Regulation of GluR1 by the A-Kinase Anchoring Protein 79 (AKAP79) Signaling Complex Shares Properties with Long-Term Depression (original) (raw)
Abstract
Second messengers regulate synaptic plasticity by influencing the balance between kinase and phosphatase activity. One target of this balance is the phosphorylation state of the AMPA receptor glutamate receptor 1 (GluR1) subunit. Hippocampal long-term depression (LTD) is a calcium-dependent downregulation of synaptic AMPA receptor currents associated with dephosphorylation of Ser845, a cAMP-dependent protein kinase (PKA) site on GluR1. Recruitment of kinases and phosphatases to the AMPA receptor might enable modulation of AMPA receptor function. The neuronal A-kinase anchoring protein AKAP79/150 interacts with PKA and the calcium-dependent protein phosphatase PP2B and is linked to the AMPA receptor GluR1 subunit by synapse-associated protein 97 (SAP97), a membrane-associated guanylate kinase family protein. Here we demonstrate that AKAP79 not only promotes basal phosphorylation of Ser845 but also confers a calcium- and PP2B-mediated downregulation to GluR1 receptor currents. This AKAP79-dependent downregulation is contingent on the local presence of PKA, Ser845 of GluR1, and a PDZ (postsynaptic density 95/Discs large/zona occludens 1)-domain interaction between GluR1 and SAP97, all of which support basal phosphorylation of the receptor. These findings suggest that the AKAP79 signaling complex is sufficient to couple intracellular calcium levels to the PKA phosphorylation state of GluR1. Thus, the integration of intracellular signals relevant for LTD may be transduced to GluR1 by the AKAP79 signaling complex.
Keywords: AKAP79, kinase, phosphatase, cAMP, calcium, AMPA receptor, synaptic plasticity
Activity-dependent alterations in AMPA receptor-mediated synaptic transmission such as long-term potentiation (LTP) and long-term depression (LTD) are thought to provide a cellular basis for information storage (Malenka and Nicoll, 1999). As reviewed recently (Winder and Sweatt, 2001), calcium entry through NMDA-type glutamate receptors triggers both forms of synaptic plasticity. High levels of intracellular calcium achieved during LTP upregulate synaptic strength by preferentially activating the calcium/calmodulin-dependent protein kinase II (CaMKII). In contrast, the lower calcium concentrations that occur during LTD may downregulate synaptic strength by favoring a phosphatase cascade involving PP2B and protein phosphatase 1 (PP1). Thus, the balance between kinase and phosphatase activity has been proposed to be a critical determinant for controlling the degree of synaptic efficacy by regulating the phosphorylation state of synaptic substrates. Recent evidence suggests that LTP and LTD are reflected in the phosphorylation state of the AMPA receptor glutamate receptor 1 (GluR1) subunit (Barria et al., 1997;Kameyama et al., 1998; Lee et al., 1998, 2000). However, the mechanisms by which GluR1 phosphorylation and dephosphorylation are orchestrated remain unclear.
A-kinase anchoring proteins (AKAPs) bind to the regulatory subunits of protein kinase A (PKA) and target PKA to various subcellular compartments, providing a mechanism to efficiently regulate the phosphorylation state of relevant substrates (Colledge and Scott, 1999). The multivalent anchoring protein AKAP79 (AKAP150 in rodents) associates with PP2B and PKC, in addition to PKA, and is enriched at postsynaptic sites of hippocampal neurons (Carr et al., 1992b; Coghlan et al., 1995; Klauck et al., 1996; Colledge et al., 2000; Sik et al., 2000). Recently, we demonstrated that AKAP79/150 and GluR1 are linked together by synapse-associated protein 97 (SAP97), facilitating basal phosphorylation of Ser845 on GluR1 (Colledge et al., 2000). Phosphorylation of Ser845 enhances native and recombinant AMPA receptor currents (Roche et al., 1996; Banke et al., 2000). Ser845 is basally phosphorylated (Mammen et al., 1997) and is selectively dephosphorylated during LTD (Kameyama et al., 1998; Lee et al., 1998,2000). Although the AKAP79/150 signaling complex may facilitate Ser845 phosphorylation, a corresponding mechanism for the dephosphorylation of this site remains to be identified.
Disruption of PKA function should favor phosphatase activity reducing AMPA receptor currents. Indeed, previous studies indicate that PKA inhibition produces synaptic depression that occludes subsequent LTD (Kameyama et al., 1998). Similarly, peptide-mediated disruption of AKAP–PKA interactions attenuates AMPA receptor currents and synaptic events (Rosenmund et al., 1994). Therefore, we sought to identify whether a phosphatase contributes to AMPA receptor downregulation when PKA anchoring is disrupted and whether the AKAP79/150 can account for this modulation. Here, we report that PP2B regulates hippocampal AMPA receptor currents in opposition to anchored PKA. Furthermore, we demonstrate that this dual PKA- and PP2B-dependent regulation of AMPA receptor currents can be reconstituted by coexpression of AKAP79 with GluR1. AKAP79-dependent regulation of GluR1 occurs at Ser845 and depends on protein–protein interactions that promote basal phosphorylation at this site. Our data suggest that the AKAP79/150 signaling complex is sufficient to support modulation of the phosphorylation state of GluR1 in a manner analogous to LTD.
MATERIALS AND METHODS
Cell cultures. Glass coverslips (22 × 22 mm) were coated overnight with 100 μg/ml poly-d-lysine and 10 μg/ml laminin. Neuronal cells were plated on a bed of glial cells. Hippocampi were removed from neonatal Sprague Dawley rats (1- to 3-d-old). Tissue was dissociated by papain treatment and trituration through Pasteur pipettes and suspended in media consisting of MEM, 10% FBS, and penicillin–streptomycin (P/S), and plated at a density of 250,000 cells/ml for glial cells. Glutamate at 100 μm was added for 15 min to kill off neuronal cells and washed with media. Glial cultures were allowed to grow for 1 week until they reached confluence. Neurons were prepared in the same manner, except that glutamate was omitted and cells were plated at 150,000 cells/ml. After 6 hr cells, were washed with Neurobasal media with B27 supplement, 0.5 mml-glutamine, and P/S added.
Human embryonic kidney 293 (HEK293) cells were obtained from American Type Culture Collection (Manassas, VA) at passage 31 and were used at passages 33–40. HEK293 cell cultures were maintained in DMEM with 10% FBS (HyClone, Logan, UT) and P/S. For electrophysiology, HEK293 cells were plated at low density (∼50,000 cells) on 35 mm culture dishes (Falcon, Franklin Lakes, NJ). For biochemistry, HEK293 cells were plated at ∼50% confluence on 10 cm dishes.
Expression constructs. GluR1flip was in the pRK5 vector, and AKAP18-green fluorescent protein (GFP) and AKAP79-GFP were in pcDNA3, as described previously (Colledge et al., 2000). Dr. Tom Soderling (Vollum Institute, Portland, OR) provided GluR1flipS831A and GluR1flipS845A in the pRK5 vector. AKAP79(1–360)-GFP in pcDNA3 was provided by Dr. Mark Dell'Acqua (University of Colorado Health Sciences Center, Denver, CO). Dr. Craig Garner (University of Alabama at Birmingham, Birmingham, AL) provided the SAP97 construct. Dr. John Adelman (Vollum Institute, Portland, OR) provided the CD4 cell surface marker in a JPA vector. GluR1flipT887A was constructed by site-directed mutagenesis and inserted into pcDNA3.1 using the TOPO TA cloning kit (Invitrogen, San Diego, CA).
Electrophysiology. Pyramidal-shaped neurons from cultures at 5–12 d in vitro were selected for recordings. HEK293 cells were transfected by the calcium phosphate method. One microgram of each construct was used per dish for electrophysiological experiments. For these experiments, the CD4 cell surface marker was also included as a transfection marker. Twenty-four hours after transfection, cells were visually selected for recording by adherence of CD4 antibody-coated beads. GFP epifluorescence confirmed the expression of the corresponding AKAP.
Whole-cell recordings were made with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Patch pipettes (2–4 MΩ) contained (in mm): 140 Cs methanesulfonate, 10 HEPES, 5 adenosine triphosphate (Na salt), 5 MgCl2, 0.2 CaCl2, and 1 or 10 of either BAPTA or EGTA, pH 7.4. Peptides were added to the pipette solution from frozen concentrated stocks. Extracellular solution contained (in mm): 150 NaCl, 5 KCl, 1.8 or 0 CaCl2, 10 HEPES, and 10 glucose, pH 7.4. TTX (1 μm) and (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) (1 μm) were included for neuronal recordings. Glutamate, cyclothiazide, domoate, SYM-2206, 8-bromo-cAMP, cyclosporin A, and okadaic acid were added from frozen stocks. Solution exchanges were accomplished through a series of flow pipes. Flow pipes were controlled by solenoid valves and moved into position by a piezoelectric bimorph. HEK293 cells were lifted off the bottom of the dish to speed the solution exchange time. Neuronal AMPA receptor currents were evoked by 1 sec applications of domoate at 30 sec intervals. The AMPA receptor-selective antagonist SYM-2206 (100 μm) inhibited current responses by 94.7 ± 1.6% (n = 3; data not shown), indicating that domoate-activated currents were predominantly mediated by AMPA receptors. GluR1 receptor currents were evoked by 500 msec application of glutamate in the presence of cyclothiazide at 30 sec intervals. Currents were digitized at 5 kHz and filtered at 1 kHz with a Digidata 1200B board and Clampex 7 software (Axon Instruments). Series resistance (85–95%) and whole-cell capacitance compensation were used and routinely monitored throughout the experiments. Only neurons with series resistance <12 MΩ and HEK293 with series resistance <6 MΩ were included for analysis. All experiments were initiated within 1 min of establishing the whole-cell configuration and were performed at a holding potential of −60 mV at 20°C.
For experiments in which phosphatase inhibitors were used, cells were pretreated with the inhibitors, and recordings were performed in the continued presence of the inhibitor. Pretreatment periods were 5 min for cyclosporin A (1 μm) and 15 min for okadaic acid (1 μm). In approximately one-half (three of seven for control and five of 10 for AKAP79) of the okadaic acid experiments in HEK293 cells, okadaic acid (1 μm) was also included in the pipette solution. Because identical results were obtained under these conditions, the data were pooled.
Current amplitudes of recombinant GluR1 receptors varied considerably for all conditions tested (range of 150–10,000 pA), likely because of differences in transfection efficiency. We observed no significant difference in current amplitudes between conditions. No correlation between the results obtained and current density were observed. For each experiment, currents were normalized to the amplitude of current from the initial agonist application. All data are expressed as means ± SEM. Statistical significance was determined by ANOVA, followed by Student's t test.
Immunoprecipitations and immunoblotting from transfected HEK293 cells. For immunoprecipitation from heterologous cells, HEK293 cells were transfected at 50–70% confluence in 10 cm plates by the calcium phosphate method, using 4 μg of each cDNA construct per plate. Twenty-four to 48 hr after transfection, cells were rinsed in PBS and lysed in 400 μl of IP buffer (10 mmphosphate, 150 mm NaCl, 5 mm EDTA, 5 mm EGTA, and 1% Triton X-100) containing protease inhibitors. For GluR1 phosphorylation experiments, phosphatase inhibitors were also included (10 mm sodium pyrophosphate, 50 mm sodium fluoride, 1 mmsodium orthovanadate, 1 μm okadaic acid, 1 μm microcystin, and 1 μm cyclosporin). Cells were incubated on ice for 30 min and then centrifuged at 40,000 × g for 30 min. Supernatants were incubated with 3 μg of antibody or control nonimmune IgG and 30 μl of prewashed protein A agarose beads. An anti-GluR1 C-terminal antibody was used to immunoprecipitate GluR1. For immunoprecipitation of endogenous SAP97 from HEK293 cells, a rabbit polyclonal antibody that specifically recognizes SAP97 but not other membrane-associated guanylate kinase (MAGUK) family member proteins was used (Valtschanoff et al., 2000). AKAP18-GFP was immunoprecipitated with affinity-purified rabbit polyclonal antibody [V064 (Fraser et al., 1998)]. AKAP79-GFP and AKAP79(1–360)-GFP were immunoprecipitated with 918I (ICOS, Bothell, WA). After overnight incubation at 4°C, the immunoprecipitates were washed three times with IP buffer. Bound proteins were eluted with SDS sample buffer and analyzed by immunoblotting.
The following primary antibodies were used for immunoblotting: rabbit polyclonal to AKAP79 (5.6 μg/ml, 1:2500; 918I; ICOS), mouse monoclonal to PKA type II regulatory subunits α and β (1:500 dilution; Transduction Laboratories, Lexington, KY), mouse monoclonal against PP2B (1:300 dilution; Transduction Laboratories), rabbit polyclonal antibody against SAP97 (Valtschanoff et al., 2000), rabbit polyclonal to C-terminal region of GluR1 (1:1000 dilution; Upstate Biotechnology, Lake Placid, NY), and rabbit polyclonal phospho-specific antibody against ser845 of GluR1 (Mammen et al., 1997) (1:250 dilution).
RESULTS
PP2B underlies rundown of hippocampal AMPA receptor current
We recorded whole-cell AMPA receptor currents from cultured hippocampal neurons in response to the nondesensitizing agonist domoate (10 μm). Under control conditions, the amplitude of AMPA receptor currents was relatively stable during the recording period (Fig. 1A, open squares) (control, 89.6 ± 4.6% of initial current remaining, n = 5). Ht31, a 24 amino acid peptide corresponding to the type II regulatory subunit (RII)-binding domain of a human thyroid AKAP, was used as a competitive antagonist of PKA anchoring (Carr et al., 1992a). As shown previously (Rosenmund et al., 1994), disrupting PKA anchoring by inclusion of Ht31 (1 μm) within the whole-cell pipette solution caused a significant time-dependent reduction of AMPA receptor currents (Fig. 1A, filled triangles) (Ht31, 61.5 ± 3.8%, n = 7; p < 0.01). This Ht31-induced rundown is calcium dependent because it was prevented when experiments were performed in the absence of extracellular calcium (Fig. 1B, open squares, filled triangles) (control, 91.7 ± 3.9%, n = 6; Ht31, 84.5 ± 3.1%, n = 6). When calcium influx through voltage-gated calcium channels was blocked by cadmium (200 μm), Ht31-induced rundown still occurred, although there was a selective attenuation of the late phase of the rundown (Fig. 1C, open squares, filled triangles) (control, 89.4 ± 2.2%, n = 5; Ht31, 73.3 ± 1.7%, n = 6). Given that NMDA receptors were blocked by the combined presence of magnesium and MK-801, these data suggest that calcium entry through calcium-permeable AMPA receptors may promote the initial phase of the rundown. Collectively, these data indicate that calcium is required for the phenomenon and that multiple sources of calcium influx contribute to the Ht31-induced rundown.
Fig. 1.

Disruption of PKA anchoring promotes a PP2B-mediated rundown of hippocampal AMPA receptor currents. Whole-cell recordings were performed from cultured hippocampal neurons. Summary of time course of AMPA receptor currents is presented. Responses were normalized to the peak amplitude from the initial current evoked in response to domoate (10 μm). A, Displacement of PKA by whole-cell dialysis of Ht31 (1 μm) leads to rundown of AMPA receptor currents. Insets show representative first and last responses from a control cell and one in which Ht31 was included in the pipette. The number of observations for each condition is indicated. B, The Ht31-induced rundown is prevented when experiments were performed in the absence of extracellular recording solution. C, Experiments performed in the presence of 200 μm cadmium indicate that voltage-gated calcium channels contribute to a late phase of the rundown. Dotted line depicts Ht31 data from_A_ for comparison. D, Bar graph summarizing the effect of phosphatase inhibitors on the rundown. Cyclosporin A (CsA; 1 μm) treatment prevents Ht31-induced rundown of AMPA receptor currents. *p < 0.01 compared with cyclosporin A alone. Inhibition of PP1/PP2A by okadaic acid (OA; 1 μm) partially reduced the Ht31 mediated rundown. **p < 0.01 compared with okadaic acid alone.
Exogenous application of the calcium- and calmodulin-dependent protein phosphatase PP2B decreases the open probability of AMPA receptors, a property regulated by the PKA phosphorylation site on GluR1 (Banke et al., 2000). Therefore, we tested whether PP2B might be an effector, downstream of calcium, for the Ht31-induced rundown. In the presence of the PP2B-selective antagonist cyclosporin A (1 μm), Ht31 no longer produced rundown (control, 89.7 ± 3.8%,n = 5; Ht31, 83.7 ± 2.9%, n = 7;p > 0.2) (Fig. 1D). In contrast, the PP1/PP2A-selective antagonist okadaic acid (1 μm) only partially reduced the rundown (control; 88.4 ± 3.1%, n = 6; Ht31, 71.8 ± 1.8%, n = 7; p < 0.01) (Fig.1D). Given that the rundown is observed only during disruption of PKA anchoring, these data suggest that PP2B is the predominant phosphatase counteracting the action of anchored PKA. The dual regulation of hippocampal AMPA receptors by anchored PKA and PP2B is consistent with the AKAP79/150-directed location of PKA and PP2B at postsynaptic sites (Coghlan et al., 1995; Colledge et al., 2000).
Coordinate regulation of GluR1 phosphorylation by the AKAP79 signaling complex
To test directly whether an AKAP79/150 complex regulates AMPA receptor currents, we attempted to reconstitute “rundown” in HEK293 cells. Homomeric GluR1 receptors were expressed alone (control) or together with one of two plasma membrane-targeted neuronal anchoring proteins: AKAP79 or AKAP18 (also referred to as AKAP15) (Dell'Acqua et al., 1998; Fraser et al., 1998; Gray et al., 1998). As shown in Figure2, both anchoring proteins associate with endogenous PKA in HEK293 cells. The RII subunit of PKA was coimmunoprecipitated with either AKAP79 or AKAP18 (Fig.2A, top panel). In contrast, endogenous PP2B copurified with AKAP79 but not with AKAP18 (Fig.2A, bottom panel). We then tested whether these AKAPs could enhance phosphorylation of Ser845, a known PKA site in the cytoplasmic tail of GluR1. Basal phosphorylation of Ser845 of GluR1 was enhanced by coexpression of AKAP18 (241 ± 47% over control, n = 4; p < 0.01) (Fig. 2B) and AKAP79 (183 ± 32% over control,n = 7; p < 0.01) (Fig.2B). These findings suggest that AKAP-mediated recruitment of PKA to the plasma membrane facilitates phosphorylation of Ser845.
Fig. 2.

Selective PP2B-mediated downregulation of GluR1 receptor currents by coexpression of AKAP79. A, Selective association of AKAP79 with endogenous PP2B. Extracts from HEK293 cells transfected with either AKAP18-GFP or AKAP79-GFP were immunoprecipitated with their respective antibodies or control IgG antibodies. Immunoprecipitates were blotted with anti-RII antibodies (top) or anti-PP2B antibodies (bottom). Although both AKAP18 and AKAP79 coimmunoprecipitated RII as expected, only AKAP79 coimmunoprecipitated PP2B. B, Basal phosphorylation of the PKA site, Ser845, of GluR1 is enhanced by coexpression of membrane-targeted AKAPs. GluR1 was expressed alone (control) or with either AKAP18 or AKAP79 in HEK293 cells. Extracts from HEK293 cells were immunoprecipitated with antibodies against the C-terminal tail of GluR1. Phosphorylation of Ser845 was determined by blotting with phospho-specific antibodies to Ser845 (top). Total amount of GluR1 was determined by blotting with the C-terminal GluR1 antibody (middle). Results from experiments are summarized (bottom). The relative amount of GluR1 phosphorylation was quantified by determining the ratio of signals from the phospho-Ser845 antibody to that from the C-terminal antibody. For each experiment, data were normalized to the GluR1 alone (group control). The number of experiments is indicated in_parentheses_. C, Whole-cell recordings were performed from transfected HEK293 cells. Summary of time course of recombinant GluR1 receptor currents. Responses were normalized to the peak amplitude from the initial current evoked in response to glutamate (1 mm) in the presence of cyclothiazide (100 μm). HEK293 cells were transfected with GluR1 alone (control) or with either AKAP18 or AKAP79. GluR1 currents selectively rundown when cotransfected with AKAP79. The pipette solution contained 1 mm BAPTA. The number of observations for each condition is indicated. Unless otherwise indicated, all subsequent figures are presented in the same manner. Insets show representative first and last responses from control, AKAP18, and AKAP79 cells.D, Blocking rises in intracellular calcium by elevating the concentration of BAPTA in the pipette solution to 10 mmprevents the AKAP79-dependent rundown of GluR1 receptor currents.E, Experiments performed in the absence of extracellular calcium indicate that the AKAP-dependent rundown is calcium dependent and suggest that calcium entry is required for the rundown.F, Bar graph summarizing effects of phosphatase inhibitors on the Ht31-induced rundown. Inhibition of PP2B activity by cyclosporin A (1 μm) but not PP1/PP2A by okadaic acid (1 μm) prevents the AKAP79-dependent rundown. *p < 0.05 compared with control. **p < 0.05 compared with okadaic acid alone.
To test the effect of AKAP coexpression on GluR1 function, we recorded nondesensitizing whole-cell GluR1 receptor currents evoked by application of glutamate (1 mm) in the presence of cyclothiazide (100 μm). Cells expressing GluR1 alone (Fig. 2C, open squares) (control, 83.8 ± 5.1%, n = 7) and those cells coexpressing AKAP18 (Fig.2C, filled circles) (AKAP18, 85.2 ± 4.8%,n = 8) exhibited relatively stable currents over the 15 min recording period. In contrast, coexpression of AKAP79 resulted in a spontaneous time-dependent reduction in GluR1 receptor currents (Fig.2C, filled triangles) (AKAP79, 60.0 ± 5.3%, n = 9; p < 0.01). The magnitude and time course of this effect was similar to the rundown observed when Ht31 was applied to hippocampal neurons (Fig.1A).
Because AKAP79 also binds to PP2B, we hypothesized that the AKAP79-dependent rundown might be attributable to dephosphorylation of GluR1 by anchored PP2B. Inclusion of 10 mm BAPTA in the whole-cell pipette prevented the AKAP79-dependent rundown (Fig.2D, open squares, filled circles, filled triangles) (control, 83.4 ± 4.9%, n = 6; AKAP18, 83.8 ± 5.6%,n = 7; AKAP79, 80.6 ± 2.7%, n = 7). Similar concentration-dependent effects were obtained when EGTA was used to buffer intracellular calcium (AKAP79 plus 1 mm EGTA, 66.6 ± 5.7%, n = 7; AKAP79 plus 10 mm EGTA, 85.9 ± 2.4%,n = 6; p < 0.05; data not shown). These data are consistent with calcium-dependent PP2B activity. In addition, the AKAP79-dependent rundown was prevented when currents were recorded in the absence of extracellular calcium (Fig.2E, open squares, filled triangles) (control, 96.4 ± 6.7%, n = 5; AKAP79, 91.7 ± 3.2%, n = 5). This suggests that calcium entry, presumably through homomeric GluR1 receptors, triggers the AKAP79-dependent rundown. Most importantly, the AKAP79-dependent rundown was blocked by the PP2B inhibitor cyclosporin A (Fig.2F) (control, 88.8 ± 6.4%, n = 6; Ht31, 92.9 ± 2.8%, n = 7). In contrast, the PP1/PP2A inhibitor okadaic acid did not block the AKAP79-dependent rundown (Fig. 2F) (control, 83.6 ± 2.4%,n = 7; Ht31, 65.1 ± 5.5%, n = 10; p < 0.05), although a reduction in the rate of rundown was observed (data not shown). These data suggest that a pool of PP2B associated with AKAP79 is required for rundown.
Regulation of GluR1 receptor currents by AKAP79-anchored PKA
We reasoned that if phosphorylated Ser845 on the AMPA receptor is the substrate for AKAP79-anchored PP2B, then expression of AKAP79 forms unable to anchor the kinase should blunt the basal phosphorylation of the channel and the concomitant rundown of GluR1 receptor currents. To test this hypothesis, we used a truncated anchoring protein, AKAP79(1–360), that lacks the PKA anchoring site. Coprecipitation experiments confirmed that the RII subunit of PKA was not present in AKAP79(1–360) immunocomplexes, whereas RII was enriched in wild-type AKAP79 immunocomplexes (Fig.3A). As anticipated, the AKAP79-dependent rundown was absent when AKAP79(1–360) was coexpressed with GluR1 (Fig. 3A) (AKAP79, 60.0 ± 5.3%,n = 9; AKAP79(1–360), 87.2 ± 4.6%,n = 5; p < 0.01). Ongoing studies indicate that the determinants for PP2B and SAP97 binding to AKAP79 are contained within the AKAP(1–360) construct (K. L. Dodge, M. Colledge, and J. D. Scott, unpublished observations). Therefore, the absence of rundown observed with AKAP79(1–360) is unlikely to be attributable to the lack of PP2B or its ability to form a complex with GluR1 but rather attributable to its inability to facilitate basal phosphorylation of GluR1. In addition, a more conservative disruption in the AKAP79-PKA interaction site by single proline mutation, which prevents cAMP-dependent phosphorylation of Ser845 (Colledge et al., 2000), also reduces the rundown (data not shown). Collectively, these data suggest that the presence of PKA in the complex is a prerequisite for the phenomenon.
Fig. 3.

GluR1 is regulated by AKAP79-anchored PKA. A, Top, Extracts from HEK293 cells transfected with AKAP79 and AKAP79(1–360) were immunoprecipitated with antibodies to AKAP79 and blotted with antibodies against the RII subunit. AKAP79(1–360) does not coimmunoprecipitate RII and thus is unlikely to bind PKA inside cells.Bottom, Parallel electrophysiology experiments demonstrate that PKA anchoring is required for the AKAP79-dependent rundown. Ex, Extract; IP, immunoprecipitate; wt, wild type.B, HEK293 cells were transfected with GluR1 alone or with either AKAP18 or AKAP79. Acute application of 8-bromo-cAMP, a cell-permeant cAMP analog, prevents the AKAP79-dependent rundown but does not affect the time course of current responses from control or AKAP18 cells (compare with Fig. 2C). C, The effect of cAMP on GluR1 receptor currents, from cells cotransfected with AKAP79, was blocked by inclusion of the PKA-anchoring inhibitory peptide Ht31 (1 μm) or the specific PKA inhibitory peptide PKI (1 μm) in the whole-cell recording pipette solution. These results indicate that prevention of AKAP79-dependent rundown by 8-bromo-cAMP was attributable to activation of a pool of anchored PKA. AKAP79 plus cAMP data from Figure 3B are shown for comparison.
We further reasoned that if PP2B activity causes channel rundown, then enhanced PKA activation might counteract the phosphatase effect. Accordingly, application of 8-bromo-cAMP (100 μm), a cell-permeable cAMP analog, prevented the AKAP79-dependent rundown, although it had little effect in control or AKAP18-expressing cells (Fig. 3B). Furthermore, the AKAP79-dependent rundown was restored in the presence of 8-bromo-cAMP when Ht31 (1 μm) or PKI (1 μm), a PKA-specific inhibitor, were included within the whole-cell pipette solution (Fig. 3C) (control, 92.4 ± 5.2,n = 7; Ht31, 65.2 ± 6.2%, n = 5;p < 0.05; PKI, 44.3 ± 5.5%, n = 5; p < 0.01). Under these conditions, rundown attributable to inhibition of PKA by PKI occurred with a faster time course and to a greater extent than rundown because of displacement of PKA by the Ht31 peptide, as might have been anticipated from previous studies (Rosenmund et al., 1994). Collectively, these data indicate that 8-bromo-cAMP prevented the AKAP79-dependent rundown by activation of a pool of anchored PKA. Thus, activation of the pool of PKA associated with AKAP79 can overcome the action of the targeted pool of PP2B. Importantly, in the presence 8-bromo-cAMP, coexpression of AKAP79 and GluR1 is sufficient to produce an Ht31-induced rundown matching the time course and extent of that observed in neurons. This reconstitution of neuronal AMPA receptor behavior further suggests that the AKAP79/150 signaling complex likely is the endogenous mediator for regulation of hippocampal AMPA receptor currents by PKA and PP2B.
Modulation of GluR1 by AKAP79 occurs at Ser845
Although PP2B inhibition or PKA activation can prevent the AKAP79-dependent rundown, it is not clear whether both enzymes act on the same site. Therefore, phosphorylation site mutants of GluR1 were used to determine the locus of regulation. Mutation of Ser831 to alanine (S831A) did not affect AKAP79-dependent rundown (Fig.4A, open squares, filled triangles) (control, 85.0 ± 4.1%, n = 6; AKAP79, 60.7 ± 8.7%,n = 6; p < 0.05), indicating that this CaMKII/PKC site is not involved. In contrast, mutation of Ser845 to alanine (S845A) abolished AKAP79-dependent rundown (Fig.4B, open squares, filled triangles) (control, 88.6 ± 2.3%, n = 7; AKAP79, 95.6 ± 7.7%, n = 8). Thus, AKAP79-mediated modulation of recombinant GluR1 receptor currents occurs selectively at Ser845, a known PKA site, consistent with the notion that basal phosphorylation of this site is a prerequisite for the rundown.
Fig. 4.
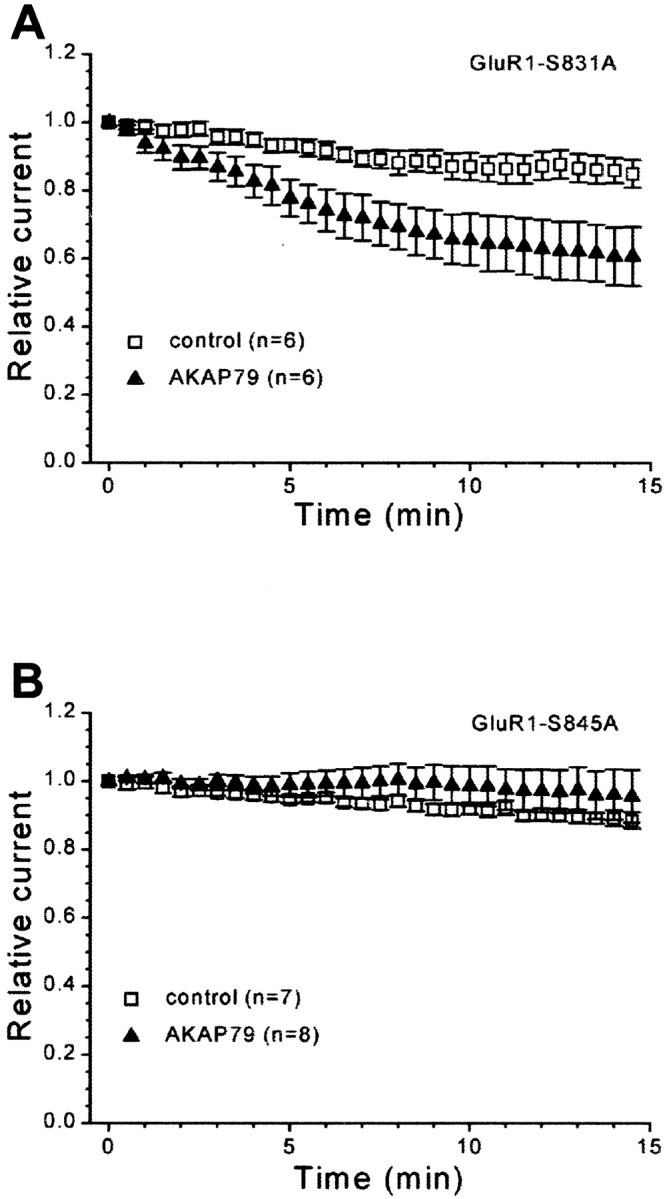
AKAP79-dependent regulation of GluR1 receptor currents occurs selectively at Ser845. HEK293 cells were transfected with GluR1 phosphorylation site mutants either alone (control) or with AKAP79. GluR1 receptor currents were evoked as in Figure2C. A, AKAP79-dependent rundown still occurs when Ser831 is mutated to alanine (S831A), indicating that Ser831 is not involved in the rundown. B, Ser845 is the site of regulation for the AKAP79-dependent rundown because GluR1 receptor currents from an alanine mutant of Ser845 (S845A) do not rundown when cotransfected with AKAP79.
A PDZ interaction at the C terminus of GluR1 is required for regulation by AKAP79
The cytoplasmic tail of GluR1 interacts with a PDZ (postsynaptic density 95/Discs large/zona occludens 1) domain on the MAGUK protein SAP97 (Leonard et al., 1998). Recently, we demonstrated that SAP97 also associates with AKAP79, facilitating basal phosphorylation of Ser845 on GluR1 by targeting PKA to GluR1 (Colledge et al., 2000). Thus, we were interested in whether SAP97 might alter the AKAP79-selective rundown. In fact, immunoblot analysis demonstrated that SAP97 is endogenously expressed in these cells (Fig.5A). Importantly, both GluR1 and AKAP79 were coimmunoprecipitated with endogenous SAP97 (Fig.5A), suggesting that a GluR1/SAP97/AKAP79 complex is formed in HEK293 cells. To test whether endogenous SAP97 contributes to AKAP79-dependent GluR1 regulation, we used a GluR1-Thr887 to alanine mutant (T887A) that does not interact with SAP97 (Colledge et al., 2000). Coexpression of AKAP79 did not cause rundown of these mutant GluR1 receptor currents (Fig. 5B, filled squares,filled triangles) (T887A, 86.2 ± 4.5%,n = 5; T887A plus AKAP79, 82.7 ± 5.6,n = 5). Thus, a PDZ-domain-mediated interaction at the C-terminal tail of GluR1 appears to be required for the AKAP79-dependent modulation of recombinant AMPA receptors. SAP97 is, at present, the only such protein that can interact with both the C-terminal tail of GluR1 and AKAP79 and is endogenously expressed in HEK293 cells. Therefore, our results are consistent with the hypothesis that formation of a GluR1/SAP97/AKAP79 complex facilitates basal phosphorylation of Ser845 and thereby provides a target for the AKAP79-anchored PP2B.
Fig. 5.
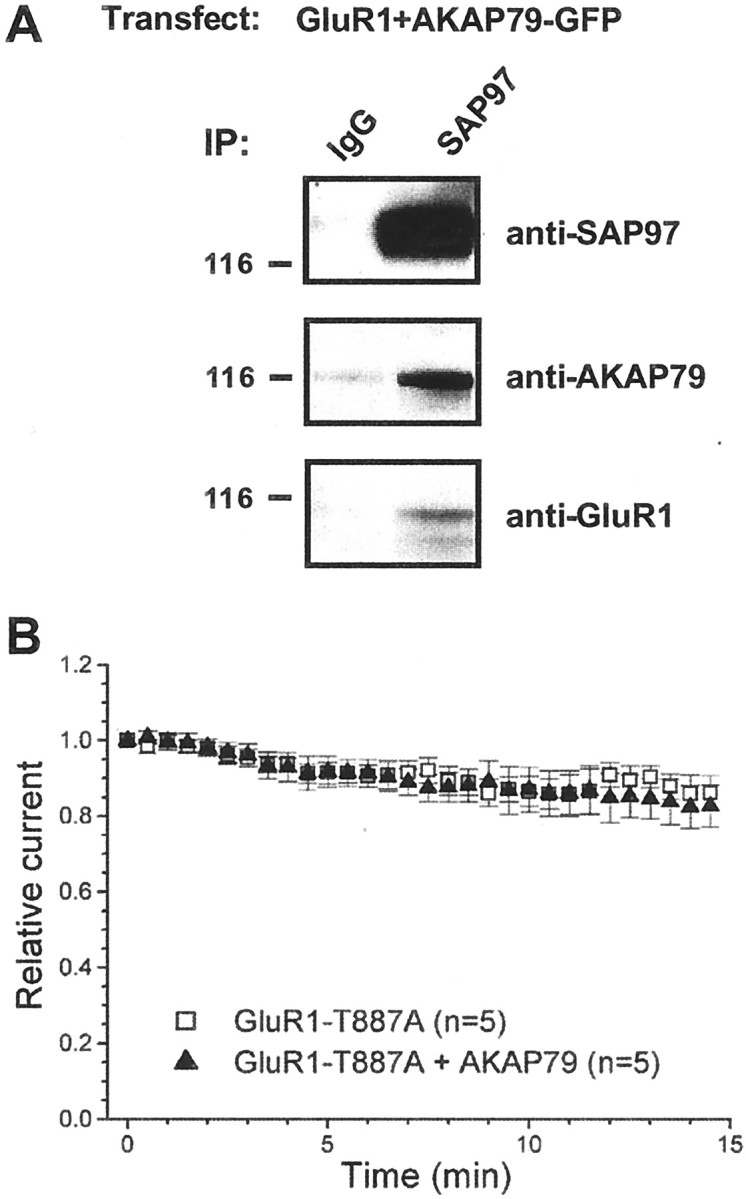
AKAP79-dependent rundown requires a PDZ-domain interaction at the C terminal of GluR1. A, HEK293 cells were transfected with GluR1 and AKAP79. Extracts were immunoprecipitated with antibodies to SAP97 and blotted for SAP97, AKAP79, and GluR1. Endogenous SAP97 is present in HEK293 cells (top). Transfected GluR1 and AKAP79 are coimmunoprecipitated with the endogenous SAP97, suggesting that a GluR1/SAP97/AKAP79 complex forms during cotransfection of GluR1 and AKAP79. IP, Immunoprecipitating antibody.B, HEK293 cells were transfected with an alanine mutant of Thr887 of GluR1 (T887A) in the presence or absence of AKAP79. Time course of GluR1 receptor currents indicate that a PDZ-domain interaction between GluR1 and endogenous SAP97 is likely required for the AKAP79-dependent rundown because it did not occur with the T887A mutant.
DISCUSSION
Recent advances suggest that multiple cellular processes contribute to long-term modifications of synaptic strength, such as LTP and LTD. However, the molecular mechanisms responsible for their orchestration remain unclear (Malenka and Nicoll, 1999; Sanes and Lichtman, 1999; Luscher et al., 2000). It is well established that kinases and phosphatases are critical in the induction and expression of synaptic plasticity (Winder and Sweatt, 2001). Indeed, regulation of the phosphorylation state of the AMPA receptor is likely to be one of the earliest events associated with LTP and LTD expression (Malenka and Nicoll, 1999; Luscher et al., 2000; Soderling and Derkach, 2000). Although considerable effort has focused on defining the kinases that mediate AMPA receptor phosphorylation, less is known about the phosphatases that catalyze the reverse reaction.
Here, we identify PP2B as the predominant phosphatase-opposing modulation of hippocampal AMPA receptors by anchored PKA. Furthermore, we show that expression of the neuronal anchoring protein AKAP79/150, which associates with PKA, PP2B, and SAP97, is sufficient to appropriately modulate recombinant GluR1 receptor currents. Our data suggest that the balance between kinase and phosphatase activity is an important determinant in the regulation of both native and recombinant AMPA receptor currents. In both systems, PP2B acts in opposition to anchored PKA. One difference between the two systems is how readily AMPA receptor downregulation occurs. In hippocampal neurons, which endogenously express AKAP150, AMPA receptors currents are relatively stable. In contrast, a spontaneous rundown of GluR1 receptor currents is observed in HEK293 cells coexpressing AKAP79. This difference can be explained if the native system possesses enhanced PKA activity and/or reduced calcium entry in response to AMPA receptor activation compared with the HEK293 cell system. Indeed, manipulation of a single variable, the extent of PKA activity by application of 8-bromo-cAMP, is sufficient to adjust the balance between kinase and phosphatase activities so that recombinant AMPA receptor currents behave exactly like their native counterparts (compare Figs. 1A,3C). Because AKAP79/150 is the major AKAP of the postsynaptic density (Carr et al., 1992b), our reconstitution of the protein–protein interactions that are present in neurons, as well as neuronal AMPA receptor behavior, strongly suggests that the endogenous AKAP79/150 signaling complex likely serves as the mediator for regulation of hippocampal AMPA receptor currents by PKA and PP2B.
Our data suggest that AKAP79/150-associated PP2B plays an important role in the regulation of GluR1 currents. However, the mechanism by which the phosphatase is activated when bound to AKAP79 has not been fully characterized. In vitro, PP2B activity is reduced to ∼25% when bound to the anchoring protein (Coghlan et al., 1995). This low level of AKAP79-associated phosphatase activity may be sufficient to dephosphorylate Ser845 given that the enzyme is precisely targeted to its substrate. Alternatively, the anchoring protein may serve a protective role. By limiting PP2B activity, AKAP79/150 may preclude inappropriate dephosphorylation and downregulation of synaptic AMPA receptors at resting calcium levels. This latter hypothesis is an attractive mechanism because previous studies have shown that PP2B has an extremely high affinity for calcium (Klee et al., 1998) and might otherwise be activated during basal synaptic transmission. More recently, we showed that PP2B may be excluded from the complex when AKAP79 is bound by postsynaptic density-95 (Colledge et al., 2000). Preliminary data suggests that PP2B may likewise be excluded from the complex when AKAP79 is bound by SAP97 (S. J. Tavalin, M. Colledge, and J. D. Scott, unpublished observations). However, our functional data indicate that endogenous PP2B and SAP97 are important for AKAP79-dependent regulation of GluR1. Indeed, both endogenous PP2B and SAP97 are found to associate with overexpressed AKAP79 in HEK293 cells. Thus, dynamic alterations in the composition of the signaling complex may give rise to functionally distinct pools of AKAP79, which contribute to GluR1 regulation. Thus, a variety of control mechanisms may ensure that only neuronal activity of the appropriate duration, intensity, and/or frequency would activate PP2B sufficiently to overcome basal PKA activity, resulting in dephosphorylation of Ser845 of the GluR1 subunit.
PP2B-mediated regulation of the AMPA receptor may be specific to the hippocampus, because recent studies suggest that PP1 is the predominant phosphatase regulating striatal AMPA receptor currents (Yan et al., 1999; Feng et al., 2000). These neurons display a spontaneous rundown of AMPA receptor currents that is prevented by PP1 inhibition, disruption of PP1 targeting, and stimulation of dopamine receptors that are coupled to cAMP production (Yan et al., 1999). Interestingly, the scaffolding protein spinophilin has been proposed to contribute to PP1-mediated regulation of striatal AMPA receptors (Yan et al., 1999;Feng et al., 2000). Thus, different brain regions may use distinct molecular scaffolds of kinases and phosphatases to regulate AMPA receptors.
Regulation of native and recombinant AMPA receptor currents by the AKAP79/150 signaling shares several properties with LTD, including calcium dependence, modulation by PP2B and PKA, and regulation of the phosphorylation state of Ser845 (Mulkey and Malenka, 1992; Mulkey et al., 1994; Kameyama et al., 1998; Lee et al., 1998, 2000). In addition, the compartmentalized nature of AKAP79-dependent GluR1 regulation is consistent with a postsynaptic and spatially restricted mechanism for LTD generation, a property that likely contributes to the input specificity of homosynaptic LTD (Kandler et al., 1998; Dodt et al., 1999). Together, our data suggest that the AKAP79/150 signaling complex may provide a mechanism to enable the calcium-dependent downregulation of these receptors during LTD.
Recent evidence suggests that GluR1 interacts with SAP97 predominantly in the biosynthetic and secretory pathway and that, after a limited time after targeting to the plasma membrane, they dissociate from one another (Sans et al., 2001). Because AKAP79/150 targets independently to the plasma membrane (Dell'Acqua et al., 1998), we propose that AKAP79-dependent regulation of GluR1 is initiated by the recruitment of GluR1 by SAP97 to AKAP79/150, resulting in formation of the previously described GluR1/SAP97/AKAP79 complex, which favors basal phosphorylation of Ser845 (Colledge et al., 2000). Once Ser845 is phosphorylated by AKAP79-anchored PKA, activity-induced elevations in intracellular calcium resulting in activation of AKAP79-anchored PP2B lead to GluR1 downregulation. Although this system may be well poised to respond to local elevations of calcium, more generalized elevations of intracellular calcium from a variety of sources of appropriate magnitude would also be expected to activate AKAP79-anchored PP2B. This would satisfy the rather loose calcium signaling requirements for LTD (Cummings et al., 1996) and contribute to the diversity of stimulation protocols that can result in AMPA receptor downregulation. Although our data suggest that putative calcium-permeable AMPA receptors may activate AKAP79/150-anchored PP2B in hippocampal neurons, it must be emphasized that this source of calcium is revealed only during disruption of PKA anchoring. In contrast, NMDA receptors and voltage-gated calcium channels are likely to represent the major physiological sources of calcium for AMPA receptor downregulation via AKAP79/150-anchored PP2B.
A prevailing view suggests that a phosphatase cascade leading to activation of PP1 underlies hippocampal LTD (Mulkey et al., 1993, 1994;Bear and Malenka, 1994; Malenka and Nicoll, 1999). In this model, the role for PP2B is to dephosphorylate the heat-stable phosphatase inhibitor protein inhibitor-1, thereby relieving inhibition of the PP1 catalytic subunit. PP1 is proposed to be the terminal phosphatase catalyzing reactions leading to LTD. In fact, we did observe noticeable alterations of the Ht31-induced rundown in neurons after inhibition of PP1/PP2A by okadaic acid. Given that the domoate-evoked currents we monitored consisted of both synaptic and extrasynaptic AMPA receptors, it is possible that this okadaic acid-sensitive component of rundown represents AKAP79-anchored PP2B regulation of the synaptic population of AMPA receptors via the phosphatase cascade model. In contrast, our data does implicate PP2B as directly controlling a substantial fraction of hippocampal AMPA receptor currents independent of PP1 activity. These data are supported by our experiments in HEK293 cells, which indicate that AKAP79-anchored PP2B directly regulates Ser845 of GluR1. Thus, our data provide an alternate signaling pathway by which PP2B may regulate the phosphorylation state of a substrate implicated in synaptic plasticity.
Recent findings support a role for PP1 and PP2B in the activity-dependent endocytosis of AMPA receptors, a process that is also thought to contribute to LTD (Beattie et al., 2000; Ehlers, 2000;Lin et al., 2000; Luscher et al., 2000; Malinow et al., 2000). Interestingly, dephosphorylation of Ser845 appears to precede AMPA receptors endocytosis in cultured neurons (Ehlers, 2000). As we demonstrated, AKAP79-dependent phosphorylation of Ser845 is controlled, in part, by a PDZ-domain interaction between the C-terminal tail of GluR1 and SAP97 (Colledge et al., 2000). Accordingly, disruption of this interaction may lead to net internalization of GluR1. Consistent with this, a recent study reported that a PDZ-domain interaction with the C-terminal of GluR1 is required for CaMKII-dependent synaptic insertion of recombinant GluR1 receptors during LTP (Hayashi et al., 2000). Importantly, the T887A mutant of GluR1 resulted in a net decrease in the AMPA receptor-mediated EPSC (Hayashi et al., 2000). Thus, it is tempting to speculate that PKA-dependent phosphorylation of Ser845 via the AKAP79/150 signaling complex may be a prerequisite for the postsynaptic insertion of GluR1 during LTP or may be required for retention of newly incorporated AMPA receptors.
Footnotes
This work was supported by grants from the Medical Research Council of Canada (M.C.) and National Institutes of Health Grants NS10202 (S.J.T.), NS35563 (J.W.H), and GM48321 (J.D.S.). We thank Ann Westphal and Jackie Soderling for preparation of the hippocampal cultures and Robert Mouton for preparation of HEK293 cells for biochemical experiments. We thank Gary Westbrook for critical review of this manuscript and Craig Jahr and members of the Scott laboratory for many critical discussions.
Correspondence should be addressed to John D. Scott, Howard Hughes Medical Institute, Vollum Institute, Oregon Health Sciences University, 3181 S.W. Sam Jackson Park Road, Portland, Oregon 97201. E-mail:scott@ohsu.edu.
S. J. Tavalin's present address: Department of Pharmacology, University of Tennessee, Health Science Center, Memphis, TN 38163.
REFERENCES
- 1.Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 3.Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 4.Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC. Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci. 2000;3:1291–1300. doi: 10.1038/81823. [DOI] [PubMed] [Google Scholar]
- 5.Carr DW, Hausken ZE, Fraser ID, Stofko-Hahn RE, Scott JD. Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J Biol Chem. 1992a;267:13376–13382. [PubMed] [Google Scholar]
- 6.Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD, Scott JD. Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins. Characterization of AKAP 79. J Biol Chem. 1992b;267:16816–16823. [PubMed] [Google Scholar]
- 7.Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD. Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science. 1995;267:108–111. doi: 10.1126/science.7528941. [DOI] [PubMed] [Google Scholar]
- 8.Colledge M, Scott JD. AKAPs: from structure to function. Trends Cell Biol. 1999;9:216–221. doi: 10.1016/s0962-8924(99)01558-5. [DOI] [PubMed] [Google Scholar]
- 9.Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 10.Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- 11.Dell'Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD. Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4, 5-bisphosphate. EMBO J. 1998;17:2246–2260. doi: 10.1093/emboj/17.8.2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodt H, Eder M, Frick A, Zieglgansberger W. Precisely localized LTD in the neocortex revealed by infrared-guided laser stimulation. Science. 1999;286:110–113. doi: 10.1126/science.286.5437.110. [DOI] [PubMed] [Google Scholar]
- 13.Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- 14.Feng J, Yan Z, Ferreira A, Tomizawa K, Liauw JA, Zhuo M, Allen PB, Ouimet CC, Greengard P. Spinophilin regulates the formation and function of dendritic spines. Proc Natl Acad Sci USA. 2000;97:9287–9292. doi: 10.1073/pnas.97.16.9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fraser ID, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV, Scott JD. A novel lipid-anchored A-kinase anchoring protein facilitates cAMP-responsive membrane events. EMBO J. 1998;17:2261–2272. doi: 10.1093/emboj/17.8.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gray PC, Johnson BD, Westenbroek RE, Hays LG, Yates JR, Scheuer T, 3rd, Catterall WA, Murphy BJ. Primary structure and function of an A kinase anchoring protein associated with calcium channels. Neuron. 1998;20:1017–1026. doi: 10.1016/s0896-6273(00)80482-1. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 18.Kameyama K, Lee HK, Bear MF, Huganir RL. Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron. 1998;21:1163–1175. doi: 10.1016/s0896-6273(00)80633-9. [DOI] [PubMed] [Google Scholar]
- 19.Kandler K, Katz LC, Kauer JA. Focal photolysis of caged glutamate produces long-term depression of hippocampal glutamate receptors. Nat Neurosci. 1998;1:119–123. doi: 10.1038/368. [DOI] [PubMed] [Google Scholar]
- 20.Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- 21.Klee CB, Ren H, Wang X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J Biol Chem. 1998;273:13367–13370. doi: 10.1074/jbc.273.22.13367. [DOI] [PubMed] [Google Scholar]
- 22.Lee HK, Kameyama K, Huganir RL, Bear MF. NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron. 1998;21:1151–1162. doi: 10.1016/s0896-6273(00)80632-7. [DOI] [PubMed] [Google Scholar]
- 23.Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- 24.Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- 25.Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci. 2000;3:1282–1290. doi: 10.1038/81814. [DOI] [PubMed] [Google Scholar]
- 26.Luscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci. 2000;3:545–550. doi: 10.1038/75714. [DOI] [PubMed] [Google Scholar]
- 27.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 28.Malinow R, Mainen ZF, Hayashi Y. LTP mechanisms: from silence to four-lane traffic. Curr Opin Neurobiol. 2000;10:352–357. doi: 10.1016/s0959-4388(00)00099-4. [DOI] [PubMed] [Google Scholar]
- 29.Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–32533. doi: 10.1074/jbc.272.51.32528. [DOI] [PubMed] [Google Scholar]
- 30.Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- 31.Mulkey RM, Herron CE, Malenka RC. An essential role for protein phosphatases in hippocampal long-term depression. Science. 1993;261:1051–1055. doi: 10.1126/science.8394601. [DOI] [PubMed] [Google Scholar]
- 32.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 33.Roche KW, O'Brien RJ, Mammen AL, Bernhardt J, Huganir RL. Characterization of multiple phosphorylation sites on the AMPA receptor GluR1 subunit. Neuron. 1996;16:1179–1188. doi: 10.1016/s0896-6273(00)80144-0. [DOI] [PubMed] [Google Scholar]
- 34.Rosenmund C, Carr DW, Bergeson SE, Nilaver G, Scott JD, Westbrook GL. Anchoring of protein kinase A is required for modulation of AMPA/kainate receptors on hippocampal neurons. Nature. 1994;368:853–856. doi: 10.1038/368853a0. [DOI] [PubMed] [Google Scholar]
- 35.Sanes JR, Lichtman JW. Can molecules explain long-term potentiation? Nat Neurosci. 1999;2:597–604. doi: 10.1038/10154. [DOI] [PubMed] [Google Scholar]
- 36.Sans N, Racca C, Petralia RS, Wang YX, McCallum J, Wenthold RJ. Synapse-associated protein 97 selectively associates with a subset of AMPA receptors early in their biosynthetic pathway. J Neurosci. 2001;21:7506–7516. doi: 10.1523/JNEUROSCI.21-19-07506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sik A, Gulacsi A, Lai Y, Doyle WK, Pacia S, Mody I, Freund TF. Localization of the A kinase anchoring protein AKAP79 in the human hippocampus. Eur J Neurosci. 2000;12:1155–1164. doi: 10.1046/j.1460-9568.2000.00002.x. [DOI] [PubMed] [Google Scholar]
- 38.Soderling TR, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- 39.Valtschanoff JG, Burette A, Davare MA, Leonard AS, Hell JW, Weinberg RJ. SAP97 concentrates at the postsynaptic density in cerebral cortex. Eur J Neurosci. 2000;12:3605–3614. doi: 10.1046/j.1460-9568.2000.00256.x. [DOI] [PubMed] [Google Scholar]
- 40.Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- 41.Yan Z, Hsieh-Wilson L, Feng J, Tomizawa K, Allen PB, Fienberg AA, Nairn AC, Greengard P. Protein phosphatase 1 modulation of neostriatal AMPA channels: regulation by DARPP-32 and spinophilin. Nat Neurosci. 1999;2:13–17. doi: 10.1038/4516. [DOI] [PubMed] [Google Scholar]