For all markers used, immunoreactivity on the contralateral (right) side is stronger than on the ipsilateral side. Insets show details of the superficial layer of the superior colliculus at higher magnification. Scale bar: a, 200 μm; inset, 50 μm.
When comparing GFAP-stained sections of CNTF-deficient and wild-type animals 3 d after an intraorbital crush of the optic nerve (3 dpl) (Fig. 2), increased staining in the CNTF–knock-out animals (Fig. 2_b_) is apparent as compared with wild-type animals (Fig. 2_a_). Astrocyte activation in wild-type animals exhibits the characteristics of the early activation response described by Raivich et al. (1999), with large GFAP-positive plaque-like cells and accentuated cell bodies. In CNTF-deficient animals on the other hand, activated astrocytes already show characteristics of the fully activated, stellate/fibrillary cells. This observation therefore clearly shows that astrocyte activation is not compromised at all in knock-out animals and further indicates that it is already more fully developed at 3 dpl in the CNTF-deficient animals. This is also supported by our densitometric analysis of GFAP-stained sections from wild-type and knock-out animals, which shows a 2.4-fold difference in staining intensity at 3 dpl (Fig. 2_e_). At later time points, differences in GFAP staining are no longer apparent between wild-type and knock-out animals (14 dpl) (Fig. 2_c_–e).
Figure 2.
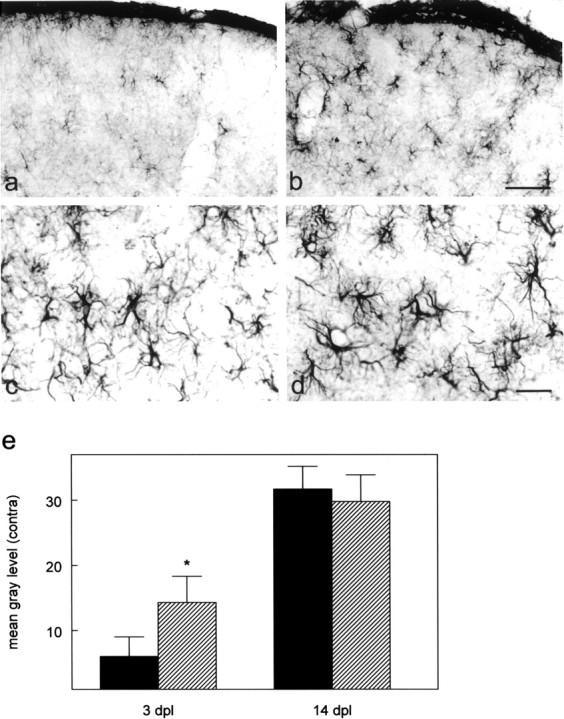
Astrocytes are more strongly activated at 3 dpl in CNTF–/– animals. a_–_d, GFAP immunostaining of cryosections through the right (contralateral) superior colliculus 3 d (a, b) and 14 d (c, d) after lesion in wild-type (a, c) and CNTF–knock-out animals (b, d). At 3 dpl astrocytes in CNTF–/–-animals (b) are more strongly stained than in wild-type animals (a). Astroglial reactivity has reached similar levels at 14 dpl (c, d). Scale bars: b, d, 50 μm. e, Densitometric analysis of GFAP-immunostained sections 3 and 14 dpl. Staining intensity was quantified by densitometric analysis as detailed in Materials and Methods (Morphological analysis). Values are means ± SD. Asterisk indicates a significant increase in staining intensity as compared with wild-type animals; p < 0.05; two-way ANOVA with post-test using the Bonferroni correction: residual df = 10; residual mean square 13.31.
With respect to activation of microglial cells, no differences could be demonstrated by immunocytochemistry between unoperated wild-type and knock-out animals with the markers used (Iba-1, F4/80, CD11b). These markers have been widely used to detect activated microglial cells after lesion (for review, see Raivich et al., 1999). Their expression is strongly up-regulated in activated microglial cells in a multitude of different lesion paradigms, reflecting their involvement in various aspects of the function of activated microglial cells like migration and phagocytosis. Differences between wild-type and knock-out animals were first detectable at 5 dpl by immunostaining with antibodies against F4/80 (Fig. 3_a,b_), whereas CD11b and Iba-1 staining was stronger at even later time points (Fig. 3_c_–f). Results from a densitometric analysis of F4/80 and Iba-1 staining at all time points examined are shown in Figure 4. Iba-1 staining was significantly stronger in CNTF-deficient animals at 8 and 14 dpl (Fig. 4_a_), indicating that although there was no significant difference in initial activation, microglial cells exhibited prolonged activation in the absence of endogenous CNTF. Similar results were obtained for another activation marker, F4/80, which was expressed more strongly in the CNTF–knock-out animals at 5, 8, and 14 dpl (Fig. 4_b_). Although differences in F4/80 staining were statistically different (p < 0.05) only at 5 dpl, values from knock-out animals tended to be higher as compared with those from wild-type animals at all time points after lesion. Taken together, these data would suggest that as was the case for astroglial activation, elimination of CNTF did not affect microglial activation per se but rather altered the time course of expression of certain microglial markers.
Figure 3.
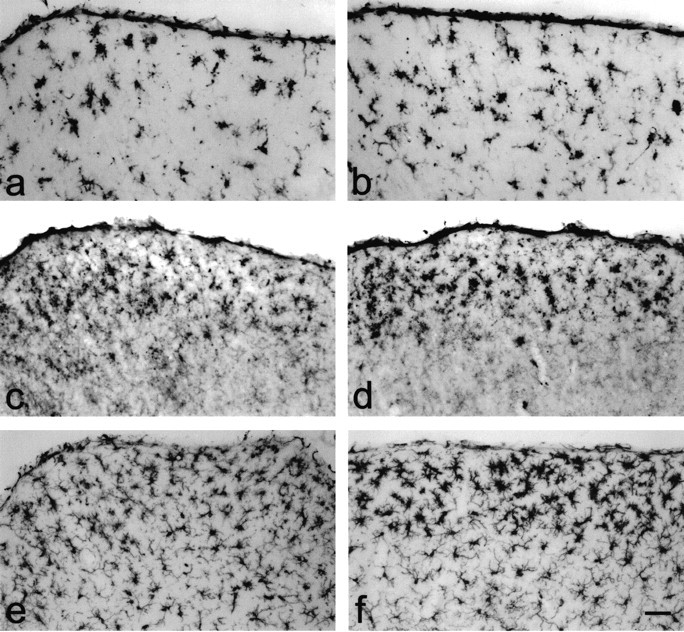
Microglial cells are more strongly activated in CNTF–/– animals. Maximal differences in microglial reactivity as shown by different activation markers could be observed at different time points after lesion. a_–_f, Immunostaining with antibodies against the microglial markers F4/80 (5 dpl; a, b), CD11b (14 dpl; b, c), and Iba-1 (8 dpl; e, f) in wild-type (a, c, e) and CNTF–knock-out animals (b, d, f). Scale bar: (in f) a_–_f, 50 μm.
Figure 4.

Densitometric analysis of Iba-1 and F4/80 staining shows stronger activation of microglial cells in CNTF–/– animals (hatched bars) as compared with wild-type animals (filled bars). Staining intensity was quantified by densitometric analysis as detailed in Materials and Methods (Morphological analysis). Results for Iba-1 (a) staining are given as optical density ratio (±SEM) between staining on the contralateral and ipsilateral sides; those for F4/80 staining (b) are expressed as the mean gray level (±SEM) on the contralateral side obtained by subtracting the mean gray level determined on the ipsilateral side from that measured on the contralateral side. Asterisk indicates significant differences in staining intensity; p < 0.05 (**p < 0.01); two-way ANOVA with post-test using the Bonferroni correction. a, Residual df = 19; residual mean square 0.02365; b, residual df = 28; residual mean square 11.06.
To determine differences in glial activation on the mRNA level, we performed a quantitative RT-PCR analysis for astroglial and microglial markers (Figs. 5, 6). Analysis of GFAP mRNA expression in unlesioned control animals revealed reduced basal expression in CNTF–knock-out as compared with wild-type animals (33.5%) (Fig. 5_a_). However, 1 d after optic nerve lesion, GFAP expression has increased 1.8-fold in knock-out animals as compared with 1.2-fold in wild-type animals (Fig. 5_b_). Statistical analysis (two-tailed, unpaired t test) showed that at this time point, GFAP expression in knock-out animals is already significantly increased (p < 0.05), whereas the increase in wild-type animals has not reached statistical significance. Thus astroglial activation has only been initiated measurably in CNTF-deficient animals at this time point. Three days after lesion, expression of GFAP mRNA has increased 8.9-fold in the knock-out animals and only 3.1-fold in the wild-type animals (Fig. 5_b_). On the basis of the observed reduced basal GFAP mRNA expression in knock-out animals, this results in similar absolute levels of GFAP mRNA at 3 dpl.
Figure 5.

Basal expression of GFAP and vimentin mRNA is reduced (a), and relative increase in expression induced by optic nerve lesion is stronger in CNTF–knock-out animals 3 dpl (b). a, b, Results from quantitation of mRNA expression by real-time quantitative PCR. Basal expression of GFAP and vimentin (a) in the superior colliculus of unoperated CNTF–/– animals (n = 4) is expressed as percentage (±SD) of the expression level determined in unoperated wild-type animals (n = 3). Lesion-induced upregulation of marker expression (b) is given as fold induction versus unoperated controls. All values are mean values (±SD) from n = 3–4 animals. Solid bars represent values from wild-type animals; hatched bars represent values from CNTF–/– animals. a, Values for GFAP and vimentin in CNTF–knock-out animals are significantly different from those obtained from wild-type animals with p < 0.01; unpaired t test, two-tailed. b, *** indicates significant differences in staining intensity between CNTF–/– and wild-type animals; p < 0.001, two-way ANOVA with post-test using the Bonferroni correction. GFAP, Residual df = 10; residual mean square 0.7089; vimentin, residual df = 10; residual mean square 0.330. c, Western blotting of protein extracts from the superior colliculus of wild-type (WT) and CNTF–/– animals (KO). Tissue samples were separated by SDS-PAGE, immunoblotted, and sequentially probed with antibodies against the indicated proteins. Bound antibodies were detected by chemiluminescence and exposure to x-ray film (Kodak-X-OMAT). d, e, Immunostaining of cryosections through the right (contralateral) superior colliculus with antibodies against the astroglial marker S100β in wild-type (d) and CNTF–knock-out animals (e) 3 dpl. Scale bar: (in e) 50 μm.
Figure 6.

Lesion-induced upregulation of microglial marker mRNAs (a) and LIFRβ, gp130, and TGF-β1 mRNA (b) in the contralateral superior colliculus 3 dpl in CNTF–knock-out animals (hatched bars) and wild-type animals (solid bars). Lesion-induced upregulation of marker expression is given as fold induction versus unoperated controls. All values are mean values (±SD) from n = 3–4 animals. Asterisk indicates significant differences between CNTF–/– and wild-type animals; p < 0.05; unpaired t test, two-tailed.
Another astroglial marker, vimentin, showed a similar expression pattern. Basal expression of vimentin mRNA is reduced to 62.7% in knock-out animals (Fig. 5_a_). Vimentin expression at 3 dpl is upregulated 4.9-fold in knock-out animals and twofold in wild-type animals. As for GFAP, increased lesion-induced up-regulation of vimentin expression is also already apparent at 1 dpl, with knock-out animals showing higher expression than wild-type animals (Fig. 5_b_). Differences in basal expression of GFAP could also be detected by immunoblotting (Fig. 5_c_). To test whether reduced basal expression of GFAP in the knock-out animals results from a reduction in astrocyte numbers, immunocytochemical staining and immunoblotting with antibodies against another astroglial marker protein, S-100β, were performed (Fig. 5_c_–e). For this marker, no differences were observed, with respect to neither total protein levels nor number of immuno-reactive astrocytes.
Quantitative analysis of the expression of microglial markers 3 dpl showed that expression of all markers studied is upregulated after lesion (Fig. 6_a_). All markers showed a consistently stronger lesion-induced upregulation in the CNTF-deficient animals (Fig. 6_a_, hatched bars) as compared with wild-type animals (Fig. 6_a_, solid bars), although statistical analysis of the data showed that only values for Iba-1 and ICAM-1 reached statistical significance. None of the microglial markers examined showed differences in basal expression between wild-type and knock-out animals (data not shown), indicating that the observed differences in lesion-induced upregulation indeed reflect stronger activation of microglial cells in the CNTF-deficient animals. This is in agreement with the immunocytochemical observations for Iba-1, CD11b, and F4/80 (Figs. 3, 4_a_).
Taken together, our results would indicate that activation of astrocytes and microglial cells in the knock-out animals is not only unimpaired, but may even be accelerated. To examine whether changes in the regulation of receptor components for CNTF and related cytokines is different in CNTF–knock-out animals or if other factors that have been shown to be involved in inducing gliosis are differentially regulated, we studied expression of candidate molecules by quantitative PCR. Figure 6_b_ summarizes these results. All expression levels are expressed as fold upregulation relative to unoperated control animals, because none of the examined genes showed differences in basal expression levels. First, we found lesion induced upregulation of two other members of the IL-6 family of cytokines, namely LIF and the recently described family member NNT-1/cardiotrophin-like cytokine. However, 3 dpl expression of both factors is upregulated to the same extent. On the other hand, expression of two of the three receptor components of the CNTF/LIF system, LIFRβ and gp130, is more strongly upregulated in the knock-out animals. Thus, signaling through LIFRβ and gp130 by other members of the IL-6 family of cytokines could be enhanced in the knock-out animals. In support of this hypothesis, we found that expression of one of the typical target genes of the CNTF/LIF signaling pathway, suppressor of cytokine signaling (SOCS)-3, is more strongly induced in CNTF–knock-out animals. SOCS proteins are a class of proteins that are negative regulators of cytokine receptor signaling via the JAK/STAT pathway. We also found strongly increased expression of TGF-β1, a member of a different family of growth factors, in the CNTF–knock-out animals. TGF-β1 has been shown previously to be involved in regulating gliosis (Logan et al., 1994). Our observation, that in the absence of endogenous CNTF lesion induces stronger upregulation of TGF-β1 expression, suggests that there is a regulatory link between these two factors and that they cooperate to induce cellular reactions after lesion.
Discussion
Previous studies had suggested a role for CNTF as a positive regulator of gliosis after lesions in the CNS. By analyzing glial activation in the SC of CNTF-deficient mice after optic nerve lesion, we show that astroglial and microglial activation are remarkably unaffected by the absence of CNTF and that differences between wild-type and knock-out animals are mainly reflected in an altered time course of activation. Specifically, we show that certain markers of glial activation are upregulated earlier in CNTF–knock-out animals. Increased lesion-induced upregulation of LIFRβ, gp130, and TGF-β1 in CNTF-deficient animals could provide an explanation for these unexpected findings.
Glial reactivity in the superior colliculus after optic nerve lesion
In unoperated animals, astrocytes in the SC are GFAP negative. Optic nerve lesion, however, leads to the appearance of strongly GFAP-stained astrocytes in the SC that exhibit the characteristics of activated cells (Fawcett and Asher, 1999). Astrocyte activation is paralleled by that of microglial cells (Raivich et al., 1999). In unoperated animals and in the ipsilateral SC after optic nerve lesion, staining for microglial markers reveals the known network of ramified cells. After lesion, expression of all microglial markers examined is upregulated in the contralateral SC, and transformation of microglia to the activated phenotype is evident (Streit et al., 1999).
In agreement with previous studies, glial activation was undetectable morphologically earlier than 2–3 dpl (Schmidt-Kastner et al., 1993; Castano et al., 1996). This is in contrast to lesions with breaching of the blood–brain barrier, where it can be detected within hours after lesioning (Streit et al., 1999). Our quantitative evaluation of F4/80 and Iba-1 staining shows that microglial activation peaks at 8 dpl. A maximum of immunostained microglial cells at 7 dpl has been found in the mouse using the same lesion paradigm (Lawson et al., 1994).
Basal expression of astroglial markers is reduced in the absence of CNTF
CNTF and LIF can control GFAP expression in cultured astrocytes and during development (Bugga et al., 1998; Koblar et al., 1998; Levison et al., 1998) and strongly promote generation of astrocytes from precursors (Richards et al., 1996; Bonni et al., 1997; Shimazaki et al., 2001). The GFAP promoter contains cytokine response elements, and CNTF stimulates transcription from this promoter (Kahn et al., 1997). Evidence that GFAP expression in astrocytes of the unlesioned CNS is regulated by CNTF is provided by our finding of reduced basal expression in CNTF–/– animals. There are two possible explanations for this finding: (1) lack of CNTF during development leads to reduced numbers of astrocytes developing from precursors (Bonni et al., 1997; but see Shimazaki et al., 2001); (2) normal numbers of astrocytes are generated, but lack of CNTF in the adult leads to reduced expression of GFAP. In support of the second possibility, we did not observe reduced numbers of astrocytes in CNTF–/– animals by immunocytochemistry with antibodies against S-100β, nor did we find reduced levels of S-100β protein by Western blotting. There were also no differences in the number of activated astrocytes in CNTF–/– animals after lesion. Reduced basal expression of GFAP and vimentin mRNA in unoperated CNTF–/– animals indicates that in wild-type animals CNTF is active in the unlesioned brain. This is also supported by findings in mice overexpressing CNTF, which show constitutive activation of astrocytes (Winter et al., 1995). Similar conclusions can be drawn for LIF (Koblar et al., 1998). In LIF-deficient animals, a 40% reduction of the number of GFAP-positive cells in the hippocampus was observed. However, similar to the situation in the unlesioned SC, where resting astrocytes are not revealed by GFAP staining, astrocytes expressing very low amounts of GFAP may have been undetectable in the aforementioned study. Results obtained by Koblar et al. (1998) and in this study thus suggest that GFAP expression in astrocytes of the unlesioned CNS is under dual control of two cytokines of the same family, namely CNTF and LIF.
None of the microglial markers examined showed differences in basal expression levels (data not shown), suggesting that unlike the situation for astrocytes, microglial cells in the unlesioned brain are not affected by the absence of CNTF.
Glial activation in CNTF–/– mice
We reasoned that elimination of CNTF would lead to reduced glial activation after lesion, thus directly proving its suggested contribution in inducing gliosis. Unexpectedly, however, glial responses, and that of astrocytes in particular, are not impaired at all in CNTF–knock-out animals. Rather, lesion-induced upregulation of the astrocytic activation markers GFAP and vimentin could be detected earlier in CNTF-deficient animals. Starting at lower basal levels, upregulation of GFAP mRNA is detectable at 1 dpl only in the mutants. At later time points, mRNAs for GFAP as well as vimentin reach the same expression levels as in wild-type mice at 3 dpl, suggesting that the absence of CNTF affects only the initial phase of activation. This was also observed morphologically by immunostaining for GFAP, which showed that at 3 dpl fully activated astrocytes were more abundant in CNTF-deficient animals. At later time points activation was indistinguishable from that in wild-type mice, indicating that the lack of CNTF, rather than leading to reduced activation, results in a subtle change of its time course.
With respect to microglial cells, CNTF elimination affected microglial markers differentially. For F4/80 staining, maximal differences were found at 5 dpl, whereas Iba-1 and CD11b staining were maximally different at 8–14 dpl. Because different activation markers probably characterize different functional states of microglial cells (for review, see Raivich et al., 1999), it may be speculated that CNTF influences only certain aspects of microglial function. At later time points after lesion, microglial cells are supposed to be phagocytotic (Raivich et al., 1999). Thus, it may be speculated that in the knock-out animals they are confronted with more material to be phagocytosed. There is no reported evidence of cell death of deafferented collicular neurons after optic nerve lesion. However, CNTF has been shown to protect oligodendrocytes from death induced by TNF-α (Louis et al., 1993; D'Souza et al., 1996). Lack of CNTF in the knock-out animals could therefore lead to increased death of oligodendrocytes, increasing the damage sensed by microglial cells. In support of this hypothesis, it was reported recently that the CNTF/LIF system is critically involved in protecting oligodendrocytes in a model of immune-mediated demyelination (Butzkueven et al., 2002; Linker et al., 2002).
Because it is assumed that microglial cells are first to detect damage (Kreutzberg, 1996) and in turn induce astroglial activation, increased activation of astrocytes seen in the CNTF–/– animals may also result from an altered communication between these cells in the absence of CNTF. Herx et al. (2000) showed that CNTF upregulation in astrocytes depends on IL-1β upregulation in microglial cells. IL-1 is a potent activator of astrocytes and induces expression of a plethora of other cytokines such as TNF-α, IL-6, and colony-stimulating factors, which feed back onto microglial cells (Lee et al., 1995).
Increased expression of CNTFRα, gp130, and TGF-β1 in CNTF–/– mice
Expression of gp130 and LIFRβ was upregulated more strongly in the knock-out animals after lesion. This could result in increased signaling by family members known to participate in regulation of astrogliosis (LIF, IL-6) (Klein et al., 1997; Sugiura et al., 2000). It has been shown recently that astroglial and microglial reactivity are significantly attenuated in LIF-deficient animals in a cortical stab wound model (Sugiura et al., 2000), suggesting that this cytokine is one of the key regulators. Our finding of a lesion-induced upregulation of LIF and the concomitant upregulation of the components of the functional LIF receptor, LIFRβ and gp130, is in accordance with these findings. Signaling via LIFRβ and gp130 would lead to activation of the JAK/STAT pathway (Takeda and Akira, 2000). This leads to increased transcription of STAT3 target genes. Our finding that SOCS-3, one of these target genes, is more strongly induced in CNTF–knock-out animals would support this hypothesis. Apart from our finding that NNT-1 expression is upregulated after lesion, nothing is known at present about the role of this cytokine in the context of CNS lesions. Our results indicate that it is not only acting during development (Elson et al., 2000), but could also play a role in the adult nervous system.
We observed a significant difference in lesion-induced up-regulation of TGF-β1 between wild-type and CNTF–/– animals. TGF-β1 is a potent gliosis-inducing molecule, in particular for astrocytes (Wu and Schwartz, 1998; Fawcett and Asher, 1999), and it has been shown that application of neutralizing antibodies inhibits GFAP expression and glial scar formation (Logan et al., 1994). There is convincing evidence that microglial cells are the source of TGF-β1 (Lehrmann et al., 1998). Increased expression of TGF-β1 mRNA in CNTF–/– animals thus could reflect the increased activation of microglial cells, which in turn could lead to the observed activation of astrocytes. We conclude from our experiments that although previous experiments had suggested a prominent role for CNTF as a potent positive regulator of glial reactivity, elimination of this factor does not lead to impaired activation of glial cells. However, our results show that disruption of the CNTF gene leads to alterations in the time course of glial responses to injury and to changes in the expression of other components involved in glial activation, thereby indicating that CNTF is part of a network of regulating signals that control this activation process.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant KI 419/2-1, 2-2 (M.K.). We thank Birgit Egle and G. Kaiser for excellent technical assistance. We are indebted to Dr. M. Sendtner for the CNTF–knock-out animals.
Correspondence should be addressed to Matthias Kirsch, Institute of Anatomy I, University of Freiburg, Albertstrasse 23, D-79104 Freiburg, Germany. E-mail: matthias.kirsch@zfn.uni-freiburg.de.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235416-09$15.00/0
References
- Acarin L, Gonzalez B, Castellano B ( 2000) STAT3 and NFkappaB activation precedes glial reactivity in the excitotoxically injured young cortex but not in the corresponding distal thalamic nuclei. J Neuropathol Exp Neurol 59: 151–163. [DOI] [PubMed] [Google Scholar]
- Balasingam V, Yong VW ( 1996) Attenuation of astroglial reactivity by interleukin-10. J Neurosci 16: 2945–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A, Frank DA, Schindler C, Greenberg ME ( 1993) Characterization of a pathway for ciliary neurotrophic factor signaling to the nucleus. Science 262: 1575–1579. [DOI] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg ME ( 1997) Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278: 477–483. [DOI] [PubMed] [Google Scholar]
- Brosnan CF, Litwak MS, Schroeder CE, Selmaj K, Raine CS, Arezzo JC ( 1989) Preliminary studies of cytokine-induced functional effects on the visual pathways in the rabbit. J Neuroimmunol 25: 227–239. [DOI] [PubMed] [Google Scholar]
- Bugga L, Gadient RA, Kwan K, Stewart CL, Patterson PH ( 1998) Analysis of neuronal and glial phenotypes in brains of mice deficient in leukemia inhibitory factor. J Neurobiol 36: 509–524. [DOI] [PubMed] [Google Scholar]
- Butzkueven H, Zhang JG, Soilu H, Hochrein H, Chionh F, Shipham KA, Emery B, Turnley AM, Petratos S, Ernst M, Bartlett PF, Kilpatrick TJ ( 2002) LIF receptor signaling limits immune-mediated demyelination by enhancing oligodendrocyte survival. Nat Med 8: 613–619. [DOI] [PubMed] [Google Scholar]
- Castano A, Lawson LJ, Fearn S, Perry VH ( 1996) Activation and proliferation of murine microglia are insensitive to glucocorticoids in Wallerian degeneration. Eur J Neurosci 8: 581–588. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Conti L, De-Fraja C ( 1999) Signaling through the JAK-STAT pathway in the developing brain. Trends Neurosci 22: 365–369. [DOI] [PubMed] [Google Scholar]
- Chiang CS, Stalder A, Samimi A, Campbell IL ( 1994) Reactive gliosis as a consequence of interleukin-6 expression in the brain: studies in transgenic mice. Dev Neurosci 16: 212–221. [DOI] [PubMed] [Google Scholar]
- Clatterbuck RE, Price DL, Koliatsos VE ( 1996) Ciliary neurotrophic factor stimulates the expression of glial fibrillary acidic protein by brain astrocytes in vivo. J Comp Neurol 369: 543–551. [DOI] [PubMed] [Google Scholar]
- Davis S, Yancopoulos GD ( 1993) The molecular biology of the CNTF receptor. Curr Opin Cell Biol 5: 281–285. [DOI] [PubMed] [Google Scholar]
- Davis S, Aldrich TH, Stahl N, Pan L, Taga T, Kishimoto T, Ip NY, Yancopoulos GD ( 1993) LIFRβ and gp130 as heterodimerizing signal transducers of the tripartite CNTF receptor. Science 260: 1805–1808. [DOI] [PubMed] [Google Scholar]
- D'Souza SD, Alinauskas KA, Antel JP ( 1996) Ciliary neurotrophic factor selectively protects human oligodendrocytes from tumor necrosis factor-mediated injury. J Neurosci Res 43: 289–298. [DOI] [PubMed] [Google Scholar]
- Elson GC, Lelievre E, Guillet C, Chevalier S, Plun-Favreau H, Froger J, Suard I, de Coignac AB, Delneste Y, Bonnefoy JY, Gauchat JF, Gascan H ( 2000) CLF associates with CLC to form a functional heteromeric ligand for the CNTF receptor complex. Nat Neurosci 3: 867–872. [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA ( 1999) The glial scar and central nervous system repair. Brain Res Bull 49: 377–391. [DOI] [PubMed] [Google Scholar]
- Giulian D, Woodward J, Young DG, Krebs JF, Lachman LB ( 1988) Interleukin-1 injected into mammalian brain stimulates astrogliosis and neovascularization. J Neurosci 8: 2485–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herx LM, Rivest S, Yong VW ( 2000) Central nervous system-initiated inflammation and neurotrophism in trauma: IL-1 beta is required for the production of ciliary neurotrophic factor. J Immunol 165: 2232–2239. [DOI] [PubMed] [Google Scholar]
- Ip NY, Wiegand SJ, Morse J, Rudge JS ( 1993) Injury-induced regulation of ciliary neurotrophic factor mRNA in the adult rat brain. Eur J Neurosci 5: 25–33. [DOI] [PubMed] [Google Scholar]
- Justicia C, Gabriel C, Planas AM ( 2000) Activation of the JAK/STAT pathway following transient focal cerebral ischemia: signaling through Jak1 and Stat3 in astrocytes. Glia 30: 253–270. [DOI] [PubMed] [Google Scholar]
- Kahn MA, Huang CJ, Caruso A, Barresi V, Nazarian R, Condorelli DF, de Vellis J ( 1997) Ciliary neurotrophic factor activates JAK/Stat signal transduction cascade and induces transcriptional expression of glial fibrillary acidic protein in glial cells. J Neurochem 68: 1413–1423. [DOI] [PubMed] [Google Scholar]
- Kirsch M, Schneider T, Lee MY, Hofmann HD ( 1998) Lesion-induced changes in the expression of ciliary neurotrophic factor and its receptor in rat optic nerve. Glia 23: 239–248. [PubMed] [Google Scholar]
- Klein MA, Moller JC, Jones LL, Bluethmann H, Kreutzberg GW, Raivich ( 1997) Impaired neuroglial activation in interleukin-6 deficient mice. Glia 19: 227–233. [DOI] [PubMed] [Google Scholar]
- Koblar SA, Turnley AM, Classon BJ, Reid KL, Ware CB, Cheema SS, Murphy M, Bartlett PF ( 1998) Neural precursor differentiation into astrocytes requires signaling through the leukemia inhibitory factor receptor. Proc Natl Acad Sci USA 95: 3178–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW ( 1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19: 312–318. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Frost L, Risbridger J, Fearn S, Perry VH ( 1994) Quantification of the mononuclear phagocyte response to Wallerian degeneration of the optic nerve. J Neurocytol 23: 729–744. [DOI] [PubMed] [Google Scholar]
- Lee MY, Deller T, Kirsch M, Frotscher M, Hofmann HD ( 1997) Differential regulation of ciliary neurotrophic factor (CNTF) and CNTF receptor α expression in astrocytes and neurons of the fascia dentata after entorhinal cortex lesion. J Neurosci 17: 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Dickson DW, Brosnan CF ( 1995) Interleukin-1, nitric oxide and reactive astrocytes. Brain Behav Immun 9: 345–354. [DOI] [PubMed] [Google Scholar]
- Lehrmann E, Kiefer R, Christensen T, Toyka KV, Zimmer J, Diemer NH, Hartung HP, Finsen B ( 1998) Microglia and macrophages are major sources of locally produced transforming growth factor-beta1 after transient middle cerebral artery occlusion in rats. Glia 24: 437–448. [DOI] [PubMed] [Google Scholar]
- Levison SW, Ducceschi MH, Young GM, Wood TL ( 1996) Acute exposure to CNTF in vivo induces multiple components of reactive gliosis. Exp Neurol 141: 256–268. [DOI] [PubMed] [Google Scholar]
- Levison SW, Hudgins SN, Crawford JL ( 1998) Ciliary neurotrophic factor stimulates nuclear hypertrophy and increases the GFAP content of cultured astrocytes. Brain Res 803: 189–193. [DOI] [PubMed] [Google Scholar]
- Linker RA, Maurer M, Gaupp S, Martini R, Holtmann B, Giess R, Rieckmann P, Lassmann H, Toyka KV, Sendtner M, Gold R ( 2002) CNTF is a major protective factor in demyelinating CNS disease: a neurotrophic cytokine as modulator in neuroinflammation. Nat Med 8: 620–624. [DOI] [PubMed] [Google Scholar]
- Logan A, Oliver JJ, Berry M ( 1994) Growth factors in CNS repair and regeneration. Prog Growth Fact Res 5: 379–405. [DOI] [PubMed] [Google Scholar]
- Louis J-C, Magal E, Takayama S, Varon S ( 1993) CNTF protection of oligodendrocytes against natural and tumor necrosis factor-induced death. Science 259: 689–692. [DOI] [PubMed] [Google Scholar]
- Masu Y, Wolf E, Holtmann B, Sendtner M, Brem G, Thoenen H ( 1993) Disruption of the CNTF gene results in motor neuron degeneration. Nature 365: 27–32. [DOI] [PubMed] [Google Scholar]
- Planas AM, Soriano MA, Berruezo M, Justicia C, Estrada A, Pitarch S, Ferrer I ( 1996) Induction of Stat3, a signal transducer and transcription factor, in reactive microglia following transient focal cerebral ischaemia. Eur J Neurosci 8: 2612–2618. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CUA, Werner A, Jones LL, Kreutzberg GW ( 1999) Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Rev 30: 77–105. [DOI] [PubMed] [Google Scholar]
- Richards LJ, Kilpatrick TJ, Dutton R, Tan SS, Gearing DP, Bartlett PF, Murphy M ( 1996) Leukaemia inhibitory factor or related factors promote the differentiation of neuronal and astrocytic precursors within the developing murine spinal cord. Eur J Neurosci 8: 291–299. [DOI] [PubMed] [Google Scholar]
- Rudge JS, Morrissey D, Lindsay RM, Pasnikowski EM ( 1994) Regulation of ciliary neurotrophic factor in cultured rat hippocampal astrocytes. Eur J Neurosci 6: 218–229. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Wietasch K, Weigel H, Eysel UT ( 1993) Immunohistochemical staining for glial fibrillary acidic protein (GFAP) after deafferentation or ischemic infarction in rat visual system: features of reactive and damaged astrocytes. Int J Dev Neurosci 11: 157–174. [DOI] [PubMed] [Google Scholar]
- Schwaiger FW, Hager G, Schmitt AB, Horvath A, Streif R, Spitzer C, Gamal S, Breuer S, Brook GA, Nacimiento W, Kreutzberg GW ( 2000) Peripheral but not central axotomy induces changes in Janus kinases (JAK) and signal transducers and activators of transcription (STAT). Eur J Neurosci 12: 1165–1176. [DOI] [PubMed] [Google Scholar]
- Shimazaki T, Shingo T, Weiss S ( 2001) The ciliary neurotrophic factor/leukemia inhibitory factor/gp130 receptor complex operates in the maintenance of mammalian forebrain neural stem cells. J Neurosci 21: 7642–7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA ( 1999) Reactive microgliosis. Prog Neurobiol 57: 563–581. [DOI] [PubMed] [Google Scholar]
- Sugiura S, Lahav R, Han J, Kou SY, Banner LR, de P, Patterson PH ( 2000) Leukaemia inhibitory factor is required for normal inflammatory responses to injury in the peripheral and central nervous systems in vivo and is chemotactic for macrophages in vitro. Eur J Neurosci 12: 457–466. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S ( 2000) STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev 11: 199–207. [DOI] [PubMed] [Google Scholar]
- Winter CG, Saotome Y, Levison SW, Hirsh D ( 1995) A role for ciliary neurotrophic factor as an inducer of reactive gliosis, the glial response to central nervous system injury. Proc Natl Acad Sci USA 92: 5865–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu VW, Schwartz JP ( 1998) Cell culture models for reactive gliosis—new perspectives. J Neurosci Res 51: 675–681. [DOI] [PubMed] [Google Scholar]
- Yong VW, Moumdjian R, Yong FP, Ruijs TC, Freedman MS, Cashman N, Antel JP ( 1991) Gamma-interferon promotes proliferation of adult human astrocytes in vitro and reactive gliosis in the adult mouse brain in vivo. Proc Natl Acad Sci USA 88: 7016–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]