Activity of Thalamic Reticular Neurons during Spontaneous Genetically Determined Spike and Wave Discharges (original) (raw)
Abstract
This study reports the first intracellular recordings obtained during spontaneous, genetically determined spike and wave discharges (SWDs) in nucleus reticularis thalami (NRT) neurons from the genetic absence epilepsy rats from Strasbourg (GAERS), a model that closely reproduces the typical features of childhood absence seizures.
A SWD started with a large hyperpolarization, which was independent of the preceding firing, and decreased in amplitude but did not reverse in polarity up to potentials ≥ −90 mV. This hyperpolarization and the slowly decaying depolarization that terminated a SWD were unaffected by recording with KCl-filled electrodes. The prolonged (up to 15 action potentials), high-frequency bursts present during SWDs were tightly synchronized between adjacent neurons, correlated with the EEG spike component, and generated by a low-threshold Ca2+ potential, which, in turn, was brought about by the summation of high-frequency, small-amplitude depolarizing potentials.
Fast hyperpolarizing IPSPs were not detected either during or in the absence of SWDs. Recordings with KCl-filled electrodes, however, showed a more depolarized resting membrane potential and a higher background firing, whereas the SWD-associated bursts had a longer latency to the EEG spike and a lower intraburst frequency. This novel finding demonstrates that spontaneous genetically determined SWDs occur in the presence of intra-NRT lateral inhibition.
The unmasking of these properties in the GAERS NRT confirms their unique association with spontaneous genetically determined SWDs and thus their likely involvement in the pathophysiological processes of the human condition.
Keywords: cortex, thalamus, burst firing, GAERS, absence epilepsy, lateral inhibition
Childhood absence epilepsy (CAE) is a generalized epilepsy of multifactorial genetic origin (Panayiotopoulos, 1997). Experimental studies (Avoli et al., 1990,2001; Snead, 1995) have demonstrated the involvement of the GABAergic neurons of the nucleus reticularis thalami (NRT) in the generation of spike and wave discharges (SWDs), the EEG hallmark of absence seizures. In particular, lesions of, or Cd2+injections in, the NRT block SWDs in genetic absence epilepsy rats from Strasbourg (GAERS) (Avanzini et al., 1993), an inbred genetic model of CAE (Marescaux et al., 1992). Moreover, NRT but not thalamocortical neurons from preseizure GAERS show a larger T-type Ca2+ current than nonepileptic control rats (Tsakiridou et al., 1995) and an increased mRNA expression of α1I (Talley et al., 2000), one of the T-type Ca2+ channel subunits (Lee et al., 1999). Finally, intra-NRT injection of GABAB agonists aggravates absence seizures in every CAE model, whereas GABAB antagonists abolish them (Hosford et al., 1992; Liu et al., 1992; Snead, 1992).
Although our understanding of the pathophysiological mechanisms operating in the NRT during SWDs has been greatly advanced by recent_in vivo_ and in vitro studies, difficulties exist in the interpretations of these results, because most of the model systems used do not fully reproduce the CAE seizure properties. In particular:
(1) The in vivo recorded, spontaneous (and electrically or bicuculline-induced) 2–4 Hz spike/polyspike–wave complexes in cats are often accompanied by “fast runs” (10–15 Hz) and postictal depression (Steriade and Contreras, 1995; Neckelmann et al., 1998;Timofeev et al., 1998), which are absent in CAE. Furthermore, the spontaneity of these 2–4 Hz paroxysms appears to be linked to repeated electrical stimulation more than to a genetic predisposition (Steriade and Contreras, 1998; Steriade et al., 1998).
(2) The in vitro studies, either in the ferret perigeniculate nucleus (PGN) (the visual segment of the NRT) after application of bicuculline (Bal et al., 1995a,b) or in the NRT of transgenic mice lacking intra-NRT GABAA-mediated inhibition (Huntsman et al., 1999), assume that the pharmacological or transgenic block of GABAA inhibition within an isolated thalamus reproduces the thalamic network activity underlying SWDs. Intrathalamic application of bicuculline, however, does not elicit SWDs in the thalamus of decorticated animals (Steriade and Contreras, 1998). Furthermore, the addition of an “artificial” corticothalamic feedback to the in vitro thalamic network (Bal et al., 2000; Blumenfeld and McCormick, 2000) produces in thalamocortical neurons a low-threshold Ca2+ potential (LTCP) at each cycle, whereas LTCPs are only occasionally observed in vivo(Steriade and Contreras, 1995; Pinault et al., 1998).
(3) The sensitivity of these in vivo and _in vitro_paroxysms to anti-absence medicines is unknown.
Thus, because some mechanisms underlying NRT neuron activity during SWDs may not have been fully elucidated because of the peculiar features of the models used, we have now made _in vivo_extracellular and intracellular recordings from NRT neurons in GAERS, a model that closely reproduces the spontaneity, EEG waveform, behavioral component, and pharmacological profile of CAE seizures (Marescaux et al., 1992). Preliminary data have been published previously (Slaght et al., 2000).
MATERIALS AND METHODS
All experiments were performed in accordance with local ethical committee and European Union guidelines (directive 86/609/EEC), and every precaution was taken to minimize suffering to the animals and the number of animals used in each experiment.
Surgery. Experiments were conducted as described previously (Pinault et al., 1998; Charpier et al., 1999). Briefly, adult (4- to 5-month-old) male and female rats from the GAERS strain were initially anesthetized with pentobarbital (Sanofi, Libourne, France) (40 mg/kg, i.p.) and ketamine (Imalgène, Rhone Mérieux, France) (100 mg/kg, i.m.), and a cannula was placed into the trachea before they were positioned in a stereotaxic frame. All wounds and pressure points were infiltrated with Xylocaine 2% (Astra, Neuilly, France) (repeated every 2 hr) while body temperature was maintained (36.5–37.5°C) with a homeothermic blanket (Harvard Apparatus Ltd, Edinbridge, UK). Once the surgical procedures had been completed, neurolept-anesthesia was initiated with an injection of Fentanyl (Janssen, Issy-Lef-Moulineiux, France) (3 μg/kg, i.v.) and haloperidol (Haldol; Janssen, France) (1 mg/kg, i.p.) that was repeated every 20–30 min (Flecknell, 1996). To obtain long-lasting stable intracellular recordings, rats were immobilized with gallamine triethiodide (Flaxedil, Specia, Paris, France) (40 mg, i.m., repeated every 2 hr) and artificially ventilated. The degree of anesthesia was assessed by continuously monitoring the EEG and heart rate, and additional doses of anesthetic were administered at the slightest change toward an awake pattern (i.e., an increase in the frequency and reduction in the amplitude of the EEG waves and/or an increase in heart rate). At the end of the experiments, animals received an overdose of pentobarbital and were transcardially perfused as described below.
Recordings and data analysis. EEG recordings were obtained with a silver monopolar electrode placed on the dura above the orofacial motor cortex (12 mm anterior to the interaural line, 3.5–4 mm lateral to the midline). A reference electrode was placed in the muscle to the side of the head. For extracellular recordings and juxtacellular labeling, glass electrodes were filled with 0.5m NaCl and 1.5% neurobiotin (Vector Laboratories, Burlingame, CA) (15–20 MΩ). Intracellular recordings were obtained with glass electrodes containing 1.5% neurobiotin and 2m KAc (45–85 MΩ) or 3 mKCl (30–40 MΩ). Stereotaxic coordinates for NRT recordings were 7–7.5 mm anterior to the interaural line, 2.4 mm lateral to the midline, and 4.5–6.5 mm ventral to the brain surface (Paxinos and Watson, 1986). NRT units showed a monosynaptic response to motor cortex stimulation, were antidromically activated by stimulation of the ventrolateral nucleus, and were characterized by short-duration action potentials and an accelerating–decelerating pattern in LTCP-elicited bursts of action potentials (Mulle et al., 1986; Spreafico et al., 1988; Avanzini et al., 1989; Bal and McCormick, 1993; Contreras et al., 1993).
Extracellular and intracellular recordings were obtained using the active bridge mode of an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA), filtered at 0.3–3 and 30 kHz, respectively, and stored on a Biologic DAT recorder (Intracel, Royston, UK). Data were subsequently digitized at 40 kHz (intracellular/extracellular) or 2 kHz (EEG) for off-line analysis with Spike2 software (Cambridge Electronic Design, Cambridge, UK).
The dominant frequency of the EEG during SWD was calculated by successive fast Fourier transforms using the Power Spectrum tool in Spike2. The start and end of a SWD in the EEG were taken to be the first and last spike–wave complexes, respectively, where the size of the spike was at least three times the peak-to-peak amplitude of the baseline EEG. Cross-correlograms of the firing between two units of a multiunit recording were obtained by first encoding the position of the peak of the action potentials into separate event channels using the memory buffer function of Spike2; the event correlation function of Spike2 (width, 2 sec; bin size, 5 msec) was then used to produce the cross-correlogram for 10 sec periods either during or between SWDs.
The apparent input resistance of NRT neurons was measured by averaging at least 10 voltage responses to hyperpolarizing current pulses (0.2–0.5 nA, 100 msec). The action potential properties (afterhyperpolarization, duration at threshold, and half-width) were obtained by averaging at least 50 action potentials recorded at resting membrane potential. The amplitude of the afterhyperpolarization was measured from the resting potential to its peak amplitude, whereas the time to peak was calculated from the point at which the downstroke of the action potential crossed the resting potential.
Statistical significance was assessed using Student's _t_test for comparison between two groups, and one-way ANOVA with all pairwise comparisons for analysis among three or more groups. Some data were fitted to a Gaussian–Laplace distribution using the Gaussian fit function of Origin 6.0 (Microcal Software Inc, Northampton, MA) after the normality of their distribution had been tested with the Kolmogorov–Smirnov test. Quantitative data are presented throughout as mean ± SD, unless stated otherwise.
Neuron visualization. Extracellularly recorded neurons were labeled using juxtacellular injection of neurobiotin (Pinault, 1996;Mailly et al., 2001). Briefly, at the end of the recording session, positive current pulses (1–8 nA, 200 msec) were applied at a frequency of 2.5 Hz through the bridge circuit of the amplifier. The current was slowly increased while the electrode was advanced toward the neuron by 1 μm steps until the cell discharge was driven by the injected current. Current pulses were applied for a 10–15 min period to obtain a reliable labeling of neuronal processes. For intracellular recordings, depolarizing current pulses (0.2–1 nA, 100–200 msec) were applied at a frequency of 2.5 Hz at the end of the recording period.
At 1–2 hr after the injection, the animal received a lethal dose of pentobarbital and was perfused via the ascending aorta with 200 ml of saline followed by 500 ml of 0.3% glutaraldehyde and 4% paraformaldehyde in phosphate buffer (PB), 0.1 m, pH 7.4. Brains were post-fixed for 2 hr in the same fixative solution without glutaraldehyde and then immersed in 20% sucrose PB at 4°C until sectioning. Frozen sections of fixed brains were cut at 50–70 μm in the frontal plane and serially collected in PB. After several rinses in PB, neurobiotin was revealed by incubation of the sections in the avidin–biotin peroxidase complex (1:100; Vector Laboratories) in PB containing 0.3% Triton X-100 for at least 12 hr at 4°C. Incubated sections were washed in PB (two times for 10 min) before immersion in a solution containing 0.05% 3,3′-diaminobenzidine tetrahydrochloride (Sigma, St. Louis, MO), 0.4% nickel-ammonium sulfate, and 0.0006% H2O2. After several washes in PB, sections were mounted on gelatin-coated slides, counterstained with safranin, and dehydrated through alcohol to xylene for light microscopic examination. The position of labeled neurons within the NRT was confirmed using the atlas of Paxinos and Watson (1986).
RESULTS
The results of this study are based on 18 extracellularly and 16 intracellularly recorded NRT neurons. Eleven (of 13 injected) neurons were recovered after intracellular (n = 7) or juxtacellular (n = 4) injection of neurobiotin. These neurons were located in the rostral portion of the NRT (see Figs.1A2, 9A2), were scattered throughout its dorsoventral extent, and had morphological features similar to those described previously (Ohara and Lieberman, 1985; Spreafico et al., 1988; Steriade et al., 1997). In particular, they presented either an ovoid (fusiform) or polygonal perikaryon and were characterized by a number of varicose dendrites (see Fig. 9A3). The axon emerged from the perikaryon or from a proximal dendrite and coursed caudally and medially, forming a well defined terminal field in ipsilateral relay nuclei (seven in the ventrobasal complex, two in the posterior nucleus, and one in the laterodorsal nucleus). These axonal projections followed a dorsoventral topography, with cells located in the dorsal portion of the NRT innervating dorsal portions of the relay nuclei.
Fig. 1.

Extracellularly recorded activity of NRT neurons during SWDs. A1, The very occasional single spike firing of this unit (bottom trace), recorded in the rostral pole of the NRT (filled circle in_A2_, schematic horizontal plane drawing), becomes a high-frequency burst pattern concomitantly with the appearance of the first spike–wave complex in the EEG (top trace). The burst firing continues for the entire duration of the SWD, matching all but one spike–wave complex. A marked single spike and burst are enlarged in C and D, below.A3, A power spectrum shows the dominant frequency (indicated) of the SWD in A1. B, The background firing of this unit (a mixture of single spikes and short bursts) is transformed to high-frequency bursts after four spike–wave complexes are visible in the EEG (top trace). Marked firing before and during the SWD is enlarged in E and_F_, below. Time calibration in B and_F_ also applies to A1 and_C–E_, respectively. AM, Anteromedial thalamic nucleus; AV, anteroventral thalamic nucleus;nRT, thalamic reticular nucleus; PT, parathenial thalamic nucleus; Re, reunions thalamic nucleus; sm, stria medullaris. Anteriority relative to the interaural line is indicated (Paxinos and Watson, 1986). In this and all following figures, the top trace in each pair is the EEG, and the bottom trace is the simultaneously recorded extracellular or intracellular voltage (unless stated otherwise).
Fig. 9.

Activity of NRT neurons recorded intracellularly with KCl-filled electrodes. A1, Compared with the recordings with KAc electrodes (Fig. 5), NRT neurons recorded with KCl electrodes had a more depolarized resting membrane potential (arrow) and a much stronger background firing (B). During SWDs, the bursts of action potentials (C) had a lower frequency than those observed with KAc electrodes. Marked periods are enlarged in B,C, D1, and E1 below.A2, Schematic horizontal plane drawing showing the position (filled circle) of the NRT neuron from which the activity in A1 was recorded.AM, Anteromedial thalamic nucleus; AV, anteroventral thalamic nucleus; nRT, thalamic reticular nucleus; VL, ventrolateral thalamic nucleus;VM, ventromedial thalamic nucleus (anteriority relative to the interaural line is indicated). A3, Photomicrograph of the neurobiotin-injected NRT neuron in_A2_. Note the typical fusiform perikaryon and numerous varicose dendrites. D1, D2, Hyperpolarization (arrowheads) could be detected at the start of a SWD during recordings with KCl electrodes, both at resting (D1) and hyperpolarized (D2) (−1.3 nA) membrane potentials. The smaller size of the hyperpolarization at potentials greater than −60 mV was not peculiar to KCl recordings.E1–E3, LTCPs and associated bursts of action potentials recorded during SWD at resting membrane potential (E1) and at two hyperpolarized levels achieved by injection of −0.5 nA (E2) and −1.3 nA (E3) (membrane potential before the SWDs is indicated by an arrow). As in KAc recordings (Fig. 8), the LTCPs become larger in amplitude with steady hyperpolarization. The resting membrane potential indicated in B (arrow) also applies to_C_ and D1. Dashed lines in_E1–E3_ correspond to the indicated membrane potentials. Voltage calibration in D2 also applies to_B_, C, and D1. Time calibration in C and D2 also applies to_B_ and D1, respectively. Calibrations in_E3_ also apply to E1 and_E2_.
The properties of the SWDs recorded by the EEG electrode were identical to those described previously in vivo under similar experimental conditions (Pinault et al., 1998; Seidenbecher et al., 1998; Charpier et al., 1999; Seidenbecher and Pape, 2001) and in freely moving GAERS (Marescaux et al., 1992). Thus, the duration of SWDs ranged from 800 msec to 3 min, and even in the same animal, we could observe from a few short SWDs per minute up to a SWD every 5 min. The intra-SWD frequency ranged from 7 to 9 Hz (Fig.1A3).
Extracellular recordings
Single NRT units showed different patterns of background firing (Fig. 1A1,B); these patterns included electrical silence, single action potentials (Fig.1A1,C), short bursts of action potentials, or a mixture of short bursts and single action potentials (Fig.1B,E). The single action potential firing frequency ranged from 1 to 43 Hz, and the bursts contained 5.6 ± 3.4 action potentials (n = 10 U), had an intraburst frequency of 216 ± 41 Hz, and recurred once every 0.01–10 sec.
When a SWD appeared in the EEG, the firing of all NRT units drastically changed as it became exclusively characterized by prolonged, high-frequency action potential bursts (Fig.1D,F), which occurred at the same frequency as the SWD in the EEG (Fig. 1A3). The start (and end) of the paroxysmal activity in NRT neurons relative to the start (and end) of the corresponding SWD in the EEG was variable. Analysis of 50 representative SWDs from 5 U indicated that the shift to the distinctive high-frequency burst firing started after the appearance of the first clearly defined spike–wave complex in the EEG in 56% of SWDs (Fig. 1B), before the first spike–wave complex in 22% of cases, and at the same time in the remaining 22% (Fig.1A1). These different possibilities could be present in successive SWDs within the same unit. When the neuron did not start to burst at the same time as the first spike–wave complex, two to six extracellular bursts preceded the first spike–wave complex in the EEG. As far as the end of a SWD was concerned, the high-frequency burst pattern of NRT units could terminate after (24%), at the same time as (36%) (Fig. 1B), or before (38%) (Fig.1A1) the last clearly defined spike–wave complex of a SWD in the EEG. In the latter case, two to five spike–wave complexes were evident in the EEG after the last extracellularly recorded high-frequency burst.
The first action potential in the high-frequency burst preceded the peak of the EEG spike component of the corresponding spike–wave complex by 18.7 ± 10.6 msec (n = 450 bursts from 9 U) (Fig. 2A1,A2). A similar analysis conducted using all action potentials in a burst showed that the latency to the peak of the spike component was 7.4 ± 12.7 msec (Fig. 2A3). The duration of the high-frequency burst was 25.9 ± 4.8 msec (n = 450 from 9 U), its mean intraburst frequency was 301 ± 48 Hz, and the number of action potentials it contained ranged from 6 to 15 (8.5 ± 1.7; n = 450) (Figs. 1D,F,2A); however, bursts present at the start and end of a SWD generally contained three to five action potentials fewer than those present in the main body of a SWD. For each burst, independent of its position within a SWD, the instantaneous firing frequency showed the accelerating–decelerating pattern that is considered characteristic of a LTCP-evoked burst in NRT neurons (Mulle et al., 1986; Spreafico et al., 1988; Avanzini et al., 1989; Bal and McCormick, 1993; Contreras et al., 1993), reaching a peak value of 443 ± 76 Hz (Fig. 2B1,B2).
Fig. 2.

Properties of extracellularly recorded high-frequency burst firing in single NRT neurons during SWDs.A1, Latency of five bursts (from the same SWD) to the peak negativity of the spike component in the EEG (superimposed records in top trace). A2 and_A3_, Histogram (gray bars) and Gaussian–Laplace distribution (black line) show the latency (Δt) of the first and all action potentials, respectively, in a burst to the peak negativity of the EEG spike (taken as time 0, see A1) (n = 450 bursts from 9 U; bin size, 5 msec). The vast majority (98%) of bursts start before the EEG spike (A2), and >73% of all action potentials occur before the EEG spike (A3).B1, Instantaneous frequency plot shows the accelerating–decelerating pattern for 50 bursts from the unit shown in_A1_. B2, Average instantaneous frequency plot for the same 50 bursts as shown in B1.
Similar firing properties, both during and outside periods of SWDs, were confirmed in the three cases of multiunit recordings encountered in this study. In addition, these recordings highlighted how the generally random firing patterns of two neighboring NRT units in the absence of SWDs (Fig. 3A1,A2) became highly correlated with each other (Fig. 3A4) and tightly time-locked to the spike–wave complexes (Fig.3A3) during SWDs. These recordings also showed that the first action potential in a burst from one unit could either precede or follow the first action potential in the concomitant burst of the other unit, and on average the delay between these two action potentials was 3.4 ± 7.7 msec (n = 150 bursts from three double units) (Fig. 3C). It is also worth noting that at times the burst firing of a single unit ceased altogether for a variable number of spike–wave complexes, although the SWDs continued unabated in the EEG. Indeed, double-unit recordings showed that although one unit might temporarily stop firing during a SWD, the other continued its characteristic high-frequency burst pattern (Fig. 3B). For three double-unit recordings, the probability of one unit not discharging on a given spike–wave complex was 0.1 ± 0.1, whereas the probability of both units not discharging simultaneously was 0.05 ± 0.05. Finally, in both single-unit and multiunit recordings, we noticed that toward the beginning of a SWD, the high-frequency burst firing could be substituted by singlets/doublets of action potentials for a time period equivalent to four to eight spike–wave complexes (see below and Fig.6B2,C2).
Fig. 3.

Firing properties in extracellular double-unit recordings during SWDs. The randomly occurring single or double action potentials in the two units (A1) become a high-frequency burst pattern, tightly correlated with the spike component of the spike–wave complexes during a SWD (A3). The synchronized firing of the two units during SWDs is evident from the comparison of the cross-correlation plots before (A2) and during (A4) SWDs (bin size, 5 msec; 10 sec sample). B, Absence of burst firing in one unit during two consecutive spike–wave complexes, whereas the other unit continues unabated to show the prolonged burst firing in correspondence to the spike–wave complexes. C, Histogram (gray bars) and Gaussian–Laplace distribution (black line) show the relative timing (Δt) between the first action potentials of concomitantly occurring bursts (see inset) in two units (n = 150 bursts from 3 double units; bin size, 2 msec). Time calibration in A3 also applies to A1. A and B are from the same double-unit recording.
Fig. 6.

Interruption of high-frequency burst firing during SWDs. Intracellular (A1) and extracellular (B1, single unit; C1, double unit) recordings during the early part of SWDs indicate that the characteristic high-frequency burst firing is replaced by periods of single/double action potentials or short bursts. The EEG before and during this interruption generally has a higher frequency and smaller amplitude spike–wave complexes than the fully developed paroxysm. The intracellular records (A1 and A2) show that these periods of tonic/short burst firing are generated by a slowly decaying depolarization similar to the one observed at the end of a SWD (compare Fig. 7A,B). The multiunit recording in_C_ shows both units to simultaneously stop and later restart their high-frequency burst firing. A2,B2, and C2 are enlargements of a portion (arrows) of A1, B1, and_C1_, respectively. Dashed lines in_A1_ and A2 correspond to the indicated membrane potentials. Voltage calibration in A2 also applies to_A1_; time calibration in C1 and_C2_ also applies to A1, B1 and A2, B2, respectively.
Intracellular recordings: firing properties during SWDs
The passive membrane properties of our sample of 12 NRT neurons recorded with KAc-filled electrodes included a resting membrane potential of −57.3 ± 6.5 mV (n = 12) and an apparent input resistance of 45 ± 11 MΩ (n = 6). Although no time-dependent inward rectification was apparent (Mulle et al., 1986; Spreafico et al., 1988; Avanzini et al., 1989; Bal and McCormick, 1993; Contreras at al., 1993), some fast inward rectification was present, as suggested by the nonlinearity of their voltage-current relationships below −85 mV.
Analysis of the intracellularly recorded firing pattern of NRT neurons confirmed and extended the observations made during extracellular recordings. In addition, the quantitative properties of the different firing patterns observed during periods with no SWDs as well as those of the high-frequency bursts during SWDs that were recorded at resting membrane potential were remarkably similar to the corresponding values measured with extracellular electrodes (compare Fig. 10). Thus, whatever their background firing [i.e., single action potentials (Fig.4A1,B), short bursts (with two to six action potentials) (Fig. 4D,E), or a mixture of single action potentials and short bursts], all intracellularly recorded NRT neurons switched to a firing pattern consisting exclusively of prolonged high-frequency bursts of 5–15 action potentials during a SWD (7.7 ± 3.1; n = 234 bursts from eight cells) (Fig. 4C,F). The first action potential in a burst preceded the EEG spike component by 25.3 ± 21.9 msec (n = 234 bursts from eight cells), whereas the burst duration was 28.8 ± 13.0 msec (compare Fig. 10B) and the mean intraburst frequency 283 ± 72 Hz. The instantaneous frequency of the intracellularly recorded action potentials in a burst was also characterized by an accelerating–decelerating pattern (compare Fig. 10E) that reached a peak value of 414 ± 69 Hz.
Fig. 10.
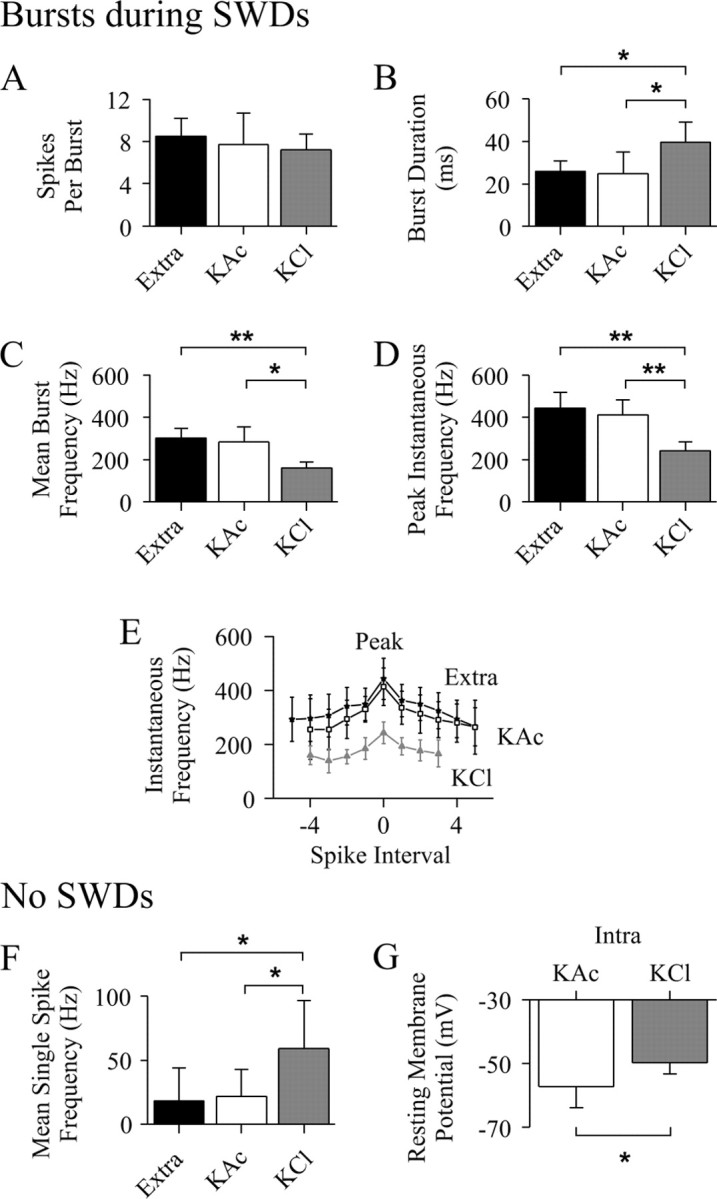
Comparison of NRT neuron firing recorded extracellularly and intracellularly with KAc- and KCl-filled electrodes. A–D, Properties (as indicated) of burst firing during SWDs (extracellular, n = 450 bursts from 9 U; KAc, n = 234 bursts from 8 U; KCl,n = 107 bursts from 3 U). E, The instantaneous burst firing frequency (plotted by aligning the peak frequency of each burst to spike interval zero) is lower in bursts from KCl recordings (gray triangles) than KAc recordings (open squares) and extracellular recordings (filled stars) (number of observations as in_A–D_). Note the greater rate of change in frequency for the intervals just before and after the peak value, in particular for the KAc and extracellular recordings. F, Single action potential firing in the absence of SWDs (extracellular,n = 9; KAc, n = 7; KCl,n = 3). G, Resting membrane potential measured in the absence of SWDs (KAc, n = 12; KCl, n = 4). *p < 0.05; **p < 0.01.
Fig. 4.

Intracellularly recorded activity of NRT neurons during SWDs. A, The occasional firing (single or double action potentials, enlarged in B) of this neuron becomes a high-frequency burst pattern (enlarged in C) during a SWD. D, The background firing of this neuron (a mixture of single action potentials and short, relatively low-frequency bursts) also changed to high-frequency bursts during the illustrated SWD. Marked firing before and during the SWD is enlarged in E_and F. Dashed lines in B,C, E, and F correspond to the indicated membrane potentials. Calibrations in A and_F also apply to D and B,C, E, respectively.
Intracellular recordings: evolution of the voltage waveform during a SWD
In the majority of SWDs (85%; n = 45 of 53 SWDs in five cells), the main intracellularly recorded component observed at the start of a SWD in the EEG was a relatively large hyperpolarization (mean, 5.6 ± 3.8 mV; n = 8 in three cells at −63 mV; range, 4–11 mV) (Fig.5A1,B1, filled arrowhead). This hyperpolarization did not appear to be linked to the firing behavior that immediately preceded it; i.e., it could follow a period of silence, a few action potentials, or a short burst, indicating an unlikely contribution by a Ca2+- and/or a Na+-dependent K+ current (Avanzini et al., 1989; Bal and McCormick, 1993; Kim and McCormick, 1998). At the peak of the hyperpolarization, the first LTCP generally appeared (see below), with or without a high-frequency burst of action potentials (Fig.5A1,B1). At more negative membrane potentials, this hyperpolarization became very small or disappeared altogether (Fig.5A2,A3). Interestingly, a hyperpolarization with properties similar to the one present at the beginning of a SWD could also be seen without the concomitant development of a SWD in the EEG (Fig.5B2,B3, open arrowheads). Similarly, this hyperpolarization was not linked to the firing pattern that immediately preceded it, because it could follow a period of silence (Fig.5B2), a few action potentials (Fig. 5B3), or a short burst. The similarities between these two hyperpolarizations are highlighted by their superposition shown in Figure 5B1+2 and_B1+3_. In those SWDs in which no clear hyperpolarization could be observed at the start, the first component of the intracellularly recorded paroxysm was a small LTCP with no action potential burst.
Fig. 5.

The start of a SWD. A1, A typical example of the start of a SWD shows a clear hyperpolarization (filled arrowhead) leading to a small-amplitude LTCP. Subsequent LTCPs quickly grow in amplitude, and prolonged bursts are evident at the same time that the first clear spike–wave complex appears in the EEG. Note that the most hyperpolarized potential during the SWD is already achieved by the trough that follows the first LTCP. Steady hyperpolarization of the neuron to −82 mV by DC injection almost abolishes this hyperpolarization (A2), whereas additional steady hyperpolarization (A3) does not produce any additional change. B1, Another example of the hyperpolarization (filled arrowhead) that is present at the start of a SWD, where strong and prolonged burst firing is reached by the second LTCP. B2, B3, In the absence of any SWD in the EEG, hyperpolarization (open arrowhead) with properties similar to the one present at the start of the SWDs is often recorded at resting membrane potential, after either a period of electrical silence (B2) or a short low-frequency burst (B3). Superimposition of the traces in_B1_ (black) and B2(gray) (B1+2) and superimposition of the traces in B1(black) and B3(gray) (B1+3) highlight the similarities between the hyperpolarization that is present at the start of a SWD and the one that does not lead to any paroxysmal activity.Arrows in B1–B3 indicate −60 mV. Action potential height in B1+2 and B1+3 has been truncated for clarity. Calibration in A1 also applies to A2, A3, and_B1–B3_.
During the early part of a SWD, and once the LTCPs and the associated high-frequency action potential bursts appeared to have been fully established, a slowly decaying depolarization could appear in the intracellular record (87% of SWDs in five neurons). This is clearly evident from the examples shown in Figures 4D, 6A1,A2, and7A. After a few large LTCPs, the neuron depolarized (often to a more positive potential than before the paroxysm), concomitantly changed its firing into singlets/doublets of action potentials or short bursts, and then slowly (1.0 ± 0.2 sec; range, 0.3–1.3 sec; i.e., up to eight spike–wave complexes) repolarized back, while the firing would gradually return to the pattern of rhythmic LTCPs (and associated prolonged high-frequency bursts) for the remainder of the SWD. This sequence of events occurred only once within a SWD (Fig. 4D). In a few cases, the SWD in the EEG became apparent only during this period of interruption in high-frequency bursts (Figs. 4D, 7A), whereas in the majority of cases, we noticed that the EEG during and before this burst firing interruption had spike–wave complexes of smaller amplitude and slightly but significantly higher frequency (before, 9.4 ± 1.3 Hz, n = 12; during, 9.4 ± 1.0 Hz, n = 18) than the fully developed, main body of the SWD (7.4 ± 0.3 Hz; p < 0.0001 for both) (Fig. 6A1). A similar change in firing pattern from high-frequency bursts to either short bursts or singlets/doublets of action potentials within the early part of a SWD was also detected in single-unit extracellular recordings (Fig. 6B1,B2) and concomitantly in both units of multiunit recordings (Fig.6C1,C2).
Fig. 7.

End of a SWD and short intracellular paroxysms in the absence of SWDs. A, B, Two SWDs from two neurons that had different resting membrane potentials (indicated) show that the slowly decaying depolarization is more clearly visible at more negative potentials. Note the higher firing rate of single action potentials at the end of the SWD shown in A.Dashed lines in A, B, and_C1_ correspond to the indicated membrane potentials.C1, A short intracellular paroxysm is depicted, whereas the EEG shows no SWDs. The evolution of the voltage waveform carries the same characteristics as those present during a SWD [i.e., the hyperpolarization (arrowhead) present at the start, the quick instatement of high-frequency bursts, and the higher-frequency, single action potential firing (between arrows) at the end]. The slowly decaying depolarization becomes visible when the neuron is hyperpolarized (C2), before becoming smaller with additional steady hyperpolarization (C3).Dashed lines in C2–C3 indicate the membrane potential before the intracellular paroxysm. Calibrations in_B_ also apply to A and_C1–C3_.
The intracellular paroxysm ended with a series of voltage and firing changes similar to those underlying the interruption of burst firing described above (Fig. 7A,B). Thus, the neuron would depolarize to a more positive membrane potential than before the SWD and then slowly repolarize back to the pre-SWD membrane potential (Fig.7A,B). This depolarization was accompanied by the abolishment of LTCPs and instatement of single or double action potentials, often at a higher frequency than that present before the SWD (Fig. 7A). In 40% of SWDs (n = 21 of 53 in five cells), this slowly decaying depolarization was hardly detectable at resting membrane potential (although its presence could be inferred from the higher firing rate) (Fig. 7A), but it was clearly visible at slightly more negative membrane potentials (Fig.7B). Thus, whereas the hyperpolarization present at the start of a SWD was at its maximum amplitude close to the resting membrane potential, the slowly decaying depolarization that ended a SWD was larger (9.8 ± 2.7 mV; n = 10 in three cells) when the neuron was slightly hyperpolarized (−67 mV). Note that the increase in tonic firing that was present at the end of a SWD was also visible in extracellular recordings (compare Fig.1B).
The full sequence of intracellular events occurring during a SWD (i.e., large hyperpolarization at the start, LTCPs plus high-frequency bursting, and slowly decaying depolarization at the end) could also be observed in the absence of any paroxysmal activity in the EEG (Fig.7C1). These intracellular paroxysms that occurred in the absence of SWDs had properties (including the interburst frequency and the voltage dependence of their different components) similar to those occurring during SWDs, except for their relatively short duration (0.3–0.6 sec) (Fig. 7C1–C3). In this respect, therefore, these intracellular paroxysms that occurred in the absence of SWDs (Fig. 7C1–C3) were strikingly similar to the sequence of events occurring at the beginning of a SWD (Figs. 4C,7A). Note that short paroxysms (i.e., two to four high-frequency burst firings) in the absence of SWDs in the EEG were also observed during extracellular recordings (data not shown).
Intracellular recordings: LTCPs and small depolarizing potentials
As described previously, one of the striking features of the intracellular voltage waveform during SWDs was the presence of large LTCPs tightly linked to each spike–wave complex in the EEG and crowned by a prolonged burst of action potentials (Fig.8A1). As expected, when NRT neurons were increasingly hyperpolarized by DC injection, the LTCPs became larger in amplitude and shorter in duration (Fig.8A2,A3). Somatic current steps that provided a time and voltage for removal of T-type Ca2+current inactivation similar to or slightly greater than those experienced by the neuron during SWDs failed to elicit a LTCP (Fig.8C2,C3), with much larger steps resulting in only the occasional generation of a LTCP (14 ± 2% of trials;n = 32 of 458 in five cells) (Fig.8C4); these results indicate that the origin of SWD-associated LTCPs is primarily dendritic (cf. Destexhe et al., 1996).
Fig. 8.

Properties of LTCPs during SWDs. A, LTCPs and associated high-frequency bursts of action potentials recorded during SWDs at resting membrane potential (A1) and at two hyperpolarized levels achieved by the injection of DC (A2, −0.5 nA; A3, −1 nA) (membrane potential before the SWDs is indicated by an arrow). Note the decreased duration and increased amplitude of the LTCPs for more negative values of membrane potential (LTCPs marked by_asterisks_ are enlarged in B1).B1, A sequence of high-frequency SDPs leads to the activation of each LTCP during SWDs. (Action potentials have been truncated for clarity.) B2, Similar groups of SDPs that do not lead, however, to the activation of LTCPs, are also observed when no SWD is present in the EEG. C1, Two successive LTCPs during a SWD recorded at resting membrane potential (arrow). C2–C4, Each_panel_ shows three superimposed voltage responses to current steps of −0.3 nA (C2), −0.5 nA (C3), and −0.8 nA (C4) in the absence of SWDs (same NRT neuron as in C1). Voltage excursions similar to (C2) or larger than (C3) the hyperpolarization achieved between two LTCPs during SWDs (C1) do not evoke any LTCP (number of trials indicated at the top), whereas much larger voltage responses (C4) evoke a LTCP in only 14 of 95 trials. Action potential height in C1–C4 has been truncated for clarity. Dashed lines in A1–A3,B1–B2, and C1–C4 correspond to the indicated membrane potentials. Calibrations in A3,B2, and C4 also apply to_A1–A2_, B1, and C1–C3, respectively.
Whereas the decay of the LTCPs occurring during a SWD had an overall smooth appearance (Fig. 8A,B1), their rising phase was invariably sculptured by the presence of three to nine small-amplitude (1–8 mV), high-frequency (200–1000 Hz) depolarizing potentials (SDPs) (Fig. 8B1). In the vast majority of cases, these SDPs started during the trough present between two successive LTCPs, and thus they basically made up the depolarizing phase leading to a LTCP and associated action potential burst. Indeed, because the SDPs often summed even up to the first action potential in a burst, it was at times difficult to establish the “true” start of the LTCP along the depolarizing waveform. Interestingly, groups of SDPs similar to those leading to LTCPs during SWDs were also observed when no SWD and none of its intracellularly recorded components were present in the EEG and the intracellular voltage trace, respectively (Fig.8B2). However, these groups of SDPs only rarely lead to the generation of a LTCP, even when recorded at hyperpolarized membrane potentials (6.3 ± 1.1%,;n = 28 of 444 SDP groups in four cells) (Fig. 8B2).
Intracellular recordings with KCl-filled electrodes
To test the possible participation of Cl−-dependent events in the activity of NRT neurons during SWDs, intracellular recordings were performed with KCl-filled electrodes. The morphological features of the neurons recorded with KCl electrodes (Fig.9A3) were similar to those of the neurons recorded extracellularly or intracellularly with KAc electrodes. The input resistance of these cells (36 ± 5 MΩ;n = 3) was similar to those recorded with KAc-filled electrodes, but the resting membrane potential was less negative (−49.8 ± 3.6 mV; n = 4; p < 0.05) (Figs. 9A1,B,10G). This was associated with a higher background firing rate (59 ± 38 Hz) (Fig.9A1,B) than in neurons recorded extracellularly or intracellularly with KAc electrodes (p < 0.05) (Fig. 10F). Moreover, neurons recorded with KCl electrodes showed a prominent afterhyperpolarization of the action potential (compare Fig. 9B with Fig.4B,E). When measured in the absence of SWDs and at a similar membrane potential (−52 mV), the amplitude of the afterhyperpolarization was 74% larger in NRT neurons recorded with KCl than KAc electrodes (KCl, 9.1 ± 0.8 mV, n = 324 from four cells; KAc, 5.3 ± 2.0 mV, n = 571 from six cells; p < 0.01), and its time to peak was shorter (KCl, 0.76 ± 0.26 msec; KAc, 1.28 ± 0.29 msec;p < 0.05).
A hyperpolarization (7.9 ± 3.1 mV; n = 7 in three cells at −63 mV; p = 0.45 compared with KAc recordings) (Fig. 9D1,D2) and a depolarization were present at the start and end, respectively, of the SWDs recorded with KCl electrodes, and the firing pattern of these neurons during SWDs was also characterized by high-frequency bursts of action potentials (Fig.9C–D2) that occurred at the same frequency as and were time-locked to the EEG spike. These high-frequency bursts (Fig.9C,D1,E1) were generated by LTCPs, which, because of the less negative resting membrane potential of KCl recordings, became clearly evident only at hyperpolarized membrane potentials (Fig.9D2,E2,E3). In addition, the waveform of the overall depolarization (i.e., SDPs plus LTCP) leading to the action potential burst (Fig. 9E1–E3) appeared broader than that in KAc recordings at similar membrane potentials (Fig. 8, compare_A1_ and A3).
The number of action potentials in a burst was 7.2 ± 1.5 (n = 107 bursts from three cells), which is similar to that observed in extracellular or KAc intracellular recordings (Fig.10A). However, the duration of a burst was longer in recordings with KCl (39.1 ± 9.6 msec; n = 107;p < 0.05) (Fig. 10B), and thus the mean intraburst frequency was lower (161 ± 27 Hz;n = 107; p < 0.05) (Fig.10C). The first action potential in a burst preceded the EEG spike by 31.2 ± 20.9 msec (n = 107 bursts) and thus occurred significantly earlier (p < 0.01) than in extracellularly or KAc intracellularly recorded neurons. The instantaneous frequency profile of a burst retained its accelerating–decelerating pattern in KCl-recorded cells, although it showed lower values for all intervals, including the peak measurement (243 ± 40 Hz; n = 107), compared with cells recorded with KAc electrodes or extracellularly (p < 0.01) (Fig. 10D,E).
DISCUSSION
The novel findings of this intracellular analysis of NRT neurons during spontaneous, genetically determined SWDs are (1) the identification of a large-amplitude hyperpolarization at the start of a SWD; (2) the presence, during the early part of a SWD, of a short interruption of burst firing that is mediated by a slowly decaying depolarization and is accompanied by a smaller-amplitude, higher-frequency EEG paroxysm; (3) the occurrence of short intracellular paroxysms in the absence of SWDs; (4) the lack of hyperpolarizing GABAA IPSPs during and in the absence of SWDs; and (5) the marked changes in background firing and SWD-associated bursts observed in recordings with KCl electrodes. The unmasking of these properties in the GAERS NRT confirms their unique association with spontaneous genetically determined SWDs and their probable involvement in the pathophysiological processes of CAE seizures.
Firing characteristics during SWDs
This study has enlarged the findings of previous studies in the GAERS NRT during SWDs (Seidenbecher et al., 1998; Pinault et al., 2001) by showing the following firing properties: (1) Because the SWD-associated change from tonic to burst firing could precede or follow the first EEG spike–wave complex, a NRT neuron does not have a fixed role in the generation/synchronization of SWDs within the thalamocortical loop but could be either leading or being recruited by other neurons in successive SWDs. (2) The firing of single NRT units started ∼19 msec earlier than the EEG spike, and thus before the sensory thalamic nuclei [Seidenbecher et al. (1998), compare their Fig. 4]. This and the higher strength of cortical EPSPs in NRT (compared with thalamocortical) neurons (Golshani et al., 2001) explain the inhibition and low firing rate of most thalamocortical neurons during SWDs (Steriade and Contreras, 1995; Pinault et al., 1998). (3) The high probability that adjacent units will produce bursts in relation to a given spike–wave complex and the low probability than one unit alone will discharge a burst indicate that the majority of closely apposed NRT neurons will discharge at each spike–wave complex and stress the high level of synchrony between adjacent NRT neurons during SWDs (Sohal et al., 2000).
Start and end of a SWD
It seems unlikely that the hyperpolarization at the start of a SWD involves the type II metabotropic glutamate receptor (mGluR)-activated K+ current that is present in young NRT neurons (Cox and Sherman, 1999), because the maximum amplitude of the hyperpolarization elicited by type II mGluR agonists is only 3 mV, and the synaptic activation of these receptors remains to be demonstrated. It is also unlikely that this hyperpolarization represents a GABAA IPSP generated by adjacent NRT neurons (Sanchez-Vives et al., 1997; Ulrich and Huguenard, 1997a), because (1) it is not composed of multiple summing events, (2) its time to peak is much longer than that of a single GABAA IPSP in NRT neurons (Zhang et al., 1997), (3) it did not reverse in polarity within the range of reversal potentials (−68 to −76 mV) of GABAA IPSPs/IPSCs in NRT/PGN neurons (Bal and McCormick, 1993; Sanchez-Vives et al., 1997; Ulrich and Huguenard 1997a,b; Bazhenov et al., 1999), and (4) it was unaffected by recording with KCl electrodes.
Conversely, the waveform of the hyperpolarization was remarkably similar to that of the hyperpolarization generated by the switching off of the window component of the T-type Ca2+current in thalamocortical [Williams et al. (1997), their Fig. 6_A_; Hughes et al. (1999), their Fig. 3_A_] and PGN [Steriade et al. (1997), their Fig. 4.8_A_] neurons. This possibility is supported by the findings that the T-type Ca2+ channel subunit (α1I), which is preferentially expressed in NRT neurons (Lee et al., 1999; Talley et al., 2000), generates a larger window current than α1G or α1H (Klockner et al., 1999), and that the T-type Ca2+ current in GAERS NRT neurons is larger than in nonepileptic control rats (Tsakiridou et al., 1995). The hyperpolarization present at the start of a SWD also shares striking similarities with GABAB IPSPs in PGN neurons [Sanchez-Vives et al. (1997), their Fig. 5_B_], although in ferrets and rats, synaptic activation of these receptors is restricted to only 40% and 17%, respectively, of PGN/NRT neurons (Ulrich and Huguenard, 1996; Sanchez-Vives et al., 1997). Because a higher expression of GABAB receptor mRNA is present in the GAERS NRT compared with in nonepileptic control rats (Depaulis et al., 2000), however, it might be possible that most GAERS NRT neurons have functional GABAB receptors that mediate this hyperpolarization.
As far as the slowly decaying depolarization observed at the end of a SWD is concerned, its properties (i.e., waveform, voltage dependence, and insensitivity to somatic Cl−injection) are similar to those of the slow afterdepolarization of guinea pig NRT neurons (Bal and McCormick, 1993), and therefore a Ca2+-activated, nonselective cation current is likely to mediate the termination of genetically determined SWDs.
Intracellular paroxysms in the absence of SWDs and interruption of high-frequency burst firing during a SWD
Because the burst firing interruption appeared only within the early part of a SWD, it might indicate that single and adjacent NRT neurons are attempting to exit from, or to stop the spreading of, a developing seizure (cf. Sohal et al., 2000). The similarities in voltage waveform and frequency between the early part of a SWD (compare Fig. 7A) and the short intracellular paroxysms occurring in the absence of SWDs (compare Fig. 7C1–C3), however, suggest an alternative possibility (i.e., that these two sequences of intracellular events represent the same NRT phenomenon, although they are associated with different levels of cortical synchrony). In other words, a short NRT paroxysm would either appear in isolation when the degree of cortical synchronization is minimal (i.e., no SWD is evident in the EEG) or develop into a full SWD when cortical synchrony is higher (i.e., a SWD is or soon becomes evident in the EEG). Note that the higher frequency in the initial stage of an EEG paroxysm is also reflected in intracellular recordings of thalamocortical and layer V cortical neurons [Charpier et al. (1999), their Figs. 1_C_, 2_A_] (together with layer III/VI cells; our unpublished observations). While all of these neurons slowly decelerate their membrane potential oscillations and firing discharges down to the main frequency of the SWD without any other change in voltage waveform or firing pattern, NRT neurons show a characteristic burst firing interruption at the time of transition to the lower frequency. This suggests that NRT neurons might be implicated in phase resetting and/or strengthening of the paroxysmal activity to achieve a higher synchronization of all elements of the thalamocortical loop to the preferred frequency of the GAERS paroxysm. Because a similarly higher EEG frequency is present in the early part of a SWD in freely moving GAERS (Pinault et al., 2001) and in absence epilepsy patients (Panayiotopoulos, 1997), our data provide the first evidence of the membrane voltage and firing changes in a neuronal element of the thalamocortical loop that are associated with the gradual development of a SWD.
Synaptic potentials
The lack of hyperpolarizing GABAA IPSPs in GAERS NRT neurons either during or in the absence of SWDs supports previous suggestions of a preferential shunting mode of intra-NRT GABAA-mediated inhibition (Sanchez-Vives et al., 1997; Ulrich and Huguenard, 1997a). In addition, because no other Cl−-dependent current but the one activated by GABAA receptors has been described in NRT neurons, the decrease in resting membrane potential (and associated increase in background firing) and the changes in SWD-associated burst firing that were observed with KCl-filled electrodes provide the first evidence that spontaneous genetically determined SWDs occur in the presence of intra-NRT shunting inhibition. Conversely, recent genetic evidence from CAE pedigrees has highlighted some GABAA subunit abnormalities (Feucht et al., 1999; Baulac et al., 2001; Wallace et al., 2001), and a transgenic model with NRT-selective impairment of the GABA system shows an increased prevalence of synchronized thalamic oscillations (Huntsman et al., 1999). Whether the lack of hyperpolarizing GABAA IPSPs and/or a putative weaker shunting inhibition represent a contributing factor in the generation of SWDs in GAERS must await the results of appropriate comparative studies in the nonepileptic control rat strain.
It is unlikely that the SDPs that lead to the generation of LTCPs are reversed GABAA IPSPs (because they did not reverse in polarity at potentials greater than −70 mV) or EPSPs originating from thalamocortical neurons (cf. Bal et al., 1995a) (because during SWDs in vivo these neurons are mainly silent or occasionally fire one to three action potentials) (Steriade and Contreras, 1995; Pinault et al., 1998). Instead, the SDPs might represent primarily cortical EPSPs, because of the higher strength of this input to NRT (compared with thalamocortical) neurons (Golshani et al., 2001) and the suggested leading role of the cortex over the thalamus in absence-like paroxysms (Neckelmann et al., 1998). Alternatively, as indirectly supported by their high frequency, the SDPs might represent gap junction potentials generated by connexin-immunopositive NRT neurons (Belluardo et al., 2000; Condorelli et al., 2000), although their presence has so far been elucidated only in young rats (Landisman et al., 2000).
Footnotes
This work was supported by Wellcome Trust Grant 37089-98, by European Union Grant 97-2093, and by the Ministere Français de la Recherche (Action Concertée d'Initiative Biologie du Développement et Physiologie Intégrative 2000). S.J.S. is a Wellcome Prize Student. We thank Dr. S. W. Hughes, S. Mahon, and Dr. H. R. Parri for critical discussions on the experiments and comments on this manuscript and A. Menetrey for assistance with the histological processing.
Correspondence should be addressed to V. Crunelli, School of Biosciences, Cardiff University, Museum Avenue, Cardiff CF10 3US, UK. E-mail: crunelli@cardiff.ac.uk.
REFERENCES
- 1.Avanzini G, de Curtis M, Panzica F, Spreafico R. Intrinsic properties of nucleus reticularis thalami neurones of the rat studied in vitro. J Physiol (Lond) 1989;416:111–122. doi: 10.1113/jphysiol.1989.sp017752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avanzini G, Vergnes M, Spreafico R, Marescaux C. Calcium-dependent regulation of genetically determined spike and waves by the reticular thalamic nucleus of rats. Epilepsia. 1993;34:1–7. doi: 10.1111/j.1528-1157.1993.tb02369.x. [DOI] [PubMed] [Google Scholar]
- 3.Avoli M, Gloor P, Kostopoulos G, Naquet R. Generalized epilepsy: neurobiological approaches. Birkhäuser; London: 1990. [Google Scholar]
- 4.Avoli M, Rogawski MA, Avanzini G. Generalized epileptic disorders: an update. Epilepsia. 2001;42:445–457. doi: 10.1046/j.1528-1157.2001.39800.x. [DOI] [PubMed] [Google Scholar]
- 5.Bal T, McCormick DA. Mechanisms of oscillatory activity in guinea-pig nucleus reticularis thalami in vitro: a mammalian pacemaker. J Physiol (Lond) 1993;468:669–691. doi: 10.1113/jphysiol.1993.sp019794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bal T, von Krosigk M, McCormick DA. Synaptic and membrane mechanisms underlying synchronized oscillations in the ferret lateral geniculate nucleus in vitro. J Physiol (Lond) 1995a;483:641–663. doi: 10.1113/jphysiol.1995.sp020612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bal T, von Krosigk M, McCormick DA. Role of the ferret perigeniculate nucleus in the generation of synchronized oscillations in vitro. J Physiol (Lond) 1995b;483:665–685. doi: 10.1113/jphysiol.1995.sp020613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bal T, Debay D, Destexhe A. Cortical feedback controls the frequency and synchrony of oscillations in the visual thalamus. J Neurosci. 2000;20:7478–7488. doi: 10.1523/JNEUROSCI.20-19-07478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud'homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E. First genetic evidence of GABAA receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- 10.Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Self-sustained rhythmic activity in the thalamic reticular nucleus mediated by depolarizing GABAA receptor potentials. Nat Neurosci. 1999;2:168–174. doi: 10.1038/5729. [DOI] [PubMed] [Google Scholar]
- 11.Belluardo N, Mud G, Trovato-Salinaro A, Le Gurun S, Charollais A, Serre-Beinier V, Amato G, Haefliger JA, Meda P, Condorelli DF. Expression of connexin36 in the adult and developing rat brain. Brain Res. 2000;865:121–138. doi: 10.1016/s0006-8993(00)02300-3. [DOI] [PubMed] [Google Scholar]
- 12.Blumenfeld H, McCormick DA. Corticothalamic inputs control the pattern of activity generated in thalamocortical networks. J Neurosci. 2000;20:5153–5162. doi: 10.1523/JNEUROSCI.20-13-05153.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charpier S, Leresche N, Deniau JM, Mahon S, Hughes SW, Crunelli V. On the putative contribution of GABAB receptors to the electrical events occurring during spontaneous spike and wave discharges. Neuropharmacology. 1999;38:1699–1706. doi: 10.1016/s0028-3908(99)00139-2. [DOI] [PubMed] [Google Scholar]
- 14.Condorelli DF, Belluardo N, Trovato-Salinaro A, Mudo G. Expression of Cx36 in mammalian neurons. Brain Res Brain Res Rev. 2000;32:72–85. doi: 10.1016/s0165-0173(99)00068-5. [DOI] [PubMed] [Google Scholar]
- 15.Contreras D, Curro Dossi R, Steriade M. Electrophysiological properties of cat reticular thalamic neurones in vivo. J Physiol (Lond) 1993;470:273–294. doi: 10.1113/jphysiol.1993.sp019858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cox CL, Sherman SM. Glutamate inhibits thalamic reticular neurons. J Neurosci. 1999;19:6694–6699. doi: 10.1523/JNEUROSCI.19-15-06694.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Depaulis A, Leonhard-Pozza S, Marescaux C, Bischoff S. Differential expression of GABAB receptor subunits mRNA in thalamic nuclei of genetic absence epilepsy rats of Strasbourg (GAERS). Soc Neurosci Abstr. 2000;26:622.3. [Google Scholar]
- 18.Destexhe A, Contreras D, Steriade M, Sejnowski TJ, Huguenard JR. In vivo, in vitro, and computational analysis of dendritic calcium currents in thalamic reticular neurons. J Neurosci. 1996;16:169–185. doi: 10.1523/JNEUROSCI.16-01-00169.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feucht M, Fuchs K, Pichlbauer E, Hornik K, Scharfetter J, Goessler R, Fureder T, Cvetkovic N, Sieghart W, Kasper S, Aschauer H. Possible association between childhood absence epilepsy and the gene encoding GABRB3. Biol Psychiatry. 1999;46:997–1002. doi: 10.1016/s0006-3223(99)00039-6. [DOI] [PubMed] [Google Scholar]
- 20.Flecknell P. Laboratory animals anaesthesia. Academic; London: 1996. [Google Scholar]
- 21.Golshani P, Liu XB, Jones EG. Differences in quantal amplitude reflect GluR4-subunit number at corticothalamic synapses on two populations of thalamic neurons. Proc Natl Acad Sci USA. 2001;98:4172–4177. doi: 10.1073/pnas.061013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hosford DA, Clark S, Cao Z, Wilson WA, Jr, Lin FH, Morrisett RA, Huin A. The role of GABAB receptor activation in absence seizures of lethargic (lh/lh) mice. Science. 1992;257:398–401. doi: 10.1126/science.1321503. [DOI] [PubMed] [Google Scholar]
- 23.Hughes SW, Cope DW, Toth TI, Williams SR, Crunelli V. All thalamocortical neurones possess a T-type Ca2+ “window” current that enables the expression of bistability-mediated activities. J Physiol (Lond) 1999;517:805–815. doi: 10.1111/j.1469-7793.1999.0805s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- 25.Kim U, McCormick DA. Functional and ionic properties of a slow afterhyperpolarization in ferret perigeniculate neurons in vitro. J Neurophysiol. 1998;80:1222–1235. doi: 10.1152/jn.1998.80.3.1222. [DOI] [PubMed] [Google Scholar]
- 26.Klockner U, Lee JH, Cribbs LL, Daud A, Hescheler J, Pereverzev A, Perez-Reyes E, Schneider T. Comparison of the Ca2+ currents induced by expression of three cloned alpha1 subunits, alpha1G, alpha1H and alpha1I, of low-voltage-activated T-type Ca2+ channels. Eur J Neurosci. 1999;11:4171–4178. doi: 10.1046/j.1460-9568.1999.00849.x. [DOI] [PubMed] [Google Scholar]
- 27.Landisman CE, Beierlein M, Connors BW. Electrical synapses between thalamic reticular neurons. Soc Neurosci Abstr. 2000;26:308.13. doi: 10.1523/JNEUROSCI.22-03-01002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci. 1999;19:1912–1921. doi: 10.1523/JNEUROSCI.19-06-01912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Z, Vergnes M, Depaulis A, Marescaux C. Involvement of intrathalamic GABAB neurotransmission in the control of absence seizures in the rat. Neuroscience. 1992;48:87–93. doi: 10.1016/0306-4522(92)90340-8. [DOI] [PubMed] [Google Scholar]
- 30.Mailly P, Charpier S, Mahon S, Menetrey A, Thierry AM, Glowinski J, Deniau J-M. Dendritic arborizations of the rat substantia nigra pars reticulata neurons: spatial organization and relation to the lamellar compartmentation of striato-nigral projections. J Neurosci. 2001;21:6874–6888. doi: 10.1523/JNEUROSCI.21-17-06874.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marescaux C, Vergnes M, Depaulis A. Genetic absence epilepsy in rats from Strasbourg: a review. J Neural Transm Suppl. 1992;35:37–69. doi: 10.1007/978-3-7091-9206-1_4. [DOI] [PubMed] [Google Scholar]
- 32.Mulle C, Madariaga A, Deschenes M. Morphology and electrophysiological properties of reticularis thalami neurons in cat: in vivo study of a thalamic pacemaker. J Neurosci. 1986;6:2134–2145. doi: 10.1523/JNEUROSCI.06-08-02134.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neckelmann D, Amzica F, Steriade M. Spike-wave complexes and fast components of cortically generated seizures. III. Synchronizing mechanisms. J Neurophysiol. 1998;80:1480–1494. doi: 10.1152/jn.1998.80.3.1480. [DOI] [PubMed] [Google Scholar]
- 34.Ohara PT, Lieberman AR. The thalamic reticular nucleus of the adult rat: experimental anatomical studies. J Neurocytol. 1985;14:365–411. doi: 10.1007/BF01217752. [DOI] [PubMed] [Google Scholar]
- 35.Panayiotopoulos CP. Absences epilepsies. In: Engel J Jr, Pedley TA, editors. Epilepsy: a comprehensive textbook. Lippincott-Raven; Philadelphia: 1997. pp. 2327–2346. [Google Scholar]
- 36.Paxinos G, Watson C. The rat brain in stereotaxic coordinates, Ed 2. Academic; London: 1986. [DOI] [PubMed] [Google Scholar]
- 37.Pinault D. A novel single-cell staining procedure performed in vivo under electrophysiological control: morpho-functional features of juxtacellularly labeled thalamic cells and other central neurons with biocytin or neurobiotin. J Neurosci Methods. 1996;65:113–136. doi: 10.1016/0165-0270(95)00144-1. [DOI] [PubMed] [Google Scholar]
- 38.Pinault D, Leresche N, Charpier S, Deniau JM, Marescaux C, Vergnes M, Crunelli V. Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. J Physiol (Lond) 1998;509:449–456. doi: 10.1111/j.1469-7793.1998.449bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinault D, Vergnes M, Marescaux C. Medium-voltage 5–9-Hz oscillations give rise to spike-and-wave discharges in a genetic model of absence epilepsy: in vivo dual extracellular recording of thalamic relay and reticular neurons. Neuroscience. 2001;105:181–201. doi: 10.1016/s0306-4522(01)00182-8. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez-Vives MV, Bal T, McCormick DA. Inhibitory interactions between perigeniculate GABAergic neurons. J Neurosci. 1997;17:8894–8908. doi: 10.1523/JNEUROSCI.17-22-08894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seidenbecher T, Pape HC. Contribution of intralaminar thalamic nuclei to spike-and-wave-discharge during spontaneous seizures in a genetic rat model of absence epilepsy. Eur J Neurosci. 2001;13:1537–1546. doi: 10.1046/j.0953-816x.2001.01537.x. [DOI] [PubMed] [Google Scholar]
- 42.Seidenbecher T, Staak R, Pape HC. Relations between cortical and thalamic cellular activities during absence seizures in rats. Eur J Neurosci. 1998;10:1103–1112. doi: 10.1046/j.1460-9568.1998.00123.x. [DOI] [PubMed] [Google Scholar]
- 43.Slaght SJ, Charpier S, Deniau J-M, Leresche N, Crunelli V. In vivo intracellular recordings in neurons of the nucleus reticularis thalami during spike and wave discharges in the GAERS genetic model of absence epilepsy. Soc Neurosci Abstr. 2000;26:274.2. [Google Scholar]
- 44.Snead OC. Evidence for GABAB-mediated mechanisms in experimental generalized absence seizures. Eur J Pharmacol. 1992;213:343–349. doi: 10.1016/0014-2999(92)90623-c. [DOI] [PubMed] [Google Scholar]
- 45.Snead OC. Basic mechanisms of generalized absence seizures. Ann Neurol. 1995;37:146–157. doi: 10.1002/ana.410370204. [DOI] [PubMed] [Google Scholar]
- 46.Sohal VS, Huntsman MM, Huguenard JR. Reciprocal inhibitory connections regulate the spatiotemporal properties of intrathalamic oscillations. J Neurosci. 2000;20:1735–1745. doi: 10.1523/JNEUROSCI.20-05-01735.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spreafico R, de Curtis M, Frassoni C, Avanzini G. Electrophysiological characteristics of morphologically identified reticular thalamic neurons from rat slices. Neuroscience. 1988;27:629–638. doi: 10.1016/0306-4522(88)90294-1. [DOI] [PubMed] [Google Scholar]
- 48.Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. J Neurosci. 1995;15:623–642. doi: 10.1523/JNEUROSCI.15-01-00623.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steriade M, Contreras D. Spike-wave complexes and fast components of cortically generated seizures. I. Role of neocortex and thalamus. J Neurophysiol. 1998;80:1439–1455. doi: 10.1152/jn.1998.80.3.1439. [DOI] [PubMed] [Google Scholar]
- 50.Steriade M, Jones EG, McCormick DA. Thalamus: organization and function. Elsevier Science; Oxford: 1997. [Google Scholar]
- 51.Steriade M, Amzica F, Neckelmann D, Timofeev I. Spike-wave complexes and fast components of cortically generated seizures. II. Extra- and intracellular patterns. J Neurophysiol. 1998;80:1456–1479. doi: 10.1152/jn.1998.80.3.1456. [DOI] [PubMed] [Google Scholar]
- 52.Talley EM, Solorzano G, Depaulis A, Perez-Reyes E, Bayliss DA. Low-voltage-activated calcium channel subunit expression in a genetic model of absence epilepsy in the rat. Brain Res Mol Brain Res. 2000;75:159–165. doi: 10.1016/s0169-328x(99)00307-1. [DOI] [PubMed] [Google Scholar]
- 53.Timofeev I, Grenier F, Steriade M. Spike-wave complexes and fast components of cortically generated seizures. IV. Paroxysmal fast runs in cortical and thalamic neurons. J Neurophysiol. 1998;80:1495–1513. doi: 10.1152/jn.1998.80.3.1495. [DOI] [PubMed] [Google Scholar]
- 54.Tsakiridou E, Bertollini L, de Curtis M, Avanzini G, Pape HC. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J Neurosci. 1995;15:3110–3117. doi: 10.1523/JNEUROSCI.15-04-03110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ulrich D, Huguenard JR. GABAB receptor-mediated responses in GABAergic projection neurones of rat nucleus reticularis thalami in vitro. J Physiol (Lond) 1996;493:845–854. doi: 10.1113/jphysiol.1996.sp021427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ulrich D, Huguenard JR. GABAA-receptor-mediated rebound burst firing and burst shunting in thalamus. J Neurophysiol. 1997a;78:1748–1751. doi: 10.1152/jn.1997.78.3.1748. [DOI] [PubMed] [Google Scholar]
- 57.Ulrich D, Huguenard JR. Nucleus-specific chloride homeostasis in rat thalamus. J Neurosci. 1997b;17:2348–2354. doi: 10.1523/JNEUROSCI.17-07-02348.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABAA receptor gamma2-subunit in childhood absence epilepsy, febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- 59.Williams SR, Toth TI, Turner JP, Hughes SW, Crunelli V. The “window” component of the low threshold Ca2+ current produces input signal amplification and bistability in cat and rat thalamocortical neurones. J Physiol (Lond) 1997;505:689–705. doi: 10.1111/j.1469-7793.1997.689ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang SJ, Huguenard JR, Prince DA. GABAA receptor-mediated Cl− currents in rat thalamic reticular and relay neurons. J Neurophysiol. 1997;78:2280–2286. doi: 10.1152/jn.1997.78.5.2280. [DOI] [PubMed] [Google Scholar]