Disruption of Glial Glutamate Transport by Reactive Oxygen Species Produced in Motor Neurons (original) (raw)
Abstract
Observations of elevated CSF glutamate in amyotrophic lateral sclerosis (ALS), together with findings that motor neurons are selectively vulnerable to glutamate receptor-mediated (“excitotoxic”) injury, support an excitotoxic contribution to the motor neuron loss in the disease. However, the basis of the apparent loss of astrocytic glutamate transport capacity in affected areas of motor cortex and spinal cord, which probably underlies the extracellular glutamate elevations, is unexplained. Here, we find that glutamate induces far greater reactive oxygen species (ROS) generation in cultured motor neurons than in other spinal neurons. In addition, we found that the ROS seem to be able to leave the motor neurons and induce oxidation and disruption of glutamate uptake in neighboring astrocytes. Correspondingly, in a transgenic mouse model of ALS, protein oxidation was increased in regions immediately surrounding motor neurons. These results provide a mechanism that can account for the localized loss of glial glutamate transport seen in the disease. Furthermore, the observations lend support for a feedforward model involving reciprocal interactions between motor neurons and glia, which may prove useful in understanding ALS pathogenesis.
Keywords: motor neuron, amyotrophic lateral sclerosis, ROS, glutamate, excitotoxicity, glutamate transport, cell culture, free radicals, SOD, nitrotyrosine, AMPA
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by the selective loss of upper and lower motor neurons (MNs). Although its cause remains elusive, many clues to pathogenesis have emerged recently; among these are observations of mitochondrial dysfunction and oxidative damage in MNs and reactive astrocytosis in ventral horn and motor cortex (Tu et al., 1996; Robberecht, 2000; Menzies et al., 2002). Although a small percentage (1–2%) of cases have been linked to mutations in the enzyme superoxide dismutase 1 (SOD1) (Rosen et al., 1993), the vast majority (90–95%) are sporadic.
Prolonged or excessive exposures to the excitatory transmitter glutamate injure neurons. Although there is considerable evidence for such an excitotoxic role in acute conditions of ischemia or epilepsy, observations of elevated CSF glutamate levels (Rothstein et al., 1990;Shaw et al., 1995; Spreux-Varoquaux et al., 2002) and indications of disturbed astrocytic glutamate transport in spinal cord and motor cortex of sporadic ALS patients (Rothstein et al., 1992, 1996; Shaw et al., 1994) support an excitotoxic contribution to this disease also. Indeed, chronic inhibition of glutamate uptake reproduces selective MN degeneration in vitro (Rothstein et al., 1993; Carriedo et al., 1996). Furthermore, clues have emerged as to the basis of this selective vulnerability. MNs seem to possess large numbers of unusual AMPA-type glutamate receptor channels that are directly permeable to Ca2+ (Carriedo et al., 1995, 1996;Williams et al., 1997; Vandenberghe et al., 2000), and activation of these channels with the selective agonists AMPA or kainate induces preferential MN injury. As has been observed with NMDA-type glutamate receptors (Lafon-Cazal et al., 1993; Dugan et al., 1995; Reynolds and Hastings, 1995), strong activation of these channels causes high intracellular Ca2+ increases and consequent mitochondrial reactive oxygen species (ROS) generation (Carriedo et al., 1998, 2000). Indeed, large intracellular Ca2+ loads in MNs are buffered substantially by mitochondria (Carriedo et al., 2000), and populations of MNs that have high levels of cytosolic Ca2+ binding proteins, which may limit mitochondrial Ca2+ uptake, seem to be spared in ALS (Alexianu et al., 1994).
Although these observations probably pertain to the high intrinsic vulnerability of MNs to excitotoxic injury, they do not explain the astrocytic pathology in affected areas of spinal cord and motor cortex. Specifically, astrocytes are the principal regulators of extracellular glutamate levels (Rothstein et al., 1996) and seem to be responsible for the loss of glutamate transport capacity that has been noted in these regions. Although this astrocytic dysfunction can explain excitotoxic MN injury in ALS and thus might be considered to be the primary defect leading to MN loss, its cause is unknown.
The present study aims to explore the connection between MN ROS generation and astroglial transport. Specifically, in light of a known susceptibility of glutamate uptake to inhibition by oxidants (Volterra et al., 1994; Sorg et al., 1997; Trotti et al., 1998) and our previous studies demonstrating that exposure to AMPA or kainate induced strong and selective ROS generation in cultured MNs (Carriedo et al., 2000), the present study seeks to address two questions. The first is to determine whether MNs produce more ROS than other spinal neurons during activation with the endogenous excitatory transmitter glutamate. In addition, we sought to examine the hypothesis that such ROS, generated within MNs in response to excitotoxic activation, can pass across the plasma membrane of the MN and damage glutamate transport in surrounding glia.
Materials and Methods
Animals. Cultured neurons were produced from embryonic Swiss-Webster mice. For spinal cord immunohistochemical studies, SOD1 G93A transgenic mice (The Jackson Laboratory, Bar Harbor, ME) were bred, and offspring were genotyped by PCR analysis (Gurney et al., 1994). All animal procedures were approved by the Institutional Animal Care and Use Committee.
Primary dissociated cultures. Spinal cord suspensions (removed from both meninges and dorsal root ganglia) were prepared from 13-d-old Swiss-Webster mouse embryos and plated at a density of 3 × 105cells/cm2 on established astrocytic monolayers, as described previously (Carriedo et al., 1996). Cultures were studied after 14–18 d in vitro. Motor neurons were identified by morphological characteristics (soma >20 μm; extensive dendritic arborization) along with labeling by the nonphosphorylated neurofilament antibody SMI-32 (Sternberger Monoclonals, Lutherville, MD). In previous studies, we found that these criteria in combination provide excellent identification of MNs in culture and slice models (Carriedo et al., 1996). In these cultures, astrocytes form a confluent monolayer, have indistinct cell borders, and possess flat oval nuclei [visible on hydroethidine (HEt) loading or labeling with the nuclear stain Hoechst 33258] that appear in a focal plane below the neurons. Control studies using the astrocyte-specific marker glial fibrillary acidic protein (GFAP) confirmed the identity of cells with these morphological characteristics (see Fig. 1B).
Fig. 1.

Characterization of cultures. A, Multifluorescence microscopy shows neurons (fluorescent Nissl stain,red), MNs (SMI-32, blue), and astrocytes (anti-GFAP, green). B, High-magnification image shows MNs (SMI-32,red), astrocytes (anti-GFAP,green), and nuclei (primarily of astrocytes; Hoechst 33258, blue). C, Under confocal microscopy, the close spatial relationship between the MN (SMI-32,red) and glial glutamate transporters (anti-GLT-1, green) is apparent. Scale bars, 50 μm.
Immunocytochemistry. Cultures were fixed in 4% paraformaldehyde, blocked, and exposed to primary antibody [SMI-32, 1:5000 and GFAP, 1:400 (Dako, Glostrup, Denmark); glutamate transporter GLT-1 (also known as EAAT2), 0.17 μg/ml (kindly supplied by Jeff Rothstein, Johns Hopkins University, Baltimore, MD); 3-nitrotyrosine, 10 μg/ml (Upstate Biotechnology, Waltham, MA)]. Labeling was visualized either by routine immunoperoxidase techniques or under fluorescence using secondary antibodies linked to fluorophores [AlexaFluor 594 or 488 (Molecular Probes, Eugene, OR); Cy3 or aminomethylcoumarin acetate (Jackson ImmunoResearch, West Grove, PA)]. Fluorescent labeling of nuclei was performed with the dye Hoechst 33258 and that of neurons with a fluorescent Nissl stain (NeuroTrace red; Molecular Probes). Digital images were acquired using routine transmitted light, fluorescence, or confocal microscopy as indicated, with appropriate emission filters. For SOD1 G93A transgenic mouse studies, spinal cords were removed from 90- to 100-d-old mice, embedded in paraffin, cut into 7 μm sections, and processed for immunocytochemistry. Nontransgenic littermates served as controls.
Imaging studies. Cultures were mounted on the stage of a inverted microscope (TE-200; Nikon, Tokyo, Japan), and agonist exposures were performed at 22°C in a static bath consisting of HEPES-buffered salt solution (HSS) (in mm: 120 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgCl2, 20 HEPES, 15 glucose, and 10 NaOH, pH 7.4). Preselected fields containing one morphologically identified presumptive MN (and typically 5–15 other neurons) were illuminated with a xenon light source, and fluorescence signals were collected using a 12-bit cooled digital CCD camera (Orca-100; Hamamatsu, Bridgewater, NJ) and analyzed after background subtraction using MetaFluor software (Universal Imaging, West Chester, PA).
ROS generation was monitored by use of the oxidation-sensitive dyes HEt (Bindokas et al., 1996; Carriedo et al., 1998) and dihydrorhodamine (DHR) (Dugan et al., 1995) (Molecular Probes). Cultures were loaded in the dark (HEt, 5 μm, 45 min, 22°C; DHR, 10 μm, 30 min, 37°C), followed by wash into a static HSS bath (for HEt experiments, 5 μm HEt was added). Cells were excited at 510–560 nm, and emission was monitored at >590 nm. For both dyes, fluorescence measurements for each cell (F_x) were normalized to the fluorescence intensity for that cell at the beginning of the experiment (F_0) to compensate for differences in cell size or dye loading. In the DHR experiments, Δ_F was monitored only in “mitochondria-rich” perinuclear regions of the soma. In HEt experiments, Δ_F was measured in the soma and nucleus of neurons, whereas astrocytic Δ_F_ was measured only over nuclei, because nuclear HEt signals are readily detectable, whereas astrocyte cell borders are difficult to discern within a confluent monolayer.
Autoradiographic [3H]glutamate uptake studies. Cultures were exposed for 15 min to sham wash or to kainate [100 μm plus (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801)] alone or in the presence of antioxidants (SOD, 100 U/ml; catalase, 400 U/ml), followed after another 10 min by incubation in [3H]glutamate (2 μCi/ml) in HSS for 5 min. Because glutamate transport is Na+ dependent, uptake was terminated by washes (three times) in Na+-free HSS with excess unlabeled glutamate. Immediately after uptake, cultures were fixed in 4% paraformaldehyde (with 0.1% glutaraldehyde to cross-link the [3H]glutamate) (Reynolds and Herschkowitz, 1987) for 30 min, followed by fluorescent SMI-32 labeling (using AlexaFluor 594 secondary antibody), coating with emulsion (NTB2; Eastman Kodak, Rochester, NY), and exposure for 2 weeks (4°C). After development, the slides were scanned under fluorescence to identify MNs (by SMI-32 labeling and morphology), and images were acquired of fields surrounding each MN. Optical density (OD) measurements were made of the transmitted light image (using MetaMorph software; Universal Imaging) in zones 0–50, 50–100, and 100–150 μm from the perimeter of the MN soma after masking out of the soma and proximal dendrites of the MN and somata of other neurons detectible by faint background fluorescence (to isolate astrocytic signal). For each field, OD values in the two closest zones were normalized to that in the distal (100–150 μm) zone.
Results
Glutamate triggered ROS generation in cultured motor neurons
MN ROS generation was examined in a dissociated cell culture model. Spinal cord suspensions were plated on established astrocytic monolayers and studied after 14–18 d in vitro. In these cultures, putative MNs can often be identified prospectively on the basis of morphological criteria (soma >20 μm; extensive dendritic arborization). We found previously that these criteria, combined with subsequent immunolabeling with the nonphosphorylated neurofilament antibody SMI-32, are confirmatory (Carriedo et al., 1996) (see Materials and Methods). The cultures recapitulate many of the key components of the in vivo spinal cord; specifically, they contain MNs and other spinal neurons on a monolayer of astrocytes in which glutamate transporters are present (Fig.1). Because each of these cell types can be distinguished readily, this model system is useful for examining interactions between them under highly controlled conditions.
In our previous studies using these cultures, we found that the ROS generation induced by exposure to AMPA or kainate was highly selective for MNs (Carriedo et al., 2000). To assess the physiological significance of these findings further, our first aim was to determine whether this restricted pattern of ROS generation still occurred with the endogenous nonselective agonist glutamate (Fig.2). Cultures were loaded with the oxidant-sensitive fluorophore HEt; in the presence of superoxide anions, HEt is oxidized to ethidium, which fluoresces strongly on intercalation in DNA (Bindokas et al., 1996). Addition of glutamate (250 μm) triggered significantly greater increases in fluorescence (Δ_F_) in MNs (most evident over the nuclei) than other spinal neurons. Additional trials using the mitochondrially sequestered oxidation-sensitive fluorophore DHR (Dugan et al., 1995) also revealed preferential fluorescence increases in MNs, but, in this case, Δ_F_ was most notable in the perinuclear mitochondria-rich regions of the neurons (Fig. 2). Additional experiments using HEt-loaded cultures were performed to examine the mechanisms of ROS generation in MNs (Fig.3). The glutamate-induced Δ_F_was dependent on the presence of Ca2+ in the extracellular medium, and, suggesting a central role for mitochondria in the ROS generation, it was blocked by addition of the electron transport blocker rotenone (10 μm).
Fig. 2.
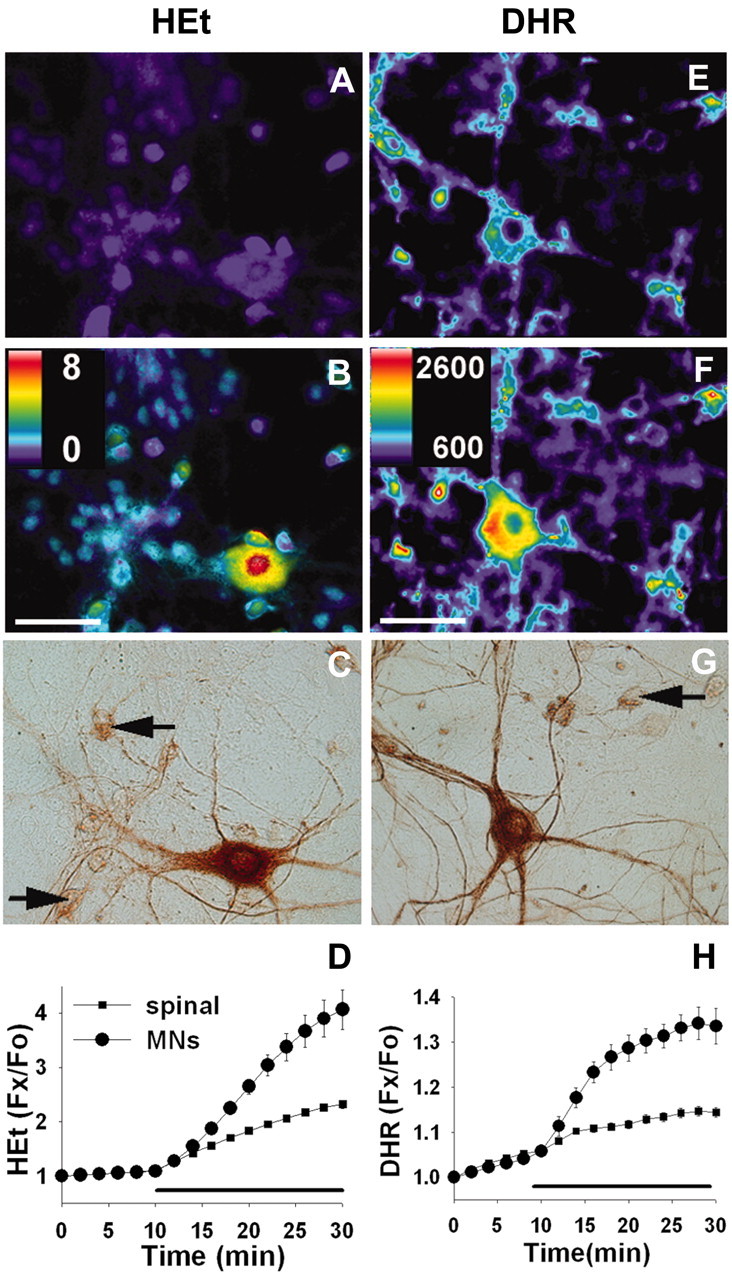
Glutamate exposure causes preferential ROS generation in cultured MNs. Spinal cultures were loaded with the oxidant-sensitive fluorophores HEt (left column) or DHR (right column) and exposed to glutamate (250 μm). Pseudocolor images depict fluorescence intensity before (A, E) and 20 min after (B, F) addition of glutamate. The pseudocolor scale depicts fluorescence ratios to baseline for HEt and raw fluorescence for DHR. Scale bars, 50 μm. Motor neuron identity was confirmed subsequently by SMI-32 immunoreactivity (C, G) and morphological criteria (arrows mark representative nonmotor neurons).Traces show time course of fluorescence changes in cultures loaded with HEt (D) or DHR (H), in MNs (circles) and other spinal neurons (squares), before and after addition of glutamate (indicated by bars). Each_trace_ represents mean ±SEM of 8–10 MNs or >80 other spinal neurons.
Fig. 3.

Mechanisms of MN ROS generation. Spinal cultures were loaded with the oxidant-sensitive fluorophore HEt and exposed to glutamate (250 μm). A, Ca2+ dependence of MN ROS generation (circles) was examined by exposing cultures to glutamate (bar) in Ca2+-free buffer before the addition of Ca2+ (1.8 mm) (arrow). B, The role of mitochondria in MN ROS generation was tested by addition of the electron transport blocker rotenone (10 μm) to cultures before and during the glutamate exposure (black circles). Fluorescence changes in MNs from matched cultures exposed to glutamate alone are shown for comparison (gray circles). Each_trace_ represents mean ± SEM of 8–10 MNs.
Motor neuron ROS generation induces oxidation in neighboring astrocytes
We next sought to determine whether ROS generated in MNs was capable of inducing oxidation in surrounding astrocytes. Spinal cultures were loaded with HEt much as above, but, in this case, instead of glutamate, we used the selective agonist kainate in the presence of the NMDA antagonist MK-801. This exposure paradigm, which we found previously to cause strong ROS generation in MNs with virtually no response in other spinal neurons (Carriedo et al., 2000), was selected to ensure that measured astrocytic oxidation was caused by only MN ROS, uncontaminated by any minimal ROS produced in other nearby neurons (Fig. 4). Because the specific fluorescence of ethidium increases markedly during interaction with DNA, examination of Δ_F_ in astrocytic nuclei permits highly sensitive ROS detection. Before imaging, a microscope field (400×) was selected that contained a visually identified putative MN. During addition of kainate (100 μm plus 10 μm MK-801), Δ_F_ was measured in astrocytes at various distances from the MN soma (<50, 50–100, and 100–150 μm) and in the MN and other spinal neurons. Because we anticipated that the extent of oxidation in astrocytes would be proportionate to the intensity of ROS generation in the central MN, we compiled results initially only from regions surrounding strongly responding MNs (Δ_F_ >3, a level of response seen in one-half of the imaged MNs).
Fig. 4.
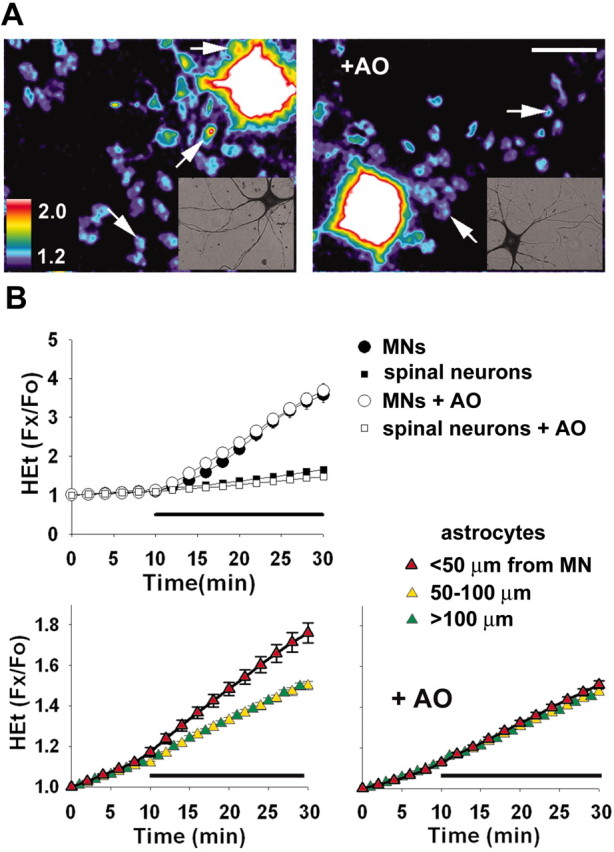
MN ROS generation induces oxidation in neighboring astrocytes. A, Pseudocolor images depict HEt fluorescence, as ratios to baseline, 20 min after addition of kainate (100 μm plus 10 μm MK-801) alone (left) or with addition of the antioxidant SOD (100 U/ml) to the bath (+AO, right).Arrows indicate HEt fluorescence signal from representative astrocyte nuclei (pseudocolor scaling to visualize astrocytic signals renders MN signals off-scale), and_insets_ show imaged MNs after SMI-32 labeling. Scale bar, 50 μm. B, Time course. In HEt-loaded cultures, fluorescence changes were measured in neurons (top) and astrocytes (bottom) before and after addition of kainate (indicated by bar). Note strong fluorescence increases in MNs (circles) in both the absence (black) or presence (white) of SOD, with minimal response in other neurons (squares). Astrocytic responses (triangles) were measured in nuclei at various distances from the MN soma (<50 μm, _red_; 50–100 μm, _yellow_; 100–150 μm, _green_) during exposure in the absence (_bottom left_) or presence (_bottom right_) of extracellular SOD (+_AO_). Each _trace_ represents mean ± SEM of 80–200 astrocytes, 11 MNs, or >70 other spinal neurons. At the end point, fluorescence increases in nearby (<50 μm) astrocytes during exposure to kainate alone were greater than astrocytic responses in all other conditions (p < 0.001, by ANOVA with Student–Newman–Keuls post hoc test).
Twenty minutes after addition of kainate, astrocytes within 50 μm of the strongly responding MNs showed significantly greater Δ_F_ than those farther away (p < 0.001). With the addition of a cell-impermeant antioxidant enzyme (SOD, 100 U/ml), despite closely matched MN Δ_F_, the astrocytic response was eliminated. This selective effect of extracellular SOD on astrocytic responses suggests strongly that the ROS generated in MNs passes into the extracellular bath (in which it can be quenched by SOD) before inducing oxidation in neighboring glia. In control studies, few or no astrocytic responses were seen surrounding weakly responding MNs or nonmotor neurons (data not shown).
Motor neuron ROS can disrupt glutamate uptake in surrounding astrocytes
The ability of this ROS to affect glutamate transport in nearby glia was assessed using autoradiographic assays of [3H]glutamate uptake (Fig.5). Spinal cultures were bathed in [3H]glutamate (2 μCi/ml for 5 min), followed by fixation, fluorescent labeling for SMI-32, and processing for autoradiography as described. Visual inspection of cultures subjected to autoradiography in the absence of agonist exposure revealed a generally uniform distribution of granules throughout the astrocyte monolayer but near absence from neurons, consistent with the dominant role of astrocytes in glutamate transport. Indicating the specificity of the assay for high-affinity Na+-dependent glutamate transport, the signal was eliminated completely if Na+was removed from the buffer.
Fig. 5.
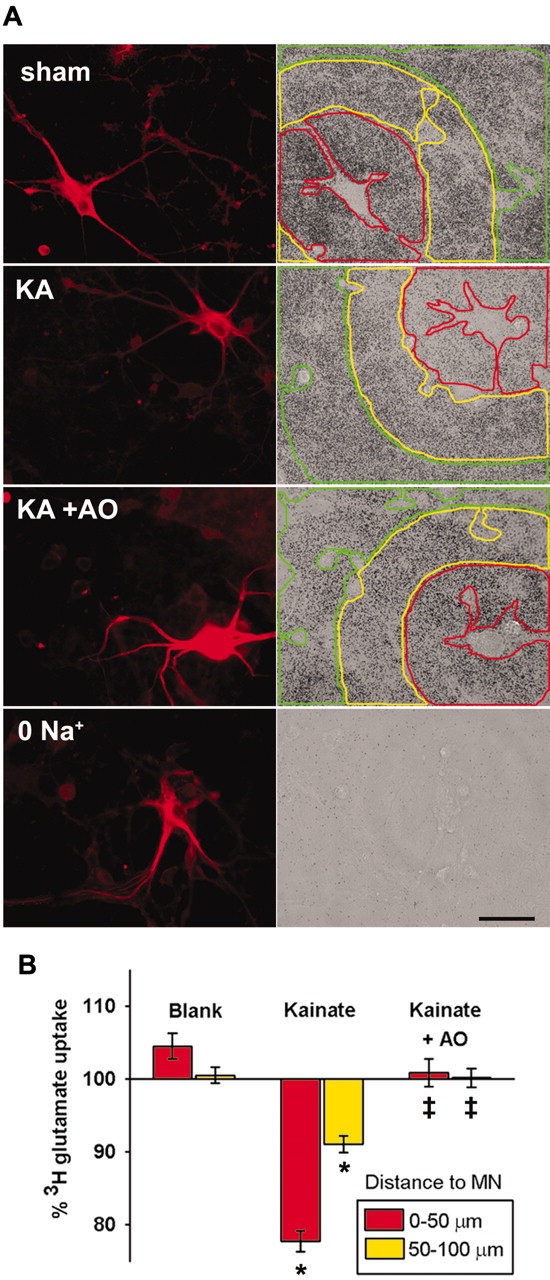
MN ROS can disrupt glutamate uptake in surrounding astrocytes. A, Spinal cultures were exposed to sham wash or to kainate (KA; 100 μm plus 10 μm MK-801) alone or with addition of the antioxidants SOD (100 U/ml) and catalase (400 U/ml) to the bath (KA+AO) before [3H]glutamate uptake assays as described. A control condition examined [3H]glutamate uptake in Na+-free buffer (0 Na +). Fluorescent images (left) show SMI-32-labeled MNs and some faintly labeled surrounding neurons. Transmitted light images (right) show autoradiographic granules corresponding to glutamate uptake.Colored lines indicate zones of increasing distance from the MN (<50 μm, red; 50–100 μm,yellow; 100–150 μm, green). Scale bar, 50 μm. B, Quantitative assessment of uptake. Relative uptake was calculated by normalizing optical density values in the two closer zones (<50, red; 50–100 μm,yellow) to that in the distal zone (100–150 μm) for each cell (mean ± SEM from zones surrounding 60–80 individual MNs from 13–14 cultures). * indicates difference from same zone in blank condition; ‡ indicates difference from same zone after kainate exposure; p < 0.001 by two-tailed_t_ test.
To quantify astrocytic uptake, optical density was assessed in zones of increasing distance from each MN generally as above (see Materials and Methods); in these experiments, uptake was analyzed in zones surrounding all identified MNs. In untreated cultures, uptake was increased slightly in the zone immediately surrounding MNs (<50 μm) compared with more distant zones. Other cultures were exposed to kainate (100 μm plus 10 μm MK-801 for 15 min) before the [3H]glutamate uptake assay was performed. Paralleling the regional HEt oxidation described above, kainate exposure induced decreases in uptake in the zone closest to MNs (<50 μm) and to a lesser extent in the next zone (50–100 μm). Furthermore, this local decrease in uptake was prevented by addition of cell-impermeant antioxidant enzymes (SOD, 100 U/ml; catalase, 400 U/ml) to the bath.
Oxidative changes surrounding motor neurons in transgenic mice
Use of the present simplified culture system enabled us to perform real-time examination of ROS generated in MNs and their effects in local glia. However, to begin to address the degree to which similar events might occur in vivo, we made use of transgenic mice expressing the G93A mutant form of SOD1, associated with familial forms of ALS (Gurney et al., 1994). These mice provide an excellent animal model, showing the oxidative MN damage, loss of glutamate uptake, and gliosis that are features of the human disease (Tu et al., 1996;Ferrante et al., 1997; Alexander et al., 2000). In ALS MNs, protein oxidative damage has been well documented by 3-nitrotyrosine immunolabeling (Abe et al., 1995; Beal et al., 1997).
The G93A mice were killed at 90–100 d (the approximate time of symptom onset in this strain), and lumbar spinal cord sections were examined for 3-nitrotyrosine immunoreactivity. In agreement with previous reports (Ferrante et al., 1997), MNs in G93A mice were labeled strongly (Fig. 6A). Furthermore, we observed consistently a previously unreported pattern of increased labeling in discrete confluent regions, incorporating the tissue surrounding and between MNs, in ventral horn of G93A mice (Fig.6B). In addition, high-power examination of fluorescent 3-nitrotyrosine labeling revealed increased immunoreactivity in a distinct zone immediately adjacent to many MNs in G93A mice but never in control mice (Fig. 6C).
Fig. 6.
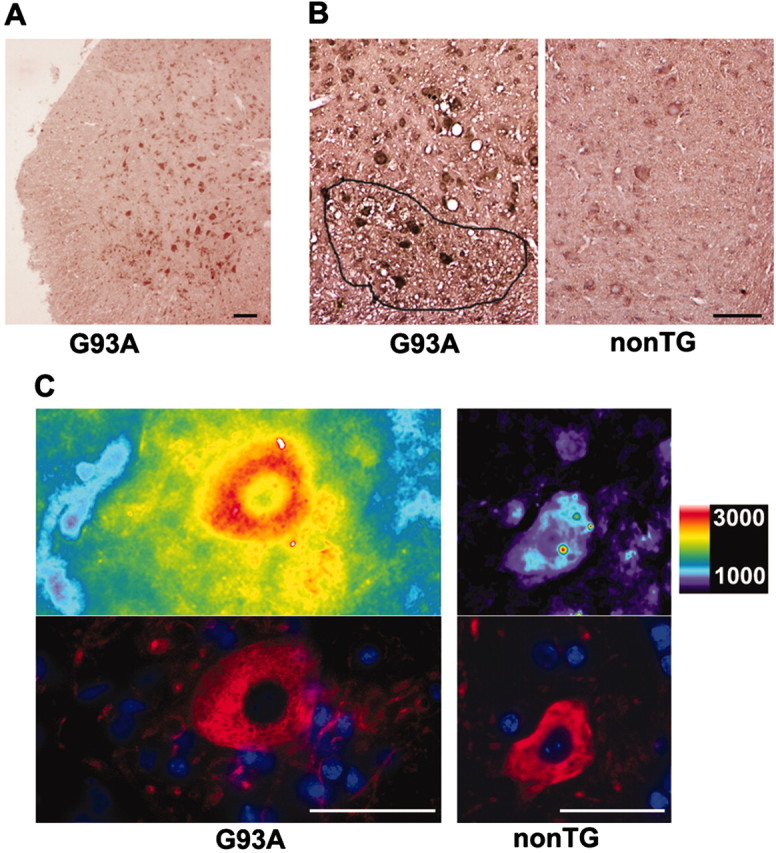
Oxidative changes in regions surrounding MNs in transgenic mouse spinal cord. Lumbar spinal cord sections from 3-month-old SOD1 transgenic mice (G93A) and nontransgenic controls (non-TG) (n = 4 of each) were examined for 3-nitrotyrosine immunoreactivity.A, Spinal cord hemisection from G93A mouse.B, Representative ventral horn details from G93A and control mice. C, Fluorescent 3-nitrotyrosine labeling of region surrounding ventral horn motor neurons from G93A and control mice displayed (top) on a pseudocolor intensity scale (in arbitrary fluorescence units). Bottom images show labeling with SMI-32 (red) and Hoechst 33258 (blue) to identify MNs and nuclei, respectively.Lines demarcate confluent regions of increased labeling around ventral horn MNs seen in G93A mice. Scale bars:A, B, 100 μm; C, 50 μm.
Discussion
Synopsis of principal findings
The present study builds on and integrates clues to possible mechanisms involved in ALS pathogenesis suggested by a number of previous studies. First, several studies have indicated that excitotoxic exposures (generally mediated through NMDA receptors) can induce ROS generation in central neurons (Lafon-Cazal et al., 1993;Dugan et al., 1995; Reynolds and Hastings, 1995); the present report is the first to indicate that the nonselective endogenous excitatory transmitter glutamate induces ROS quite selectively, with strong generation in MNs, and minimal ROS production in other spinal neurons. In addition, although previous studies indicated that glutamate transporters are sensitive to inhibition by exogenously applied ROS (Trotti et al., 1998), this is the first to demonstrate that ROS generated within MNs (or within any neuron, in fact) in response to excitotoxic exposure can pass across the plasma membrane and disrupt transport in neighboring astrocytes. Finally, previous studies found evidence for oxidative changes in MNs and in spinal cord astrocytes in ALS (Abe et al., 1995; Beal et al., 1997; Ferrante et al., 1997). The present observation that nitrotyrosine staining is often seen in an annular pattern immediately surrounding MNs in the SOD mouse model of ALS is novel and lends support to the hypothesis that the oxidation results from ROS emanating directly out of MNs. Thus, together, these results provide direct evidence for the feasibility of a previously untested mechanism in which MN ROS contributes directly to transporter dysfunction in surrounding astrocytes.
Experimental model and discussion of results
For this study, we elected to make primary use of a dissociated spinal cord coculture system containing MNs along with other spinal neurons growing on a monolayer of astrocytes. Although this system is highly simplified compared with animal models, we believe that it is ideally suited for testing the present hypothesis, which cannot be examined readily in intact spinal cord or slices. Indeed, in the dense three-dimensional array of cells in intact spinal cord, it is not possible to isolate ROS emanation to individual neurons or to assess its effects on surrounding cells. The present primarily two-dimensional culture system permits ready examination of fields of astrocytes surrounding single large MNs under highly controlled conditions before and after stimulation. Furthermore, considerable evidence supports the idea that MNs in mixed cultures behave similarly to those in situ, with both seeming to possess large numbers of Ca2+-permeable AMPA channels, to show poor intracellular Ca2+ buffering, and to be selectively vulnerable to injury caused by glutamate receptor activation (O'Brien and Fischbach, 1986; Hugon et al., 1989; Rothstein et al., 1993; Carriedo et al., 1996, 2000; Williams et al., 1997;Vandenberghe et al., 1998; Palecek et al., 1999). Thus, the relatively selective ROS generation we observe in cultured MNs in response to glutamate receptor activation and the ability of this ROS to escape the cell and affect the surrounding microenvironment is likely to model events that can occur in vivo.
Although we reported previously that AMPA or kainate exposures cause selective ROS generation in MNs, it is striking that such a high degree of selectivity persists during activation with the nonselective endogenous transmitter glutamate. Indeed, glutamate is an effective agonist at widely expressed and highly Ca2+-permeable NMDA channels and thus might be expected to induce a more uniform pattern of ROS production. As in the case of AMPA or kainate exposures, this highly selective ROS generation is probably best explained by rapid Ca2+ entry through the Ca2+-permeable AMPA channels that are expressed strongly on motor neurons. In addition, as with AMPA or kainate exposures, the glutamate-triggered ROS generation appears to require Ca2+ influx and is inhibited by electron transport blockers, suggesting a mitochondrial origin.
Subsequent experiments using the ROS-sensitive dye HEt revealed that excitotoxic exposures causing strong ROS generation within MNs also induce increases in fluorescence of astrocyte nuclei closely surrounding the MN. Because the oxidized product ethidium is charged and is minimally permeable through membranes (Aeschbacher et al., 1986), this observation suggests that the ROS passes across two plasma membranes, both exiting the MN and entering the astrocyte, before interacting with HEt. Indeed, the observed block of this oxidation by an extracellular antioxidant (SOD) provides strong support for the idea that the ROS passes through the extracellular space, in which it can be inactivated before interacting with astrocytes. Additional experiments examined the ability of ROS generated in MNs to affect the function of nearby glutamate transporters. The technique used here of [3H]glutamate uptake autoradiography is advantageous in that it permits simultaneous comparison of uptake capacity in all cells at various distances from a central MN. Indeed, beyond being selective producers of ROS, the present observations suggest that MNs, if stimulated strongly, can produce ROS in sufficient quantities to induce a rapid disruption of glutamate transport in surrounding astrocytes. In studies of glutamate transporters in ALS and SOD mutant mouse models, there has been discussion as to whether alterations in glutamate transport are more reflective of a loss of or a dysfunction in transporter protein (Trotti et al., 1998; Deitch et al., 2002). The present findings of glutamate transport deficiencies occurring within minutes after an excitotoxic exposure are most compatible with oxidative modification of transporters and provide support to the idea that similar oxidative dysfunction might contribute to transport loss in ALS.
Although determination of the precise forms of ROS that may mediate disruption of glutamate transport is beyond the scope of this project, it is likely that excitotoxic exposures cause a range of species to be produced. Specifically, our data suggest that mitochondrial Ca2+ overload can result in superoxide production (which can be converted readily to hydrogen peroxide or hydroxyl radical) (Fridovich, 1998) and that Ca2+-dependent nitric oxide generation provides a substrate to react with superoxide, with consequent peroxynitrite formation (Beckman, 1991). Indeed, findings of increased nitrotyrosine labeling in motor neurons and ventral horn in ALS and/or SOD mutant mouse models (Abe et al., 1995; Beal et al., 1997; Ferrante et al., 1997) and our findings of increased nitrotyrosine staining in an annular pattern around MNs in SOD mutant mice suggest that this species may be involved.
Novel support for a feedforward mechanism contributing to ALS pathogenesis
In principle, the loss of astrocytic glutamate transport that has been suggested to underlie excitotoxic damage to MNs in ALS could reflect a primary astrocytic deficit. However, the present observation suggesting that disruption of astrocytic transport is induced specifically by ROS generated within MNs provides support for an alternative model, in which reciprocal interactions between MNs and adjacent astrocytes underlie disease propagation. This model provides an attractive explanation for the close physical proximity of affected MNs and astrocytes in spinal cord and motor cortex with little pathology elsewhere. Indeed, in contrast to the local changes predicted by the present model and observed in ALS, a primary global loss of astrocytic glutamate transport capacity induces a distinct phenotype of lethal seizures (Tanaka et al., 1997). Also supporting a critical involvement of both cell types in the mutant SOD1 mouse models of ALS, development of disease seems to require the mutant enzyme to be present in astrocytes and neurons (Gong et al., 2000; Pramatarova et al., 2001).
In addition to providing a basis for the close association of affected MNs and astrocytes in ALS, the present model presents a mechanistic framework that can explain other diverse features in the disease. First, the apparent ability of glutamate to cause selective mitochondrial ROS generation in MNs is compatible with the pronounced oxidative damage and mitochondrial abnormalities seen in ALS. In addition, the suggestion that the ROS can exit MNs and affect surrounding astrocytes provides an explanation for previous reports of oxidative modifications of the GLT-1 transporter in ALS (Pedersen et al., 1998; Trotti et al., 1999; Deitch et al., 2002). Indeed, this transporter is present in astrocytic foot processes directly abutting MNs (Sasaki et al., 2000) and thus should be readily accessible to MN ROS. Finally, because ROS-induced disruption of glutamate transport would be expected to cause additional elevations of ambient extracellular glutamate, which, in turn, would result in increased excitotoxic activation and ROS generation in the local population of MNs, the present observations provide the framework for a feedforward cycle contributing to disease progression. Such a cycle could in principle be triggered at different steps and thus might be compatible with a multiplicity of inciting mechanisms (e.g., mutant SOD) leading into a common self-propagating disease pathway. Additional elucidation of the nature of reciprocal interactions between MNs and astrocytes in ALS could powerfully impact the development of new therapeutic strategies.
Footnotes
This work was supported by National Institutes of Health Grants NS-36548 and AG-00836 (J.H.W.) and F31-NS-42570 (S.R.). We thank Simin Amindari and Dien Ton-That for expert assistance with cell cultures and Jeff Rothstein for sharing antibodies.
Correspondence should be addressed to John H. Weiss, 2101 Gillespie Building, University of California at Irvine, Irvine, CA 92697-4292. E-mail: jweiss@uci.edu.
References
- 1.Abe K, Pan LH, Watanabe M, Kato T, Itoyama Y. Induction of nitrotyrosine-like immunoreactivity in the lower motor neuron of amyotrophic lateral sclerosis. Neurosci Lett. 1995;199:152–154. doi: 10.1016/0304-3940(95)12039-7. [DOI] [PubMed] [Google Scholar]
- 2.Aeschbacher M, Reinhardt CA, Zbinden G. A rapid cell membrane permeability test using fluorescent dyes and flow cytometry. Cell Biol Toxicol. 1986;2:247–255. doi: 10.1007/BF00122693. [DOI] [PubMed] [Google Scholar]
- 3.Alexander GM, Deitch JS, Seeburger JL, Del Valle L, Heiman-Patterson TD. Elevated cortical extracellular fluid glutamate in transgenic mice expressing human mutant (G93A) Cu/Zn superoxide dismutase. J Neurochem. 2000;74:1666–1673. doi: 10.1046/j.1471-4159.2000.0741666.x. [DOI] [PubMed] [Google Scholar]
- 4.Alexianu ME, Ho BK, Mohamed AH, La Bella V, Smith RG, Appel SH. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol. 1994;36:846–858. doi: 10.1002/ana.410360608. [DOI] [PubMed] [Google Scholar]
- 5.Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW, Brown RH., Jr Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997;42:644–654. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- 6.Beckman JS. The double-edged role of nitric oxide in brain function and superoxide-mediated injury. J Dev Physiol. 1991;15:53–59. [PubMed] [Google Scholar]
- 7.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carriedo SG, Yin HZ, Lamberta R, Weiss JH. In vitro kainate injury to large, SMI-32(+) spinal neurons is Ca2+ dependent. NeuroReport. 1995;6:945–948. doi: 10.1097/00001756-199504190-00030. [DOI] [PubMed] [Google Scholar]
- 9.Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carriedo SG, Yin HZ, Sensi SL, Weiss JH. Rapid Ca2+ entry through Ca2+-permeable AMPA/kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J Neurosci. 1998;18:7727–7738. doi: 10.1523/JNEUROSCI.18-19-07727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carriedo SG, Sensi SL, Yin HZ, Weiss JH. AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro. J Neurosci. 2000;20:240–250. doi: 10.1523/JNEUROSCI.20-01-00240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deitch JS, Alexander GM, Del Valle L, Heiman-Patterson TD. GLT-1 glutamate transporter levels are unchanged in mice expressing G93A human mutant SOD1. J Neurol Sci. 2002;193:117–126. doi: 10.1016/s0022-510x(01)00656-6. [DOI] [PubMed] [Google Scholar]
- 13.Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrante RJ, Shinobu LA, Schulz JB, Matthews RT, Thomas CE, Kowall NW, Gurney ME, Beal MF. Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann Neurol. 1997;42:326–334. doi: 10.1002/ana.410420309. [DOI] [PubMed] [Google Scholar]
- 15.Fridovich I. Oxygen toxicity: a radical explanation. J Exp Biol. 1998;201:1203–1209. doi: 10.1242/jeb.201.8.1203. [DOI] [PubMed] [Google Scholar]
- 16.Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng H-X, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 18.Hugon J, Vallat JM, Spencer PS, Leboutet MJ, Barthe D. Kainic acid induces early and delayed degenerative neuronal changes in rat spinal cord. Neurosci Lett. 1989;104:258–262. doi: 10.1016/0304-3940(89)90585-5. [DOI] [PubMed] [Google Scholar]
- 19.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 20.Menzies FM, Ince PG, Shaw PJ. Mitochondrial involvement in amyotrophic lateral sclerosis. Neurochem Int. 2002;40:543–551. doi: 10.1016/s0197-0186(01)00125-5. [DOI] [PubMed] [Google Scholar]
- 21.O'Brien RJ, Fischbach GD. Modulation of embryonic chick motoneuron glutamate sensitivity by interneurons and agonists. J Neurosci. 1986;6:3290–3296. doi: 10.1523/JNEUROSCI.06-11-03290.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palecek J, Lips MB, Keller BU. Calcium dynamics and buffering in motoneurones of the mouse spinal cord. J Physiol (Lond) 1999;520:485–502. doi: 10.1111/j.1469-7793.1999.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pedersen WA, Fu W, Keller JN, Markesbery WR, Appel S, Smith RG, Kasarskis E, Mattson MP. Protein modification by the lipid peroxidation product 4-hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann Neurol. 1998;44:819–824. doi: 10.1002/ana.410440518. [DOI] [PubMed] [Google Scholar]
- 24.Pramatarova A, Laganiere J, Roussel J, Brisebois K, Rouleau GA. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci. 2001;21:3369–3374. doi: 10.1523/JNEUROSCI.21-10-03369.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reynolds R, Herschkowitz N. Simultaneous immunofluorescence and autoradiography: a useful technique for investigating neurotransmitter uptake by neurons and glia in primary central nervous system culture. Brain Res. 1987;433:1–11. doi: 10.1016/0165-3806(87)90060-5. [DOI] [PubMed] [Google Scholar]
- 27.Robberecht W. Oxidative stress in amyotrophic lateral sclerosis. J Neurol. 2000;247 [Suppl 1]:I1–I6. doi: 10.1007/s004150050551. [DOI] [PubMed] [Google Scholar]
- 28.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 29.Rothstein JD. Excitotoxicity hypothesis. Neurology. 1996;47:S19–S25. doi: 10.1212/wnl.47.4_suppl_2.19s. [DOI] [PubMed] [Google Scholar]
- 30.Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- 31.Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- 32.Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA. 1993;90:6591–6595. doi: 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 34.Sasaki S, Komori T, Iwata M. Excitatory amino acid transporter 1 and 2 immunoreactivity in the spinal cord in amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 2000;100:138–144. doi: 10.1007/s004019900159. [DOI] [PubMed] [Google Scholar]
- 35.Shaw PJ, Chinnery RM, Ince PG. [3H]d-aspartate binding sites in the normal human spinal cord and changes in motor neuron disease: a quantitative autoradiographic study. Brain Res. 1994;655:195–201. doi: 10.1016/0006-8993(94)91614-4. [DOI] [PubMed] [Google Scholar]
- 36.Shaw PJ, Forrest V, Ince PG, Richardson JP, Wastell HJ. CSF and plasma amino acid levels in motor neuron disease: elevation of CSF glutamate in a subset of patients. Neurodegeneration. 1995;4:209–216. doi: 10.1006/neur.1995.0026. [DOI] [PubMed] [Google Scholar]
- 37.Sorg O, Horn TF, Yu N, Gruol DL, Bloom FE. Inhibition of astrocyte glutamate uptake by reactive oxygen species: role of antioxidant enzymes. Mol Med. 1997;3:431–440. [PMC free article] [PubMed] [Google Scholar]
- 38.Spreux-Varoquaux O, Bensimon G, Lacomblez L, Salachas F, Pradat PF, Le Forestier N, Marouan A, Dib M, Meininger V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J Neurol Sci. 2002;193:73–78. doi: 10.1016/s0022-510x(01)00661-x. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 40.Trotti D, Danbolt NC, Volterra A. Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol Sci. 1998;19:328–334. doi: 10.1016/s0165-6147(98)01230-9. [DOI] [PubMed] [Google Scholar]
- 41.Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:427–433. doi: 10.1038/8091. [DOI] [PubMed] [Google Scholar]
- 42.Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA. 1996;93:3155–3160. doi: 10.1073/pnas.93.7.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vandenberghe W, Van Den Bosch L, Robberecht W. Glial cells potentiate kainate-induced neuronal death in a motoneuron-enriched spinal coculture system. Brain Res. 1998;807:1–10. doi: 10.1016/s0006-8993(98)00569-1. [DOI] [PubMed] [Google Scholar]
- 44.Vandenberghe W, Ihle EC, Patneau DK, Robberecht W, Brorson JR. AMPA receptor current density, not desensitization, predicts selective motoneuron vulnerability. J Neurosci. 2000;20:7158–7166. doi: 10.1523/JNEUROSCI.20-19-07158.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Volterra A, Trotti D, Tromba C, Floridi S, Racagni G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J Neurosci. 1994;14:2924–2932. doi: 10.1523/JNEUROSCI.14-05-02924.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams TL, Day NC, Ince PG, Kamboj RK, Shaw PJ. Calcium-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors: a molecular determinant of selective vulnerability in amyotrophic lateral sclerosis. Ann Neurol. 1997;42:200–207. doi: 10.1002/ana.410420211. [DOI] [PubMed] [Google Scholar]